DOI:
10.1039/C8NH00415C
(Communication)
Nanoscale Horiz., 2019,
4, 658-666
A novel strategy based on a ligand-switchable nanoparticle delivery system for deep tumor penetration†
Received
13th November 2018
, Accepted 20th December 2018
First published on 20th December 2018
Abstract
The poor tumor penetration of many nanomedicines severely limits their therapeutic efficiency. Although the combination of cell-penetrating peptides (CPPs) has been reported as a promising strategy to improve the penetration capability of nanoparticles, the nonspecific interactions of CPPs may reduce nanoparticles’ circulation time and increase their side effects, making them inapplicable in many in vivo settings. Herein, we report a ligand-switchable strategy to inhibit CPPs’ nonspecific interactions in the bloodstream and quickly reactivate their functions when entering into tumor sites. Due to the rapid phase transition of PAE, the CPPs can be completely hidden under the PEG corona in plasma, avoiding direct interaction with the immune cells and normal tissues. Once extravasation from blood vessels occurs, the acidic tumor microenvironment would trigger the exposure of the CPPs, promoting the diffusion of antitumor agents deep into the tumor sites. With this ligand-switchable property, nanocarriers are capable of achieving long durations of circulation, preferential accumulation and efficient tumor penetration simultaneously, giving rise to high antitumor activity and low adverse reactions, and also providing a universal approach to enable in vivo applications of CPPs as well as other nonspecific ligands.
Conceptual insights
Developing antitumor agents with excellent tumor penetration remains one of the formidable challenges for medical researchers. Although CPPs are promising candidates for improving their penetrative capabilities, the rapid immune clearance due to the nonspecific interactions still severely represses the drug delivery efficiency of many CPP-based nanomedicines in cancer therapy. To address this dilemma, we demonstrate a ligand-switchable strategy to develop smart CPP-based nanoparticles that can hide CPPs in the bloodstream and quickly expose them on the surface of nanoparticles when entering tumor sites. These smart nanoparticles not only prolong the circulation time in blood but also promote tissue penetration in tumors, giving rise to high antitumor activity. More importantly, this strategy is achieved by the specially designed shell structure of the micelle and is compatible with a large variety of functional ligands, providing a general approach to enable in vivo applications of those nonspecific ligands in the treatment of cancer as well as other diseases.
|
Introduction
Nanomedicine has emerged as a promising strategy to improve the efficacy of anticancer therapeutics and reduce their adverse effects.1–6 However, the use of many clinically approved nanoparticles has not always translated into improved clinical outcomes, in great part due to their poor tumor penetration.7 After extravasation from tumor vessels, nanocarriers are mainly limited to the adjacent regions because of the high interstitial fluid pressure (IFP), dense interstitial matrix, and abnormal vasculature of tumors, leading to large tumor areas remaining untouched by the carried therapeutic agents and thus greatly limiting their therapeutic effect.8–10 Recently, evidence has suggested that there are many types of cells that play a critical role in the initiation and progression of tumors that are fed by defective blood vessels in the solid tumor.11–14 Targeting these tumor-associated cells has been considered a more effective strategy to alleviate tumor growth and metastases.15,16 Nevertheless, despite decades of intensive research, most of these strategies have still focused on the intractable obstacle that traditional nanocarriers are incapable of reaching targeted cells, especially those distant from blood vessels, significantly impairing the drug delivery efficacy and overall performance of cancer treatments. To exacerbate matters further, the residual cells may uncontrollably proliferate as soon as the treatment is withdrawn, leading to severe relapse. Thus, development of novel nanocarriers with more preferential penetration activity and the ability to delivery intratumorally to achieve more comprehensive treatment is still an urgent problem for nanotherapeutics.
To improve the penetration properties, a variety of different strategies have been proposed, including decreasing particle size,17–20 increasing positive charge,21,22 and combining the carriers with penetrating ligands.23–27 In particular, the conjugation of cell-penetrating peptides (CPPs), which approach and attach to the membrane of cancer cells because of their unique cationic structures, has been reported as a potential strategy to achieve deep tumor penetration and has shown significantly improved delivery efficiency of nanoparticles.28–32 Unfortunately, the full clinical potential of CPPs nanoparticles (CPP-NPs) has not yet been realized due to nonspecific tissue interaction of CPPs, as well as toxicity issues associated with their inherent cationic nature, which limits the dose of drug that can be safely administered to cancer patients.7 An additional undesired feature of conventional CPP-NPs is that they will be rapidly cleared from circulation because of immune system recognition or that they may readily penetrate into most organs, thus dramatically increasing the adverse effects and off-setting the therapeutic efficacy.
Recently, many studies have suggested that there are many conflicting requirements for nanomedicines in different stages of the cancer drug delivery process. For example, the ideal anticancer agent is expected to show inferior penetration into the bloodstream to avoid extensive diffusion, and demonstrate targeted penetration of the tumor. With this understanding, the design of effective CPP-NPs must take the immune clearance aspects into consideration. In fact, many strategies have been attempted to inhibit the nonspecific interactions of CPPs to overcome the intrinsically conflicting attributes between extended periods of circulation and deep penetration. Several typical representatives include shielding the cationic charges of CPPs with polyanions or modifying them with responsive molecules. With respect to charge shielding, the CPPs were fused with an anionic peptide by intramolecular electrostatic interactions. Once in the targeted tissues such as a tumor, proteolysis of the linker facilitated the disassociation of the two domains, activating the CPP's functions.33 However, the main concern with this method is the in vivo stability of the complex because of the weak electrostatic interactions and complexity of blood.26 For surface modifications, the conversion of protecting groups can inactivate the CPPs in the bloodstream and reactivate them in the tumor by microenvironmental response. For example, Shen's group used TAT to demonstrate an acid-sensitive molecular modification approach for effectively inhibiting nonspecific interactions of CPPs in the bloodstream while reactivating their functions in the targeted tissues or cells.26 Unfortunately, this method may be inapplicable for arginine-rich CPPs such as D-octaarginine (R8), which lack suitable protecting ligands but show higher activity in cellular uptake and tumor penetration.34 Moreover, these processes are usually irreversible, resulting in the exposure of ligands to the immune system again once the nanoparticles flow back into the bloodstream.35
Herein, we report the development of ligand-switchable nanocarriers (LSNs) to overcome the trade-off between long circulation and deep penetration efficiency to improve the overall delivery effect. These micelles have a core–shell structure with a hydrophobic core of poly(ε-caprolactone) (PCL) and a pH-responsive shell composed of poly(β-amino ester) (PAE) and poly(ethylene glycol) (PEG).36–38 By conjugating to the terminal position of the PAE chains, CPPs can be completely hidden under the PEG corona in the neutral environment (e.g., the bloodstream), while quickly exposing the surface in tumor sites as a result of the PAE's phase transition upon exposure to the tumor's acidic microenvironment. Due to the shielding effect of PEG, the CPPs cannot directly interact with the immune cells and normal tissues, thus inhibiting their nonspecific interactions, leading to the enhancement in circulation stability of the CPP-conjugated micelles in the bloodstream (pH 7.4). In this case, a higher portion of the micelles are able to accumulate into tumor sites via the EPR effect. Once extravasation from blood vessels occurs, the acidic tumor microenvironment (pH 6.5) triggers the exposure of the CPPs on the micelles’ surface by the protonation of PAE, reactivating their penetrative capability to promote deep diffusion into the tumor, achieving efficient drug delivery and enhancing the overall anticancer effect (Scheme 1). With the help of such smart nanocarriers, the drug delivery efficiency was improved significantly. In this study, doxorubicin (DOX) and Pexidartinib (PLX-3397), which inhibit the intratumoral accumulation of immunosuppressive macrophages by blockading the CSF-1 receptor (CSF-1R),39–42 were employed to respectively target cancer cells as well as M2-Tumor Associated Macrophages (M2-TAMs) for improving antitumor performance and preliminarily investigating the feasibility of immunochemotherapy.
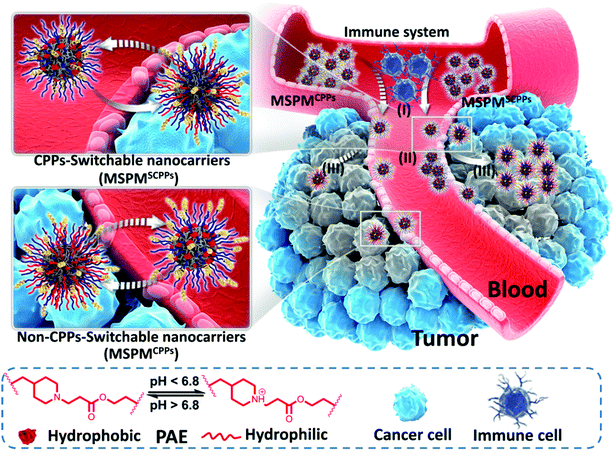 |
| | Scheme 1 Illustration of the ligand-switchable capacity of LSNs for overcoming the trade-off between circulation and penetration. (I) Evasion from the immune clearance in blood circulation system; (II) Tumor accumulation; (III) Tumor penetration. LSNs (CPPs-switchable mixed-shell polymeric micelles, denoted as MSPMSCPPs) hide CPPs under the PEG corona in the bloodstream for reducing immune recognition and clearance, achieving preferential accumulation via the EPR effect and deep penetration at tumor sites due to the exposed CPPs. Non-ligand-switchable nanocarriers (non-switchable mixed-shell polymeric micelles, denoted as MSPMCPPs) exposed CPPs on the surface, resulting in quick immune clearance, insufficient accumulation, and inappreciable penetration. | |
Results and discussions
Fabrication and characterization of CPP-micelles
To prepare CPP-switchable micelles, all block copolymers, including PCL-b-PEG, PCL-b-PAE, PCL-b-PEG-TAT, PCL-b-PAE-TAT, PCL-b-PEG-R8, and PCL-b-PAE-R8, were synthesized.42 Importantly, the CPPs (TAT and R8) were conjugated to the end of PEG and PAE accordingly via the click reaction of maleimide and sulfhydryl. As shown in the 1H NMR spectra, the disappearance of the peak at 6.7 ppm, which is attributed to the double bond of maleimide, indicated the successful conjugation of CPPs (TAT and R8) to block copolymers (Fig. 1B and Fig. S4B, ESI†). With a specific feeding ratio (Table S1, ESI†), five types of micelles, including mixed-shell-polymeric-micelles (MSPM), CPPs-non-switchable mixed-shell-polymeric-micelles with TAT or R8 at the terminal end of PEG (MSPMTAT or R8), and CPPs-switchable mixed-shell-polymeric-micelles with TAT or R8 at the terminal end of PAE (MSPMSTAT or SR8), were constructed through self-assembly (Fig. 1A). Dynamic light scattering (DLS) indicated that the diameters of these micelles were around 72 ± 10 nm (Fig. 1C), which was consistent with the transmission electron microscopy (TEM) observations (Fig. 1D and Fig. S6, ESI†). Zeta potential analyses were performed using a Brookheaven ZetaPALS. As shown in Fig. 1E, all micelles were negatively charged (∼−8 mV) at pH 7.4 due to the presence of PEG, and quickly converted to a positive charge (∼+5 mV) at pH 6.5 because of PAE's protonation, implying a robust phase transformation at different pH conditions. When the pH was adjusted from 7.4 to 6.5, the PAE quickly became hydrophilic and stretched to the surface from the PEG corona. By conjugating to the terminal end of PAE, CPPs were shielded by the PEG in the bloodstream since PAE is hydrophobic and compacted, while quickly stretching out when exposed to the acidic tumor microenvironment.
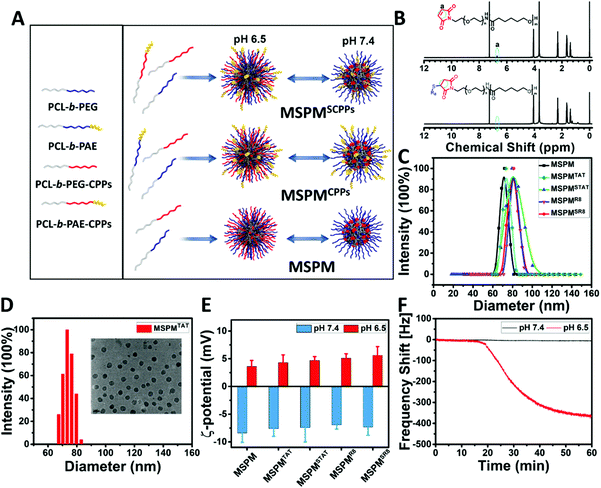 |
| | Fig. 1 Fabrication and characterization of LSNs. (A) Schematic of the self-assembly procedure of LSNs. (B) 1H NMR spectra of PCL-b-PEG-MAL and PCL-b-PEG-R8 in CDCl3. (C) The hydrodynamic diameters of five types of micelles (MSPM, MSPMTAT, MSPMSTAT, MSPMR8 and MSPMSR8) at pH 7.4. (D) The hydrodynamic diameter and TEM image of MSPMTAT at pH 7.4. (E) Zeta potentials of five types of micelles under different pH conditions (pH 7.4 and 6.5). Error bars indicate s.d. (n = 8). (F) Quartz crystal microbalance (QCM) analysis of sulfhydryl-switchable micelles (MSPMSSH) at pH 7.4 and pH 6.5. | |
Characterization of in vitro ligand-switchable capacity of MSPMSCPPs
For MSPMSCPPs, the phase transition is capable of hiding/exposing the CPPs on the surface when the pH is higher than 7.4 or lower than 6.5, respectively, because the CPPs were conjugated covalently on the terminal end of PAE. To prove this hypothesis, we demonstrated the reversible phase transition and charge conversion of PAE by zeta potential analyses and 1H NMR spectroscopy in a previous study.42 Here, QCM analyses was employed to further confirm the ligand-switchable process. By conjugating with sulfhydryl, the LSNs can function in the gold chips of QCM. In this work, the sulfhydryl was conjugated to PAE's terminal end to replace CPPs for investigating the ligand-switchable capacity of LSNs. Since the sulfhydryl was hidden within the PEG micelles at pH 7.4 due to the hydrophobic effect of PAE, the micelles failed to interact with gold chips, resulting in a negligible reduction of the frequency shift. In contrast, the frequency shift declined rapidly as the pH decreased to 6.5, implying a strong interaction because the sulfhydryl returned to the micelles’ surface under acidic conditions (Fig. 1F). In the following cytotoxicity assessment, CLSM and FCM were employed to investigate the ligand-switchable feature of LSNs at different pH conditions (pH 7.4 and 6.5). As depicted in Fig. 2A, due to the low ratios of TAT or R8, all micelles showed negligible cytotoxicity for HepG2 cells. In addition, similar drug release kinetics were observed from all micelles, with less than 15% of DOX released over 48 h incubation at pH 7.4, and more than 40% of DOX released at pH 5.0, indicating that the location of CPPs had an inapparent effect on drug release. Whereafter, the DOX-loaded micelles (MSPMCPPs/DOX, and MSPMSCPPs/DOX) were incubated with HepG2 cells. Compared with MSPMCPPs/DOX, MSPMSCPPs/DOX showed lower cell apoptosis at pH 7.4 and significantly enhanced cytotoxicity at pH 6.5, implying the CPPs had returned to the surface of the micelles (Fig. 2C and Fig. S7, ESI†). This result was likely caused by the different cellular internalization efficiency due to the different interactions between CPP with cells. The LSNs were capable of effectively inactivating/reactivating the CPPs at different pH (pH 7.4/6.5) for reducing or enhancing the internalization. This result was then confirmed by CLSM observations (Fig. 2D), indicating that the mean fluorescence of cells treated with MSPMSCPPs/DOX was similar to that of the MSPM-treated cells but significantly lower than the MSPMCPPs group. With the decrease of pH, the internalizations of DOX in all groups were enhanced because of the charge conversion of PAE. However, the CPP-conjugated micelles including MSPMTAT or R8 as well as MSPMSTAT or SR8 showed higher cellular internalization than MSPM under normal culture conditions, owing to the strong interaction between the cell and CPPs. Further quantification of the cell uptake was confirmed using flow cytometry analysis (Fig. 2E and F), demonstrating the effectiveness of the switchable structure of MSPMSCPPs in hiding and exposing surface ligands in response to the different microenvironments. With the ligand-switchable features, LSNs showed the potential to reduce interaction with normal tissues as well as the immune system by concealing CPP in the PEG corona under physiological conditions and then revealing the CPPs on the surface to selectively facilitate cellular internalization and apoptosis when reaching the targeted tissues such as a tumor.
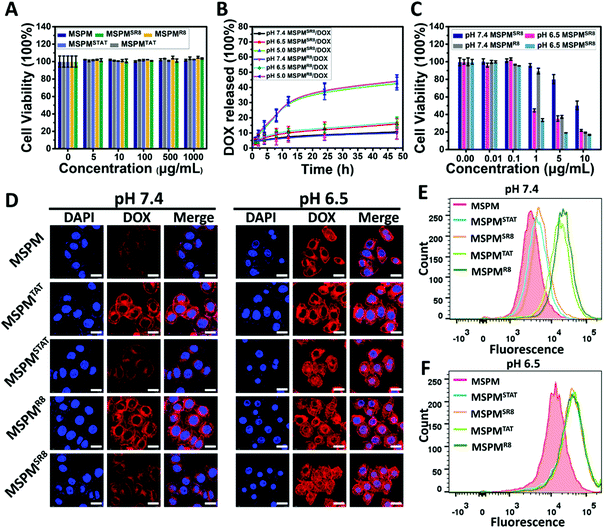 |
| | Fig. 2 Characterization of ligand-switchable capacity in HepG2 cells. (A) Cytotoxicity of five types of micelles (MSPM, MSPMTAT, MSPMSTAT, MSPMR8 and MSPMSR8) at pH 7.4. (B) In vitro DOX release from MSPMR8 and MSPMSR8 under different conditions (pH 7.4, 6.5, and 5.0). Error bars indicate s.d. (n = 3). Micelles at 1 mg mL−1 were shaken at 37 °C in dialysis against PBS, and the released DOX was measured via its fluorescence intensity. (C) Cytotoxicity of DOX-loaded micelles (MSPMR8/DOX and MSPMSR8/DOX) under different conditions. (D) Confocal microscopy images of HepG2 cells treated with DOX-loaded micelles (10 μg mL−1) under different conditions (scale bar = 20 μm). (E and F) Flow cytometry of HepG2 cells cultured with DOX-loaded micelles for 2 hours at pH 7.4 and pH 6.5, respectively. | |
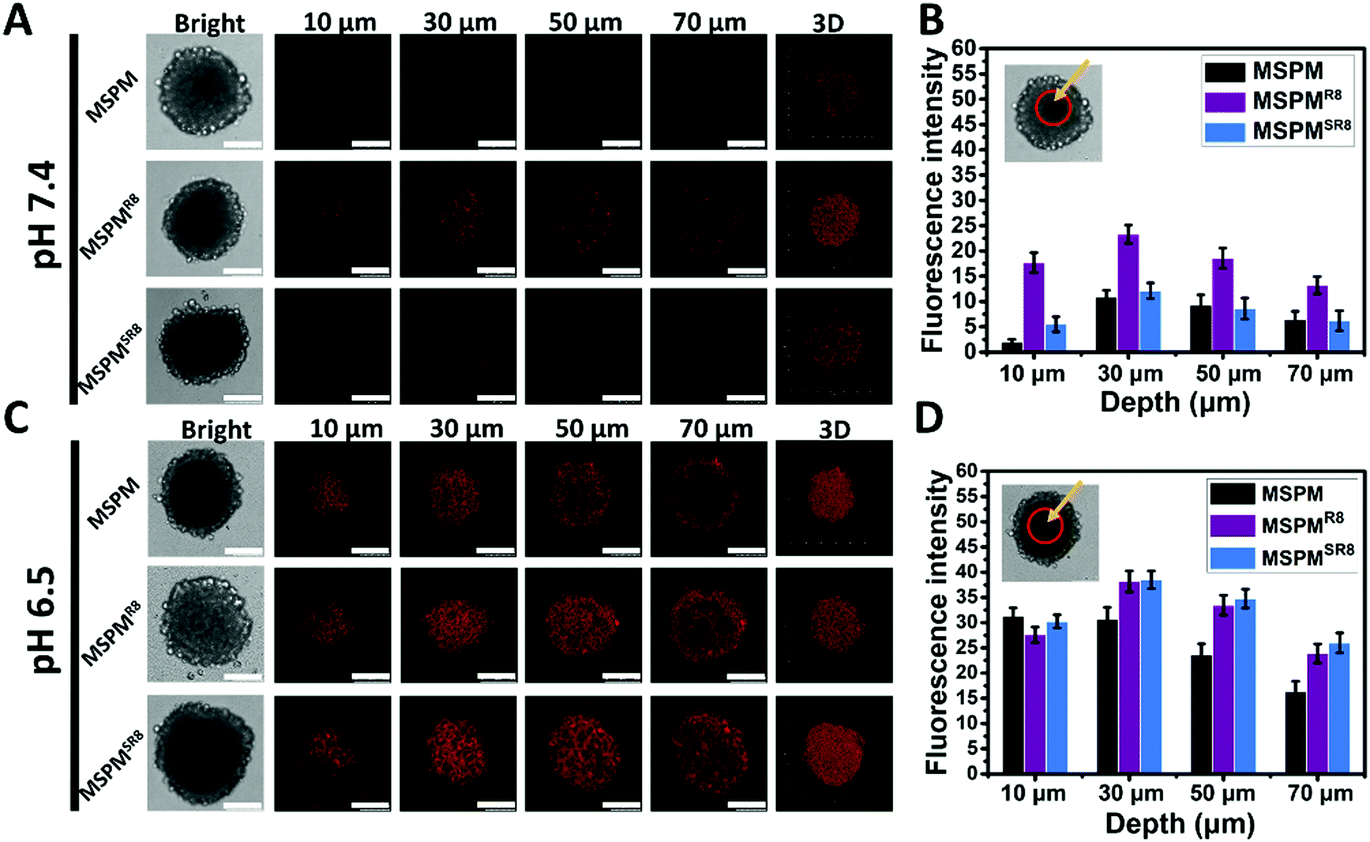
Acidic-responsive penetration of MSPMSR8 in HepG2 multicellular tumor spheroids
To further investigate its ligand-switchable ability, R8-conjugated micelles were employed to assess their penetration capacity in HepG2 multicellular spheroids (MCSs). After incubating with three types of micelles (MSPM/DOX, MSPMR8/DOX and MSPMSR8/DOX), the penetration behaviors of all micelles were assessed using CLSM with Z-stacking scanning. The surface of the MCSs was defined as 0 μm (bottom). As shown in Fig. 3A and C, the fluorescence intensity from the MSPMSR8/DOX group was very similar to that of the control group (MSPM), and much weaker than the MSPMR8/DOX group at pH 7.4, implying that the MSPMSR8 could not penetrate deep into the tumor spheroids because of the inactivation of R8. In contrast, the diffusion region of MSPMSR8 was significantly enlarged at pH 6.5. Compared with MSPM/DOX, more strong signals were observed in the tumor spheroids treated with both MSPMSR8/DOX and MSPMR8/DOX, indicating that the function of R8 was only activated in acidic conditions, which could potentially enhance the efficiency of tumor-targeting and reduce harmful side effects. Quantitative analysis of the CLSM images confirmed these results, indicating a more efficient penetration of MSPMSR8 under acidic conditions. These in vitro results suggested that the CPP-switchable micelles can selectively expose the CPPs to facilitate penetration under acidic conditions. Thus, such micelles were expected to achieve penetration only in tumors when circulating within the body, providing a prospect to improve the drug delivery efficiency.
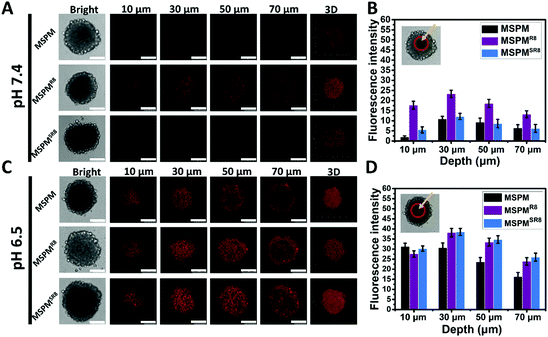 |
| | Fig. 3 Penetration in HepG2 tumor spheroids. (A and C) Confocal microscopy images of tumor spheroids of MSPM/DOX, MSPMR8/DOX, and MSPMSR8/DOX at pH 7.4 and pH 6.5, respectively (scale bar = 100 μm). (B and D) Quantification of fluorescence intensity in the center of tumor spheroids at different depths. The fluorescence signals were analyzed by image J. Error bars indicate s.d. (n = 3). | |
Pharmacokinetics, tumor accumulation efficacy, biodistribution, and penetration of MSPMSR8in vivo
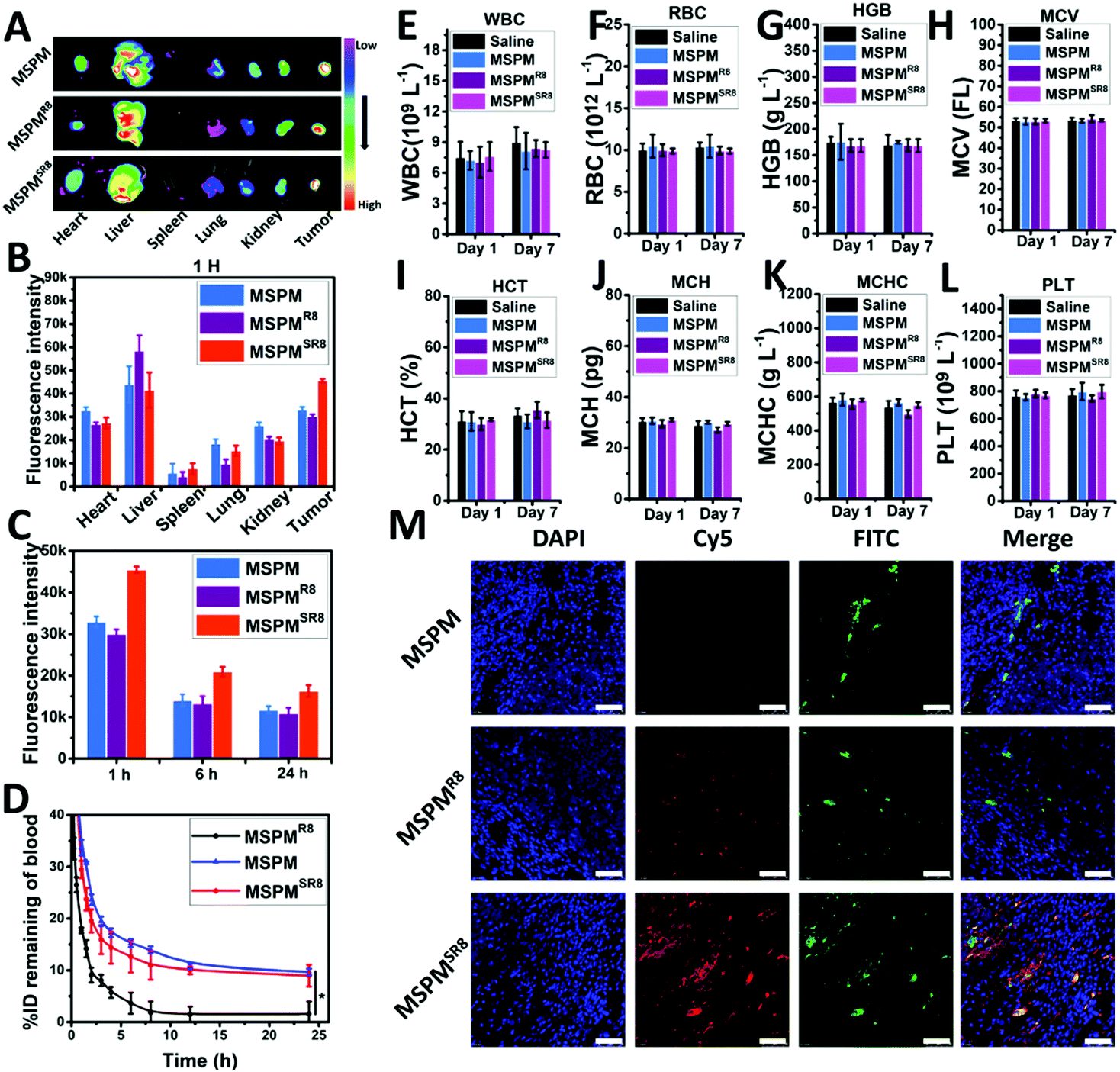
The ligand-switchable capacity of MSPMSR8 in response to tumor extracellular pH would selectively expose R8 in the tumor site, motivating us to investigate its in vivo potential for drug delivery. In this work, the pharmacokinetics of MSPM, MSPMR8, and MSPMSR8 were evaluated in tumor-free rats. According to the results, Cy5-MPSMSR8 exhibited prolonged circulation time (T1/2 = 8.1 ± 1.2 h, AUC = 305.6% h) in the bloodstream, which was similar to that of Cy5-MSPM (T1/2 = 9.7 ± 1.5 h, AUC = 349.3% h). In contrast, Cy5-MPSMR8 was eliminated rapidly (T1/2 = 2.7 ± 0.3 h, AUC = 90.6% h), implying that concealing CPPs when circulating in the bloodstream was an effective strategy for the construction of CPP-based drug delivery systems without compromising the circulation time (Fig. 4D). With sufficient circulation time, MSPMSR8 was anticipated to accumulate in tumors via the EPR effect and penetrate into deep tumor tissue, attributed to the function of R8. To verify this hypothesis, we studied the tumor accumulation and penetration of Cy5-MSPM, Cy5-MSPMR8 and Cy5-MSPMSR8 in HepG2 tumor-bearing nude mice. After the injection of the formulations, the mice were sacrificed at each time point of 1 h, 6 h and 24 h to harvest the tumor, as well as major organs (heart, liver, spleen, lung, and kidney), followed by observation with a near-infrared optical imager. As shown in Fig. 4A, it was clearly seen the Cy5-MSPMSR8 group showed the highest accumulation efficiency in tumors compared to those of both Cy5-MSPM and Cy5-MSPMR8 groups, again indicating that the switchable structure of MSPMSR8 in hiding and exposing CPPs is capable of inhibiting the immune clearance in the bloodstream and exposing them to enhance the deposition of MSPMSR8 upon contact with tumor tissues. In addition, quantification of the biodistributions also revealed that the accumulation of MSPMSR8 in tumors was higher than those of the other two groups at all the time points (Fig. 4C and Fig. S8, ESI†). Moreover, the fluorescence intensity of Cy5 in liver treated with Cy5-MSPMSR8 was very similar to that of Cy5-MSPM, and significantly lower than that of Cy5-MSPMR8, implying a lower retention in the reticuloendothelial system (RES), and further verifying the long circulation property of MSPMSR8 (Fig. 4A and B). Furthermore, considering the effective penetration of Cy5-MSPMSR8, a CLSM was employed to observe the penetration behavior of three types of micelles (Cy5-MSPM, Cy5-MSPMR8 and Cy5-MSPMSR8) in tumor sections after staining with DAPI and anti-CD31-FITC. At 1 h after administration with Cy5-MSPMSR8, the Cy5 signal was scattered in the tumor slice, whereas minor Cy5 fluorescence was detected in both Cy5-MSPM and Cy5-MSPMR8 groups at the same depth. More importantly, Cy5-MSPMSR8 was distributed further away from the tumor vessels than other groups (Fig. 4M). Combined with the results of the 3D tumor spheroid findings and pharmacokinetic study, we found that the penetration ability of the micelles appeared to be greatly enhanced in the tumors by conjugating with R8. However, owing to rapid immune clearance in vivo, only a limited amount of MSPMR8 can reach the tumor, resulting in an inapparent accumulation and ineffective penetration in tumor tissues. In contrast, MSPMSR8 was not limited by this issue because of its ligand-switchable capacity. The function of R8 would be inhibited in blood circulation and reactivated in the tumor, overall enhancing the efficiency of drug delivery. Finally, considering the in vivo safety of the material, the standard hematology markers were also analyzed after treating the mice with 20 mg kg−1 per dose of three type of micelles. According to the results (Fig. 4E–L), all of the hematology markers show no significant differences as compared to those of the mice with PBS treatment on Day 1 and Day 7, implying a good blood compatibility and again demonstrating the negligible toxicity of LSNs in vivo. Altogether, these in vivo results indicated that the ligand-switchable property of LSNs could improve the tumor penetration without reducing the circulation time, providing an alternative and safe strategy to overcome the trade-off between circulation and accumulation as well as penetration for achieving high-efficiency cancer therapy.
 |
| | Fig. 4 Blood clearance, tumor accumulation and tumor penetration of the micelles in vivo. (A) Ex vivo fluorescence imaging of the tumor and organs (heart, liver, spleen, lung, kidney) harvested from the HepG2 tumor-bearing nude mice at 1 h post-injection. (B) Quantitative analysis of fluorescent signals from the tissues and the tumor from the ex vivo fluorescence images. Error bar indicates s.d. (n = 3). (C) Quantitative analysis of fluorescent signals from ex vivo fluorescence imaging of the tumor treated with different micelles (Cy5-MSPM, Cy5-MSPMR8, Cy5-MSPMSR8) at 1 h, 6 h and 24 h. Error bar indicates s.d. (n = 3). (D) Blood clearance profiles of Cy5-MSPM, Cy5-MSPMR8 and Cy5-MSPMSR8. Error bars indicate s.d. (n = 3) t-test (P* < 0.05, P** < 0.01). (E–L) Hematology results of the mice injected with MSPM, MSPMR8, and Cy5-MSPMSR8. Error bar indicated s.d. (n = 4). (E) White blood cells (WBC), (F) red blood cells (RBC), (G) hemoglobin (HGB), (H) mean corpuscular volume (MCV), (I) hematocrit (HCT), (J) mean corpuscular hemoglobin (MCH), (K) mean corpuscular hemoglobin concentration (MCHC) and (L) platelet (PLT). (M) Confocal microscopy images (40×) of Cy5-MSPM, Cy5-MSPMR8 and Cy5-MSPMSR8 after penetrating into the tumor tissue for 1 h (blue: nucleus stained by DAPI; green: anti-CD31 FITC; red: Cy5.), (scale bar = 50 μm). | |
In vivo antitumor growth effect
The prolonged circulation time, enhanced tumor accumulation, and improved intratumoral penetration provided us with a strong rationale to evaluate the antitumor activity of MSPMSR8in vivo. In this study, DOX and PLX-3397 were employed to achieve a better treatment effect and validate the feasibility of their immunochemotherapy. The antitumor activity was evaluated on HepG2-bearing BALB/c nude mice. As depicted in Fig. 5A and B, the tumor volume increased rapidly for the free drugs (DOX, PLX-3397) groups, with only approximately a 30.5% and 8.4% inhibition rate versus PBS treatment. By contrast, the tumor growth of mice administrated with drug-loaded micelles (MSPMSR8/DOX and MSPMSR8/PLX-3397) (denoted as M/D and M/P, respectively) was much slower, and this difference became more significant after day 21. Particularly, the most effective inhibition of the tumor growth was observed from the mice treated with M/D + M/P, indicating that combining DOX with PLX-3397 could achieve a better therapeutic effect. At the end of the treatment, we measured the weight of each tumor mass. Delivering via MSPMSR8 increased the inhibition rates of DOX and PLX-3397 from 30.5% (free DOX) and 8.4% (free PLX-3397) to 60.1% and 39.5%, respectively. In addition, the mice treated with M/D had no body weight loss and negligible differences in physical activity levels compared to PBS-treated tumor-bearing mice, while the treatment of free DOX resulted in obvious body weight loss. These results indicate better biocompatibility and much lower adverse reactions of M/D in comparison with free DOX. Finally, the antitumor mechanisms of DOX and PLX-3397-loaded MSPMSR8 were further analyzed by H&E and TUNEL staining. Analysis of the H&E tumor sections suggested considerably enhanced necrosis in the M/D + M/P, M/D, and M/P groups (∼80%, ∼60%, and ∼50% necrotic area, respectively) compared with the control groups (saline: ∼20%, DOX: ∼40%, PLX-3397: ∼30%, and DOX + PLX-3397: 45%). Histological analyses also indicated that drug-loaded MSPMSR8 (M/D + M/P, M/D, and M/P) were able to markedly increase cell apoptosis, again confirming that ligand-switchable MSPMSR8 can significantly enhance the anti-tumor efficacy of DOX and PLX-3397 (Fig. 5E and F).
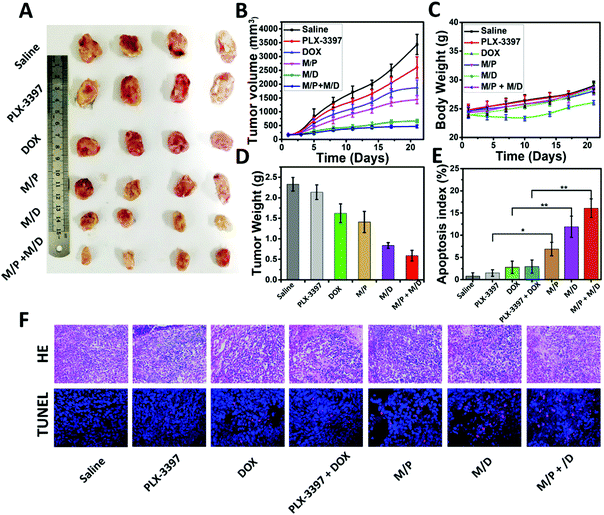 |
| | Fig. 5 Antitumor growth effect. (A) Images of tumor at day 21. (B) HepG2 tumor growth curves after injection with saline, free DOX, free PLX-3397, M/D, M/P and M/D + M/P at a dose of 5 mg (DOX) kg−1 and 5 mg (PLX-3397) kg−1 body weight, respectively. Error bars indicate s.d. (n = 5). (C) Relative body weight during treatment for 21 days. Error bars indicate s.d. (n = 5). (D) Tumor weight at day 21. (E) Quantification of apoptotic cells in tumor tissue after the treatment, which was obtained through terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end (TUNEL) staining. Error bars indicate s.d. (n = 5) t-test (P* < 0.05, P** < 0.01) (F). Histological analysis of tumor sections stained with H&E (20×) and TUNEL (40×). | |
Conclusions
In summary, we have demonstrated an efficient strategy to overcome the trade-off between extended durations of circulation and deep tumor penetration based on LSNs, enabling the in vivo application of CPPs. Using R8 as an example, such micelles are capable of hiding the R8 during circulation in the bloodstream to inhibit the unexpected immune clearance, and can expose them back to the surface for reactivating their penetrating ability on acidic tumor tissues. With this ligand-switchable capacity, MSPMSR8 not only facilitates tumor extravasation and accumulation by taking advantage of the long-circulating benefit of mixed-shell-polymeric-micelles but also enables deep tumor penetration by making use of the penetrating superiority of CPPs, achieving a higher overall drug delivery efficiency. More importantly, the unique and quick ligand-switching process is accomplished by the special design of the responsive micelles, which allows for the conjugation of a wide scope of functional ligands for more effective drug delivery. Without compromising circulation time and increasing side effects in normal tissues, a higher portion of ligand-conjugated LSNs are capable of reaching targeted tissues to improve drug delivery efficiency, enabling the in vivo application of those nonspecific ligands. In addition, this strategy is not only appropriate for anticancer but also for other diseases such as bacterial infections, which need to be treated with intravenous injections, providing a universal approach for the development of novel nanomedicines with better therapeutic effects.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51390483, 91527306, 21620102005 and 51603231), National Key Research and Development Programs of China (2018YFA0209700), Thousand Talents Program for Young Professionals, PCSIRT (IRT1257), the Fundamental Research Funds for the Central Universities, and National Natural Science Foundation for Excellent Young Scholars of China (81722026).
Notes and references
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2014, 13, 813–827 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKHzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- Y. Zhang, F. Wang, M. Q. Li, Z. Q. Yu, R. G. Qi, J. X. Ding, Z. Y. Zhang and X. S. Chen, Adv. Sci., 2018, 5, 1700821 CrossRef PubMed.
- N. Chen, Y. Han, Y. Luo, Y. Zhou, X. Hu, Y. Yu, X. Xie, M. Yin, J. Sun, W. Zhong, Y. Zhao, H. Song and C. Fan, Mater. Horiz., 2018, 5, 1204–1210 RSC.
- Y. Li, X. Zhang, Z. Zhang, H. Wu, X. Xu and Z. Gu, Mater. Horiz., 2018, 5, 1047–1057 RSC.
- Z. Yang, Y. Dai, L. Shan, Z. Shen, Z. Wang, B. C. Yung, O. Jacobson, Y. Liu, W. Tang, S. Wang, L. Lin, G. Niu, P. Huang and X. Chen, Nanoscale Horiz., 2019 10.1039/c8nh00307f.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, D. R. Greenwald and E. Ruoslahti, Science, 2010, 328, 1031–1035 CrossRef CAS PubMed.
- W. Wu, W. Driessen and X. Jiang, J. Am. Chem. Soc., 2014, 136, 3145–3155 CrossRef CAS PubMed.
- R. K. Jain, Annu. Rev. Biomed. Eng., 1999, 1, 241–263 CrossRef CAS PubMed.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- Y. Dai, C. Xu, X. Sun and X. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- J. E. Visvader and G. J. Lindeman, Nat. Rev. Cancer, 2008, 8, 755–768 CrossRef CAS PubMed.
- H. Clevers, Nat. Med., 2011, 17, 313–319 CrossRef CAS PubMed.
- Z. Liu, M. Xiong, J. Gong, Y. Zhang, N. Bai, Y. Luo, L. Li, Y. Wei, Y. Liu, X. Tan and R. Xiang, Nat. Commun., 2014, 5, 4280 CrossRef CAS PubMed.
- S. M. Pyonteck, L. Akkari, A. J. Schuhmacher, R. L. Bowman, L. Sevenich, D. F. Quail, O. C. Olson, M. L. Quick, J. T. Huse, V. Teijeiro, M. Setty, C. S. Leslie, Y. Oei, A. Pedraza, J. A. Zhang, C. W. Brennan, J. C. Sutton, E. C. Holland, D. Daniel and J. A. Joyce, Nat. Med., 2013, 19, 1264 CrossRef CAS PubMed.
- H. J. Li, J. Z. Du, X. J. Du, C. F. Xu, C. Y. Sun, H. X. Wang, Z. T. Cao, X. Z. Yang, Y. H. Zhu, S. M. Nie and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4164–4169 CrossRef CAS PubMed.
- H. Liang, X. Ren, J. Qian, X. Zhang, L. Meng, X. Wang, L. Li, X. Fang and X. Sha, ACS Appl. Mater. Interfaces, 2016, 8, 10136–10146 CrossRef CAS PubMed.
- H. J. Li, J. Z. Du, J. Liu, X. J. Du, S. Shen, Y. H. Zhu, X. Wang, X. Ye, S. Nie and J. Wang, ACS Nano, 2016, 10, 6753–6761 CrossRef CAS PubMed.
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848–8855 CrossRef CAS PubMed.
- C. Ju, R. Mo, J. Xue, L. Zhang, Z. Zhao, L. Xue, Q. Ping and C. Zhang, Angew. Chem., Int. Ed., 2014, 1–7 Search PubMed.
- X. Guan, Z. Guo, L. Lin, J. Chen, H. Tian and X. Chen, Nano Lett., 2016, 16, 6823–6831 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, J. Li, Z. H. Peng, Y. Sheinin, J. Zhou and D. Oupicky, ACS Nano, 2017, 11, 2227–2238 CrossRef CAS PubMed.
- Z. Krpetic, S. Saleemi, P. A. Prior, V. See, R. Qureshi and W. Brust, ACS Nano, 2011, 5, 5195–5201 CrossRef CAS PubMed.
- H. Zhou, Z. Fan, J. Deng, P. K. Lemons, D. C. Arhontoulis, W. B. Bowne and H. Cheng, Nano Lett., 2016, 16, 3268–3277 CrossRef CAS PubMed.
- E. Jin, B. Zhang, X. Sun, Z. Zhou, X. Ma, Q. Sun, J. Tang, Y. Shen, E. Van Kirk, W. J. Murdoch and M. Radosz, J. Am. Chem. Soc., 2013, 135, 933–940 CrossRef CAS PubMed.
- X. He, X. L. Chen, L. S. Liu, Y. Zhang, Y. F. Lu, Y. J. Zhang, Q. J. Chen, C. H. Ruan, Q. Guo, C. Li, T. Sun and C. Jiang, Adv. Sci., 2018, 5, 1701070 CrossRef PubMed.
- M. Yan, J. Du, Z. Gu, M. Liang, Y. Hu, W. Zhang, S. Priceman, L. Wu, Z. H. Zhou, Z. Liu, T. Segura, Y. Tang and Y. Lu, Nat. Nanotechnol., 2010, 5, 48–53 CrossRef CAS PubMed.
- I. Nakase, H. Akita, K. Kogure, A. Graslund, U. Langel, H. Harashima and S. Futaki, Acc. Chem. Res., 2012, 45, 1132–1139 CrossRef CAS PubMed.
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Acc. Chem. Res., 2013, 46, 2944–2954 CrossRef CAS PubMed.
- J. Ding, T. X. Z. Liang, Y. Zhou, Z. W. He, Q. H. Min, L. P. Jiang and J. J. Zhu, Nano Res., 2017, 10, 690–703 CrossRef CAS.
- W. Jiang, J. L. Wang, J. B. Yang, Z. W. He, Z. H. Hou, Y. L. Luo, L. Wang, J. Liu, H. B. Zhang, Y. Y. Zhao, G. Q. Zhang, F. Huang, X. C. Zhou, L. F. Yan, X. Z. Yang, Y. C. Wang and J. Wang, Nano Res., 2018, 11, 5716–5734 CrossRef CAS.
- S. Ruan, Q. He and H. Gao, Nanoscale, 2015, 7, 9487–9496 RSC.
- S. Futaki and I. Nakase, Acc. Chem. Res., 2017, 50, 2449–2456 CrossRef CAS PubMed.
- E. A. Sykes, J. Chen, G. Zheng and W. C. W. Chan, ACS Nano, 2014, 8, 5696–5706 CrossRef CAS PubMed.
- H. Ou, T. Cheng, Y. Zhang, J. Liu, Y. Ding, J. Zhen, W. Shen, Y. Xu, W. Yang, P. Niu, J. Liu, Y. An, Y. Liu and L. Shi, Acta Biomater., 2018, 65, 339–348 CrossRef CAS PubMed.
- H. J. Gao, T. J. Cheng, J. F. Liu, J. J. Liu, C. H. Yang, L. P. Chu, Y. M. Zhang, R. J. Ma and L. Q. Shi, Biomacromolecules, 2014, 15, 3634–3642 CrossRef CAS PubMed.
- Y. Liu, H. J. Busscher, B. R. Zhao, Y. F. Li, Z. K. Zhang, H. C. van der Mei, Y. J. Ren and L. Q. Shi, ACS Nano, 2016, 10, 4779–4789 CrossRef CAS PubMed.
- V. Chitu, V. Nacu, J. F. Charles, W. M. Henne, H. T. McMahon, S. Nandi, H. Ketchum, R. Harris, M. C. Nakamura and E. R. Stanley, Blood, 2012, 120, 3126–3135 CrossRef CAS PubMed.
- D. G. DeNardo, D. J. Brennan, E. Rexhepaj, B. Ruffell, S. L. Shiao, S. F. Madden, W. M. Gallagher, N. Wadhwani, S. D. Keil, S. A. Junaid, H. S. Rugo, E. S. Hwang, K. Jirstrom, B. L. West and L. M. Coussens, Cancer Discovery, 2011, 1, 54–67 CrossRef CAS PubMed.
- S. J. Coniglio, E. Eugenin, K. Dobrenis, E. R. Stanley, B. L. West, M. H. Symons and J. E. Segall, Mol. Med., 2012, 18, 519–527 CAS.
- T. Cheng, Y. Zhang, J. Liu, Y. Ding, H. Ou, F. Huang, Y. An, Y. Liu, J. Liu and L. Shi, ACS Appl. Mater. Interfaces, 2018, 10, 5296–5304 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00415c |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Yumin
Zhang
b,
Xue
Li
a,
Hanlin
Ou
a,
Tangjian
Cheng
a,
Lin
Ma
b,
Yingli
An
a,
Jianfeng
Liu
b,
Fan
Huang
*b,
Yang
Liu
*a and
Linqi
Shi
b,
Yumin
Zhang
b,
Xue
Li
a,
Hanlin
Ou
a,
Tangjian
Cheng
a,
Lin
Ma
b,
Yingli
An
a,
Jianfeng
Liu
b,
Fan
Huang
*b,
Yang
Liu
*a and
Linqi
Shi