
Pd@Rh core–shell nanocrystals with well-defined facets and their enhanced catalytic performance towards CO oxidation†
Sang-Il
Choi
 ab,
Allison
Young
c,
Sujin R.
Lee
d,
Cheng
Ma
e,
Ming
Luo
a,
Miaofang
Chi
e,
Chia-Kuang
Tsung
*c and
Younan
Xia
*adf
ab,
Allison
Young
c,
Sujin R.
Lee
d,
Cheng
Ma
e,
Ming
Luo
a,
Miaofang
Chi
e,
Chia-Kuang
Tsung
*c and
Younan
Xia
*adf
aThe Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia 30332, USA. E-mail: younan.xia@bme.gatech.edu
bDepartment of Chemistry and Green-Nano Materials Research Center, Kyungpook National University, Daegu 41566, Republic of Korea
cDepartment of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, Massachusetts 02467, USA. E-mail: tsungc@bc.edu
dSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
eMaterials Science and Technology Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37830, USA
fSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
First published on 23rd July 2019
Abstract
Here we report a facile synthesis of Pd@Rh core–shell nanocrystals with octahedral and cubic shapes. Under optimized conditions, Rh atoms can be deposited on Pd octahedral or cubic seeds in a layer-by-layer fashion to generate core–shell nanocrystals with a well-controlled shape. We then use CO oxidation as a probe to evaluate the catalytic performance of the core–shell nanocrystals with reference to a number of commercial catalysts. When supported on mesoporous silica, both the octahedral and cubic Pd@Rh nanocrystals show CO to CO2 conversion levels similar to that of a commercial Pt/Al2O3 catalyst while the two catalysts based on pure Rh (commercial Rh/C and Rh nanocubes/silica) needed much higher temperatures to reach the same level of conversion. In terms of ignition temperature, the Rh nanocubes show a value of 260 °C while those of the octahedral and cubic Pd@Rh nanocrystals are as low as 140 and 150 °C, respectively. Our results suggest that there is no significant difference between the octahedral and cubic Pd@Rh nanocrystals in terms of performance towards CO oxidation while both of them are advantageous over Rh nanocubes or Rh/C.
New conceptsRhodium-based nanocrystals are interesting for a variety of catalytic applications such as CO oxidation, NO reduction, and hydrogenations. In this work, we demonstrated a facile synthesis of Pd@Rh core–shell nanostructures via seed-mediated growth, in which Pd octahedra and cubes were utilized as seeds for the conformal deposition of Rh atoms. The deposition of Rh atoms on Pd could be switched from an island to a layer-by-layer mode by optimizing the experimental conditions, including reaction temperature, Rh precursor concentration, and the size of the Pd nanocrystals, to achieve a smooth surface for the Rh shell. The as-obtained Pd@Rh core–shell octahedra and cubes were then compared as catalysts towards CO oxidation to evaluate the catalytic performance of the core–shell nanocrystals with reference to a number of commercial catalysts. The Pd@Rh core–shell octahedra exhibited the lowest ignition temperature, followed by the Pd@Rh cubes, indicating significant enhancement of reaction kinetics compared to the monometallic catalysts. These results indicate that the structural changes arising from the core–shell structure give more significant improvement towards CO oxidation relative to the effect of facets. |
Introduction
Bimetallic nanocrystals with a core–shell structure have received considerable interest in recent years owing to their advantages over their monometallic counterparts in catalytic and electrocatalytic applications.1,2 Depending on the subsurface species of core–shell nanocrystals, one can manipulate the surface elemental and electronic structures and thus tune their catalytic activity and/or selectivity.3–8 As another advantage of the core–shell structure, one can achieve highly active catalysts through the ligand and/or strain effects.9 To this end, Pd@Pt core–shell nanocrystals have been fabricated through conformal, layer-by-layer deposition of Pt atoms on Pd nanocrystals to enhance the electrocatalytic activity of Pt-based catalysts towards the oxygen reduction reaction key to the operation of proton-exchange membrane fuel cells.10–12 Computational studies have established that the oxygen reduction kinetics on the Pd@Pt nanocrystals can be enhanced through a combination of ligand and strain effects.6,10Core–shell nanocrystals are often synthesized through seed-mediated growth in a solution phase. By controlling experimental parameters such as temperature and feeding amount/rate of the metal precursor, the reduction rate and thus the deposition mode of the surface metal on the seed can be optimized, leading to the conformal deposition of surface metal as a uniform shell on the seed.10–13 Compared to Ag, Au, Pd, and Pt, it has been more challenging to produce Rh- and Ir-terminated core–shell nanocrystals through seed-mediated growth because the energy barrier to homogeneous nucleation is lower than that for heterogeneous nucleation for these two metals, favoring the formation of separate nanoparticles (<4 nm) rather than a conformal shell on the surface of the seed.14–19 To address this issue, one needs to control the diffusion rate of the adatoms relative to the deposition rate while eliminating self-nucleation. In one example, our group demonstrated seed-mediated growth of Pd@Ir core–shell nanocrystals with cubic and octahedral shapes by templating with Pd cubic and octahedral seeds, respectively. Using a combination of slow injection and relatively high temperatures, the Ir atoms could be conformally deposited on the surfaces of Pd cubic and octahedral seeds, generating well-defined Ir{100} and Ir{111} facets.20
In this work, we extend the concept of core–shell structure to control Rh overgrowth owing to its superior performance in catalyzing reactions such as CO oxidation, NO reduction, and hydrogenations.21–26 In addition, Rh has been used as a powerful catalyst for automotive three-way-conversion reactions.27 Several groups including our own have actively pursued conformal coating of Rh on Pd nanocrystals by leveraging seed-mediated growth. However, we always observed the island growth mode (Volmer–Weber) due to the susceptibility of Rh to homogeneous nucleation.28–30 As a result, there is still no report on the synthesis of Rh-terminated core–shell nanocrystals with an octahedral or cubic shape despite the successful syntheses of Rh solid nanocrystals with many controlled shapes, including cubes, decahedra, icosahedra, plates, and concave cubes.31–36 Herein, we demonstrate a facile route to the synthesis of Pd@Rh core–shell nanocrystals enclosed by well-defined Rh{111} and Rh{100} facets, respectively. In our synthesis, the island growth of Rh was eliminated by optimizing the experimental conditions, including reaction temperature, Rh precursor concentration, and the size of the Pd seeds. The Pd@Rh core–shell octahedra and cubes were then applied as catalysts toward a CO oxidation reaction in comparison with Rh cubes, commercial Rh/C, and commercial Pt/Al2O3 catalysts. Our catalytic measurements indicate that the Pd@Rh core–shell octahedra exhibited the lowest ignition temperature at 140 °C, followed by Pd@Rh cubes at 150 °C, suggesting significant enhancement of the reaction kinetics relative to all the monometallic catalysts. Overall, the CO oxidation reaction did not show any strong dependence on the type of Rh facet expressed on the surface of the catalyst.
Results and discussion
We synthesized Pd octahedra with an average edge length of 6.4 nm by following a protocol in the literature (Fig. S1A, ESI†).37 We then deposited Rh shells on the Pd octahedra to generate well-defined Rh{111} facets in benzyl alcohol that contained Rh(OAc)3, tetraethylene glycol (TTEG), and poly(vinyl pyrrolidone) (PVP). The synthesis was conducted at 185 °C for 3 h under magnetic stirring. Fig. 1A and B show typical transmission electron microscopy (TEM) images of the as-obtained Pd@Rh core–shell octahedra, which had an average edge length of 7.4 nm. The Rh shells had an average thickness of 0.5 nm, corresponding to about two atomic layers based on an inter-planar distance of 0.22 nm for the {111} planes of face-centered cubic (fcc) Rh. The number of Rh atomic layers was also analyzed using inductively-coupled plasma mass spectrometry (ICP-MS). As shown in Table S1 (ESI†), the average number of Rh atomic overlayers was about 2.1. Fig. S2 (ESI†) shows a TEM image at a lower magnification, confirming a uniform size and shape for the as-prepared Pd@Rh octahedra. Fig. 1C shows a high-resolution TEM image taken from a single Pd@Rh octahedron along the [011] zone axis, indicating a single-crystal structure. The Fast Fourier Transform (FFT) pattern in the inset confirms the formation of {111} facets on the side faces. The Rh shell deposited on a Pd octahedron might have atomic steps but it was smooth almost down to the atomic scale without clustering. Fig. 1D shows a high-angle annular dark-field scanning TEM (HAADF-STEM) image and the corresponding energy-dispersive X-ray spectroscopy (EDX) mapping of an individual Pd@Rh octahedron, revealing the Pd core and Rh shell. | ||
| Fig. 1 (A and B) TEM and (C) atomic resolution TEM images of Pd@Rh core–shell octahedra synthesized using the standard protocol. The inset in (C) shows the corresponding FFT pattern. (D) HAADF-STEM image and the corresponding EDX elemental mapping of Pd and Rh, confirming a core–shell structure. | ||
There are a number of prior reports on Pd@M core–shell octahedra (M = Pt, Rh, and Ir), and the island growth mode was often observed for the surface metals because of the higher inter-atomic (M–M) bond energy than that of the Pd–M bond.20,28,30,38 We reported a synthesis of Pd@Pt core–shell octahedra where the island growth of the Pt overlayers was mitigated by adding the precursor into a reaction mixture containing Pd seeds at a slow injection rate, together with the use of a relatively high temperature to help break the strong Pt–Pt bond (307 kJ mol−1).11 Consequently, the surface diffusion of Pt adatoms across the surface of the Pd substrate was promoted, resulting in a layer-by-layer growth for the Pt atoms. Based on the fact that the Rh–Rh bond energy (236 kJ mol−1) is lower than that of the Pt–Pt bond, we expected to obtain a smooth Rh(111) surface on the Pd octahedral seeds. However, in previous work, Rh islands were still observed on the surface of Pd nanocrystals because of the reduction in homogeneous nucleation energy barrier for Rh relative to that for Pt.28,30
A careful control over the reaction temperature, concentration of Rh atoms, and size of the Pd seeds is necessary to achieve a smooth surface for the Rh coating. According to previous experiments, the diameters of Rh islands were typically 3–7 nm in size.28,30 Therefore, we initially chose Pd octahedral seeds with an edge length of 6.4 nm to supply Pd faces with dimensions comparable to those of the Rh islands. We then explored the effect of seed size on the overgrowth of Rh by increasing the edge length of the Pd octahedral seeds to 18 and 37 nm, respectively. As shown in Fig. 2, the deposited Rh took an island growth mode when the standard protocol was used. In these two cases, it should be pointed out that the amount of Rh precursor was adjusted to ensure that the resultant Rh atoms would be just adequate to form a shell of two atomic layers in thickness when the coating was smooth and uniform. Our ICP-MS data indicated that essentially all the Rh precursors were reduced to Rh atoms for all the syntheses involving Pd seeds of different sizes. As such, the dispersion of Rh in Pd was more or less the same for all these samples, except for the difference in morphology.
 | ||
| Fig. 2 TEM images showing the dominance of an island growth mode when Rh was deposited on Pd octahedral seeds with edge lengths of (A) 18 and (B) 37 nm while the other parameters were kept the same as the standard protocol. | ||
For the 6.4 nm Pd seeds, an appropriate amount of Rh(OAc)3 solution (4.98 wt%Rh, 27 μL) had to be used in order to achieve a thin coating of two atomic layers. It is worth pointing out that further increasing the volume of Rh(OAc)3 from 27 to 81 μL for the standard protocol did not result in a thicker shell on the octahedra. Instead, the products contained both Pd@Rh octahedra and small Rh nanoparticles (Fig. S3, ESI†). This observation indicates that the increased Rh atoms might not be deposited on the surface of the Pd@Rh octahedra once their surface has been completely covered by Rh atoms with well-defined {111} facets. Instead, the additional Rh atoms favored the formation of Rh nuclei due to the low energy barrier to homogeneous nucleation, which is in agreement with previous observations.11 To this end, the reaction temperature was optimized to eliminate the formation of Rh islands. Under milder conditions at 160 °C for 3 h, Pd octahedra and Rh icosahedra were observed (Fig. 3A). As discussed in the previous work, the formation of Rh icosahedra lined with twin defects is favored by a slow reduction of Rh precursor relative to their single-crystal counterparts.36 When the reaction temperature was increased to 190 °C, both Rh islands on Pd seeds and small Rh nanoparticles were observed (Fig. 3B). Taken together, a fine tuning of the reaction temperature is necessary in order to generate a well-defined Rh(111) surface on the core–shell octahedra.
 | ||
| Fig. 3 TEM images of two samples prepared using the standard protocol except for the change of reaction temperature from 185 °C to (A) 160 and (B) 190 °C, respectively. The sample in (A) shows a mixture of Pd octahedra and Rh icosahedra (indicated by arrows) and the sample in (B) contains Pd@Rh core–shell nanocrystals formed through island growth and small Rh particles derived from self-nucleation. | ||
Next, we investigated the effects of a molecular ligand in the precursor and the shape of Pd seeds on the formation of a well-defined core–shell structure. The ligand in the precursor could affect the reaction kinetics. As demonstrated in a previous study involving one-pot synthesis, a bi-dentate ligand resulted in the formation of Rh icosahedra whereas a mono-dentate ligand led to the formation of single-crystal Rh nanocrystals.36 However, our present work indicates that the octahedral shape of the Pd@Rh core–shell nanocrystals was maintained for both mono-dentate (Cl− and NO3−) and bi-dentate (acetylacetonate) ligands (Fig. S4, ESI†). Therefore, we demonstrate that the layer-by-layer growth of Rh on a Pd nanocrystal seed was not affected by the type of ligand during the overgrowth of Rh. In addition, we found that the layer-by-layer deposition of Rh atoms could also lead to the formation of octahedral shape even when Pd seeds with different morphologies were used. The synthesis was conducted using the standard protocol with the exception of using Pd cubes rather than Pd octahedra as the seeds. In this case, we obtained a mixture of Pd@Rh core–shell octahedra and truncated octahedra (Fig. S5A, ESI†). The driving force for the evolution from a cube to an octahedron lies in the fact that the growth rate along the 〈100〉 direction is faster than that along the 〈111〉 direction. This trend stems from the fact that the {100} facets are typically higher in free energy than the {111} facets for metals with an fcc structure. At a slightly lower temperature (185 °C) than what was used for Rh island growth at 190 °C, the Rh shells are predominantly terminated by {111} facets in an effort to minimize the total surface energy. Similarly, we also obtained Pd@Rh octahedra when a Pd cuboctahedral seed was used (Fig. S5B, ESI†).
In order to compare the catalytic properties of different facets, we also prepared Pd@Rh core–shell cubes by following the protocol previously developed for the synthesis of Pd@Pt core–shell cubes.10 The successful deposition of Rh atoms in a layer-by-layer fashion on Pd cubic seeds (Fig. S1B, ESI†) relies on the use of a slow injection rate for the Rh precursor, along with fast reduction kinetics, to allow all the Rh atoms to be deposited on the Pd surface. Typically, the synthesis of Pd@Rh core–shell cubes was conducted by injecting the Rh(OAc)3 solution at a rate of 2.0 mL h−1 into the reaction mixture containing Pd cubes and ascorbic acid (a reducing agent). Fig. 4A and B show typical TEM images of the as-synthesized Pd@Rh cubes with an average edge length of 8.5 nm. Based on the inter-planar distance of 0.19 nm for the Rh{200} planes, it was estimated that about 3.4 atomic layers of Rh were deposited on the Pd cubes. From the ICP-MS analysis, the number of atomic layers of Rh on the Pd@Rh cubes was 2.9 (Table S1, ESI†). Fig. 4C and D show a high-resolution TEM image and the ED pattern, as well as an HAADF-STEM image and the corresponding EDX mapping recorded from an individual Pd@Rh cube.
 | ||
| Fig. 4 (A and B) TEM and (C) atomic-resolution TEM images of Pd@Rh core–shell cubes synthesized using the standard protocol. (D) HAADF-STEM image and the corresponding EDX mapping of Pd and Rh, confirming a core–shell structure. | ||
Carbon monoxide oxidation by O2 is not only an important reaction for the automobile industry but also a well-established model reaction system for evaluating catalysts.39–41 For this reaction, it is a thermodynamically favorable process with a negative free energy of conversion from CO to CO2 but it has a high activation energy barrier due to the involvement of O2 dissociation.42 With the use of transition metal catalysts and certain oxide supports,43–49 the dissociation energy of O2 can be reduced to generate O atoms for reaction with CO on the catalytically active surface. Previously, it was thought to be a surface insensitive reaction when CO is the predominant species in an oxygen rich environment.21,50 However, some aspects of catalytic sensitivity have demonstrated structure- and facet-dependence for nanocatalysts by tailoring the surface electronic structure of the metal nanocrystals and thus the adsorption, desorption, and related energies in a more favorable fashion.51,52 One strategy for tuning the electronic structure of a metal surface is to coat a catalytically active metal on the surface of a different metal, generating a core–shell structure.10 In this study, the deposition of ultrathin shells of Rh on Pd cores can impose the ligand effect and/or lattice strain on the catalytic surface comprised of Rh.53
To test the catalytic properties of the as-synthesized Pd@Rh core–shell nanocrystals, we deposited them on mesoporous silica to obtain Pd@Rh/SBA15 catalysts. Carbon monoxide oxidation was performed over different catalysts with the same metal (Pd + Rh) loading using a gas phase micro-reactor. The use of monometallic cubic Rh/SBA15 provides a comparison between the solid and core–shell structured catalysts elucidating the influence of the core. To benchmark the samples, two commercially available catalysts, Rh/C and Pt/Al2O3, were included. We also repeated the measurement of CO oxidation on each sample at least five times. By comparing the catalytic activity in terms of CO to CO2 conversion (Fig. 5), it can be seen that the Pd@Rh core–shell structures had a performance similar to the commercially available Pt/Al2O3 catalyst while the two catalysts based on pure Rh (commercial Rh/C and Rh nanocubes/SBA15) needed much higher temperatures to reach the same degree of conversion.
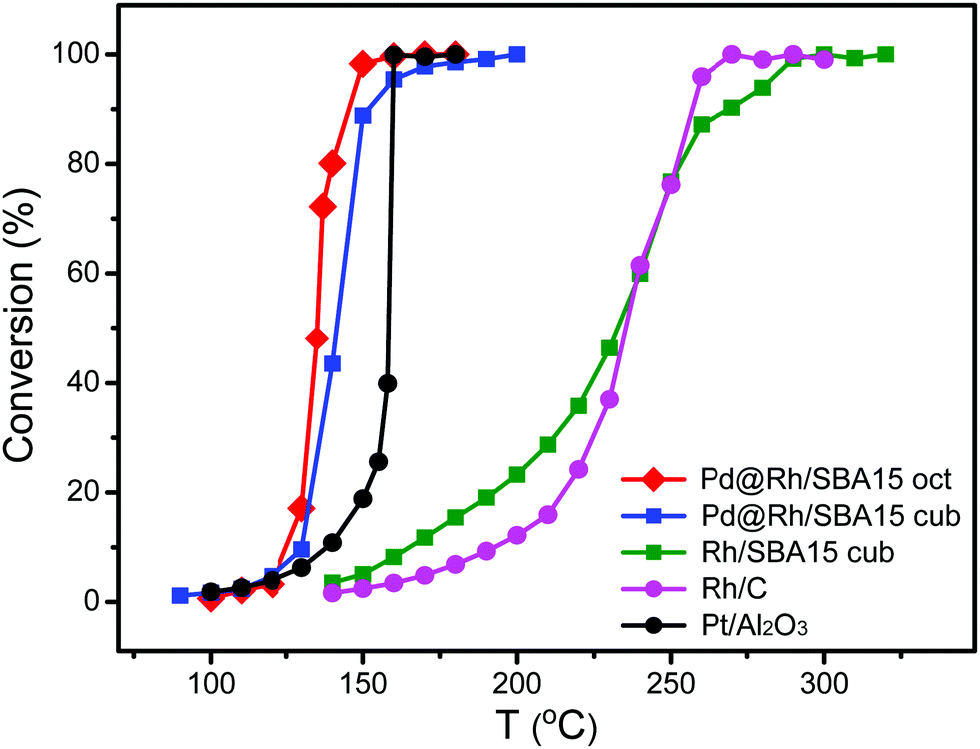 | ||
| Fig. 5 Comparison of the catalytic performances of octahedral Pd@Rh/SBA15, cubic Pd@Rh/SBA15, cubic Rh/SBA15, and the commercial Rh/C and Pt/Al2O3 catalysts towards CO oxidation. | ||
Typically, CO oxidation reaction has to be performed at an increased temperature first until reaching a point where the exothermic conversion process becomes self-sustainable and additional heating is longer needed. The temperature at which this transition occurs is known as the ignition point or ignition temperature (IT). When the surface electronic structure of a catalyst is changed, the IT will be altered. The IT of a catalyst can be determined from the Arrhenius plot. The point at which the activation energy (Ea) changes gives the IT of the catalyst. As shown in Fig. S6 (ESI†) and Table 1, the ITs of the octahedral and cubic Pd@Rh/SBA15 were 140 and 150 °C, respectively. In comparison, the IT of the cubic Rh/SBA15 was 260 °C, indicating a much higher activation energy barrier for the CO to CO2 conversion when conducted on Rh cubes than on Pd@Rh core–shell cubes. The core–shell structures also showed a slightly lower IT than that of the traditionally used Pt catalyst. The difference in Ea can be attributed to the variation in surface electronic structure.54 These results demonstrate that the CO oxidation reaction was affected by the core–shell structure. In addition, it can be concluded that the changes caused by a core–shell configuration give more significant improvement towards CO oxidation relative to the effect of facets.
| Samples | IT (°C) | E a prior to IT (kcal mol−1) | E a after IT (kcal mol−1) |
|---|---|---|---|
| Octahedral Pd@Rh/SBA15 | 140 | 56.8 (±3.4) | 1.43 (±0.27) |
| Cubic Pd@Rh/SBA15 | 150 | 34.7 (±2.5) | 0.62 (±0.13) |
| Cubic Rh/SBA15 | 260 | 11.5 (±0.8) | 1.52 (±0.34) |
| Commercial Rh/C | 260 | 15.1 (±0.2) | 0.40 (±0.18) |
| Commercial Pt/Al2O3 | 160 | — | — |
We also examined the shape stability of the core–shell structures during the CO oxidation reaction. As shown by the TEM and high-resolution TEM images in Fig. S7 (ESI†), the octahedral and cubic morphologies of the core–shell nanocrystals in Pd@Rh/SBA15 were well-retained after going through the catalytic reaction held at 200 °C, together with 40 TorrCO, 100 TorrO2, and a balance of He. These results demonstrate good stability of the shape-controlled, core–shell nanocrystals towards an environment involving both thermal stress55 and CO oxidation. However, the surface of the Pd@Rh core–shell structure might evolve into an alloy in the presence of CO, promoting the CO oxidation reaction through a different mechanism.1 To this end, it is necessary to conduct a systematic, in situ TEM study in order to resolve this issue.
Conclusions
In summary, we have demonstrated the synthesis of Pd@Rh core–shell octahedra and cubes with well-defined Rh{111} and Rh{100} facets on the surface. The deposition of Rh could be switched from an island mode to a layer-by-layer mode by optimizing the experimental conditions, including reaction temperature, Rh precursor concentration, and the size of the Pd nanocrystal seeds, to achieve a smooth surface for the Rh shell. We then used CO oxidation as a model reaction to probe the structure-dependent surfaces of the catalytic particles. The CO to CO2 conversion indicated that the Pd@Rh octahedra exhibited the lowest IT of 140 °C, followed by the Pd@Rh cubes at 150 °C, demonstrating enhancement in reaction kinetics relative to the monometallic counterparts such as Rh cubes and commercial Rh/C catalysts. It is also worth pointing out that one can scale up the production of the Pd@Rh nanocrystals using recently reported methods such as continuous flow or droplet reactors.56–58Experimental
Chemicals and materials
Sodium tetrachloropalladate(II) (Na2PdCl4), L-ascorbic acid (AA), potassium bromide (KBr), potassium chloride (KCl), benzyl alcohol, PVP (MW ≈ 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), citric acid (CA), and TTEG (99.0%) were purchased from Sigma-Aldrich while EG (99.0%) was ordered from J. T. Baker. Rh(OAc)3 was obtained from BASF. All the chemicals were used as received. Deionized (DI) water with a resistivity of 18.2 MΩ cm was used for all experiments.
000), citric acid (CA), and TTEG (99.0%) were purchased from Sigma-Aldrich while EG (99.0%) was ordered from J. T. Baker. Rh(OAc)3 was obtained from BASF. All the chemicals were used as received. Deionized (DI) water with a resistivity of 18.2 MΩ cm was used for all experiments.
Synthesis of Pd nanocrystals
The synthesis and characterization of Pd nanocrystals can be found elsewhere.37 In a typical synthesis of Pd octahedra with an average edge length of 6.4 nm, Na2PdCl4 (57 mg), CA (180 mg), and PVP (105 mg) were dissolved in a mixture containing 3 mL of ethanol and 8 mL of water. The mixture was heated at 80 °C in air under magnetic stirring for 3 h and then cooled down to room temperature. The Pd octahedra were collected by centrifugation and dispersed in 11 mL of benzyl alcohol after washing with water twice. In a typical synthesis of Pd cubes with an average edge length of 7.2 nm, 11 mL of an aqueous solution containing Na2PdCl4 (57 mg), AA (60 mg), PVP (105 mg), KBr (5 mg), and KCl (185 mg) was placed in a 20 mL vial and then heated at 80 °C in air under magnetic stirring for 3 h. After cooling down to room temperature, the Pd cubes were collected by centrifugation and dispersed in 11 mL of EG after washing with water twice.Synthesis of Pd@Rh core–shell structures
In a typical synthesis of Pd@Rh octahedra with an average edge length of 7.4 nm, 1 mL of benzyl alcohol suspension of the Pd octahedra, PVP (15 mg), and 27 μL of Rh(OAc)3 solution (4.98 wt%Rh,) were dissolved in 5 mL of TTEG. The mixture was heated at 185 °C in air under magnetic stirring for 3 h and cooled down to room temperature. In a typical synthesis of Pd@Rh cubes with an average edge length of 8.5 nm, 1 mL of EG suspension of the Pd cubes was added into a mixture of PVP (25 mg), AA (25 mg), KBr (25 mg), and 1.5 mL of EG, and preheated at 160 °C for 10 min. The deposition of an Rh shell was conducted by titrating 1 mL of EG containing 38 μL of Rh(OAc)3 solution (4.98 wt%Rh) into the reaction solution at a relatively slow rate of 2.0 mL h−1. The mixture was heated at 160 °C in air under magnetic stirring for 3 h and cooled down to room temperature. The final solid samples were collected by centrifugation and washed with water three times.Preparation of Pd@Rh/SBA15 catalysts
The synthesis of mesoporous silica followed the previous reports.59,60 In a typical preparation, 4 g of 1,3,5-trimethylbenzene (TMB) was added into 75 mL of an aqueous solution that contained 4 g of triblock copolymer Pluronic P123 and 10 mL of concentrated HCl. The mixture was magnetically stirred at 40 °C for 2 h and 9.2 mL of tetraethoxysilane (TEOS) was then added. After stirring for 5 min, the mixture was aged at 40 °C for 20 h under a quiescent condition. Then, 46 mg of NH4F was added and the solution was transferred into an autoclave and aged at 100 °C for another 24 h. The solid products were filtered, washed with water and ethanol, and calcined in air at 600 °C for 6 h. The white powder was kept in a desiccator for further use. Colloidal suspension of the Pd@Rh nanocrystals was diluted to 0.1 mg mL−1. A certain amount of the suspension (0.25 wt%metal) was added into the suspension of mesoporous silica and stirred for 3 h at room temperature. The brown precipitates were separated by centrifugation (3000 rpm, 20 min), thoroughly washed with ethanol twice, and dried in an oven at 100 °C overnight.Morphological, structural, and elemental characterization
TEM images were taken using a HT-7700 microscope (Hitachi, Japan) operated at 120 kV by drop casting the nanoparticle dispersions on carbon-coated copper grids and drying under ambient conditions. High-resolution TEM, HAADF-STEM, and EDS mapping analyses were performed using a JEOL JEM 2200FS STEM/TEM microscope at 200 kV. Concentrations of the suspensions and elemental ratios of the nanoparticles were determined using inductively-coupled plasma mass spectrometry (ICP-MS, PerkinElmer, NexION 300Q).CO oxidation reaction
The catalytic rates under different conditions for CO oxidation were measured for the catalysts in a laboratory-scale flow reactor operated at atmospheric pressure. Gas flows were regulated using calibrated mass flow controllers. Temperature was controlled using a type-K thermocouple and a PID controller. Catalysts were diluted with low surface area quartz sand (acid washed, water rinsed, and calcined before use) and then loaded into quartz reactors. CO oxidation rates were acquired at 40 TorrCO and 100 TorrO2 with a balance of He. Activation energies and ignition temperatures were determined using the Arrhenius plots in the temperature range of 120 to 340 °C, with a step of 10 °C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by start-up funds from the Georgia Institute of Technology. S.-I. C. was partially supported by the National Research Foundation (NRF) of Korea under grant no. NRF2018R1C1B6004272.Notes and references
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932 CrossRef CAS PubMed.
- K. D. Gilroy, X. Yang, S. Xie, M. Zhao, D. Qin and Y. Xia, Adv. Mater., 2018, 30, 1706312 CrossRef PubMed.
- N. J. Divins, I. Angurell, C. Escudero, V. Pérez-Dieste and J. Llorca, Science, 2014, 346, 620 CrossRef CAS PubMed.
- M. Zhou, H. Wang, A. Elnabawy, Z. Hood, M. Chi, P. Xiao, Y. Zhang, M. Mavrikakis and Y. Xia, Chem. Mater., 2019, 31, 1370 CrossRef CAS.
- J. Park, M. K. Kabiraz, H. Kwon, S. Park, H. Baik, S.-I. Choi and K. Lee, ACS Nano, 2017, 11, 10844 CrossRef CAS PubMed.
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 2013, 46, 1816 CrossRef CAS PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410 CrossRef CAS PubMed.
- Y.-J. Deng, V. Tripkovic, J. Rossmeisl and M. Arenz, ACS Catal., 2016, 6, 671 CrossRef CAS.
- S. Xie, S.-I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570 CrossRef CAS PubMed.
- J. Park, L. Zhang, S.-I. Choi, L. T. Roling, N. Lu, J. A. Herron, S. Xie, J. Wang, M. J. Kim, M. Mavrikakis and Y. Xia, ACS Nano, 2015, 9, 2635 CrossRef CAS PubMed.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412 CrossRef CAS PubMed.
- S.-I. Choi, M. Shao, N. Lu, A. Ruditskiy, H.-C. Peng, J. Park, S. Guerrero, J. Wang, M. J. Kim and Y. Xia, ACS Nano, 2014, 8, 10363 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS PubMed.
- N. Zettsu, J. M. McLellan, B. Wiley, Y. Yin, Z.-Y. Li and Y. Xia, Angew. Chem., Int. Ed., 2006, 45, 1288 CrossRef CAS PubMed.
- S. M. Humphrey, M. E. Grass, S. E. Habas, K. Niesz, G. A. Somorjai and T. D. Tilley, Nano Lett., 2007, 7, 785 CrossRef CAS PubMed.
- A. M. Watson, X. Zhang, R. A. de la Osa, J. M. Sanz, F. González, F. Moreno, G. Finkelstein, J. Liu and H. O. Everitt, Nano Lett., 2015, 15, 1095 CrossRef CAS PubMed.
- F. Bonet, V. Delmas, S. Grugeon, R. H. Urbina, P.-Y. Silvert and K. Tekaia-Elhsissen, Nanostruct. Mater., 1999, 11, 1277 CrossRef CAS.
- C. A. Stowell and B. A. Korgel, Nano Lett., 2005, 5, 1203 CrossRef CAS PubMed.
- X. Xia, L. Figueroa-Cosme, J. Tao, H.-C. Peng, G. Niu, Y. Zhu and Y. Xia, J. Am. Chem. Soc., 2014, 136, 10878 CrossRef CAS PubMed.
- Y. Zhang, M. E. Grass, W. Huang and G. A. Somorjai, Langmuir, 2010, 26, 16463 CrossRef CAS PubMed.
- R. Wang, H. He, J. N. Wang, L. C. Liu and H. X. Dai, Catal. Today, 2013, 201, 68 CrossRef CAS.
- J. R. Renzas and Y. W. Zhang, Catal. Lett., 2009, 132, 317 CrossRef CAS.
- Y. Yuan, N. Yan and P. J. Dyson, ACS Catal., 2012, 2, 1057 CrossRef CAS.
- J. H. Holles, M. A. Switzer and R. J. Davis, J. Catal., 2000, 190, 247 CrossRef CAS.
- W. Wang, Z. Cao, K. Liu, J. Chen, Y. Wang and S. Xie, Nano Lett., 2017, 17, 7613 CrossRef CAS PubMed.
- U. Lassi, R. Polvinen, S. Suhonen, K. Kallinen, A. Savimäki, M. Härkönen, M. Valden and R. L. Keiski, Appl. Catal., A, 2004, 263, 241 CrossRef CAS.
- B. T. Sneed, C. H. Kuo, C. N. Brodsky and C.-K. Tsung, J. Am. Chem. Soc., 2012, 134, 18417 CrossRef CAS PubMed.
- S. Xie, N. Lu, Z. Xie, J. Wang, M. J. Kim and Y. Xia, Angew. Chem., Int. Ed., 2012, 51, 10266 CrossRef CAS PubMed.
- S. Xie, H.-C. Peng, N. Lu, J. Wang, M. J. Kim, Z. Xie and Y. Xia, J. Am. Chem. Soc., 2013, 135, 16658 CrossRef CAS PubMed.
- Y. Zhang, M. E. Grass, J. N. Kuhn, F. Tao, S. E. Habas, W. Y. Huang, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 5868 CrossRef CAS PubMed.
- S. Yao, Y. Yuan, C. Xiao, W. Li, Y. Kou, P. J. Dyson, N. Yan, H. Asakura, K. Teramura and T. Tanaka, J. Phys. Chem. C, 2012, 116, 15076 CrossRef CAS.
- K. Jang, H. J. Kim and S. U. Son, Chem. Mater., 2010, 22, 1273 CrossRef CAS.
- H. Duan, N. Yan, R. Yu, C.-R. Chang, G. Zhou, H.-S. Hu, H. Rong, Z. Niu, J. Mao, H. Asakura, T. Tanaka, P. J. Dyson, J. Li and Y. Li, Nat. Commun., 2014, 5, 3093 CrossRef PubMed.
- S. Xie, H. Zhang, N. Lu, M. Jin, J. Wang, M. J. Kim, Z. Xie and Y. Xia, Nano Lett., 2013, 13, 6262 CrossRef CAS PubMed.
- S.-I. Choi, S. R. Lee, C. Ma, B. Oliy, M. Luo, M. Chi and Y. Xia, ChemNanoMat, 2016, 2, 61 CrossRef CAS.
- M. Shao, T. Yu, J. H. Odell, M. Jin and Y. Xia, Chem. Commun., 2011, 47, 6566 RSC.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302 CrossRef CAS PubMed.
- Y. Lu, J. Wang, L. Yu, L. Kovarik, X. Zhang, A. S. Hoffman, A. Gallo, S. R. Bare, D. Sokaras, T. Kroll, V. Dagle, H. Xin and A. M. Karim, Nat. Catal., 2018, 2, 149 CrossRef.
- D. Widmann, A. Krautsieder, P. Walter, A. Brückner and R. J. Behm, ACS Catal., 2016, 6, 5005 CrossRef CAS.
- J. Gustafson, O. Balmes, C. Zhang, M. Shipilin, A. Schaefer, B. Hagman, L. R. Merte, N. M. Martin, P.-A. Carlsson, M. Jankowski, E. J. Crumlin and E. Lundgren, ACS Catal., 2018, 8, 4438 CrossRef CAS.
- A. Alavi, P. Hu, T. Deutsch, P. L. Silvestrelli and J. Hutter, Phys. Rev. Lett., 1998, 80, 3650 CrossRef CAS.
- N. Lopez and J. K. Nørskov, J. Am. Chem. Soc., 2002, 124, 11262 CrossRef CAS PubMed.
- M. M. Kappes and R. H. Staley, J. Phys. Chem., 1981, 85, 942 CrossRef CAS.
- L. C. Grabow, B. Hvolbæk and J. K. Nørskov, Top. Catal., 2010, 53, 298 CrossRef CAS.
- E. Eichler, Surf. Sci., 2002, 498, 314 CrossRef.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113 CrossRef CAS.
- G. R. Bamwenda, S. Tsubota, T. Nakamura and M. Haruta, Catal. Lett., 1997, 44, 83 CrossRef CAS.
- N. Lopez, T. V. W. Janssens, B. S. Clausen, Y. Xu, M. Mavrikakis, T. Bligaard and J. K. Nørskov, J. Catal., 2004, 223, 232 CrossRef CAS.
- D. W. Goodman and C. H. F. Peden, J. Phys. Chem., 1986, 90, 4839 CrossRef CAS.
- M. J. P. Hopstaken and J. W. Niemantsverdriet, J. Chem. Phys., 2000, 113, 5457 CrossRef CAS.
- M. E. Grass, Y. Zhang, D. R. Butcher, J. Y. Park, Y. Li, H. Bluhm, K. M. Bratlie, T. Zhang and G. A. Somorjai, Angew. Chem., Int. Ed., 2008, 47, 8893 CrossRef CAS PubMed.
- J. R. Renzas, W. Huang, Y. Zhang, M. E. Grass, D. T. Hoang, S. Alayoglu, D. R. Butcher, F. Tao, Z. Liu and G. A. Somorjai, Phys. Chem. Chem. Phys., 2011, 13, 2556 RSC.
- M. Valden, S. Pak, X. Lai and D. W. Goodman, Catal. Lett., 1998, 56, 7 CrossRef CAS.
- N. Lu, J. Wang, S. Xie, Y. Xia and M. J. Kim, Chem. Commun., 2013, 49, 11806 RSC.
- G. Niu, M. Zhou, X. Yang, J. Park, N. Lu, J. Wang, M. J. Kim, L. Wang and Y. Xia, Nano Lett., 2016, 16, 3850 CrossRef CAS PubMed.
- L. Zhang, Y. Wang, L. Tong and Y. Xia, Nano Lett., 2014, 14, 4189 CrossRef CAS PubMed.
- G. Niu, L. Zhang, A. Ruditskiy, L. Wang and Y. Xia, Nano Lett., 2018, 18, 3879 CrossRef CAS PubMed.
- D. Zhao, J. Sun, Q. Li and G. D. Stucky, Chem. Mater., 2000, 12, 275 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00360f |
| This journal is © The Royal Society of Chemistry 2019 |