
On the advancement of polymeric bicontinuous nanospheres toward biomedical applications
Sean D.
Allen†
 a,
Sharan
Bobbala†
b,
Nicholas B.
Karabin†
b and
Evan A.
Scott
*abcde
a,
Sharan
Bobbala†
b,
Nicholas B.
Karabin†
b and
Evan A.
Scott
*abcde
aInterdisciplinary Biological Sciences, Northwestern University, Evanston, USA. E-mail: evan.scott@northwestern.edu
bDepartment of Biomedical Engineering, Northwestern University, Evanston, USA
cChemistry of Life Processes Institute, Northwestern University, Evanston, USA
dSimpson Querrey Institute, Northwestern University, Chicago, USA
eRobert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, USA
First published on 14th November 2018
Abstract
Self-assembled soft nanocarriers that are capable of simultaneous encapsulation of both lipophilic and water soluble payloads have significantly enhanced controlled delivery applications in biomedicine. These nanoarchitectures, such as liposomes, polymersomes and cubosomes, are primarily composed of either amphiphilic polymers or lipids, with the polymeric variants generally possessing greater stability and control over biodistribution and bioresponsive release. Polymersomes have long demonstrated such advantages over their lipid analogs, liposomes, but only recently have bicontinuous nanospheres emerged as a polymeric cubic phase alternative to lipid cubosomes. In this review, we summarize the current state of the field for bicontinuous nanosphere formulation and characterization and suggest future directions for this nascent delivery platform as it is adopted for biomedical applications.
 Sean D. Allen | Sean Allen received his BA in Molecular and Cell Biology from the University of California, Berkeley in 2009 and his MS in Cellular and Molecular Biology from San Francisco State University in 2013. He is currently a PhD candidate in the Scott Lab at Northwestern University, where he plans to receive his PhD in early 2019. His research interests primarily focus on the formulation and translational application of nanomaterials, particularly relating to immunomodulation and sustained-release. |
 Sharan Bobbala | Dr Bobbala is currently a Postdoctoral Fellow in Evan Scott's lab at Northwestern University. He obtained his PhD in Pharmaceutical Sciences from the University of Otago, New Zealand and the BPharm and MPharm (Industrial Pharmacy) from the Kakatiya University, India. His research interests are in the areas of nanobiomaterials, novel drug delivery systems and single-shot vaccine formulations. Sharan was the recipient of Prestigious University of Otago Doctoral Scholarship and Northwestern Postdoctoral Professional Development awards. |
 Nicholas B. Karabin | Nicholas Karabin received his BS in Chemical Engineering with an additional major in Biomedical Engineering from Carnegie Mellon University in 2013 and his MS in Biomedical Engineering from Northwestern University in 2017. Currently, he is a PhD candidate in the Scott Lab at Northwestern University. His research focuses on designing and synthesizing surface functionalized nanomaterials for both targeted and sustained delivery applications relating to immunomodulation. |
 Evan A. Scott | Dr Scott has been as an Assistant Professor of Biomedical Engineering at Northwestern University since the fall of 2013. He respectively received a BS and PhD in Biomedical Engineering from Brown University in 2002 and Washington University in St. Louis in 2009. As a Whitaker International Scholar, he spent four years in Switzerland at the EPFL performing postdoctoral research in the laboratories of Prof. Jeffrey Hubbell and Prof. Melody Swartz. Dr Scott is a recipient of the 2015 NIH Director's New Innovator Award, the 2015 National Science Foundation CAREER Award and the 2014 American Heart Association Scientist Development Grant. He was selected as a 2017 BMES Young Innovator of Cellular and Molecular Bioengineering and a 2018 Nano Research Young Innovator in Nanobiotechnology. His laboratory is currently focused on the development of bioresponsive soft nanomaterials for applications in controlled drug delivery. |
1. Introduction
Nanocarriers employed for biomedical applications typically exhibit dimensions between 1 and 1000 nm and are capable of stably transporting therapeutics and diagnostics, either individually or in combinations, to specific biological targets.1 Their size, morphology, charge and surface chemistry are frequently engineered to achieve strategic bioavailability and biodistributions.2,3 As a result, nanocarriers are classified depending on their physiochemical properties, method of formation and nature of cargo loading.4,5 Hard or solid core nanocarriers, such as metallic nanoparticles6 and quantum dots,7 necessitate loading of cargoes onto their surfaces either through passive adsorption or chemical conjugation. Alternatively, soft nanocarriers, a designation that includes lipid- and polymer-based systems, allow partitioning of cargoes within their lipophilic and hydrophilic domains and can therefore encapsulate payloads without additional steps of chemical modification.8 This versatility for loading diverse cargoes along with their biocompatibility have allowed soft nanocarriers to become staples of current nanomedicine platforms.9,10Nanomedicine strategies frequently demand simultaneous delivery of both hydrophilic and hydrophobic payloads.11,12 For example, subunit vaccine formulations often require synchronous delivery of both hydrophilic protein/peptide antigens and either hydrophilic or hydrophobic adjuvants that could be in the form of lipids, small molecules or nucleic acids.13,14 Solid core self-assembled soft nanocarriers like micelles and filomicelles (cylindrical micelles) are not ideal for these types of applications due to their inability to load hydrophilic cargoes without surface conjugation. Therefore, self-assembled vesicular nanocarriers like liposomes and polymersomes have been of great interest within the field due to their ability to load hydrophilic and hydrophobic compounds in their aqueous core and bilayer membrane, respectively. Furthermore, large conformationally sensitive biologics, like cytokines and growth factors, can be stably loaded within the lumen of vesicular nanocarriers, which both provides protection from opsonization and enzymatic degradation as well as allows transport without modifying the nanocarrier surface chemistry.15,16
Given the expansive applications that can be addressed through dual loading platforms, alternative self-assembled nanocarrier systems have been developed for the delivery of hydrophilic payloads. In addition to liposomes and polymersomes, lipid-based liquid crystalline nanostructures with an internal bicontinuous cubic phase (cubosomes) or reversed hexagonal phase (hexosomes) have been investigated as drug and vaccine delivery vehicles.17Fig. 1 illustrates their internal organization: cubosomes are inverse bicontinuous cubic systems formed by a three-dimensional folding of lipid bilayers to create non-intersecting, continuous aqueous channels;18,19 whereas hexosomes are hexagonal packed arrangements of rod-shaped inverse micelles with closed aqueous channels.20 Glyceryl monooleate- and phytantriol-based liquid crystalline assemblies are most commonly reported to possess such internal structures.21–23 These highly organized nanostructures have demonstrated enhanced loading of hydrophilic, amphiphilic and hydrophobic payloads along with unique sustained release capabilities, suggesting potential use as controlled delivery vehicles and alternatives to liposomes.24–26
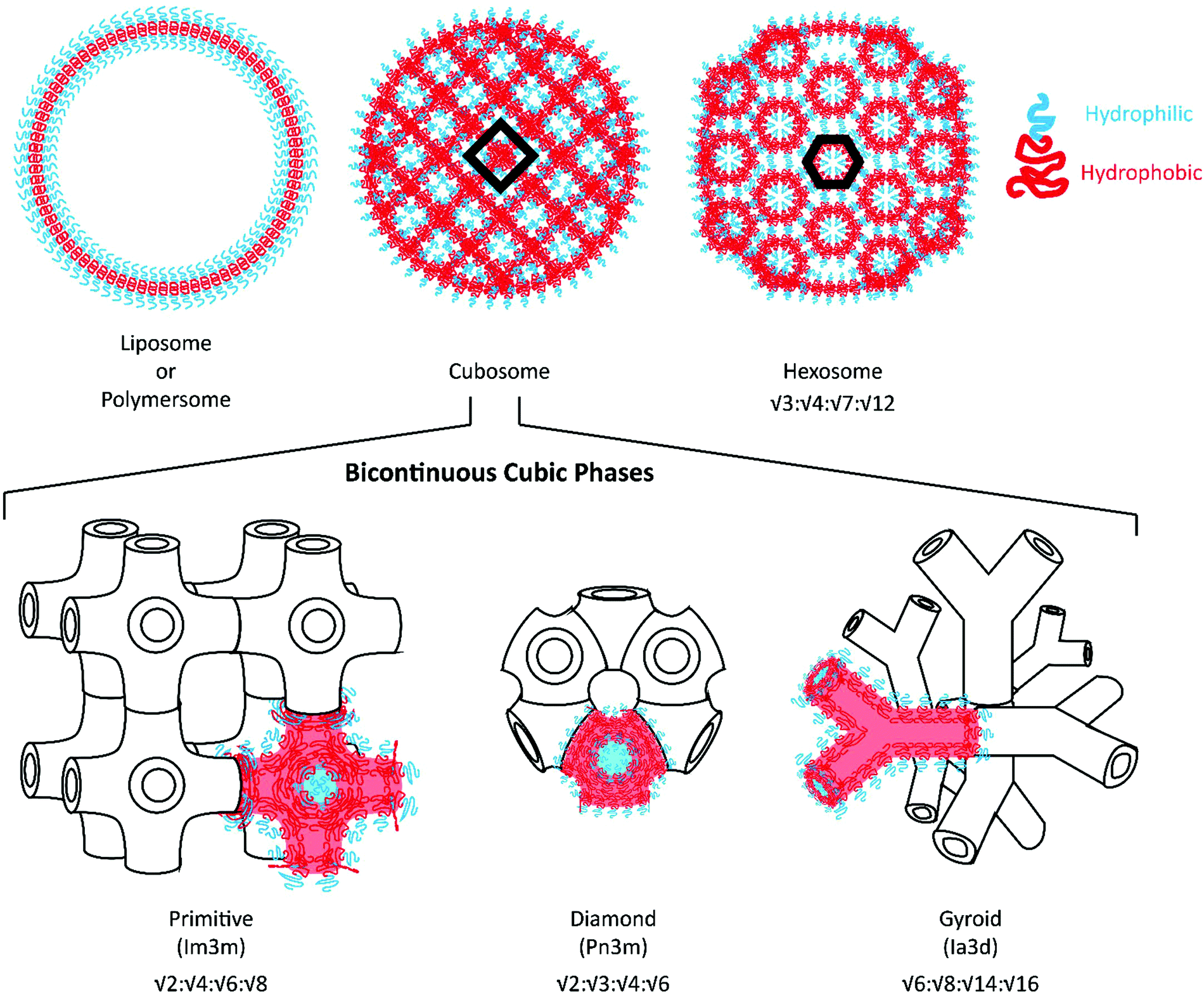 | ||
| Fig. 1 Structure of cubosomes and hexosomes with a liposome/polymersome shown for comparison. Nanoarchitectures are drawn as polymeric nanocarriers, but lipid nanocarriers possess analogous internal structures. Blue regions are hydrophilic, while red regions are hydrophobic. Schematic representations of the different bicontinuous cubic phases are included with typical Bragg peak ratios listed below. | ||
While the pioneering work to develop self-assembled cubic phase nanocarriers has primarily relied on lipids, alternative materials have been explored to address key disadvantages of lipid systems. Lipid-based nanocarriers are hindered in their application due to a variety of stability issues. They are susceptible to aggregation in vitro and often require additives to enhance their stability.19 Furthermore, the in vivo susceptibility of these structures to hydrolysis by esterases has been documented and limits control over bioresponsive release.27 To mitigate these stability-related issues, many lipid nanocarriers incorporate polymers for stabilization. Alternatively, some have eschewed these lipid/polymer composite systems and developed fully polymeric platforms for preparing both vesicular and bicontinuous nanocarriers. Self-assembled polymeric nanocarriers are composed of block copolymers (BCPs) that, like lipids, exhibit distinct hydrophobic and hydrophilic regions. Nanocarriers assembled from BCP amphiphiles exhibit significant advantages with respect to their mechanical and chemical stability. This enhanced stability in part stems from the BCP's increased molecular weight, which in turn increases the nanocarrier membrane thickness and alters the mechanisms through which neighbouring amphiphiles interact.9 A more detailed discussion of these mechanisms can be found in a previous review.9 In addition to their enhanced stability, BCPs can be more easily tailored for specific applications, as they can be synthesized from an expansive pool of established monomers, are highly amenable to functionalization, and can display well-defined molecular architectures. To capitalize on these advantages for drug delivery applications, polymeric equivalents to cubosomes and hexosomes have garnered significant interest.
Polymeric bicontinuous nanospheres (BCNs) can be considered polymeric analogues to lipid cubosomes28 and have been recently found to outperform lipid-based systems in terms of stability and ease of manufacturability.29–31 Similar to cubosomes, BCNs have been categorized into gyroid (Ia3d), diamond (Pn3m) and primitive (Im3m) phases depending on their internal cubic organization32 (see Fig. 1 for an illustration of these phases). Self-assembly of BCNs in aqueous solutions often requires polymers with complex polymeric structures that include comb-like, semi-crystalline, dendrimeric, and dendritic BCPs. These complex and often difficult syntheses have posed challenges for the scalable fabrication of BCNs. Furthermore, structural confirmation of the BCN internal organization requires analysis by electron microscopy and Small angle X-ray scattering (SAXS), neither of which are easily accessible techniques. As a result of this prior difficulty in uniform scalable fabrication, polymeric BCNs have not been explored for various medical and non-medical applications in contrast to their lipid-analogues. However, self-assembly of BCNs is highly dependent on the method of formation, concentration of the polymer, manufacturing conditions and solvents employed. Several recent studies have shown that simple amphiphilic linear BCPs could also be utilized to form BCNs.29,33,34
Recent advances in both scalable techniques of nanocarrier self-assembly and in the development of simple amphiphilic linear BCPs that can form BCNs now make these versatile nanocarriers compelling options for controlled delivery applications. In this review, we focus on the different BCP chemistries known to assemble BCNs, advantages and disadvantages of BCN fabrication techniques, current methods of BCN characterization, and future directions essential for employing BCNs as a new platform for nanomedicine.
2. Polymers employed for BCN self-assembly
Amphiphilic BCP systems have been exploited to prepare a variety of nanoparticle morphologies including spherical micelles, cylindrical filomicelles, and vesicular polymersomes in addition to the cubic structures discussed here. The morphology assumed by the BCP system is dictated by a reduction in free energy realized through the stretching of the hydrophobic block that comprises the nanoparticle core, the interfacial tension that exists between the core and surrounding solvent, and the repulsion between hydrophilic polymer blocks that comprise the outer corona.35 These parameters are influenced by the method of nanostructure formation, solution conditions, as well as the polymer chemistry and architecture.35Drawing from the extensive body of work describing the behaviour of lipid amphiphiles in solution, researchers have utilized the packing parameter or critical packing factor (p) to ascertain why specific BCPs self-assemble into certain morphologies. The packing parameter is defined as p = V/aolc, where V represents the volume of the hydrophilic domain, ao is the surface area of the hydrophilic moiety, and lc the critical length of the hydrophobic segment.36–38 In lipid systems, defined value ranges of p correspond to the formation of spheres, cylinders, lamellae/vesicles, and inverted structures.37 The formation of inverted or cubic structures with lipid amphiphiles typically occurs when p exceeds unity and similar principles apply for amphiphilic BCP systems as well. BCPs designed for the formation of bicontinuous structures often exhibit significant block asymmetry, with a hydrophobic block much larger than its corresponding hydrophilic block, to meet the criteria defined by p (see Fig. 2a for illustrations of p in the case of vesicles and inverted structures). While p has been useful in providing a starting point in the design of BCPs for the formation of bicontinuous structures, the block ratio (f), defined as the ratio of the molecular weight of the hydrophilic block to the molecular weight of the hydrophobic block, is a characteristic that has been more widely applied to define the phase behaviour of polymer systems including their assembly into cubic structures.36 It should be noted that f is alternatively used to represent the hydrophilic mass fraction.
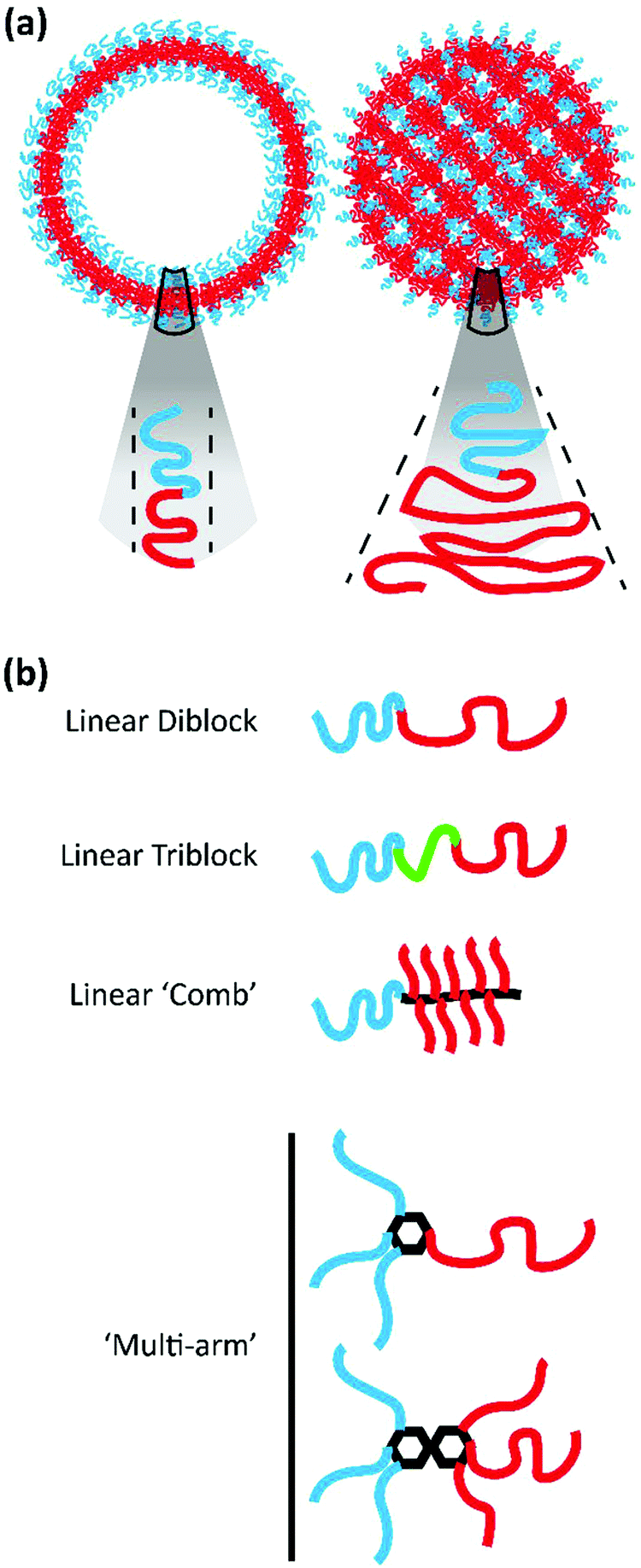 | ||
| Fig. 2 Polymers used for BCN formation. (a) Schematic illustrating the difference in packing parameter between polymers forming lamellar bilayer structures and bicontinuous structures. (b) Example illustrations of the different polymer architectures used for the formation of BCNs. | ||
A variety of bicontinuous nanostructures have been prepared from BCPs that vary in both their block chemistries and defining architecture. Here, we will explore the recent BCP systems confirmed to form BCNs organized by polymer architecture (Fig. 2b).
2.1 Comb-like block copolymers
Comb-like BCPs possess densely branched architectures wherein the branch chains differ in composition from the polymer backbone (Fig. 2b).37 Given its commercial availability and solubility in both aqueous and organic solvents, it is unsurprising that poly(ethylene glycol) (PEG), or poly(ethylene oxide) (PEO) at MW > 20k g mol−1, is commonly utilized as the hydrophilic block within self-assembling BCP systems. A variety of comb-like BCPs incorporating PEG/PEO have been described for the formation of bicontinuous structures. McKenzie et al. investigated the feasibility of using PEO with a variety of hydrophobic blocks for forming BCNs.30,33,34,39 Initially, their work focused on amphiphilic AB(C) PEO-block-poly(octadecyl methacrylate) (PEO39-b-PODMA17).34 The semicrystalline comb-like BCP, exhibiting a f = 0.25, formed complex micelles with bicontinuous internal structures in aqueous dispersions, and the PODMA block permitted temperature responsive behaviour. As the temperature falls below the transition point, crystalline packing of the octadecyl chains drives the formation of the internally ordered BCNs. McKenzie et al. further explored the PEO-b-PODMA system by preparing a series of BCPs that varied in molecular weight and f.30 While BCPs ranging in f from 0.07 to 0.25 all produced BCNs, the value of f was inversely related to the internal pore size of the assemblies. This work highlighted how polymer chemistry could be used to tailor the physical characteristics of bicontinuous structures. Holder et al. demonstrated how PEO-b-PODMA BCNs could be used for the delivery and sustained release of a hydrophobic active in an aqueous environment.40 The authors used PEO45-b-PODMA20 to encapsulate the hydrophobic dye pyrene for release in vitro to highlight the potential application of BCNs as delivery vehicles. Building from this work, Monaghan et al. explored a variant of the semicrystalline, comb-like PEO-b-PODMA for controlling release from BCNs.41 The authors prepared PEO-b-PODMA along with PEO-b-poly(docosyl methacrylate) (PEO-b-PDSMA) and PEO-b-(poly(octadecyl methacrylate)-co-poly(docosyl methacrylate)) (PEO-b-(PODMA-co-PDSMA)). Designing their BCPs with the understanding that alkyl side-chain length directly impacts the melting temperature (Tm) of the poly(methacrylate), they sought to vary the BCP Tm to control the release of ibuprofen and successfully highlighted their ability to tailor the BCP platform for controlled release in vitro.Parry et al. synthesized a novel double-comb diblock copolymer composed of a norborene backbone with oligo(ethylene glycol) and tripeptide branches to assemble a variety of structures, including complex BCNs.42 This BCP system exploited the incorporated peptide sequence rather than the hydrophobic/hydrophilic balance to dictate the primary self-assembled structure and internal organization. Barnhill et al. also employed norborene as the backbone for a library of amphiphilic BCPs.35 While the hydrophobic block was composed of norborene–phenyl, the hydrophilic component was prepared from either norborene–PEG, norborene–ethanolamide, or norborene–glycine. Of the BCPs described, only those with hydrophilic blocks composed of norborene–PEG and norborene–ethanolamide were capable of producing bicontinuous micelles.
2.2 Linear diblock copolymers
In addition to the comb-like BCPs described, linear diblock BCPs have been regularly utilized to form BCNs. McKenzie et al. prepared BCNs from a simple amphiphilic diblock copolymer composed of PEO and poly(n-butyl methacrylate) (PEO-b-PBMA) with f ≤ 0.17.33 These results demonstrated that neither crystallinity nor complex polymer architecture were prerequisites for BCN formation. Chen et al. utilized an alternative hydrophobic block for BCN assembly.28 In their work, the authors prepared PEO-b-poly(3-(trimethoxysilyl)propyl methacrylate) (PEO-b-PTMSPMA), a BCP that had been shown to form vesicles in mixtures of methanol and water when containing a significantly larger hydrophobic block (PEO45-b-PTMSPMA180) than previously reported. PEO45-b-PTMSPMA180, which exhibits a f of approximately 0.04, formed perforated vesicles with an internal bicontinuous structure that the authors initially believed to be metastable and trapped via the BCP's high molecular weight. Alternatively, Haluska et al. synthesized PEO-b-poly(butadiene) (PEO-b-PB), which formed giant micron length vesicular structures that exhibited a tubular network of channels with hexagonal symmetry in a solution of glucose.43Linear diblock copolymers incorporating polystyrene (PS) in conjunction with a range of hydrophilic blocks have been reported to form BCNs. Lin et al. prepared simple linear diblocks composed of PEG and polystyrene (PS) for the assembly of BCNs. The authors synthesized a series of PEG-b-PS BCPs that exhibited f ranging from 0.057 to 0.091.37 By varying the initial concentration of BCP along with f, they could produce a variety of BCNs with surface poration corresponding to the Schoen gyroid surface (Ia3d), the Schwarz diamond surface (Pn3m), and the Schwarz primitive surface (Im3m). Yu et al. used PS-b-poly(acrylic acid) (PS-b-PAA) to produce micron-sized BCNs with nanometer sized pores.44 PS-b-PAA produced both Schoen Gyroid and Schwarz P periodic structures, and interestingly, the PAA block permitted tuning of the nanopores without any additional modification of the material. Due to pH induced changes in PAA swelling and electrostatic repulsion, PS-b-PAA bicontinuous structures exhibited stimuli responsive gating that could be particularly useful for applications in controlled release. Zhang et al. relied on polymer induced self-assembly and reorganization to form nanostructures with highly ordered internal organization, dubbed hexagonally packed hollow hoops and rods, from PS-b-poly(2-dimethylaminoethyl methacrylate) (PS1190-b-PDMAEMA37).45
A variety of linear BCPs rely on less commonly used polymers for the formation of bicontinuous structures. Recently, Allen et al.46 and Bobbala et al.29 reported a simple linear diblock polymer composed of PEG-block-poly(propylene sulfide) (PEG-b-PPS) to form BCNs. This study found that PEG17-b-PPS75 (fPEG = 0.118) diblock copolymers can self-assemble into BCNs with primitive (Im3m) cubic organization. Due to the PPS block, these BCNs showed on-demand payload release in the presence of reactive oxygen species, making it a promising system for controlled release and intracellular delivery. He et al. synthesized amphiphilic poly(ionic liquid) (PIL) diblock copolymers composed of PAA-block-poly(4-vinylbenzyl)-3-butyl imidazoliumbis (trifluoromethylsulfonyl)imide (PAA45-b-PIL23) that are capable of assembling into a unique cuboidal morphology exhibiting an internal bicontinuous structure.47 These cuboidal nanostructures are potentially the result of the conglomeration of intra- and inter-molecular interactions that occur within the PAA45-b-PIL23 BCP system, and their thermosensitivity make them an interesting candidate for drug delivery applications. Kang et al. reported a unique system for the formation of bicontinuous nanostructures that incorporated poly nucleobases.48 These amphiphilic nucleobase-containing BCPs utilize poly(oligo(ethylene glycol)methyl ether methacrylate) as their hydrophilic block with hydrophobic blocks of adenine and thymine, either alone or in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture. Interestingly, the BCP composed of the adenine/thymine mixture (POEGMA70-b-(PAMA0.5-co-PTMA0.5)102), which exhibited a relatively high f of approximately 0.40, formed bicontinuous micelles.
:1 mixture. Interestingly, the BCP composed of the adenine/thymine mixture (POEGMA70-b-(PAMA0.5-co-PTMA0.5)102), which exhibited a relatively high f of approximately 0.40, formed bicontinuous micelles.
2.3 Linear triblock copolymers
In addition to the linear diblock BCPs described, linear triblock BCPs have also been utilized to form BCNs. Gao et al. prepared PS-b-PAA-b-poly(sodium 4-styrenesulfonate) (PS83-b-PAA33-b-PSSNa12) that self-assembled into micellar structures that can be coerced into forming more complex bicontinuous nanostuctures.49 The triblock initially self-assembled into core–shell–corona micelles, and self-limited aggregation of the micelles into superparticles was triggered through the addition of water. Phase transition within the superparticles promoted the formation of interconnected nanocylinders and resulted in their bicontinuous organization. Hales et al. utilized the triblock copolymer PAA-block-poly(methyl acrylate)-block-PS (PAA-b-PMA-b-PS) that can be complexed with di- or triamine counterions to form nanostructures with either bicontinuous-like or lamellar internal phases.50 Löbling et al. investigated the complex structures that can be formed through the self-assembly of linear ABC triblock terpolymers.51 Utilizing the PS-block-PB-block-poly(tert-butyl methacrylate) (PS-b-PB-b-PT) BCP system, the authors were able to produce nanostructures ranging from spherical micelles to vesicles with unique and well-defined surface patches. Notably they demonstrated the ability to produce vesicular structures with bicontinuous membranes.2.4 Multiarm and dendrimeric block copolymers
Both multiarm and dendrimeric BCPs have been explored for BCN assembly. Kyoung Taek Kim's group has done extensive work exploring how branched PEG-b-PS (bPEG–PS) and its derivatives can be used to form bicontinuous micro- and nanostructures. La et al. first presented an elegant means of isolating the role of BCP architecture in dictating self-assembled morphology through synthesis of a series of dendritic-linear PEG–PS variants.32 The BCPs utilized second generation benzyl ether dendrons modified with six PEG chains and all exhibited comparable f, which ranged between 0.08 and 0.11. The BCPs varied only in the number of outer phenyl groups and the position of their extending PEG chains. These slight variations in BCP architecture permitted the formation of cubic structures displaying Im3m, Pn3m, and Ia3d symmetries and led the authors to conclude that BCP architecture was a central factor in achieving bicontinuous structures.An et al. explored the intersection of BCP architecture and f by preparing bPEG–PS that exhibited three hydrophilic PEG branches.52 By varying the length of the PS block and the molecular weight of the PEG branches, the authors were able to prepare a variety of BCPs that ranged in f from 0.054 to 0.197. Interestingly, bicontinuous cubic microstructures were produced with both molecular weights of PEG, but the f at which said structures formed varied. For bPEG–PS with PEG branches of Mn = 550 g mol−1, bicontinuous cubic structures formed when f ranged from 0.071 to 0.078. This varied from the cubic structures formed from bPEG–PS with Mn = 750 g mol−1 PEG branches, which required an f of approximately 0.05. The bicontinuous structures formed with the lower molecular weight PEG branches exhibited both lm3m and Pn3m symmetries while those formed with higher molecular weight PEG exhibited only Pn3m symmetry.
La et al. further explored the use of bPEG–PS prepared with PEG branches of Mn = 550 g mol−1 for the formation of bicontinuous structures by varying the solvent from aqueous to a mixture of dioxane and dimethylformamide.36 The authors established that hexosomes could be produced when f was less than 0.056. Further exploring this space, Cho et al. investigated how binary blends of bPEG–PS could be used to tailor nanostructure morphology in a nonsynthetic fashion.53 Mixing bPEG–PS42 and bPEG–PS228, both prepared with PEG branches of Mn = 550 g mol−1, permitted the preparation of a variety of nanostructures including bicontinuous cubic structures with both Im3m and Pn3m symmetries and hexosomes with P6mm symmetry as the percentage of incorporated bPEG–PS228 was increased. Jeong and Kim described a bPEG–PS derivative that incorporated photodimerizable indene groups into the hydrophobic block using trimethylsilylindanolylstyrene as a monomer.54 The indene pendant groups can undergo [2π+2π]-cycloaddition through irradiation with UV light, permitting morphologic stability in a variety of solvent conditions. Out of the series of BCPs prepared, those exhibiting PEG branches with Mn = 550 g mol−1 and a f = 0.10 self-assembled into spherical bicontinuous structures displaying Pn3m symmetry. Excitingly, this internal symmetry was maintained even after crosslinking, demonstrating the potential for producing stable structures that can withstand a variety of solvent conditions.
Cho et al., in addition to their work with bPEG–PS, also prepared a variety of bicontinuous structures with bPEG and branched PS (bb-BCPs).38 The branched BCPs, which all displayed three PEG brushes with Mn = 750 g mol−1, could be controlled for f while differing in the number of PS branches, the position of the PS branches, or the length of said branches. This allowed the authors to gauge the impact of BCP architectural characteristics, such as branch number and position, on the self-assembly. Adjusting the aforementioned characteristics allowed the authors to prepare bicontinuous cubic structures, exhibiting Im3m and Pn3m symmetries, as well as hexosomes. Ju et al. prepared a dendritic block terpolymer containing polyisoprene, PS, and poly(tert-butyl methacrylate) that self-assembled into nanospheres with internal bicontinuous organization when formed in tetrahydrofuran (THF) and ethanol.56
2.5 General polymer design considerations for BCN formation
Given the diverse range of polymers described above that can assemble bicontinuous structures, it is apparent that polymer design is not restricted to a specific architecture. While this architectural diversity inhibits the establishment of a well-defined set of generalizable rules, a rough starting point for polymer design can be drawn from the packing parameter. As a starting point, polymers synthesized for the preparation of a bicontinuous structure should exhibit significant block asymmetry, where the molecular weight of the hydrophobic block or component greatly exceeds that of the hydrophilic block. For many BCP systems, the desired range of f will fall below 0.25, but the specific range of f will vary based on the chemical composition and architecture of the BCP utilized.Of equal importance for consideration is the solvent system selected during BCN formation. The self-assembled morphology is influenced by the solubility of each block within the employed cosolvent, which impacts the degree of swelling achieved by the respective blocks.33 McKenzie et al. provided a valuable discussion about the importance of solvent selection in their work with PEO-b-PBMA. When highly asymmetric BCPs, exhibiting a large hydrophobic mass fraction, are dissolved in a cosolvent system that favors the solubilization of the hydrophilic block, the increased swelling of the hydrophilic block can suppress the inverse curvature that would typically be imparted by the block asymmetry. As such, selecting a cosolvent system that favors the solubilization of the hydrophobic block may be integral in BCN formation.33
Understanding how to tailor BCP composition and architecture to modulate the pore size of BCNs will be of significant interest moving forward. McKenzie et al. provided some valuable insight concerning the impact of polymer composition on BCN porosity.30 In their work with PEO-b-PODMA, increasing f from 0.11 to 0.25 increased the internal pore size from 10 ± 2 nm to 19 ± 3 nm without significantly changing the pore wall thickness. Interestingly, the increased pore size was determined solely by the hydrophilic block's relative content instead of its absolute molecular weight, presenting a potentially advantageous capability to control the pore size simply by specifying the degree of polymerization of the hydrophilic block. This conclusion was reached by comparing the pore sizes of BCNs formed from BCPs that exhibited the same f but varied in the molecular weight of their hydrophilic block. While these design characteristics may be dependent on the BCP used, they at least provide a starting point for researchers working with previously unexplored materials.
3. Formulation of BCNs
Lipid cubosomes, sometimes stabilized with polymers such as pluronic, are commonly formed through high energy formulation processes by adding polymer and lipid directly into water and sonicating,57–59 often followed by high-pressure homogenization.60 These high energy processes may disrupt the crystalline organization of cubosomes (although in some cases the energy appears to be useful for generating more monodisperse cubosomes19), and thus nanoprecipitation techniques have also been explored as an alternative.61,62 These nanoprecipitation processes have been subsequently used to fabricate lipid-free polymeric BCNs developed more recently. To date, nearly all formulations of BCNs have been formed through the use of nanoprecipitation, though there has been considerable variation in the choice of solvent, the rate of non-solvent introduction, and the final solvent:non-solvent ratio. The formation techniques used with particular polymers are listed in Table 1 and are illustrated in Fig. 3.
| Polymer | Solvent | Non-solvent | w% non-solvent | mL h−1 | Ref. |
|---|---|---|---|---|---|
| a Poly(ethylene oxide)-block-poly(3-(trimethoxysilyl)propyl methacrylate). b Poly(acrylic acid)-block-poly(methyl acrylate)block-polystyrene. c Poly(norborene-oligo(ethylene glycol))-poly(norborene–lysine–leucine–phenylalanine). d Poly(ethylene oxide)-block-poly(octadecyl methacrylate). e Poly(ethylene oxide)-block-poly(n-butyl methacrylate). f Branched poly(ethylene glycol)-block-polystyrene. g Norbornene block copolymer. h Branched poly(ethylene glycol)-block-branched polystyrene. i Poly(oligo(ethylene glycol)methyl ether methacrylate)-block-poly(nucleobase). j Branched poly(ethylene glycol)-block-trimethylsilylindanolylstyrene. k Polystyrene-block-poly(ethylene glycol). l Poly(ethylene oxide)-block-(poly(octadecyl methacrylate)-co-poly(docosyl methacrylate)). m Poly(acrylic acid)-block-poly(4-vinylbenzyl)-3-butyl imidazoliumbis(trifluoromethylsulfonyl)imide. n Poly(ethylene glycol)-block-poly(propylene sulfide). o Tetrahydrofuran. p Dimethyl sulfoxide. q Dimethylformamide. r Phosphate buffered saline. | |||||
| PEO-b-PTMSPMAa | Methanol | Water | 39 | n.s. | 28 |
| PAA-b-PMA-b-PSb | THFo | Water | 16 to 44 | 15 | 50 |
| PNOEG–PNGLFc | DMSOp | Water | 70 | 5.6 | 42 |
| PEO-b-PODMAd | THFo | Water | 60 | 4 | 34 |
| PEO-b-PBMAe | THFo | Water | 60 | 4 | 33 |
| PEO-b-PODMAd | THFo | Water | 20 | 1.3 | 40 |
| 3xbPEG-b-PSf | Dioxane | Water | 50 | 1 | 32 |
| 3xbPEG-b-PSf | Dioxane | Water | 50 | 1 | 52 |
| Norbornene block copolymerg | DMSOp | Water | 50 | 1 | 35 |
| 3xbPEG–PSf | Dioxane | Water | 50 | 1 | 36 |
| PEO-b-PODMAd | THFo | Water | n.s. | 4 | 30 |
| 3xbPEG–3xbPSh | Dioxane | Water | 50 | 1 | 38 |
| POEGMA-b-poly(nucleobase)i | DMF,q DMSOp | Water | 89 | 1 | 55 |
| PEO-b-POMDAd | THFo | Water | 5 to 63 | 4 | 39 |
| 3xbPEG-b-PSf | Dioxane | Water | 50 | 1 | 53 |
| 3xbPEG-b-P(styrene-ran-TMS-indanolylstyrene)j | THFo | Water | n.s. | 0.5 | 54 |
| PS209-b-PEG45k | Dioxane/DMFq | Water | 50 | 1 | 37 |
| PEG-b-(PODMA-co-PDSMA)l | THFo | Water | 40 | 5.15 | 41 |
| PAA-b-P4VBm | THFo | Water | 62 | 288 | 47 |
| PEG-b-PPSn | THFo | Water and PBSr | 86 | 1800 | 46 |
| PEG-b-PPSn | THFo | Water and PBSr | 86 | 1800 | 29 |
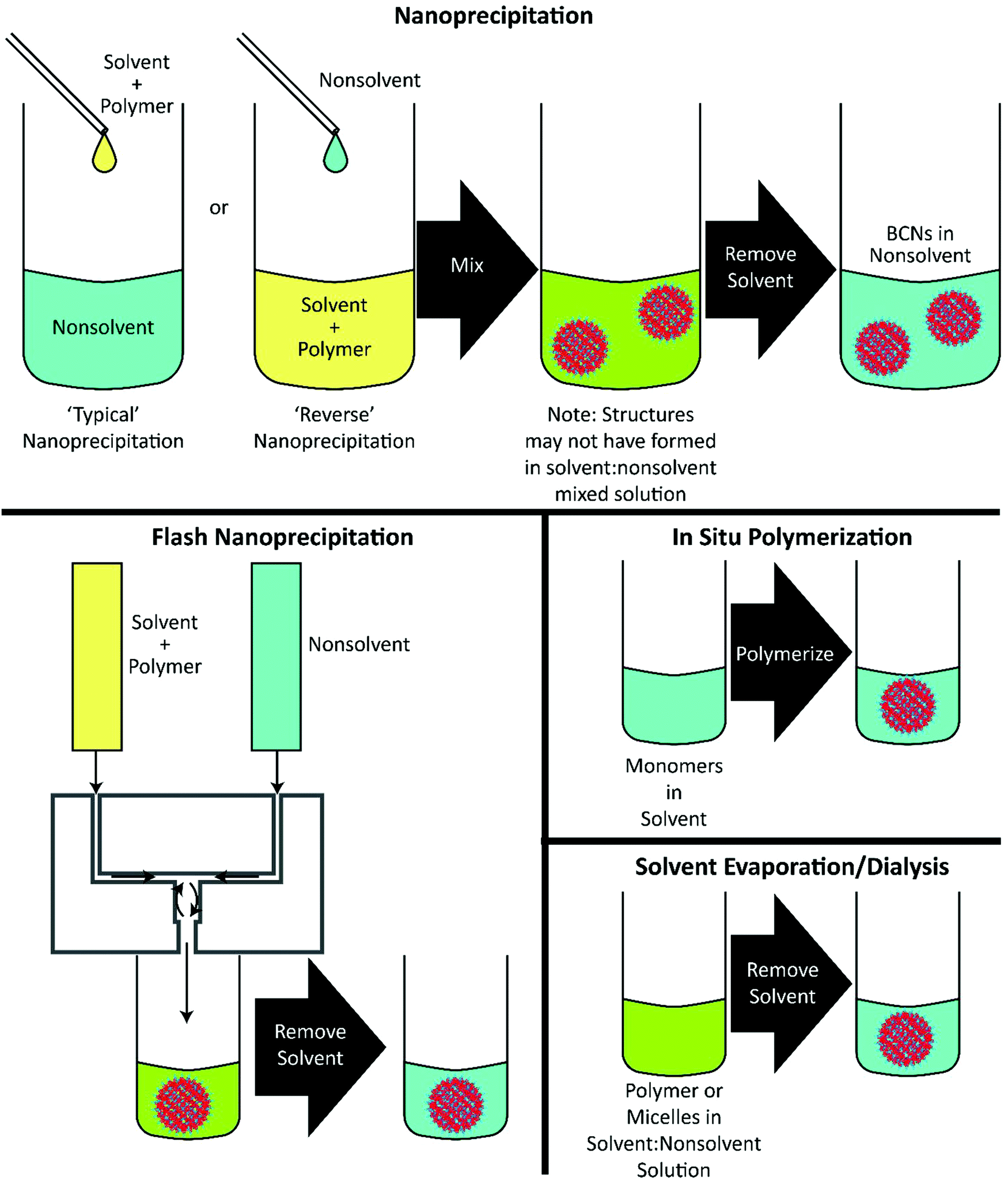 | ||
| Fig. 3 Schematic of formulation techniques for polymeric BCNs. In all sections, yellow represents polymer dissolved in solvent (typically organic), and blue represents nonsolvent (typically, but not always, aqueous). Green color represents solvent:nonsolvent mixtures. | ||
3.1 BCN formulation via nanoprecipitation
In the case of a typical nanoprecipitation, a polymer/solvent solution is added, often dropwise, to stirring non-solvent. This results in rapid dilution of the solvent and introduction of the polymer to non-solvent, leading to particle nucleation and subsequent growth. This process, while common for many other nanoparticle morphologies, is not the most typical nanoprecipitation method used for forming BCNs. Indeed, the more common method for forming BCNs is to reverse the addition process, i.e. to add the non-solvent gradually to a stirring polymer/solvent solution.28,30–42,47,50,52–56 In most cases, the solvent is removed from the final formulation, either through dialysis or solvent exchange through column filtration.The most recently employed method for BCN formation is flash nanoprecipitation.63,64 This highly scalable protocol involves the rapid impingement and controlled microsecond mixing of the polymer/solvent and non-solvent, followed by dilution of the solvent in a non-solvent reservoir.29,46 Flash nanoprecipitation is currently the only method available for scalable assembly of monodisperse BCNs in quantities necessary for clinical evaluation of new drug formulations. In contrast to traditional nanoprecipitation techniques, flash nanoprecipitation fosters uniform nucleation and growth of precipitating solutes through rapid and thorough mixing of the solvent and non-solvent, generally on the order of 1 millisecond. Flash nanoprecipitation is rapid and scalable but had been previously restricted to the formation of solid-core and micellar nanoparticle formulations. Use of flash nanoprecipitation for forming complex soft nanoarchitectures had been largely unexplored until recently, in work that demonstrated the formation of both vesicular and bicontinuous nanospheres.29,46 The parameters for flash nanoprecipitation are similar to those for traditional nanoprecipitation in terms of polymer concentration and solvent/non-solvent selection,65 but has so far only been demonstrated with the PEG-b-PPS BCP system.29,46 To assemble BCNs from PEG-b-PPS, Allen et al. used a confined impingement jets (CIJ) mixer, which is one of the simplest devices for performing flash nanoprecipitation. CIJ mixers are somewhat limited in that the initial impingement of solvent and non-solvent must occur in equal volumes to ensure proper mixing of the streams. Dilution of the organic solvent occurs immediately after mixing within a non-solvent reservoir. Multiple inlet vortex (MIV) mixers have also been developed, which overcome this limitation of CIJ mixers by permitting multiple streams to be impinged simultaneously for adjustment of the solvent to non-solvent ratio.66 However, to date MIV mixers have not been explored for use in the formation of BCNs.
Choice of solvent and non-solvent requires consideration of the solubility of the different blocks of the copolymer. As BCN formation is rare and likely exists in a narrow phase of metastable morphologies, small changes to the packing parameter of the copolymer can result in shifts in the aggregate morphology. In work by McKenzie et al.,33 shifting the solvent used in nanoprecipitation from THF to dioxane was sufficient to alter the aggregate morphology from BCNs to vesicles, likely due to the distinct solubility parameters of the different blocks of the copolymer and the two organic solvents. Similarly, Kang et al. found that their PEG-b-poly(nucleobase) copolymers formed BCNs when using dimethyl sulfoxide (DMSO) as the solvent, but formed other morphologies when N,N′-dimethylformamide (DMF) was used.55 This matches well with work by Barnhill et al. demonstrating that polynorbornene amphiphilic diblock copolymers were able to form bicontinuous structures in DMSO, while the same polymer formed different aggregate morphologies in acetonitrile and DMF.35 Further evidence of this trend was provided by La et al. with their work on branched PEG-b-PS. When either dioxane or THF were used as the single organic solvent, a bPEG5503-PS177 polymer formed vesicles. However, when a dioxane/DMF mixture was used, BCNs were formed using the same polymer.36 The hydration of the hydrophilic portion of the block copolymer may play a role in aggregate properties as well. Work by Bobbala et al. demonstrated that using phosphate buffered saline as the non-solvent resulted in increased polydispersity of the BCN formulation, compared to pure water.29 It is clear that a significant amount of work is needed to explore the potential solvent/non-solvent space with regards to BCN formation by nanoprecipitation.
The rate of non-solvent addition and final ratio of solvent to non-solvent prior to solvent removal via dialysis are both variables that have been empirically explored, as summarized in Table 1. Non-solvent addition is typically slow, ranging from 0.5 to 15 mL h−1, although He et al. performed nanoprecipitation with the rapid (288 mL h−1) addition of water,47 and the Bobbala et al. flash nanoprecipitation protocol used even more rapid (∼1800 mL h−1) addition of aqueous solution.29,46 It is notable that He et al. found that formation of BCNs with their polymer was incompatible with faster and slower rates of non-solvent addition, while Bobbala et al. found that their polymer formed BCNs by both flash nanoprecipitation and a slower standard nanoprecipitation process.29 Thus, while some polymers may only form BCNs when kinetically trapped by rapid introduction of the non-solvent, others may form BCNs under both rapid and slow non-solvent addition conditions.
In most reported nanoprecipitation methods of BCN formation, the ratio of solvent to non-solvent typically remained around 1:1. Hales et al. explored a range from 16 to 44 wt% water50 and McKenzie et al. explored a range from 5 to 63 wt% water.39 These two examples are notable due to their disparate conclusions. Hales et al. found that changing the ratio of THF:water resulted in the formation of BCNs with different internal structures. Importantly, they did not remove the THF by dialysis after particle formation prior to analysis via TEM. In contrast, McKenzie et al. found with their polymer system that the THF:water ratio did not result in BCN formation until the THF had been removed by dialysis, and that the addition of water during the nanoprecipitation process did not result in the production of BCNs. While the explanation of the differences in results undoubtedly lies in the differences of the block copolymers used in the two studies, this example illustrates the complex interplay of variables in the formation of BCNs.
3.2 Formulation methods beyond nanoprecipitation
In most cases, the non-solvent used for BCN formulations has been pure water. However, in some cases organic non-solvents have been used when appropriate for the given block copolymer system. Bicontinuous nanostructures, along with a large number of other aggregate morphologies, have been formed by first creating precursor micelles of PS-b-PB-b-PT terpolymer in N,N-dimethylacetamide and subsequently dialyzing against various acetone/isopropanol mixtures.51 Bicontinuous microparticles were also formed in a toluene/methanol mixture using PS-b-PAA, with their formation quenched by the addition of more methanol. Particles were collected by centrifugation, lyophilized, and redispersed in aqueous buffers.44 In one case, sonication of dissolved PTFEP-b-PS polymer in THF was sufficient for forming bicontinuous nanostructures, without the use of an additional non-solvent.67A new area of BCN formation utilizes polymerization techniques that can occur in solutions that contain a non-solvent for one of the blocks of the copolymer. Reversible addition-fragmentation chain transfer (RAFT) polymerization of styrene in ethanol using a PDMAEMA macroinitiatior resulted in the formation of bicontinuous nanostructures, as assessed via electron microscopy of crude nanoparticles in diluted ethanol, directly from the polymerization reaction.45 A similar reaction using a PEG macroinitiator for RAFT polymerization in an 80/20 ethanol/water solution has also been documented to form a variety of complex nanostructures, including BCNs.68
Solvent evaporation has been used to form BCNs and other complex nanoarchitectures using PS-b-poly(4-vinylpyridine) (PS-b-P4VP) polymer. The copolymer was dissolved in toluene, which was then emulsified in an aqueous SDS solution. The toluene was allowed to gradually evaporate, resulting in nano- and microparticles. This process, which involves a water-immiscible solvent (toluene), emulsification, and gradual evaporation is notably different from the far more common nanoprecipitation methods presented above.69
4. Characterization of BCNs
Employment of BCNs for both biomedical and non-medical applications will require structural reproducibility and uniformity. Techniques to confirm and characterize the BCN structure and monodispersity under different storage conditions and when loaded with diverse compounds are therefore critical to validate BCNs as a nanoplatform. Characterization of BCNs comprises analysis of size, shape, internal organization and type of cubic geometry, surface area and porosity. This section focuses on various techniques used to understand polymeric BCN structures.4.1 Dynamic light scattering and nanoparticle tracking analysis
Monodispersity is considered to be a prerequisite for achieving nanoparticle formulations with reproducible properties and functions. Dynamic light scattering (DLS) has been widely utilized to measure size and polydispersity as well as zeta potential of nanoparticles, including BCNs67,70 (Fig. 4a). Depending on the type of BCP, self-assembled BCNs were in the size range of 100 nm to greater than a micron.29,37 DLS has also been employed to track size and polydispersity changes seen in BCNs formed using semi-crystalline block copolymers at different transition temperatures.39,41 Since DLS is prone to errors stemming from higher light scattering of larger nanoparticles, usage of models requiring spherical geometry, and difficulty analysing solutions containing multiple distinct nanoparticle populations, secondary techniques are necessary to complement and confirm DLS measurements. Furthermore, DLS cannot confirm the internal organization of BCNs, which requires characterization by electron microscopy or small angle X-ray scattering. | ||
| Fig. 4 Examples of different characterization techniques for BCNs. (a) DLS data of nanoparticle size distribution. (b) CryoTEM micrograph of polymeric BCNs, scale bar = 100 nm. (c) SEM micrograph of polymeric bicontinuous microparticles, scale bar = 1 μm. (d) SAXS scattering of polymeric BCNs shown in (b), with labelled Bragg's peaks demonstrating a cubic internal organization. (a), (b) and (d) Reproduced with permission from ref. 29. Copyright 2018, The Royal Society of Chemistry and (c) Reproduced with permission from ref. 32. Copyright 2014, Nature publishing group. | ||
Nanoparticle tracking analysis (NTA) is used to measure particle size and concentration in liquid suspensions based on the properties of both light scattering and Brownian motion. During an NTA measurement, individual nanoparticles are tracked optically to assess their diffusion coefficients and resulting size. This provides NTA with several advantages over DLS, including the abilities to distinguish multiple nanoparticle populations in a single solution and determine nanoparticle concentration.46,71 For the first time, Allen & Bobbala et al., utilized NTA to measure and compare particle concentrations for PEG-b-PPS-based BCNs and polymersomes per each microgram of polymer utilized.72 As expected, at the same polymer concentration, the high density internal organization of BCNs resulted in significantly less nanoparticles per volume of solution than similarly sized vesicles possessing large aqueous lumens.
4.2 Electron microscopy
Electron microscopy is commonly used to confirm the shape and internal organization of nanoscale structures.73 Scanning electron microscopy (SEM, Fig. 4c) and transmission electron microscopy (TEM) are the two most widely used techniques to confirm bicontinuous morphologies.36,56 In one study, a high-resolution scanning electron microscopy (HR-SEM) technique was used to showcase the bicontinuous internal structure of BCNs.32 However, conventional TEM and SEM can be challenging due to dehydration during sample preparation and its ability to clearly visualize smaller internal aqueous channels of BCNs. To overcome these issues, cryogenic-transmission electron microscopy (cryo-TEM, Fig. 4b) and cryogenic-field emission scanning electron microscopy (cryo-FESEM) have been used to capture high-resolution images of BCNs in aqueous environments. This allows visualization and measurement of BCN internal aqueous channels.40,74 Recently, small and superimposing features of BCNs have been studied using cryo-electron tomography (3D-cryoTEM). McKenzie et al. studied internal pore connectivity of poly(ethylene oxide)-b-poly(octadecyl methacrylate) (PEO-b-PODMA) BCNs using cryo-electron tomography.30 In this study, slices of BCN structure captured at different tilt angles were used to construct a 3D tomogram to analyse the internal channels. Similarly, Bobbala et al. performed cryo-electron tomography studies on PEG-b-PPS BCNs to confirm the aqueous channels and internal connectivity.29 Electron microscopy studies may not allow assessment of the type of internal cubic geometry involved but can be complementary to more quantitative assessment by X-ray scattering techniques.4.3 Small angle X-ray scattering
SAXS is an important characterization tool for assigning the type of internal cubic symmetry within BCNs. In SAXS experiments, an X-ray beam is passed through a sample and the resulting scattering intensity is recorded at smaller scattering angles (typically 0.1–10°) and used to examine structure at the mesoscale.75 The modulus of the scattering vector (q) is defined as q = 4πsinθ/λ, where θ and λ are the scattering angle and X-ray wavelength, respectively. The internal geometries (Im3m, Pn3m and Ia3d) of bicontinuous cubic structures are assigned using characteristic Bragg peak spacing ratios, which is reviewed elsewhere18 (Fig. 4d). The interlayer spacings, d, for BCNs is calculated using d = 2π/q and the mean lattice parameter, a, can be calculated from d.22
SAXS has been used frequently to characterize internal geometries of polymeric BCNs.29,38,39 SAXS can also allow calculation of the radii of aqueous channels within BCNs.70 In addition, time- and temperature-resolved SAXS has been employed to understand the evolution of the BCNs during formation. As one example, McKenzie et al., utilized temperature resolved SAXS to understand the evolution of complex PEO-b-PODMA BCNs in aqueous solution.39 In this study, temperature resolved SAXS performed at 5–50 °C showed characteristic high intensity peaks corresponding to Im3m geometry at higher temperatures (>20 °C) and broad diffused peaks at lower temperatures (10 °C) that is attributed to the crystallinity of PODMA side chains. However, future studies involving usage of other variables like salt, polymer concentration and type of solvent may help in further understanding the evolution of BCN structure during self-assembly.
4.4 Nitrogen adsorption–desorption isotherm analysis
Nitrogen adsorption–desorption isotherm analysis has been applied to characterize BCN surface area and pore size distributions.76 Park et al. analysed BCNs composed of PEG–PS dendritic block copolymers using the Brunauer–Emmett–Teller (BET) and Barrett, Joyner and Halenda (BJH) methods.77 In this study, nitrogen adsorption–desorption isotherms of bicontinuous structures with Pn3m symmetry showed a large surface area (73 m2 g−1) and pore volume (0.45 cm3 g−1) with a pore size distribution of 32 nm. Similarly, La et al. analysed surface area, pore volume and pore size distributions of dendritic PEG-b-PS bicontinuous mesophases with different cubic geometries.36 Although nitrogen adsorption–desorption isotherm analysis hasn’t been explored widely to characterize polymeric BCNs to date, future application of this technique may allow routine estimation of the loading and release kinetics of BCN payloads.4.5 Differential scanning calorimetry
Differential scanning calorimetry (DSC) is a thermoanalytic technique commonly used to characterize thermal transitions of polymeric materials.78 Monaghan et al. recently utilized DSC to characterize BCNs composed of poly[ethylene oxide]-block-(poly[octadecyl methacrylate]-random-poly[docosyl methacrylate]), (PEO-b-[PODMA-co-PDSMA]). PODMA/PDSMA served as a semi-crystalline block responsible for the thermoresponsive nature of these BCNs.41 In this study, the melting transition temperature (Tm) of the hydrophobic block in BCN dispersions showed similar Tm as that of bulk samples, indicating successful retention of hydrophobic block crystallinity after self-assembly in solution. In another study, McKenzie et al., used DSC to demonstrate that the BCNs formed from the semi-crystalline block copolymer PEO39-b-PODMA17 retained the crystalline nature of the hydrophobic block below the phase transition temperature.79 DSC measurements of these BCNs above the transition temperature indicated that an amorphous transition caused a change in the morphology of BCNs to highly disordered microphase-separated structures.4.6 Energy-dispersive X-ray spectroscopy
Energy-dispersive X-ray spectroscopy is generally employed to analyse elements present within a sample.80 An energy-dispersive X-ray spectroscopic analytic technique has been utilized recently by He et al. to characterize BCNs that are formed using the amphiphilic diblock copolymer poly(acrylic acid)-block-poly(4-vinylbenzyl)-3-butyl imidazolium bis(trifluoromethylsulfonyl)imide.47 In this study, X-rays emitted through BCNs deposited on silicon wafers were analysed by subtracting the silicon peak from the emission spectrum. The emission spectrum showed the uniform distribution of ionic liquid moieties within the BCNs, thereby paving a new path to characterize BCNs formed using amphiphilic poly(ionic liquid) BCP. This technique may also be beneficial in the future to determine the distribution of payloads in BCNs on the basis of elemental composition.5. Future directions and potential for biomedical applications
5.1 Drug loading and release
The structural organization of BCNs permits the loading of diverse payloads. Recently, Bobbala et al. reported that PEG-b-PPS-based BCNs fabricated using flash nanoprecipitation were able to effectively encapsulate a diverse range of both hydrophobic and hydrophilic payloads.29 The bilayer membranes of BCNs revealed greater than 80% encapsulation efficiency of hydrophobic molecules whereas degree of entrapment of hydrophilic molecules inside the aqueous channels was found to be dependent on molecular weight of the molecules. In a subsequent study, Allen & Bobbala et al., reported superiority of BCNs in loading high molecular weight hydrophilic molecules as compared to polymersomes bearing similar surface chemistry.72 Furthermore, PEG-b-PPS BCNs were found to encapsulate higher amounts of hydrophobic molecules without losing their structural integrity when compared to PEG-b-PPS vesicles, which was attributed to the availability of higher internal hydrophobic volume within BCNs.The release behaviour of hydrophobic and hydrophilic molecular payloads from BCNs has been reported by several groups. Monaghan et al., modulated the release of the common hydrophobic non-steroidal anti-inflammatory drug ibuprofen using BCNs made up of semi-crystalline BCP, (PEO-b-[PODMA-co-PDSMA]).41 This report verified the relationship between the crystallinity of hydrophobic block and the rate of payload release, where higher crystallinity favoured lower release rates of ibuprofen from BCNs. For the first time, Bobbala et al. reported release kinetics of both hydrophobic and hydrophilic molecules from polymeric BCNs.29 In this study, the controlled release of a wide range of hydrophobic and hydrophilic molecules from PEG-b-PPS BCNs was assessed for up to 2 weeks, finding the release of hydrophilic molecules to be relatively faster than that of the hydrophobic counterparts. Furthermore, the release rate was slower for high molecular weight compared to lower molecular weight hydrophilic molecules. These studies provide early demonstrations of the potential of BCNs as delivery vehicles for low and high molecular weight therapeutics, biologics, vaccines and hydrophobic drugs. However, the loading and release of low molecular weight hydrophilic drugs remains challenging, and either controlling the internal pore size of aqueous channels or employing electrostatic retention within the aqueous channels may present interesting strategies to address this issue.
5.2 In vitro cellular delivery
Efficient cellular uptake and intracellular payload release are crucial benchmarks for nanoplatforms intended to deliver cell-targeted therapeutics. Polymeric BCNs have not been widely investigated for cellular applications. Recent work by Bobbala et al. found a linear increase in cellular uptake with time when fluorophore loaded PEG-b-PPS BCNs were incubated with a macrophages cell line.29 Confocal microscopy verified an endolysosomal co-localization of BCNs within these cells, which is a critical step for releasing payloads into the cell cytosol. To demonstrate the application of this ability for vaccination, PEG-b-PPS BCNs were then dual loaded with the model protein antigen ovalbumin (OVA) and lipophilic adjuvant monophosphoryl lipid-A (MPLA) for in vitro incubation with dendritic cells (Fig. 5a). Presentation of the OVA peptide SIINFEKL on the surface of these cells was verified, indicating effective intracellular delivery, processing and cellular stimulation. Such concurrent intracellular delivery of both hydrophobic and hydrophilic payloads is a vital aspect in vaccine delivery, and this ability was further characterized for PEG-b-PPS BCNs by Allen and Bobbala et al.72 These early in vitro examinations of cellular delivery verify that BCNs possess great potential as well as several advantages for biomedical applications compared to other polymeric nanocarrier platforms. It is in this area, the transition from material and nanoparticle characterization to in vitro and in vivo application, that BCN research has the most capacity to grow.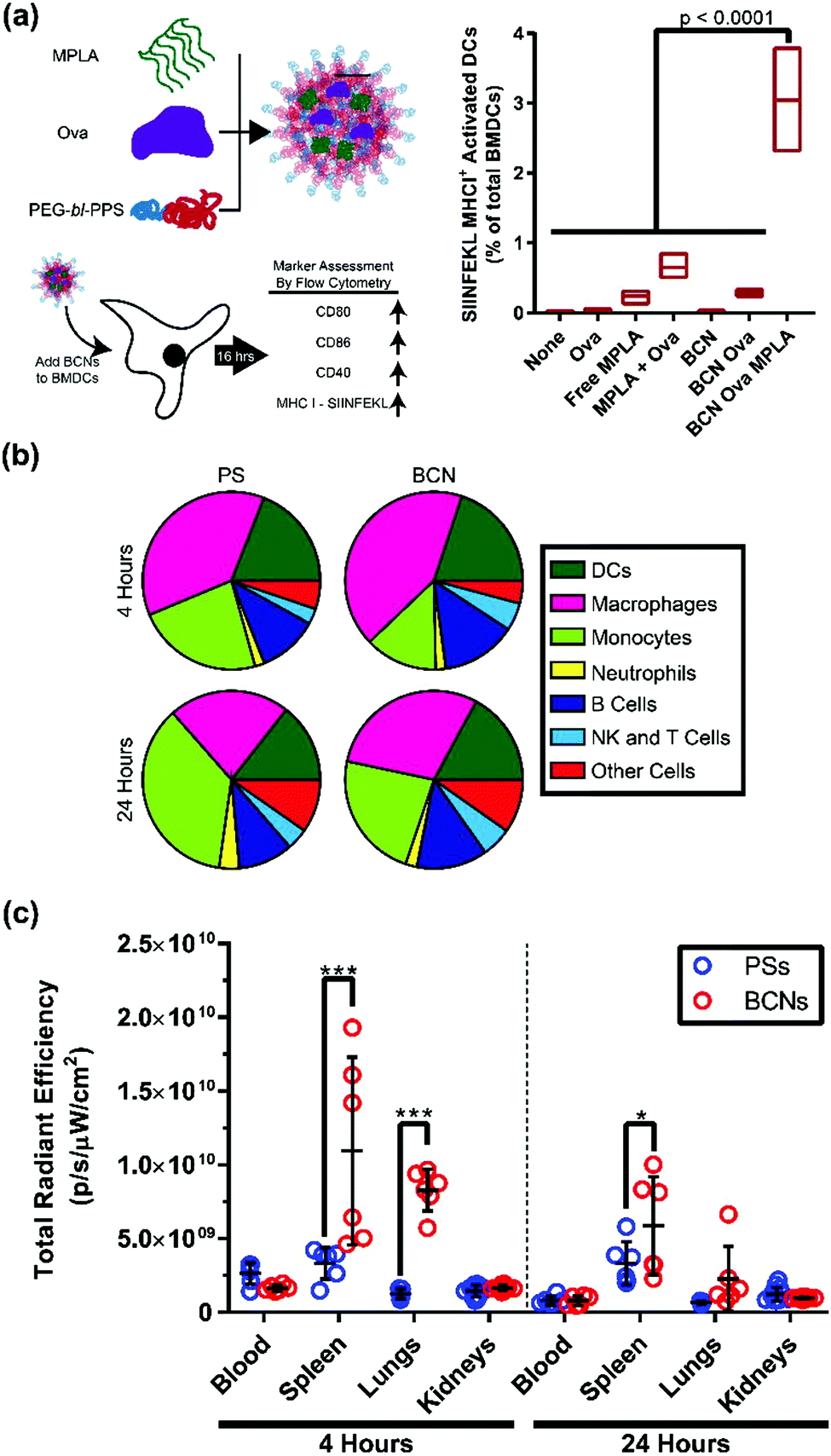 | ||
| Fig. 5 In vitro and in vivo application of BCNs. (a) In vitro delivery of adjuvant and model antigen to bone marrow derived dendritic cells and flow cytometric analysis of cell activation and antigen display. Cell (b) and organ (c) level distribution comparison between polymersomes (PSs) and BCNs 4 or 24 hours after intravenous administration. (a) Reproduced with permission from ref. 29. Copyright 2018, The Royal Society of Chemistry and (b and c) reproduced with permission from ref. 72. Copyright 2018, American Chemical Society. | ||
5.3 In vivo biodistribution
Lipid cubosomes have been administered orally, parenterally, transdermally, and intravenously in murine models. To the best of our knowledge, there is only a single report on the use of polymeric BCNs in vivo: the recent study by Allen & Bobbala et al., which compared the biodistribution and cellular uptake of BCNs to polymersomes.72 This investigation revealed differences in both cellular uptake and organ-level biodistribution after intravenous injection of these nanostructures in mice. Both polymersomes and BCNs were internalized primarily by phagocytic antigen presenting cells such as macrophages and dendritic cells (Fig. 5b). However, BCNs had significantly reduced liver uptake and higher splenic uptake compared to polymersomes (Fig. 5c). This finding suggests that BCNs have high potential for the delivery of cargo for immunomodulatory purposes, as the spleen is a critical secondary lymphoid organ. Furthermore, a reduction in liver clearance compared to polymersomes was observed, indicating that PEG-b-PPS BCNs may result in reduced non-specific clearance of therapeutics from the body during controlled delivery. This study is a significant step forward toward the application of BCNs to address biomedical challenges.Next steps for BCN development as a nanocarrier platform will likely involve the loading and delivery of therapeutic payloads in vivo. As BCNs are capable of the simultaneous loading of hydrophilic and hydrophobic compounds, they will be particularly useful for applications where two or more compounds need to arrive at the same location for specific biological stimulation. In addition to the aforementioned applications in vaccine formulations, BCNs may serve as advantageous nanocarriers for theranostic strategies by allowing controlled dual delivery of therapeutic and diagnostic agents. Longer term goals for the advancement of BCNs may involve the engineering of BCN diameter, surface functionalization, and pore size to address diverse in vivo challenges. BCNs are relatively large nanoparticles currently, and a smaller diameter could alter their biodistribution significantly. Pore size alterations, including stimuli-responsive changes to pore diameter, could allow for more nuanced release of hydrophilic payloads in vivo.
Surface functionalization could impart differential cellular uptake and clearance rates. The interaction of nanoparticles with biological components can procure several adsorbates such as a ‘protein corona’, lipids, polysaccharides, natural organic matter and adsorbed ions, each of which affects the cellular uptake and biodistribution of nanoparticles.81 Designing BCNs with PEG on their surface could prevent protein corona formation and enhance systemic circulation time, however, the presence of PEG can reduce the uptake of nanoparticles by intended cells.82,83 Recent studies also suggest that PEG density on nanoparticle surfaces can play an important part in dictating the protein corona formation, cellular uptake and circulation times.84,85 Furthermore, surface functionalization with antifouling polymers and cell-specific peptides and antibodies can improve nanoparticle targeting and will likely enhance the specificity of cellular uptake by BCNs as well.86 Modulation of BCN surface charge will also be critical for cellular interactions and toxicity. In general, cationic nanoparticles are efficiently taken up by cells when compared to anionic and neutral nanoparticles, however, a positive surface charge can increase both toxicity and non-specific cellular interactions.87,88 Further, anionic and cationic nanoparticles have been reported to exhibit faster clearance rates as compared to their neutral and zwitterionic counterparts.89 Thus, development of BCNs with neutral or zwitterionic surfaces could enhance circulation times and safety while surface conjugated targeting moieties may improve the cellular specificity of delivered therapeutics.
The mechanical properties of nanoparticles play a key role during cellular internalization and circulation in vivo.90,91 For example, several studies found that soft nanoparticles have lower cellular internalization and longer circulation times as compared to hard nanoparticles, which was attributed to the amenability of soft nanoparticles to deformation during phagocytosis.90,92 There is no standard relationship established between the degree of nanoparticle stiffness and cellular internalization in the literature, as results vary extensively between different BCP systems. However, a study reported by Banquy et al. suggests that nanoparticles with an intermediate elastic modulus (Young's moduli ∼35 and 136 kPa) have greater cellular uptake than lower (∼18 kPa) or higher stiffness nanoparticles (∼211 kPa).93 The highly organized cubic internal structure of BCNs suggests that they may have a higher stiffness compared to vesicular structures, however, a detailed analysis of the elastic modulus of BCNs has not been performed. An interesting future experiment would be to modulate BCN elastic modulus by controlling pore size and composition, possibly providing an additional means of specifying BCN biodistribution and cellular interactions.
6. Conclusions
Here, we have comprehensively reviewed the current status of the polymer systems, preparation methods and characterization tools that are available for the development of polymeric BCNs. The literature has shown that the self-assembly of BCNs is possible with a wide range of polymer chemistries and architectures of varying complexity. Given the difficulty of synthesizing many complex BCP architectures, recent developments demonstrating that simple linear diblock polymers can form BCNs are promising. As the number of BCN-forming BCP systems continues to expand and enhance both the chemical and structural diversity that BCNs can achieve, focus will need to shift from simply selecting BCPs that can consistently form BCNs to designing BCPs systems that impart enhanced functionality. With regard to their application within the biomedical field, synthesizing BCPs that can dynamically alter BCN structure in response to environmental stimuli will become a primary objective. Utilizing BCPs that respond to either passive stimuli (pH, reactive oxygen species, etc.) or induced stimuli (light, ultrasound, etc.) would present a multitude of design opportunities that include dynamic pore sizes and on-demand BCN degradation or payload release.Further development of polymeric BCNs is also dependent on the advancement of scalable manufacturing techniques. The recently reported flash nanoprecipitation method may have potential to manufacture polymeric BCNs on a commercial scale. However, this method has only been demonstrated for BCNs assembled from PEG-b-PPS copolymers. Additional work will be necessary to validate and optimize this methodology for other BCP systems.
Characterization tools like DLS, cryo-TEM and SAXS have shown great promise for confirming and analysing BCN monodispersity and internal structure. Additionally, utilization of techniques like energy-dispersive X-ray spectroscopy, atomic force microscopy and nitrogen adsorption–desorption isotherm analysis present new options to better understand BCN assembly and suitability for different applications.
Polymeric BCNs have been applied in vivo in only a single instance and have only had limited testing in vitro. However, there is little that limits the in vivo use of polymeric BCNs, as they can be made of non-toxic and bioresponsive polymeric materials, making them as safe and versatile as lipid cubosomes, liposomes and host of other nanoparticles currently employed for biomedical applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Institutes of Health (Grant No. 1DP2HL132390-01) and the National Science Foundation (CAREER award No. 1453576).References
- V. Wagner, A. Dullaart, A.-K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211 CrossRef CAS PubMed.
- E. A. Scott, N. B. Karabin and P. Augsornworawat, Annu. Rev. Biomed. Eng., 2017, 19, 57–84 CrossRef CAS PubMed.
- S. Allen, Y. G. Liu and E. Scott, Regen. Eng. Transl. Med., 2016, 2, 37–50 CrossRef PubMed.
- A. Sangtani, O. K. Nag, L. D. Field, J. C. Breger and J. B. Delehanty, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1466 Search PubMed.
- M. J. Sailor and J. H. Park, Adv. Mater., 2012, 24, 3779–3802 CrossRef CAS PubMed.
- N. T. Thanh and L. A. Green, Nano Today, 2010, 5, 213–230 CrossRef CAS.
- D. Granada-Ramírez, J. Arias-Cerón, P. Rodriguez-Fragoso, F. Vázquez-Hernández, J. Luna-Arias, J. Herrera-Perez and J. Mendoza-Álvarez, Nanobiomaterials, Elsevier, 2017, pp. 411–436 Search PubMed.
- J. Callejas-Fernández, J. Estelrich, M. Quesada-Pérez and J. Forcada, Soft nanoparticles for biomedical applications, Royal Society of Chemistry, 2014 Search PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- G. Zhao, S. Chandrudu, M. Skwarczynski and I. Toth, Eur. Polym. J., 2017, 93, 670–681 CrossRef CAS.
- P. Parhi, C. Mohanty and S. K. Sahoo, Drug Discovery Today, 2012, 17, 1044–1052 CrossRef CAS PubMed.
- S. Bobbala, B. Gibson, A. B. Gamble, A. McDowell and S. Hook, Immunol. Cell Biol., 2018, 96, 656–665 CrossRef CAS PubMed.
- E. A. Scott, A. Stano, M. Gillard, A. C. Maio-Liu, M. A. Swartz and J. A. Hubbell, Biomaterials, 2012, 33, 6211–6219 CrossRef CAS PubMed.
- S. Bobbala and S. Hook, Pharm. Res., 2016, 33, 2078–2097 CrossRef CAS PubMed.
- S. Nema, D. M. Kiick and K. E. Avis, PDA J. Pharm. Sci. Technol., 1994, 48, 231–235 CAS.
- D. A. Balazs and W. Godbey, J. Drug Delivery, 2011, 2011, 326497 Search PubMed.
- I. D. Azmi, S. M. Moghimi and A. Yaghmur, Ther. Delivery, 2015, 6, 1347–1364 CrossRef CAS PubMed.
- S. B. Rizwan and B. J. Boyd, in Subunit Vaccine Delivery, ed. C. Foged, T. Rades, Y. Perrie and S. Hook, Springer New York, New York, NY, 2015, pp. 125–140, DOI:10.1007/978-1-4939-1417-3_7.
- J. Barauskas, M. Johnsson, F. Joabsson and F. Tiberg, Langmuir, 2005, 21, 2569–2577 CrossRef CAS PubMed.
- I. Amar-Yuli, E. Wachtel, E. B. Shoshan, D. Danino, A. Aserin and N. Garti, Langmuir, 2007, 23, 3637–3645 CrossRef CAS PubMed.
- S. Engelskirchen, R. Maurer and O. Glatter, Colloids Surf., A, 2011, 391, 95–100 CrossRef CAS.
- Y. D. Dong, I. Larson, T. Hanley and B. J. Boyd, Langmuir, 2006, 22, 9512–9518 CrossRef CAS PubMed.
- X. Shi, T. Peng, Y. Huang, L. Mei, Y. Gu, J. Huang, K. Han, G. Li, C. Hu, X. Pan and C. Wu, Pharm. Dev. Technol., 2017, 22, 322–329 CrossRef CAS PubMed.
- A. Yaghmur and O. Glatter, Adv. Colloid Interface, 2009, 147–148, 333–342 CrossRef CAS PubMed.
- A. Chemelli, M. Maurer, R. Geier and O. Glatter, Langmuir, 2012, 28, 16788–16797 CrossRef CAS PubMed.
- M. Malmsten, Soft Matter, 2006, 2, 760–769 RSC.
- W. Leesajakul, M. Nakano, A. Taniguchi and T. Handa, Colloids Surf., B, 2004, 34, 253–258 CrossRef CAS PubMed.
- Y. Chen, J. Du, M. Xiong, H. Guo, H. Jinnai and T. Kaneko, Macromolecules, 2007, 40, 4389–4392 CrossRef CAS.
- S. Bobbala, S. D. Allen and E. A. Scott, Nanoscale, 2018, 10, 5078–5088 RSC.
- B. E. McKenzie, H. Friedrich, M. J. Wirix, J. F. de Visser, O. R. Monaghan, P. H. Bomans, F. Nudelman, S. J. Holder and N. A. Sommerdijk, Angew. Chem., Int. Ed., 2015, 54, 2457–2461 CrossRef CAS PubMed.
- B. E. McKenzie, S. J. Holder and N. A. Sommerdijk, Curr. Opin. Colloid Interface Sci., 2012, 17, 343–349 CrossRef.
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534–541 CrossRef CAS PubMed.
- B. E. McKenzie, J. F. de Visser, H. Friedrich, M. J. M. Wirix, P. H. H. Bomans, G. de With, S. J. Holder and N. A. J. M. Sommerdijk, Macromolecules, 2013, 46, 9845–9848 CrossRef CAS.
- B. E. McKenzie, F. Nudelman, P. H. Bomans, S. J. Holder and N. A. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256–10259 CrossRef CAS PubMed.
- S. A. Barnhill, N. C. Bell, J. P. Patterson, D. P. Olds and N. C. Gianneschi, Macromolecules, 2015, 48, 1152–1161 CrossRef CAS.
- Y. La, T. H. An, T. J. Shin, C. Park and K. T. Kim, Angew. Chem., Int. Ed., 2015, 54, 10483–10487 CrossRef CAS PubMed.
- Z. Lin, S. Liu, W. Mao, H. Tian, N. Wang, N. Zhang, F. Tian, L. Han, X. Feng and Y. Mai, Angew. Chem., Int. Ed., 2017, 56, 7135–7140 CrossRef CAS PubMed.
- A. Cho, Y. La, T. J. Shin, C. Park and K. T. Kim, Macromolecules, 2016, 49, 4510–4519 CrossRef CAS.
- B. E. McKenzie, J. De Visser, G. Portale, D. Hermida-Merino, H. Friedrich, P. Bomans, W. Bras, O. R. Monaghan, S. J. Holder and N. A. Sommerdijk, Soft Matter, 2016, 12, 4113–4122 RSC.
- S. J. Holder, G. Woodward, B. McKenzie and N. A. Sommerdijk, RSC Adv., 2014, 4, 26354–26358 RSC.
- O. Monaghan, P. Bomans, N. A. Sommerdijk and S. J. Holder, Polym. Chem., 2017, 8, 5303–5316 RSC.
- A. L. Parry, P. H. Bomans, S. J. Holder, N. A. Sommerdijk and S. C. Biagini, Angew. Chem., Int. Ed., 2008, 47, 8859–8862 CrossRef CAS PubMed.
- C. K. Haluska, W. T. Gozdz, H. G. Dobereiner, S. Forster and G. Gompper, Phys. Rev. Lett., 2002, 89, 238302 CrossRef PubMed.
- H. Yu, X. Qiu, S. P. Nunes and K. V. Peinemann, Nat. Commun., 2014, 5, 4110 CrossRef CAS PubMed.
- W. J. Zhang, C. Y. Hong and C. Y. Pan, Macromol. Rapid Commun., 2015, 36, 1428–1436 CrossRef CAS PubMed.
- S. Allen, O. Osorio, Y. G. Liu and E. Scott, J. Controlled Release, 2017, 262, 91–103 CrossRef CAS PubMed.
- H. He, K. Rahimi, M. Zhong, A. Mourran, D. R. Luebke, H. B. Nulwala, M. Moller and K. Matyjaszewski, Nat. Commun., 2017, 8, 14057 CrossRef PubMed.
- Y. Kang, A. Pitto-Barry, M. S. Rolph, Z. Hua, I. Hands-Portman, N. Kirby and R. K. O'Reilly, Polym. Chem., 2016, 7, 2836–2846 RSC.
- Y. Gao, Y. F. Wang, M. Jiang and D. Y. Chen, ACS Macro Lett., 2012, 1, 1312–1316 CrossRef CAS.
- K. Hales, Z. Chen, K. L. Wooley and D. J. Pochan, Nano Lett., 2008, 8, 2023–2026 CrossRef CAS PubMed.
- T. I. Lobling, O. Borisov, J. S. Haataja, O. Ikkala, A. H. Groschel and A. H. Muller, Nat. Commun., 2016, 7, 12097 CrossRef PubMed.
- T. H. An, Y. La, A. Cho, M. G. Jeong, T. J. Shin, C. Park and K. T. Kim, ACS Nano, 2015, 9, 3084–3096 CrossRef CAS PubMed.
- A. Cho, Y. La, S. Jeoung, H. R. Moon, J. H. Ryu, T. J. Shin and K. T. Kim, Macromolecules, 2017, 50, 3234–3243 CrossRef CAS.
- M. G. Jeong and K. T. Kim, Macromolecules, 2017, 50, 223–234 CrossRef CAS.
- Y. Kang, A. Pitto-Barry, M. S. Rolph, Z. Hua, I. Hands-Portman, N. Kirby and R. K. O'Reilly, Polym. Chem., 2016, 7, 2836–2846 RSC.
- Z. Ju and J. He, Chem. Commun., 2014, 50, 8480–8483 RSC.
- A. Yaghmur, L. de Campo, L. Sagalowicz, M. E. Leser and O. Glatter, Langmuir, 2005, 21, 569–577 CrossRef CAS PubMed.
- J. Y. T. Chong, X. Mulet, L. J. Waddington, B. J. Boyd and C. J. Drummond, Soft Matter, 2011, 7, 4768–4777 RSC.
- J. Y. T. Chong, X. Mulet, A. Postma, D. J. Keddie, L. J. Waddington, B. J. Boyd and C. J. Drummond, Soft Matter, 2014, 10, 6666–6676 RSC.
- B. Siekmann, H. Bunjes, M. M. H. Koch and K. Westesen, Int. J. Pharm., 2002, 244, 33–43 CrossRef CAS PubMed.
- P. T. Spicer, K. L. Hayden, M. L. Lynch, A. Ofori-Boateng and J. L. Burns, Langmuir, 2001, 17, 5748–5756 CrossRef CAS.
- M. Wadsater, J. Barauskas, T. Nylander and F. Tiberg, ACS Appl. Mater. Interfaces, 2014, 6, 7063–7069 CrossRef PubMed.
- B. K. Johnson and R. K. Prud'homme, Aust. J. Chem., 2003, 56, 1021–1024 CrossRef CAS.
- J. Han, Z. X. Zhu, H. T. Qian, A. R. Wohl, C. J. Beaman, T. R. Hoye and C. W. Macosko, J. Pharm. Sci., 2012, 101, 4018–4023 CrossRef CAS PubMed.
- M. O. Jara, J. Catalan-Figueroa, M. Landin and J. O. Morales, Drug Delivery Transl. Res., 2017, 8, 1797–1806 CrossRef PubMed.
- H. Shen, S. Y. Hong, R. K. Prud'homme and Y. Liu, J. Nanopart. Res., 2011, 13, 4109–4120 CrossRef CAS.
- D. Presa-Soto, G. A. Carriedo, R. de la Campa and A. Presa Soto, Angew. Chem., Int. Ed., 2016, 55, 10102–10107 CrossRef CAS PubMed.
- Z. Ding, C. Gao, S. Wang, H. Liu and W. Zhang, Polym. Chem., 2015, 6, 8003–8011 RSC.
- K. H. Ku, J. M. Shin, D. Klinger, S. G. Jang, R. C. Hayward, C. J. Hawker and B. J. Kim, ACS Nano, 2016, 10, 5243–5251 CrossRef CAS PubMed.
- V. Miceli, V. Meli, M. Blanchard-Desce, T. Bsaibess, M. Pampalone, P. G. Conaldi, C. Caltagirone, M. Obiols-Rabasa, J. Schmidt and Y. Talmon, RSC Adv., 2016, 6, 62119–62127 RSC.
- P. P. Wibroe, D. Ahmadvand, M. A. Oghabian, A. Yaghmur and S. M. Moghimi, J. Controlled Release, 2016, 221, 1–8 CrossRef CAS PubMed.
- S. D. Allen, S. Bobbala, N. B. Karabin, M. Modak and E. A. Scott, ACS Appl. Mater. Interfaces, 2018, 10, 33857–33866 CrossRef CAS PubMed.
- V. Mittal and N. B. Matsko, Analytical Imaging Techniques for Soft Matter Characterization, Springer, 2012, pp. 13–29 Search PubMed.
- S. Rizwan, Y.-D. Dong, B. Boyd, T. Rades and S. Hook, Micron, 2007, 38, 478–485 CrossRef CAS PubMed.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS PubMed.
- K. Sing, Colloids Surf., A, 2001, 187, 3–9 CrossRef.
- C. Park, Y. La, T. H. An, H. Y. Jeong, S. Kang, S. H. Joo, H. Ahn, T. J. Shin and K. T. Kim, Nat. Commun., 2015, 6, 6392 CrossRef PubMed.
- P. Gill, T. T. Moghadam and B. Ranjbar, J. Biomol. Tech., 2010, 21, 167 Search PubMed.
- B. E. McKenzie, F. Nudelman, P. H. Bomans, S. J. Holder and N. A. Sommerdijk, J. Am. Chem. Soc., 2010, 132, 10256–10259 CrossRef CAS PubMed.
- D. Shindo and T. Oikawa, Analytical Electron Microscopy for Materials Science, Springer, 2002, pp. 81–102 Search PubMed.
- A. A. Saei, M. Yazdani, S. E. Lohse, Z. Bakhtiary, V. Serpooshan, M. Ghavami, M. Asadian, S. Mashaghi, E. C. Dreaden, A. Mashaghi and M. Mahmoudi, Chem. Mater., 2017, 29, 6578–6595 CrossRef CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214 CrossRef CAS PubMed.
- B. Pelaz, P. del Pino, P. Maffre, R. Hartmann, M. Gallego, S. Rivera-Fernández, J. M. de la Fuente, G. U. Nienhaus and W. J. Parak, ACS Nano, 2015, 9, 6996–7008 CrossRef CAS PubMed.
- N. Bertrand, P. Grenier, M. Mahmoudi, E. M. Lima, E. A. Appel, F. Dormont, J.-M. Lim, R. Karnik, R. Langer and O. C. Farokhzad, Nat. Commun., 2017, 8, 777 CrossRef PubMed.
- A. N. Ilinskaya and M. A. Dobrovolskaia, Polymer Nanoparticles for Nanomedicines, Springer, 2016, pp. 505–520 Search PubMed.
- C.-F. Wang, E. M. Mäkilä, C. Bonduelle, J. Rytkönen, J. Raula, S. Almeida, A. Närvänen, J. J. Salonen, S. Lecommandoux, J. T. Hirvonen and H. A. Santos, ACS Appl. Mater. Interfaces, 2015, 7, 2006–2015 CrossRef CAS PubMed.
- R. R. Arvizo, O. R. Miranda, M. A. Thompson, C. M. Pabelick, R. Bhattacharya, J. D. Robertson, V. M. Rotello, Y. S. Prakash and P. Mukherjee, Nano Lett., 2010, 10, 2543–2548 CrossRef CAS PubMed.
- J. Lin, H. Zhang, Z. Chen and Y. Zheng, ACS Nano, 2010, 4, 5421–5429 CrossRef CAS PubMed.
- R. R. Arvizo, O. R. Miranda, D. F. Moyano, C. A. Walden, K. Giri, R. Bhattacharya, J. D. Robertson, V. M. Rotello, J. M. Reid and P. Mukherjee, PLoS One, 2011, 6, e24374–e24374 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Adv. Drug Delivery Rev., 2017, 108, 51–67 CrossRef CAS PubMed.
- X. Yi, X. Shi and H. Gao, Phys. Rev. Lett., 2011, 107, 098101 CrossRef PubMed.
- J. Sun, L. Zhang, J. Wang, Q. Feng, D. Liu, Q. Yin, D. Xu, Y. Wei, B. Ding, X. Shi and X. Jiang, Adv. Mater., 2015, 27, 1402–1407 CrossRef CAS PubMed.
- X. Banquy, F. Suarez, A. Argaw, J.-M. Rabanel, P. Grutter, J.-F. Bouchard, P. Hildgen and S. Giasson, Soft Matter, 2009, 5, 3984–3991 RSC.
Footnote |
| † All authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |