
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Azulenes with aryl substituents bearing pentafluorosulfanyl groups: synthesis, spectroscopic and halochromic properties†
Stephen J.
Webster
 a,
Carlos M.
López-Alled
b,
Xinxing
Liang
a,
Claire L.
McMullin
a,
Gabriele
Kociok-Köhn
c,
Catherine L.
Lyall
c,
Tony D.
James
ab,
Jannis
Wenk
bd,
Petra J.
Cameron
ab and
Simon E.
Lewis
*ab
a,
Carlos M.
López-Alled
b,
Xinxing
Liang
a,
Claire L.
McMullin
a,
Gabriele
Kociok-Köhn
c,
Catherine L.
Lyall
c,
Tony D.
James
ab,
Jannis
Wenk
bd,
Petra J.
Cameron
ab and
Simon E.
Lewis
*ab
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: s.e.lewis@bath.ac.uk
bCentre for Sustainable Chemical Technologies, University of Bath, Bath, BA2 7AY, UK
cMaterials and Chemical Characterisation (MC2), University of Bath, Bath, BA2 7AY, UK
dDepartment of Chemical Engineering & Water Innovation & Research Centre: WIRC @ Bath, University of Bath, Bath, BA2 7AY, UK
First published on 7th December 2018
Abstract
Four regioisomeric azulenes bearing pentafluorosulfanylphenyl substituents have been prepared and characterised by various spectroscopic techniques. The absorption spectra are qualitatively similar in the visible region for all isomers, but upon protonation exhibit pronounced variation dependent on the connectivity within each molecule.
Introduction
Azulene (1) is a non-alternant, bicyclic aromatic compound that has an intense blue colour. Whilst it is isomeric with naphthalene, it has appreciably different properties such as an unusually large dipole for a hydrocarbon (1.08 D) and a HOMO to LUMO transition in the visible region.1 By introducing substituents onto the azulene ring, the absorption spectrum may be perturbed such that a wide range of colours can be produced.2 Thus, azulene derivatives have been employed as colorimetric sensors for a variety of analytes.3 Azulene derivatives have also found application in photovoltaics4 and more broadly in organic electronics.5 The azulene motif is also encountered in the context of medicinal chemistry.6 Many azulene derivatives are reported to exhibit halochromism,7 whereby the colour change on protonation may be rationalised in terms of protonation of either a substituent on the azulene, or of the five-membered ring of the azulene itself (Scheme 1). The protonated form contains a 6π e− tropylium cation, hence the seven-membered ring remains aromatic. | ||
| Scheme 1 The structure of azulene and its protonated form. | ||
Although azulenes have been reported bearing a wide range of substituents (and hence exhibiting a wide range of colours to the naked eye), the pentafluorosulfanyl group remains unexplored in the context of azulene chemistry. Specifically, to our knowledge there have been no reports of azulenes with pentafluorosulfanyl-containing substituents. The SF5 group, whilst known for many years, has been the focus of increasing research interest in recent years.8 This may be ascribed in part to the commercial availability of SF5-containing building blocks that were previously hard to access. In the life sciences, SF5 substituents have been investigated as an alternative to CF3. The unique combination of characteristics that an SF5 group can impart includes high thermal, chemical and metabolic stability, a highly electron-withdrawing nature and a significant increase in lipophilicity. An SF5 group also presents a larger steric demand compared to a CF3 group. Aryl SF5 groups in particular are inert to a variety of reaction conditions, including Brønsted acids and bases, hydrogenation and some organometallic reagents. From an environmental standpoint, it has been shown that aryl SF5 compounds are susceptible to photodegradation9 and can undergo microbial metabolism.10 The SF5 group has also been investigated in the context of materials chemistry, with reports on liquid crystals,11 polymers,12 and photophysical properties such as triboluminescence and fluorescence.13 Theoretical studies on the SF5 group have also been disclosed.14
Results and discussion
In the present study, we have synthesised and characterised four novel isomeric azulenes bearing pentafluorosulfanylphenyl substituents; these have been accessed using cross coupling methodology. Azulene cross coupling methods that employ the azulene-containing substrate as either the nucleophilic15 or electrophilic16 cross coupling partner have been developed, as well as C–H activation approaches.17,18 We intended to employ commercially available SF5-aryl bromides 2 and 3 as the electrophilic cross coupling components. Thus, we used the reported Ir-catalysed C–H borylation of azulene19 to prepare substrates 4 and 5. These underwent Suzuki–Miyaura coupling with 2 and 3, to give target SF5-phenylazulenes 6–9 (Scheme 2).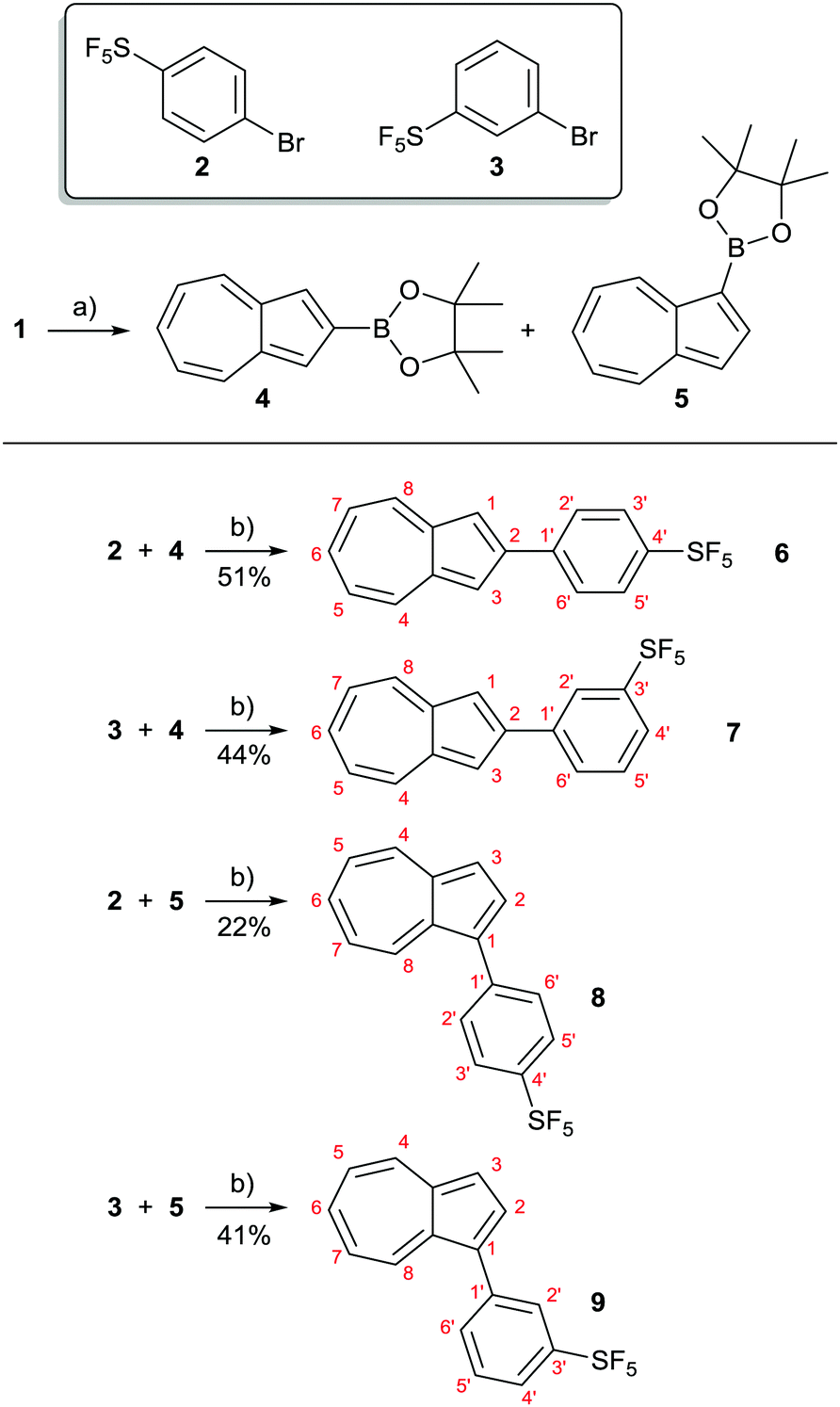 | ||
| Scheme 2 Synthesis of azulenes 6–9 bearing SF5-containing substituents. Reagents and conditions: (a) 2.2 eq. 1, 1.0 eq. B2pin2, 10 mol% bipy, 5 mol% [Ir(cod)Cl]2, cyclohexane, Δ, 14 h, 70% (4), 10% (5); see ref. 19a. (b) 1.0 eq. 4 or 5, 2 eq. 2 or 3, 5 mol% Pd(OAc)2, 10 mol% SPhos, 1.5 eq. K2CO3, dioxane, 80 °C, 16-22 h, 51% (6), 44% (7), 22% (8), 41% (9). | ||
Initial attempts at the Suzuki–Miyaura coupling to form 6 employed PPh3 as the ligand and were unsuccessful. Instead, use of Buchwald's bulky biaryl monodentate “SPhos” ligand20 allowed the synthesis of target azulenes 6–9. The products were characterised by various spectroscopic techniques, and the findings are described below.
NMR spectra were acquired for 6–9, and the aromatic region of the 1H-NMR spectra for the four products are shown in Fig. 1. All spectra have been fully assigned based on data from 2D NMR experiments (see ESI†). The plane of symmetry in isomer 6 gives rise to the simplest of the four spectra. The four proton environments on the azulene ring (H1/3, H4/8, H5/7 and H6, see Scheme 2 for numbering) are observed as a 2H singlet, 2H doublet, 2H double doublet and 1H triplet respectively, this being typical for a 2-substituted azulene. While isomer 7 no longer possesses a plane of symmetry, it is nevertheless still a 2-substituted azulene, and the same multiplicities for the azulene protons are observed as for 6. Indeed, the chemical shifts are near-identical, despite the differing location of the –SF5 group. For isomers 8 and 9, substitution at the azulene 1-position means the internal plane of symmetry of the azulene core is broken, so 7 distinct azulene proton environments are observed. H4 and H8 were differentiated by means of NOESY correlations between H8 and H2′ for both 8 and 9. For meta-disubstituted isomers 7 and 9, 4JHH coupling is present for H2′, which is observed as a double doublet (apparent triplet).
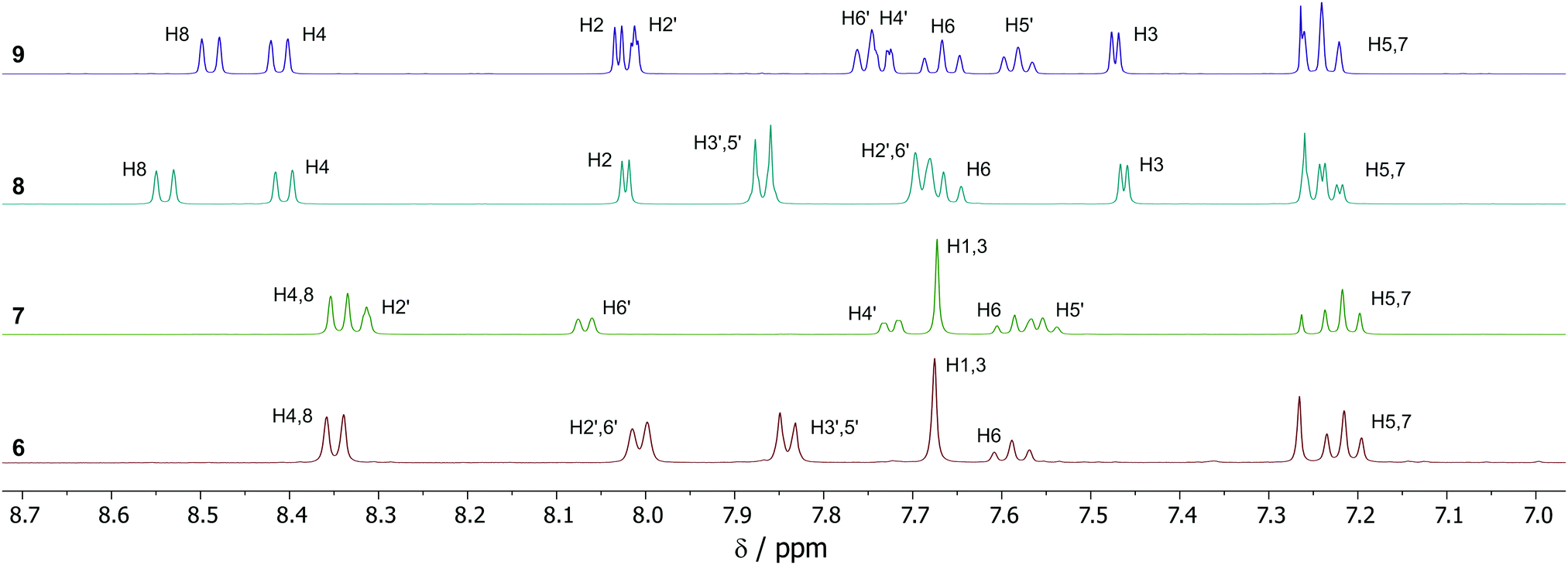 | ||
| Fig. 1 1H-NMR spectra (aromatic region) for 6–9, with assignments. See Scheme 2 for atom numbering. | ||
In the 19F NMR spectra for all four isomers 6–9, the fluorines are observed as a 4F doublet and a 1F quintet. As –SF5 groups have octahedral geometry at sulfur, the two distinct fluorine environments correspond to the four equatorial and one axial (i.e. trans to carbon) fluorines, respectively. The observed equivalence of the four equatorial fluorines indicates free rotation of the C–S bond on the NMR timescale.
In the {1H}–13C-NMR spectra, the SF5ipso carbons (C4′ for 6 and 8; C3′ for 7 and 9) exhibit 2JCF coupling, and the SF5ortho carbons (C3′/5′ for 6 and 8; C2′/4′ for 7 and 9) exhibit 3JCF coupling. Due to the presence of four equatorial and one axial fluorine, such coupling would be expected to lead to a quintet of doublets. In fact, all signals in the 13C spectra that exhibit splitting are simply quintets, i.e. only coupling to the equatorial fluorines is observed. The 2JCF couplings are in the range 16.5–16.9 Hz, whereas the 3JCF couplings are 4.2–4.9 Hz. A survey of 13C-NMR data for reported SF5-containing compounds shows that such quintet splittings are commonly observed. The lack of doublet splitting is often not discussed; it has been stated21 that for the axial fluorine, values of 2JCFax < 2 Hz and 3JCFax < 1 Hz are typical, and so such coupling is often not observable.
Of the four isomers, we were able to grow crystals of 7 of sufficient quality for analysis by X-ray diffraction. The structure obtained is shown in Fig. 2. Azulene 7 crystallised in the monoclinic space group P21/c. The dihedral angles between the azulene and the phenyl ring are appreciably different for the two molecules in the unit cell, being 8.3(5)° and 22.6(5)° respectively. The Feq–S–C–C dihedral angles are 38.4(3)° and 44.5(3)°, with the equatorial fluorines staggered with respect to the phenyl ring. C–S bond lengths are 1.802(3) Å and 1.804(3) Å. S–Feq bond lengths are in the range 1.580(2) Å to 1.591(2) Å, whereas S–Fax bond lengths are 1.585(2) Å and 1.592(2) Å. The geometry at sulfur is slightly distorted away from a perfect octahedron; the equatorial fluorine atoms of the SF5 group adopt an “umbrella” shape, canting toward the axial fluorine with Feq–S–Fax angles less than perpendicular (in the range 87.2(1)° to 87.7(1)°). All these observations are in keeping with those previously reported for other aryl-SF5 molecules.22
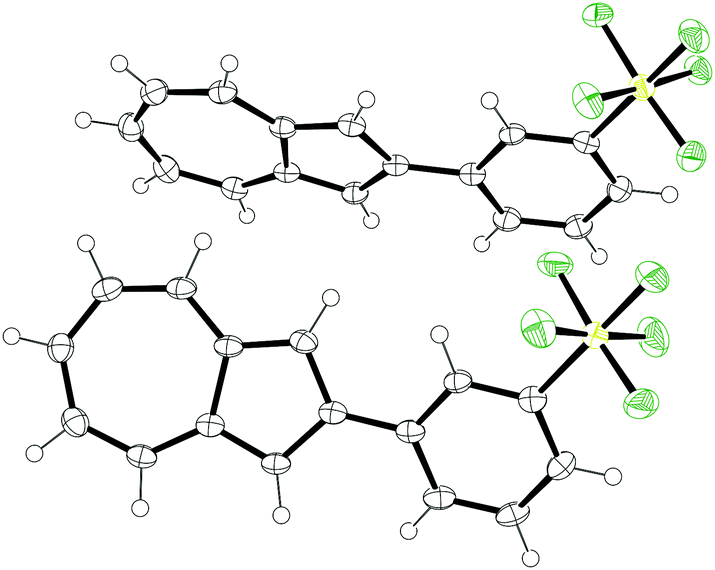 | ||
| Fig. 2 ORTEP representation of the X-ray structure of SF5-phenylazulene 7. Ellipsoids are shown at 50% probability. Hydrogens are shown as spheres of arbitrary radius. | ||
Fig. 3 shows the packing arrangement in the unit cell to be of a herringbone pattern. The two molecules in the unit cell are aligned in a head-to-head fashion, but they are not coplanar. Rather the two planes defined by the seven-membered rings of the two azulenes intersect at an angle of 60.0° (see ESI†).
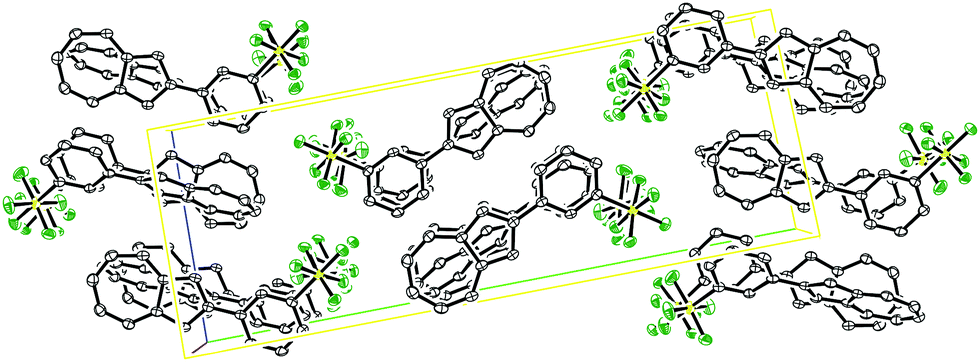 | ||
| Fig. 3 ORTEP representation of the expanded unit cell of SF5-phenylazulene 7. Ellipsoids are shown at 50% probability. Hydrogens have been omitted for clarity. | ||
UV-visible region absorption spectra for 6–9 were acquired in CH2Cl2, and the data for the visible region are shown in Fig. 4 (see ESI† for full data). The solutions of the four isomers are very similar in colour. In the visible region 6–9 all have the greatest absorbance in the green-to-red region, hence appear to be blue. Azulenes often exhibit halochromic behaviour, and upon adding excess trifluoroacetic acid to the solutions of 6–9, spectroscopic changes were observed. For the azulenes substituted at the 1-position (8 and 9), an increase in absorbance between 600–700 nm was noted; this was most pronounced for 9, with the appearance of a new absorbance maximum at λmax = 636 nm. This resulted in only a subtle colour change (Fig. 5). In contrast, protonation of 6 and 7 resulted in pronounced colour changes, the solutions turning yellow-brown and red, respectively. This may be attributed to the appearance of new absorbance peaks in the blue region (peak at λmax = 409 nm for 6, and a shoulder approximately the same wavelength for 7).
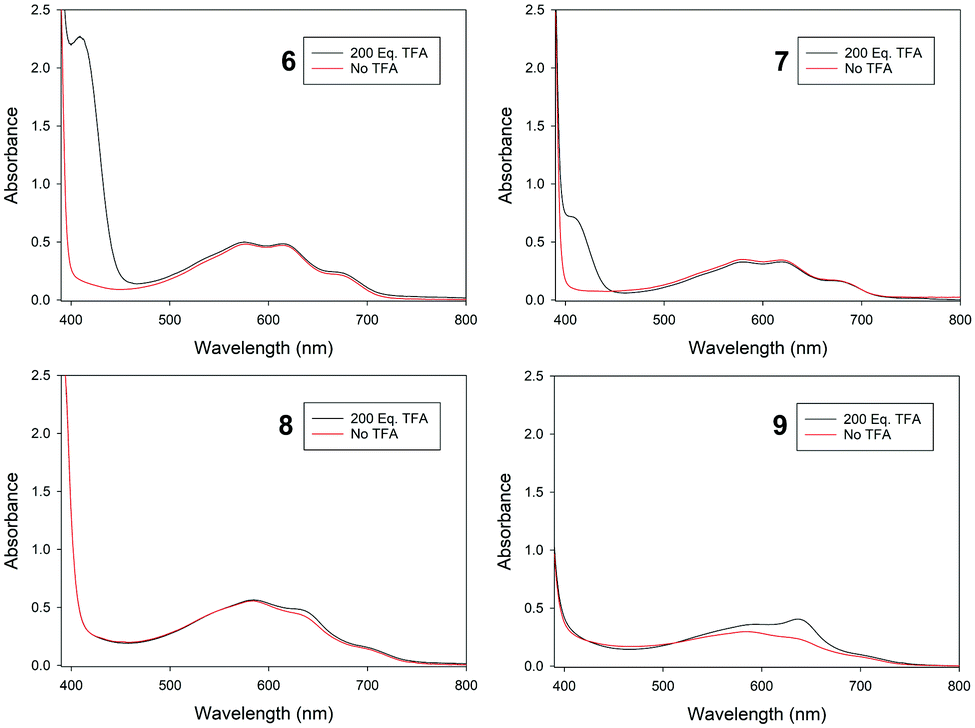 | ||
| Fig. 4 Visible region absorbance spectra for 6–9, recorded in CH2Cl2. | ||
 | ||
| Fig. 5 Solutions of 6–9 in CH2Cl2, with (right) and without (left) TFA. | ||
Although 6–9 differ only in their positions of substitution (that is to say, they are isomers), they differ markedly in their spectroscopic responses to Brønsted acid. There are precedents for this phenomenon. Wang, He and co-workers have studied azulenes bearing thienyl substituents;23 Hawker and co-workers have studied azulenes bearing diketopyrrolopyrrole substituents;24 and Murai, Takai and co-workers have studied analogues of 6 and 8 with a –CF3 group in the place of the –SF5 group.25 In each case a more significant change in absorption maxima (and hence colour) is observed for the isomer with the substituent(s) aligned with (instead of orthogonal to) the dipole of the azulene core, i.e. at the azulene 2-position.
To gain further insight into the properties of 6–9, the structures were modelled by DFT, using the BP86 functional. Both the neutral and protonated structures were modelled in the gas phase and then corrected for dispersion and a dichloromethane solvent environment (see ESI† for full computational details). For [8 + H]+ and [9 + H]+, protonation at either the azulene 1-position or 3-position would give rise to a tropylium cation, as per Scheme 1. Both regioisomeric cations were modelled, and for both [8 + H]+ and [9 + H]+, the isomer protonated at the 3-position was found to be the more stable by 3.9 kcal mol−1. The alternative of protonation at the 1-position involves rehybridisation of C1 to sp3, and hence conjugation between the seven membered ring and the phenyl substituent is lost (see ESI†). The cases of [6 + H]+ and [7 + H]+ are more straightforward, since the site of protonation is unambiguous. Of the four (neutral) isomers 6–9, the most stable were 2-substituted isomers 6 and 7, having the same free energy, whereas 8 and 9 were higher in free energy by 1.0 and 1.1 kcal mol−1, respectively. For the corresponding protonated structures, larger free energy differences were calculated. [7 + H]+ was found to be the most stable isomer, with [6 + H]+ higher in free energy by 0.6 kcal mol−1. In contrast, the 1-substituted isomers [8 + H]+ and [9 + H]+ were +4.1 and +3.9 kcal mol−1 above [7 + H]+, respectively.
The calculated frontier molecular orbitals for 6–9 are shown in Fig. 6. It can be seen that for the 2-substituted azulenes 6 and 7, the HOMO is localised entirely on the azulene ring system; the positions of the orbital lobes and nodes are extremely similar to those of azulene (1) itself.26 For the 1-substituted azulenes 8 and 9, the HOMO also extends onto the phenyl ring. In contrast, the LUMOs of 7, 8 and 9 are localised primarily on the SF5 substituent. The only isomer having C2 symmetry, 6, has a LUMO that deviates from this trend, with the orbital lobes and nodes on the azulene rings of 6 again resembling those of the corresponding MO of the parent unsubstituted azulene 1. The MOs of the protonated forms were also determined (see ESI†). The calculated HOMO and LUMO energies for 6–9 and their protonated forms, as well as their calculated dipole moments, are shown in Table 1.
 | ||
| Fig. 6 Frontier molecular orbital plots for azulenes 6–9, calculated at the BP86/6-31G**&SDDALL level of theory. | ||
| E HOMO [eV] | E LUMO [eV] | ΔELUMO–HOMO [eV] | Dipole [D] | ||
|---|---|---|---|---|---|
| Neutral | 6 | −5.093 | −3.157 | 1.936 | 6.30 |
| 7 | −5.051 | −3.170 | 1.880 | 5.43 | |
| 8 | −5.016 | −3.087 | 1.929 | 6.39 | |
| 9 | −4.980 | −3.113 | 1.867 | 6.13 | |
| Protonated | [6 + H]+ | −9.547 | −7.525 | 2.022 | 20.63 |
| [7 + H]+ | −9.553 | −7.472 | 2.081 | 17.64 | |
| [8 + H]+ | −9.603 | −7.735 | 1.869 | 21.15 | |
| [9 + H]+ | −9.613 | −7.719 | 1.894 | 19.39 | |
The electrochemical behaviour of 6–9 was studied using cyclic voltammetry to determine the oxidation potentials (Fig. 7). All cyclic voltammograms (CVs) were acquired in dry acetonitrile with 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) as supporting electrolyte, at a scan rate of 100 mV s−1 (see ESI† for full details). Three scans were taken for each run (the CVs in Fig. 7 show plots for the first and third scans only). The small quantities of 6 and 8 available necessitated that these CVs were acquired at a lower concentration than for 7 and 9. All four isomers exhibited an oxidation peak in the first scan; key electrochemical parameters derived from the CVs are given in Table 2. When comparing the data, it can be seen that the 2-substituted azulenes 6 and 7 have very similar peak oxidation potentials (666 and 657 mV, respectively). The value for 1-substituted azulene 8 is lower, at 630 mV, and that for 1-substituted azulene 9 is lower again, at 571 mV. The same trend is discernible in the onset potential of the oxidation.
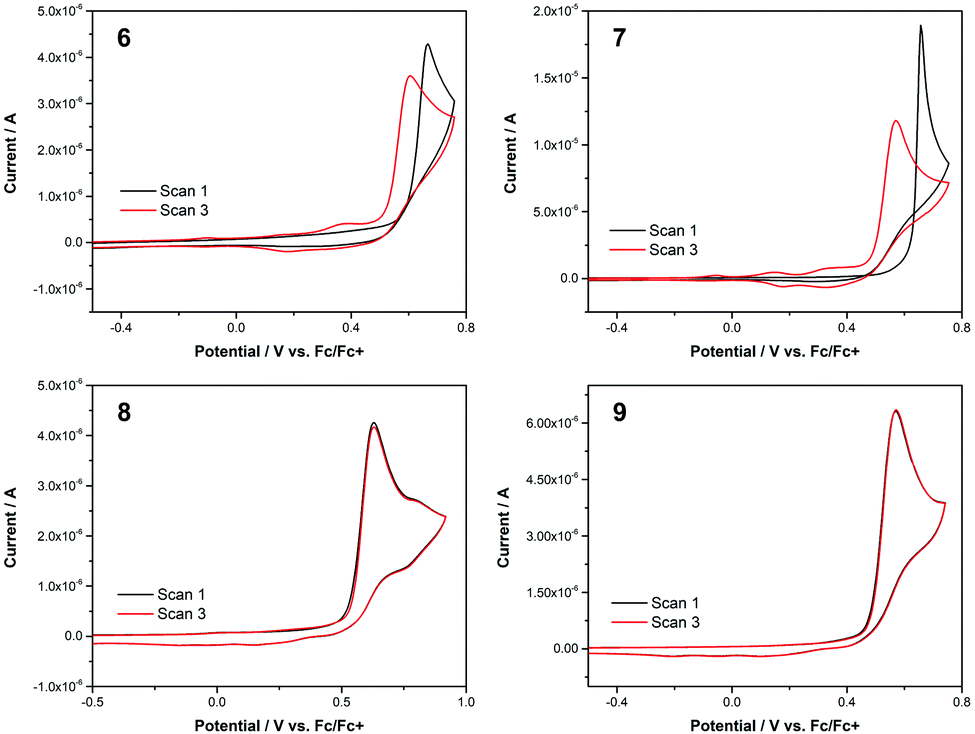 | ||
| Fig. 7 Cyclic voltammograms for 6–9 in acetonitrile. Supporting electrolyte 0.1 M Bu4NPF6. Scan rate 100 mV s−1. [6] = [8] = 0.1 mM. [7] = [9] = 0.5 mM. | ||
| Substrate | Concentration/mM | E peak/mV | I peak/μA | E onset/mV |
|---|---|---|---|---|
| 6 | 0.1 | 666 | 4.29 | 595 |
| 7 | 0.5 | 657 | 18.9 | 624 |
| 8 | 0.1 | 630 | 4.26 | 531 |
| 9 | 0.5 | 571 | 6.31 | 481 |
The most striking observation that can be made on comparing the CVs for 6–9 is that for 8 and 9 the first and third scans are superimposable, but this is not the case for 6 and 7. In the CVs for both 6 and 7, whereas in scan 1 no redox processes occur below the onset potential for the main oxidation peak, in scan 3 additional oxidation peaks can be seen at lower potential. The peak oxidation potential and current are also significantly lower for scan 3 in both cases. The CVs for 6 and 7 suggest that the oxidised substrate (an azulene radical cation) is undergoing polymerisation, as 6 and (in particular) 7 exhibit characteristic nucleation loops in scan 1. Electropolymerisation of azulene 1 has been extensively studied.27 In addition, the polymerisation of 2-substituted azulenes in particular (i.e. the substitution pattern of 6 and 7) has been studied by Daub and co-workers.28 Interestingly, under comparable experimental conditions the authors report peak oxidation potentials for a series of 2-phenylazulenes between 340 and 780 mV, depending on electronic nature (electron donating/withdrawing) of the substituents on the phenyl ring. For unsubstituted 2-phenyl azulene, the reported value is Epeak = 660 mV, extremely similar to the values of 666 and 657 mV for 6 and 7, respectively. This is despite the electron-withdrawing nature of the SF5 group, as demonstrated in various other experimental contexts.29
In contrast to the above, the CVs for 8 and 9 show scans 1 and 3 to be essentially identical. Being substituted at the azulene 1-position, 8 and 9 would not be expected to be good substrates for electropolymerisation, as the azulene 1- and 3-positions are where the union of monomers usually occurs.30 Nevertheless, it seems the oxidised forms of 8 and 9 also do not decompose to give other electrochemically active products. We have previously studied the characteristics of azulenes that exhibit such superimposable CVs,4a and have found that some substituents at the azulene 1-position can confer stability on the radical cation.16d,31 Such a characteristic is potentially beneficial for the application of azulene derivatives in various contexts such as photovoltaics and organic electronics. However, this effect is not general for all groups at the azulene 1-position, so the identification of m- and p-pentafluorosulfanylphenyl as substituents having this effect may be significant.
It is illuminating to compare the data we have gathered for 6–9 with literature data on analogues of 6–9 bearing other electron-withdrawing groups in the place of the –SF5 group. As described in the Introduction, in many cases SF5-containing compounds have been compared with their CF3 analogues, with various differences in their properties being noted. In the present case, –CF3 analogues have previously been reported for 6,257,16b8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7c,16a,25 and 9.15a,16b The halochromic properties have been studied for the –CF3 analogues of 6 and 8, with the same trend being observed in both cases: no significant colour change upon protonation of 8 or its –CF3 analogue, but a significant colour change upon protonation of 6 or its –CF3 analogue. For 6 the new peak with λmax = 409 nm leads to a brown colour, whereas for its –CF3 analogue a reported new peak with λmax = 427 nm leads to a yellow colour.25 The only data reported for all four –CF3 analogues are NMR data, which may be compared with the data for 6–9. For instance, the 1H-NMR spectrum of 6 and its –CF3 analogue are almost identical, with the only significant difference being that the –SF5ortho protons are more deshielded than those in the –CF3 analogue (Δδ 0.12 ppm). The same trend is observed when comparing 7–9 with their respective –CF3 analogues – the chemical shifts of the azulenyl protons hardly vary, and only the protons on the phenyl ring show any significant changes. This indicates that while the –SF5 group exerts a greater inductive electron-withdrawing effect than the –CF3 group, and hence perturbs the chemical shifts of the phenylene protons, it is too far removed to influence significantly the shielding of the azulenyl protons. A second informative comparison is between 6 and 8 and their nitro analogues.15e,18i,32 (Comparisons cannot be made for 7 and 9 as their nitro analogues have not been reported.) Here, the 1H-NMR spectra of the nitro analogues exhibit significantly more deshielded phenyl protons, as a consequence of the mesomeric and inductive electron-withdrawing effect of the nitro group. Some minor changes to the chemical shifts of the azulenyl protons are discernible, the largest being for the proton(s) on the azulene five-membered ring that are adjacent to the phenyl substituent. For these, Δδ = 0.04 ppm in each case. A final NMR comparison to be considered is that between 6 and 8 and their analogues lacking the SF5 substituent entirely (i.e. 2-phenylazulene and 1-phenylazulene, respectively24). In these cases, while the phenyl proton resonances differ significantly in the presence or absence of the SF5 group, the variations in the chemical shifts of the azulenyl protons are again minor (Δδ values between 0.01 and 0.08 ppm are observed for the various positions on the azulene rings). Considering the above observations as a whole, it appears that for phenylazulenes the presence of an electron withdrawing group on the phenyl ring greatly influences the electronics of the phenyl ring, but its effect on the electronics of the azulene ring is attenuated in comparison.
7c,16a,25 and 9.15a,16b The halochromic properties have been studied for the –CF3 analogues of 6 and 8, with the same trend being observed in both cases: no significant colour change upon protonation of 8 or its –CF3 analogue, but a significant colour change upon protonation of 6 or its –CF3 analogue. For 6 the new peak with λmax = 409 nm leads to a brown colour, whereas for its –CF3 analogue a reported new peak with λmax = 427 nm leads to a yellow colour.25 The only data reported for all four –CF3 analogues are NMR data, which may be compared with the data for 6–9. For instance, the 1H-NMR spectrum of 6 and its –CF3 analogue are almost identical, with the only significant difference being that the –SF5ortho protons are more deshielded than those in the –CF3 analogue (Δδ 0.12 ppm). The same trend is observed when comparing 7–9 with their respective –CF3 analogues – the chemical shifts of the azulenyl protons hardly vary, and only the protons on the phenyl ring show any significant changes. This indicates that while the –SF5 group exerts a greater inductive electron-withdrawing effect than the –CF3 group, and hence perturbs the chemical shifts of the phenylene protons, it is too far removed to influence significantly the shielding of the azulenyl protons. A second informative comparison is between 6 and 8 and their nitro analogues.15e,18i,32 (Comparisons cannot be made for 7 and 9 as their nitro analogues have not been reported.) Here, the 1H-NMR spectra of the nitro analogues exhibit significantly more deshielded phenyl protons, as a consequence of the mesomeric and inductive electron-withdrawing effect of the nitro group. Some minor changes to the chemical shifts of the azulenyl protons are discernible, the largest being for the proton(s) on the azulene five-membered ring that are adjacent to the phenyl substituent. For these, Δδ = 0.04 ppm in each case. A final NMR comparison to be considered is that between 6 and 8 and their analogues lacking the SF5 substituent entirely (i.e. 2-phenylazulene and 1-phenylazulene, respectively24). In these cases, while the phenyl proton resonances differ significantly in the presence or absence of the SF5 group, the variations in the chemical shifts of the azulenyl protons are again minor (Δδ values between 0.01 and 0.08 ppm are observed for the various positions on the azulene rings). Considering the above observations as a whole, it appears that for phenylazulenes the presence of an electron withdrawing group on the phenyl ring greatly influences the electronics of the phenyl ring, but its effect on the electronics of the azulene ring is attenuated in comparison.
Conclusions
In summary, we have synthesised and characterised the first azulene derivatives containing pentafluorosulfanylphenyl groups. These were accessed in one step from known azulenylboron species 4 and 5, by coupling with commercially available SF5-containing bromoarenes 2 and 3. Ordinarily, the absorption spectra of azulene derivatives are usually profoundly influenced by the nature and position of the substituents on the azulene rings. However, in the present case the absorption spectra of isomers 6–9 are all very similar in the visible region. The spectroscopic response of isomers 6–9 to protonation varies appreciably, with significant absorbances in the blue region for [6 + H]+ and [7 + H]+ leading to observable changes in colour. The fact that 6 and 7 exhibit a response to a stimulus (increase in [H+]) is suggestive of possible applications in chemical sensing. The pattern of the 2-substituted azulenes 6–7 having differing characteristics to the 1-substituted azulenes 8–9 was also observed in the electrochemical study, where 6 and 7 underwent electropolymerisation, but 8 and 9 did not. We anticipate that the commercial availability of an increasing number of SF5-containing building blocks will allow for the preparation of further SF5-containing azulenes with time.Experimental
General synthetic methods
Reactions were carried out under an atmosphere of nitrogen unless stated otherwise. Petrol refers to petroleum ether, bp 40–60 °C. TLCs were performed using aluminium-backed plates precoated with Alugram®SIL G/UV or aluminium backed plates precoated with Alugram®ALOX N/UV 254 nm and visualised by UV light (254 nm) and/or curcumin followed by gentle warming. All solvents used in Suzuki–Miyaura couplings were degassed by sparging with nitrogen. Flash column chromatography was carried out using Davisil LC 60 Å silica gel (35–70 micron) purchased from Sigma Aldrich. 3-Bromophenylsulfur pentafluoride and 4-bromophenylsulfur pentafluoride were purchased from Fluorochem Ltd. All other reagents were purchased from Sigma-Aldrich Chemical Co. or Fisher Scientific Ltd.; all reagents were used as received without further purification. SPhos refers to 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl. IR spectra were recorded on a Perkin-Elmer Spectrum 1600 FT IR spectrometer with universal ATR sampling accessory, with absorbances quoted as ν in cm−1. NMR spectra were run on an Agilent ProPulse 500 MHz instrument or Bruker Avance 500 MHz instruments at 298 K, unless otherwise specified. In tabulated NMR data, “p” refers to a pentet/quintet. UV/vis experiments were conducted using an Agilent Technologies Carry 60 UV/vis spectrometer. Mass spectra were acquired at the EPSRC National Mass Spectrometry Service Centre in Swansea, using an Atmospheric Solids Analysis Probe (Positive mode, Waters Xevo). For λabs and molar extinction coefficient determination, 0.125 mM solutions of 6–9 were prepared in MeCN, THF and DCM and spectra were acquired. For the Brønsted acid response test, a 1.0 M solution of TFA in DCM was prepared. 100 μL of the 0.125 mM solution of (one of) 6–9 was added to 100 μL of 1 M TFA, 800 μL of DCM was added and the resultant solution was analysed spectroscopically.General procedure for Suzuki–Miyaura couplings
To a degassed solution of azulenylboronate 4 or 5 (1.0 eq.)18 and pentafluorosulfanyl aryl bromide 2 or 3 (2.0 eq.) in dioxane (5.0 mL), were added K2CO3 (1.5 eq.), Pd(OAc)2 (5 mol%) and SPhos (10 mol%). The resulting mixture was heated at 80 °C under an N2 atmosphere. The reaction mixture was allowed to cool and CH2Cl2 (50 mL) and water (25 mL) were added, transferred to a separating funnel and shaken. The organic layer was separated, washed with water (2 × 25 mL), and then the combined aqueous extracts were back extracted with CH2Cl2 (50 mL). The combined organic layers were washed with brine (25 mL), and passed through phase separating filter paper, and the solvent removed under reduced pressure. Purification of the crude product was carried out using silica column chromatography with petrol and ethyl acetate (9:1) as eluent. In some instances additional chromatography was required, eluting with pentane, then ethyl acetate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for PhD funding to C. M. L.-A. from the EU Horizon 2020 research and innovation programme under grant agreement H2020-MSCA-CO-FUND, #665992. The Centre for Sustainable Chemical Technologies is supported by EPSRC under grant EP/L016354/1. NMR and X-ray crystallography facilities were provided through the Chemical Characterisation and Analysis Facility (CCAF) at the University of Bath. We thank the EPSRC National Mass Spectrometry Service Centre, Swansea, for sample analysis. This research also made use of the Balena High Performance Computing (HPC) Service at the University of Bath.References
- R. S. H. Liu, J. Chem. Educ., 2002, 79, 183 CrossRef CAS.
- R. S. H. Liu and A. E. Asato, J. Photochem. Photobiol., C, 2003, 4, 179 CrossRef CAS.
- For selected recent examples, see: (a) D. Lichosyt, S. Wasiłek, P. Dydio and J. Jurczak, Chem. – Eur. J., 2018, 24, 11683 CrossRef CAS PubMed; (b) G.-O. Buica, I.-G. Lazar, L. Birzan, C. Lete, M. Prodana, M. Enachescu, V. Tecuceanu, A. B. Stoian and E.-M. Ungureanu, Electrochim. Acta, 2018, 263, 382 CrossRef CAS; (c) C. M. López-Alled, A. Sanchez-Fernandez, K. J. Edler, A. C. Sedgwick, S. D. Bull, C. L. McMullin, G. Kociok-Köhn, T. D. James, J. Wenk and S. E. Lewis, Chem. Commun., 2017, 53, 12580 RSC; (d) S. Wakabayashi, M. Uchida, R. Tanaka, Y. Habata and M. Shimizu, Asian J. Org. Chem., 2013, 2, 786 CrossRef CAS.
- For selected recent examples, see: (a) P. Cowper, A. Pockett, G. Kociok-Köhn, P. J. Cameron and S. E. Lewis, Tetrahedron, 2018, 74, 2775 CrossRef CAS; (b) H. Xin, C. Ge, X. Jiao, X. Yang, K. Rundel, C. R. McNeill and X. Gao, Angew. Chem., Int. Ed., 2018, 57, 1322 CrossRef CAS PubMed; (c) H. Nishimura, N. Ishida, A. Shimazaki, A. Wakamiya, A. Saeki, L. T. Scott and Y. Murata, J. Am. Chem. Soc., 2015, 137, 15656 CrossRef CAS PubMed; (d) E. Puodziukynaite, H.-W. Wang, J. Lawrence, A. J. Wise, T. P. Russell, M. D. Barnes and T. Emrick, J. Am. Chem. Soc., 2014, 136, 11043 CrossRef CAS PubMed.
- For selected examples, see: (a) H. Xin, J. Li, C. Ge, X. Yang, T. Xue and X. Gao, Mater. Chem. Front., 2018, 2, 975 RSC; (b) H. Xin, C. Ge, X. Jiao, X. Yang, K. Rundel, C. R. McNeill and X. Gao, Angew. Chem., Int. Ed., 2018, 57, 1322 CrossRef CAS PubMed; (c) H. Xin and X. Gao, ChemPlusChem, 2017, 82, 945 CrossRef CAS; (d) H. Xin, C. Ge, L. Fu, X. Yang and X. Gao, Chin. J. Org. Chem., 2017, 37, 711 CrossRef CAS; (e) J.-X. Dong and H.-L. Zhang, Chin. Chem. Lett., 2016, 27, 1097 CrossRef CAS; (f) H. Xin, C. Ge, X. Yang, H. Gao, X. Yang and X. Gao, Chem. Sci., 2016, 7, 6701 RSC; (g) Y. Yamaguchi, M. Takubo, K. Ogawa, K.-I. Nakayama, T. Koganezawa and H. Katagiri, J. Am. Chem. Soc., 2016, 138, 11335 CrossRef CAS PubMed; (h) Y. Yamaguchi, K. Ogawa, K.-I. Nakayama, Y. Ohba and H. Katagiri, J. Am. Chem. Soc., 2013, 135, 19095 CrossRef CAS PubMed; (i) P. H. Wöbkenberg, J. G. Labram, J.-M. Swiecicki, K. Parkhomenko, D. Sredojevic, J.-P. Gisselbrecht, D. M. de Leeuw, D. D. C. Bradley, J.-P. Djukic and T. D. Anthopoulos, J. Mater. Chem., 2010, 20, 3673 RSC; (j) E. C. P. Smits, S. Setayesh, T. D. Anthopoulos, M. Buechel, W. Nijssen, R. Coehoorn, P. W. M. Blom, B. deBoer and D. M. deLeeuw, Adv. Mater., 2007, 19, 734 CrossRef CAS.
- (a) T. Wada, R. Maruyama, Y. Irie, M. Hashimoto, H. Wakabayashi, N. Okudaira, Y. Uesawa, H. Kagaya and H. Sakagami, In Vivo, 2018, 32, 479 CAS; (b) J. Peet, A. Selyutina and A. Bredihhin, Bioorg. Med. Chem. Lett., 2016, 24, 1653 CrossRef CAS PubMed; (c) K. Ikegai, M. Imamura, T. Suzuki, K. Nakanishi, T. Murakami, E. Kurosaki, A. Noda, Y. Kobayashi, M. Yokota, T. Koide, K. Kosakai, Y. Okhura, M. Takeuchi, H. Tomiyama and M. Ohta, Bioorg. Med. Chem., 2013, 21, 3934 CrossRef CAS PubMed; (d) S. Löber, H. Hübner, A. Buschauer, F. Sanna, A. Argiolas, M. R. Melis and P. Gmeiner, Bioorg. Med. Chem. Lett., 2012, 22, 7151 CrossRef PubMed; (e) C.-H. Chen, O. Lee, C.-N. Yao, M.-Y. Chuang, Y.-L. Chang, M.-H. Chang, Y.-F. Wen, W.-H. Yang, C.-H. Ko, N.-T. Chou, M.-W. Lin, C.-P. Lai, C.-Y. Sun, L.-M. Wang, Y.-C. Chen, T.-H. Hseu, C.-N. Chang, H.-C. Hsu, H.-C. Lin, Y.-L. Chang, Y.-C. Shih, S.-H. Chou, Y.-L. Hsu, H.-W. Tseng, C.-P. Liu, C.-M. Tu, T.-L. Hu, Y.-J. Tsai, T.-S. Chen, C.-L. Lin, S.-J. Chiou, C.-C. Liu and C.-S. Hwang, Bioorg. Med. Chem. Lett., 2010, 20, 6129 CrossRef CAS PubMed; (f) Y. Tanaka and K. Shigenobu, Cardiovasc. Drug Rev., 2001, 19, 297 CrossRef CAS PubMed; (g) A. E. Asato, A. Peng, M. Z. Hossain, T. Mirzadegan and J. S. Bertram, J. Med. Chem., 1993, 36, 3137 CrossRef CAS PubMed; (h) T. Tomiyama, M. Yokota, S. Wakabayashi, K. Kosakai and T. Yanagisawa, J. Med. Chem., 1993, 36, 791 CrossRef CAS PubMed.
- For selected examples, see: (a) M. Murai, S. Iba, H. Ota and K. Takai, Org. Lett., 2017, 19, 5585 CrossRef CAS PubMed; (b) Y. M. Poronik, L. M. Mazur, M. Samoć, D. Jacquemin and D. T. Gryko, J. Mater. Chem. C, 2017, 5, 2620 RSC; (c) M. Murai, M. Yanagawa, M. Nakamura and K. Takai, Asian J. Org. Chem., 2016, 5, 629 CrossRef CAS; (d) A. W. Woodward, E. H. Ghazvini Zadeh, M. V. Bondar and K. D. Belfield, R. Soc. Open Sci., 2016, 3, 160373 CrossRef PubMed; (e) E. H. Ghazvini Zadeh, S. Tang, A. W. Woodward, T. Liu, M. V. Bondar and K. D. Belfield, J. Mater. Chem. C, 2015, 3, 8495 RSC; (f) K. Ninomiya, Y. Harada, T. Kanetou, Y. Suenaga, T. Murafuji and R. Tsunashima, New J. Chem., 2015, 39, 9079 RSC; (g) K. Tsurui, M. Murai, S.-Y. Ku, C. J. Hawker and M. J. Robb, Adv. Funct. Mater., 2014, 24, 7338 CrossRef CAS; (h) M. Koch, O. Blacque and K. Venkatesan, J. Mater. Chem. C, 2013, 1, 7400 RSC; (i) X. Wang, J. K.-P. Ng, P. Jia, T. Lin, C. M. Cho, J. Xu, X. Lu and C. He, Macromolecules, 2009, 42, 5534 CrossRef CAS.
- For reviews, see: (a) P. Das, E. Tokunaga and N. Shibata, Tetrahedron Lett., 2017, 58, 4803 CrossRef CAS; (b) P. R. Savoie and J. T. Welch, Chem. Rev., 2015, 115, 1130 CrossRef CAS PubMed; (c) C. N. von Hahmann, P. R. Savoie and J. T. Welch, Curr. Org. Chem., 2015, 19, 1592 CrossRef CAS; (d) S. Altomonte and M. Zanda, J. Fluorine Chem., 2012, 143, 57 CrossRef CAS; (e) G. L. Gard, Chim. Oggi, 2009, 27, 10 CAS.
- D. A. Jackson and S. A. Mabury, Environ. Toxicol. Chem., 2009, 28, 1866 CrossRef CAS PubMed.
- (a) M. Saccomanno, S. Hussain, N. K. O’Connor, P. Beier, M. Somlyay, R. Konrat and C. D. Murphy, Biodegradation, 2018, 29, 259 CrossRef CAS PubMed; (b) C. D. Murphy, Appl. Microbiol. Biotechnol., 2016, 100, 2617 CrossRef CAS PubMed; (c) E. Kavanagh, M. Winn, C. N. Gabhann, N. K. O’Connor, P. Beier and C. D. Murphy, Environ. Sci. Pollut. Res., 2014, 21, 753 CrossRef CAS PubMed.
- For reviews, see: (a) P. Kirsch and M. Bremer, Chimia, 2014, 68, 363 CrossRef CAS; (b) P. Kirsch, M. Bremer, M. Heckmeier and K. Tarumi, Angew. Chem., Int. Ed., 1999, 38, 1989 CrossRef CAS.
- P. Kenyon and S. Mecking, J. Am. Chem. Soc., 2017, 139, 13786 CrossRef CAS PubMed.
- (a) P. Gautam, C. P. Yu, G. Zhang, V. E. Hillier and J. M. W. Chan, J. Org. Chem., 2017, 82, 11008 CrossRef CAS PubMed; (b) H. Nakayama, J.-i. Nishida, N. Takada, H. Sato and Y. Yamashita, Chem. Mater., 2012, 24, 671 CrossRef CAS.
- For selected examples, see: (a) G. L. Borosky and K. K. Laali, J. Fluorine Chem., 2017, 197, 118 CrossRef CAS; (b) X.-H. Li, H.-L. Cui, W.-W. Ju, T.-W. Li, R.-Z. Zhang and Y.-L. Yong, J. Chem. Sci., 2014, 126, 1163 CrossRef CAS; (c) P. R. Savoie, S. Higashiya, J.-H. Lin, D. V. Wagle and J. T. Welch, J. Fluorine Chem., 2012, 143, 281 CrossRef CAS; (d) O. Exner and S. Böhm, New J. Chem., 2008, 32, 1449 RSC.
- For selected examples, see: (a) J. Dubovik and A. Bredihhin, Synthesis, 2015, 2663 CAS; (b) S. Ito, T. Shoji and N. Morita, Synlett, 2011, 2279 CrossRef CAS; (c) M. Fujinaga, K. Suetake, K. Gyoji, T. Murafuji, K. Kurotobi and Y. Sugihara, Synthesis, 2008, 3745 CAS; (d) T. Shibasaki, T. Ooishi, N. Yamanouchi, T. Murafuji, K. Kurotobi and Y. Sugihara, J. Org. Chem., 2008, 73, 7971 CrossRef CAS PubMed; (e) S. Ito, T. Terazono, T. Kubo, T. Okujima, N. Morita, T. Murafuji, Y. Sugihara, K. Fujimori, J. Kawakami and A. Tajiri, Tetrahedron, 2004, 60, 5357 CrossRef CAS; (f) K. Kurotobi, H. Tabata, M. Miyauchi, A. F. M. M. Rahman, K. Migita, T. Murafuji, Y. Sugihara, H. Shimoyama and K. Fujimori, Synthesis, 2003, 30 CAS.
- (a) For selected examples, see: P. Cowper, Y. Jin, M. D. Turton, G. Kociok-Köhn and S. E. Lewis, Angew. Chem., Int. Ed., 2016, 55, 2564 CrossRef CAS PubMed; (b) J. Dubovik and A. Bredihhin, Synthesis, 2015, 538 CAS; (c) M. Koch, O. Blacque and K. Venkatesan, Org. Lett., 2012, 14, 1580 CrossRef CAS PubMed; (d) T. Shoji, S. Ito, K. Toyota, T. Iwamoto, M. Yasunami and N. Morita, Eur. J. Org. Chem., 2009, 4307 CrossRef CAS.
- For a review, see: X. Shi, A. Sasmal, J.-F. Soulé and H. Doucet, Chem. – Asian J., 2018, 13, 143 CrossRef CAS PubMed.
- For selected examples, see: (a) J. Carreras, Y. Popowski, A. Caballero, E. Amir and P. J. Perez, J. Org. Chem., 2018, 83, 11125 CrossRef CAS PubMed; (b) J. Liu, H. Li, R. Dehren, J. Liu, A. Spannenberg, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11976 CrossRef CAS PubMed; (c) A. Székely, A. Péter, K. Aradi, G. L. Tolnai and Z. Novák, Org. Lett., 2017, 19, 954 CrossRef PubMed; (d) L. Gu, L. M. Wolf, A. Zielinski, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2017, 139, 4948 CrossRef CAS PubMed; (e) J. C. Timmerman, W. W. Schmitt and R. A. Widenhoefer, Org. Lett., 2016, 18, 4966 CrossRef CAS PubMed; (f) X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636 CrossRef CAS PubMed; (g) L. Zhao, C. Bruneau and H. Doucet, Chem. Commun., 2013, 49, 5598 RSC; (h) J. Liu, E. Muth, U. Floerke, G. Henkel, K. Merz, J. Sauvageau, E. Schwake and G. Dyker, Adv. Synth. Catal., 2006, 348, 456 CrossRef CAS; (i) G. Dyker, S. Borowski, J. Heiermann, J. Körning, K. Opwis, G. Henkel and M. Köckerling, J. Organomet. Chem., 2000, 606, 108 CrossRef CAS.
- (a) K. Kurotobi, M. Miyauchi, K. Takakura, T. Murafuji and Y. Sugihara, Eur. J. Org. Chem., 2003, 3663 CrossRef CAS; (b) M. Fujinaga, T. Murafuji, K. Kurotobi and Y. Sugihara, Tetrahedron, 2009, 65, 7115 CrossRef CAS.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
- C. Zarantonello, A. Guerrato, E. Ugel, R. Bertani, F. Benetollo, R. Milani, A. Venzo and A. Zaggia, J. Fluorine Chem., 2007, 128, 1449 CrossRef CAS.
- J. Du, G. Hua, P. Beier, A. M. Z. Slawin and J. D. Woollins, Struct. Chem., 2017, 28, 723 CrossRef CAS.
- T. Tang, T. Lin, F. Erden, F. Wang and C. He, J. Mater. Chem. C, 2018, 6, 5153 RSC.
- M. Murai, S.-Y. Ku, N. D. Treat, M. J. Robb, M. L. Chabinyc and C. J. Hawker, Chem. Sci., 2014, 5, 3753 RSC.
- M. Murai, K. Takami, H. Takeshima and K. Takai, Org. Lett., 2015, 17, 1798 CrossRef CAS PubMed.
- (a) J. Michl and E. W. Thulstrup, Tetrahedron, 1976, 32, 205 CrossRef CAS; (b) R. S. H. Liu, R. S. Muthyala, X.-S. Wang and A. E. Asato, Org. Lett., 2002, 2, 269 CrossRef.
- (a) G. Tourillon and F. Garnier, J. Electroanal. Chem., 1982, 135, 173 CrossRef CAS; (b) R. J. Waltman, A. F. Diaz and J. Bargon, J. Electrochem. Soc., 1984, 131, 1452 CrossRef CAS; (c) Y.-B. Shim and S.-M. Park, J. Electrochem. Soc., 1997, 144, 3027 CrossRef CAS; (d) G. Nie, T. Cai, S. Zhang, J. Hou, J. Xu and X. Han, Mater. Lett., 2007, 61, 3079 CrossRef CAS.
- A. Mirlach, M. Feuerer and J. Daub, Adv. Mater., 1993, 5, 450 CrossRef CAS.
- (a) J. E. True, T. D. Thomas, R. W. Winter and G. L. Gard, Inorg. Chem., 2003, 42, 4437 CrossRef CAS PubMed; (b) L. J. Saethre, N. Berrah, J. D. Bozek, K. J. Boerve, T. X. Carroll, E. Kukk, G. L. Gard, R. Winter and T. D. Thomas, J. Am. Chem. Soc., 2001, 123, 10729 CrossRef CAS PubMed; (c) P. Brant, A. D. Berry, R. A. DeMarco, F. L. Carter, W. B. Fox and J. A. Hashmall, J. Electron Spectrosc. Relat. Phenom., 1981, 22, 119 CrossRef CAS; (d) W. A. Sheppard, J. Am. Chem. Soc., 1962, 84, 3072 CrossRef CAS.
- K. Iwasaki, K. Matsumoto, S. Hino and M. Yasunami, Synth. Met., 1993, 55, 1062 CrossRef CAS.
- F. Gerson, M. Scholz, H.-J. Hansen and P. Uebelhart, J. Chem. Soc., Perkin Trans. 2, 1995, 215 RSC.
- V. A. Nefedov, N. A. German, A. I. Lutsenko and G. I. Nikishin, Zh. Org. Khim., 1987, 23, 172 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational data, electrochemical procedures, NMR spectra, X-ray crystallographic data. CCDC 1850019. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8nj05520c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2019 |