
Transition metal (Fe, Co, Ni) fluoride-based materials for electrochemical energy storage
Nannan
Zhang†
,
Xiao
Xiao†
and
Huan
Pang
 *
*
School of Chemistry and Chemical Engineering, Institute for Innovative Materials and Energy, Yangzhou University, Yangzhou, 225009, Jiangsu, P. R. China. E-mail: huanpangchem@hotmail.com; panghuan@yzu.edu.cn; Web: http://huanpangchem.wix.com/advanced-material
First published on 6th September 2018
Abstract
The improvement of advanced battery performance has always been a key issue in energy research. Therefore, it is necessary to explore the applications of excellent materials in advanced batteries. Transition-metal (Fe, Co, Ni) fluoride-based materials exhibit excellent chemical tailorability due to their different functional groups, and they have attracted wide research interest for use in next-generation electrochemical energy storage. This review introduces methods to synthesize transition metal (Fe, Co, Ni) fluoride materials and their applications in batteries and supercapacitors. We also present the current challenges and future opportunities of iron fluoride in electrochemistry, including processing techniques, composite properties, and prospective applications. It is believed that in the future, the research and influence of iron fluoride and its composites will be more far-reaching and lasting.
 Huan Pang | Huan Pang received his PhD degree from Nanjing University in 2011. He is now a university distinguished professor at Yangzhou University. In the past 10 years, his group has been engaged in the design and synthesis of functional nanomaterials, especially for MOF-based materials. He is on the editorial board of FlatChem and managing editor of EnergyChem, Elsevier. He has published more than 200 papers in peer-reviewing journals including Chemical Society Reviews, Energy Environ. Sci., and Advanced Materials, with 5300 citations (H-index = 40). His research interests include the development of inorganic nanostructures and their applications in nanoelectrochemistry with a focus on energy devices. |
1. Introduction
Energy conversion and storage are a major global challenge due to problems associated with fossil fuels, such as pollution and non-sustainability.1,2 Environmental pollution caused by energy consumption has become increasingly prominent, making the discovery of renewable energy sources to relieve the current situation an urgent topic. Next-generation energy storage devices that satisfy the requirements of larger capacity, better security, faster recharge, and lower cost are in high demand. Consequently, several new energy storage device concepts have been developed, including lithium-ion batteries (LIBs),3,4 sodium-ion batteries (SIBs),5 Zn–air batteries,6 many other rechargeable batteries,7 and supercapacitors (SCs).8 Among various available energy storage technologies, development of low-cost and earth-abundant materials into outstanding electrodes is considered to be the most rational way to overcome current issues of energy storage applications.9–15In traditional LIBs, cathode materials such as lithium cobalt oxide, lithium nickel oxide, and lithium manganese oxide have unfortunate drawbacks as follows: (1) they possess intrinsically slow ionic diffusion; (2) they exhibit structural decomposition/reconstruction during repeated conversion reactions; (3) the theoretical gravimetric capacities of commercial cathode materials are still insufficient for practical applications despite several breakthroughs in cathode material chemistry; and (4) their high cost is still a major obstacle for the successful commercialization of fluoride-based cathodes in LIBs.16–18 And SIBs also have obvious disadvantages. The sodium-ion radius is larger than that of lithium-ion, making it more difficult to intercalate and remove sodium-ions in the battery material. And in the case of electrode materials, SIBs depend mostly on intercalated metal oxide cathode materials.19–21 As a result of using expensive zinc-, copper- and vanadium-containing materials, which also cause serious environmental problems, the adoption of SIBs in large-scale production is limited.22 Furthermore, the problems of Zn–air batteries are also obvious, for example, the requirement of ventilation is high, and the phenomenon of leakage exists, and the flow of electrolyte is not uniform. Although SCs have many advantages compared with current commercially available batteries, electrolyte leakage caused by improper use of these SCs must still be improved.
Under these circumstances, as mentioned, using a fluoride material as an alternative cathode material for LIB applications is possible because fluoride is used as a dopant to improve the stability and safety of LIBs.16,23,24 These fluoride species have many obvious advantages, such as relatively high energy densities when used in rechargeable power sources and low costs when compared to its oxide and sulfide counterparts.25 The discovery of iron-based fluorides has drawn considerable focus from researchers because of their predominant performance. Furthermore, iron-based fluorides are especially popular as fluoride materials. When compared with other materials, iron-based fluoride shows an excellent chemical tailorability due to their distinct functional groups. Previous studies by researchers confirm the great progress in this field.26–28 To meet the increasingly ambitious requirements for more energy efficient and environmentally friendly materials, great efforts must still be put forth.
When examining the data from previous years (2014.1–2017.12) using Fig. 1, we find that in electrochemical energy storage devices, among all of these papers, the majority of articles are about LIBs and the number of iron-based fluoride research articles has increased and remained at a high level. We speculate that iron fluoride materials have been widely studied due to their advantages of simple preparation and high iron salt content. In this review, the strategies employed to synthesize electrode materials of LIBs, SIBs, Zn–air batteries and SCs are firstly introduced, the including chemical precipitation method,25–42 vapor–solid method,30 hydrothermal method,48,52 solvothermal method,31,42,55–64 microwave synthesis method,23,32,42,65–68 ion synthesis method,30,71,74–81 chemical ball-milling method,41 sol–gel method71 and others.73–77 Second, the performance and applications are also discussed. Finally, the present challenges and future electrochemical opportunities of iron fluoride are provided. We conduct this review to provide a comprehensive summary of the most recent advances in the rational design of iron fluoride materials.
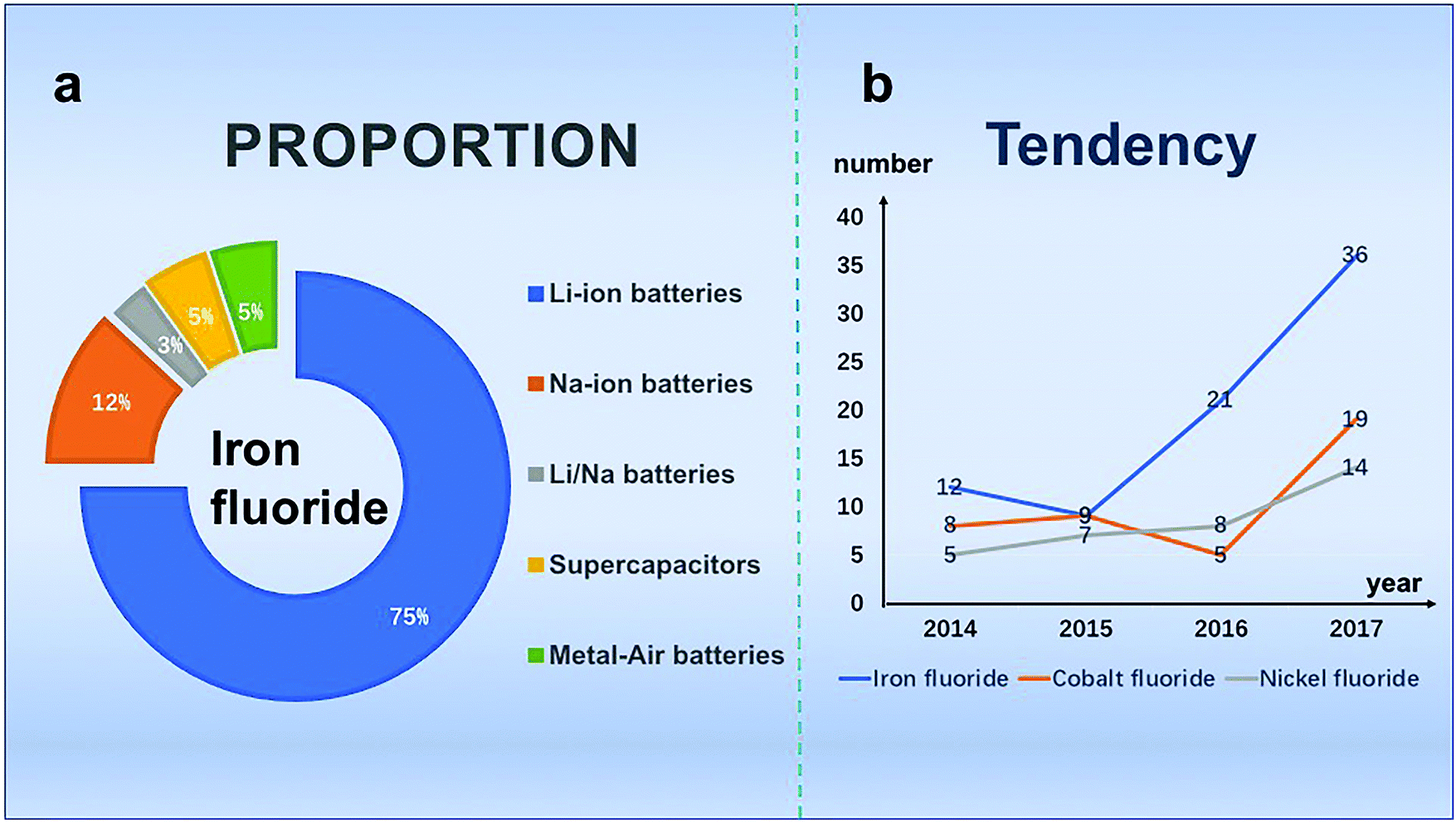 | ||
| Fig. 1 (a) The proportion of iron fluorides in electrochemical storage materials; (b) the number of publications on iron-based materials and their composites during the past years. Data were collected from Web of science. | ||
2. Synthesis of iron fluoride
2.1 Chemical precipitation method
Chemical precipitation is the treatment of water by adding a chemical agent to the water so that the dissolved substances that need to be removed in the water are converted into difficult to dissolve substances and precipitated. It provides a suitable aqueous environment that can be easily introduced into the lattice of the as-prepared iron fluoride so the chemical precipitation method is an excellent synthesis approach. Although in conventional methods there are a lot of options for fluoride sources, for example, HF is well known as the most commonly used fluoride source, but the use of it can lead to safety issues.33–41 In addition, ionic liquids (ILs) have also been applied as fluoride sources, but their high cost is a concern for industry applications.4,42–44However, the use of chemical precipitation has promoted the use of many innovative materials, which has effectively evaded the use of these materials. These new materials improve the properties of electrode materials by changing the structure and morphology of the fluorine source. So far, much work has focused on the synthesis of fluoride sources for the sake of better performance. Great improvements to the capacity, charge and discharge rates, and cycling performance during charging and discharging have been made by Zhang et al.29 They applied unique morphologies by fabricating ternary composites, displaying the important effects of morphology on electrochemical performance. As shown in Fig. 2a, there were three important stages in the synthesis of three-dimensional (3D) architecture SnO2/Ni/polyvinylidene fluoride (PVDF) composites. They firstly synthesized a PVDF fiber membrane. When coating conductive Ni metal on PVDF fibers with the chemical precipitation method, the membrane developed into a conductive 3D porous membrane that could be employed as a current collector. Finally, the SnO2 was firmly attached to the conductive Ni/PVDF fibers to prepare the pores and the 3D architecture SnO2/Ni/PVDF fiber membrane. In order to get a bigger breakthrough on this basis, Zhang et al. used the same method to produce electrode materials. A MnO2 nanosheet network anchored on a Ni/PVDF coaxial fiber membrane was successfully fabricated using a three-step route. Similar to the above, they prepared a PVDF fiber membrane; then, the Ni (shell)/PVDF (core) coaxial fiber membrane, which has a core–shell structure, was achieved by chemical precipitation; and last, they obtained the manganese dioxide nanosheet network, which was grown on a Ni/PVDF in-line fiber membrane. In addition, their product was observed by scanning electron microscopy (SEM). Fig. 2b shows a SEM image of the entire PVDF fiber membrane. The as-prepared PVDF nanofibers exhibit a long and round morphology and have different diameters ranging from 500 to 700 nm. Fig. 2c exhibits the morphology of a Ni/PVDF coaxial fiber. After coating with Ni, it could be seen that the average fiber diameter increased from 700 to 1000 nm. As shown in Fig. 2c, the PVDF fiber is evenly coated by Ni metal to prepare the Ni (shell)/PVDF (core) in-line fiber. From the inset of Fig. 2d, the average fiber diameter after coating with MnO2 increased compared with the Ni/PVDF in-line fibers. Furthermore, as shown in the high magnification image of the MnO2/Ni/PVDF coaxial fiber membrane, the MnO2 nanosheets may be interconnected to generate an enhanced porous network (Fig. 2e). The as-prepared porous PVDF fiber membrane configuration is revealed in Fig. 2f and g, showing a polyporous conductive Ni/PVDF fiber membrane and the SnO2/Ni/PVDF composite with 3D structure, respectively. Representative SEM images of the SnO2 nanosheets/Ni/PVDF ternary composite under different magnifications are shown in Fig. 2f and g. The SnO2 nanosheets are suitably developed on the surface of the conductive Ni/PVDF fibers on a large scale (Fig. 2f). The high magnification image (Fig. 2g) shows that the SnO2 nanosheets mainly developed in an upright random orientation on the conductive Ni/PVDF fiber current collector, and many pores existed randomly within the SnO2 network.
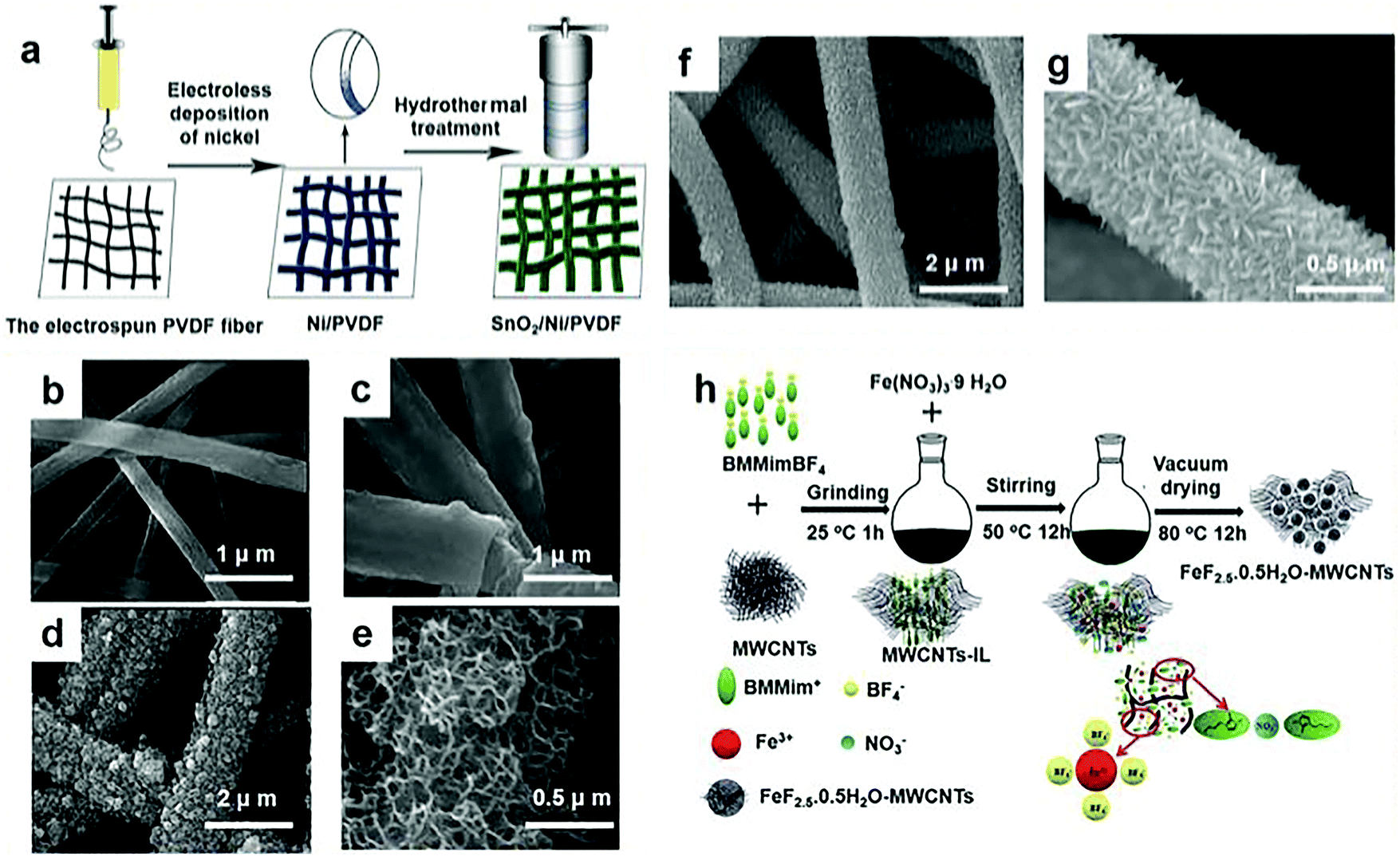 | ||
| Fig. 2 (a) Schematic diagram of the preparation of three element composites; (b) the SEM images of the pure PVDF fiber membrane; (c) the in-line Ni/PVDF fiber membrane after coating with Ni; (d and e) the in-line MnO2/Ni/PVDF fiber membrane at various magnifications; (f and g) the SEM images of the freestanding SnO2/Ni/PVDF ternary composite at different magnifications; (h) schematic illustration of the synthetic process for FeF2.5·0.5H2O–(multi-walled carbon nanotubes) MWCNT cathode materials; BMMimBF4 represents 1-butyl-2,3-dimethylimidazolium tetrafluoroborate, BMMim+ represents 1-butyl-2,3-dimethylimidazole ion, and BF4− represents tetrafluoroboric acid ion. (a–e) Reproduced with permission.29 Copyright 2015, Elsevier-Ltd; (f and g) reproduced with permission.48 Copyright 2015, Elsevier-Ltd; (h) reproduced with permission.47 Copyright 2016, The Royal Society of Chemistry. | ||
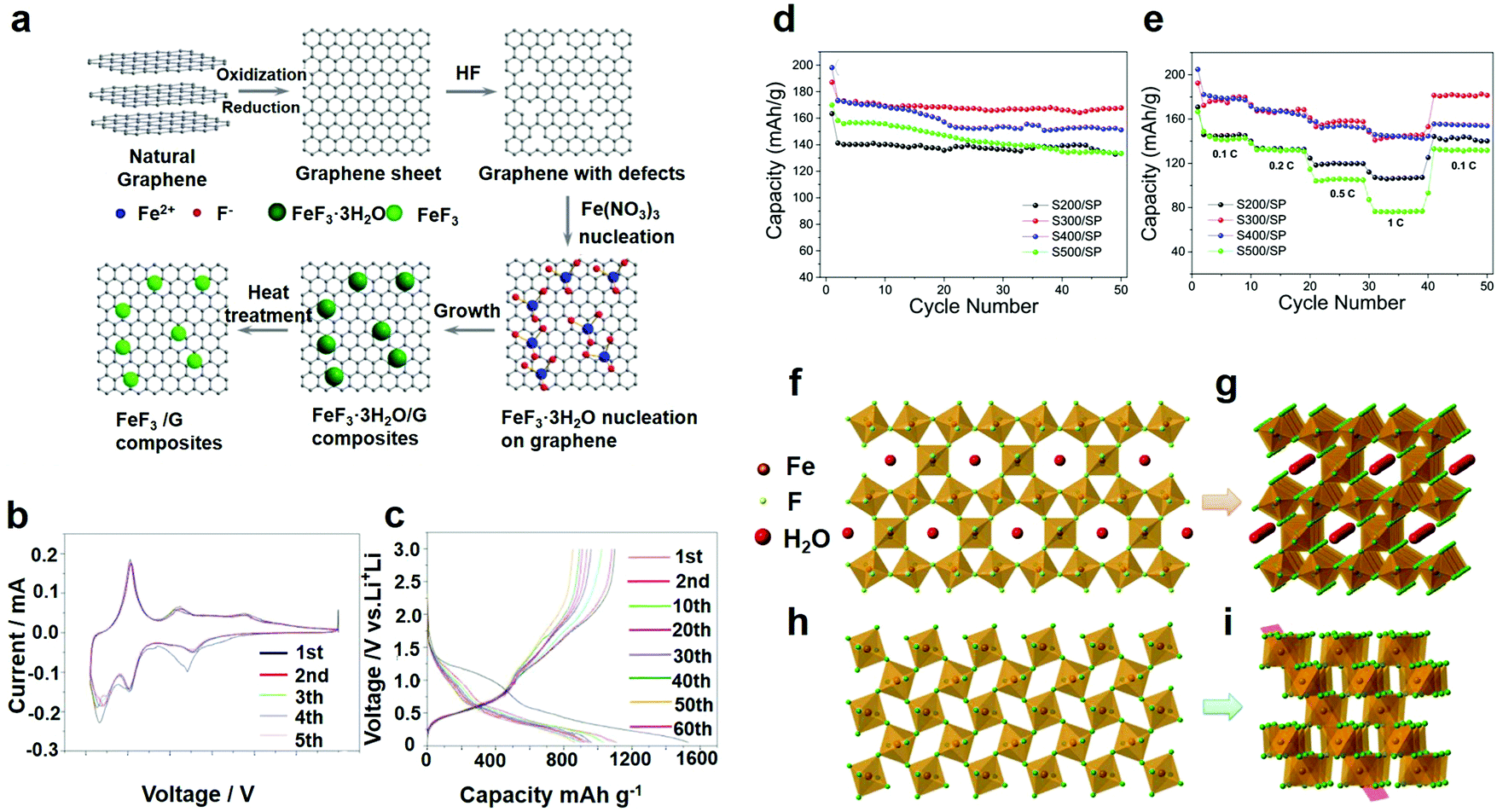
The mesoporous nanostructure provided easy permeability for the electrolyte and enabled the electrolyte to closely contact the inner surface, which contributed to a shorter transfer pathway for Li/Na-ions and prioritized the chemical conversion reactions. Furthermore, this structure provided a good buffer for the volume change in the Li or Na-ion insertion/extraction process, therefore optimizing the cycle performance.46 The fluoride composite particle sol can be fixed on the surface of graphene to further form the layered portion of the FeF3·xH2O/graphene (G) nanocomposite. In other words, FeF3·xH2O/G was composed of a large number of fluoride nanoparticles attached to the graphene surface or embedded in graphene. Therefore, very recently, Shen et al. made a kind of mesoporous material with a high specific surface area and good cycle performance by chemical precipitation.45 In addition to changing the appearance and structure of the materials, researchers also start with the methods of processing, and adopt different processing methods to achieve better performance. Using vacuum drying instead of heat treatment, Wei et al. proposed another possible reaction in 2016, in which the properties of the product were improved from the point of view of network structure as shown in Fig. 2h.47
As an important branch of chemical precipitation, the liquid-phase method has been widely adopted by researchers. As electrode materials, some researchers recently did a lot of testing and synthesized FeF3·3H2O and a FeF3·3H2O precursor assembly by the liquid-phase method and the growth processes were recorded. The slow assembling process occurred as a two-step procedure involving self-templating and self-aggregation. Shi et al. obtained FeF3·3H2O crystals with high crystallinity by the liquid-phase method.49 After ball-milling, the synthesized FeF3·3H2O particles were dispersed well in the carbon black matrix and obtained large active material aggregates. At the end of stirring, the precipitate exhibited an irregular morphology. In the process of mixing, polyethylene glycol (PEG) played an important role. By adding PEG to the mixture solvent, the viscosity increased and the fluidity decreased, which affected the reaction kinetics. As a result, the reactive interface increased, and the reaction time was reduced,50 which benefited the growth and formation of the FeF3·3H2O crystals. Of all these experiments, the research of Bai et al.49 was one of the most significant references. Regarding the kinetically controlled growth, the seeds form a thermodynamically unfavorable plate-like shape, as shown for the product in Fig. 3a–f. Fig. 3a shows the SEM of the product with no further alterations. With increasing standing time, the nearby plate crystals orient themselves to be close to each other and spontaneously form ordered interpenetration twins, as shown in Fig. 3c and d. This step is driven by the oriented attachment mechanism. The longer the standing time is, the better the morphology will be. After 12 h, well-developed flower-like particles were obtained. In addition, with increasing standing time, the edges of the particles became thinner, which favored a tightly knit structure. Minimal difference between Fig. 3e and f was observed, indicating that the 12 h standing time was sufficient. The above examples show that the technology of preparing an electrode material by a chemical precipitation method is relatively mature, the degree of automation is high, and the investment is less and the cost is low in economic terms, but it is necessary to control the use of the precipitate well, otherwise there will be the formation of additional metal complex.
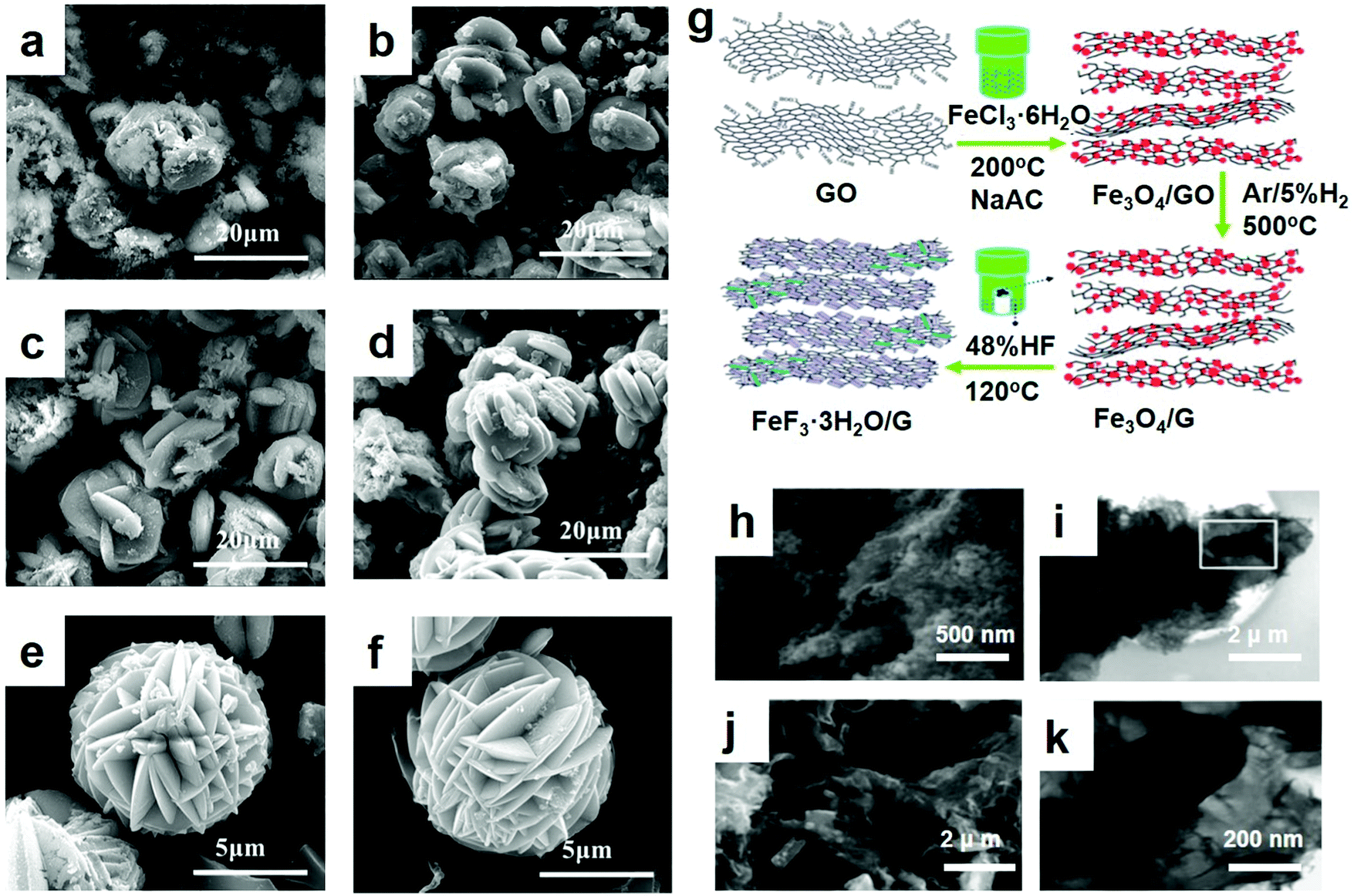 | ||
| Fig. 3 SEM diagrams of the FeF3·3H2O after standing for (a) 0 h, (b) 3 h, (c) 6 h, (d) 9 h, (e) 12 h, and (f) 15 h; (g) schematic illustration of the fabrication procedure, GO stands for graphene oxide; SEM images of (h) Fe3O4/G and (i) FeF3/G; (j) overview; (k) transmission electron microscopy (TEM) image of FeF3/G. (a–f) Reproduced with permission.49 Copyright 2016, Elsevier-Ltd; (g–k) reproduced with permission.16 Copyright 2013, The Royal Society of Chemistry. | ||
2.2 Vapor–solid method
FeF3/G was produced by thermal conversion from Fe3O4/G via a vapor–solid method, which possessed several significant advantages: (1) it can prevent the increase in the FeF3 particle sizes through a vapor–solid reaction instead of a solution reaction. Due to the high electronegativity of iron ions, the particles of the material grow rapidly after nucleation in solution. (2) The preparation of Fe3O4/G or the conversion from Fe3O4/G to FeF3/G can be scaled up to slightly increase production. (3) The synthesis strategy is versatile and can be conveniently expanded to other iron oxides/hydroxides to prepare FeF3 and other metal oxides to fabricate MxFy or MxFy/G composites as cathode materials for LIBs.51 A cathode material using the as-prepared Fe/Fe2O3/C nanocomposite which was wrapped was successfully fabricated by Ma et al.33 They place the wrapped powder in a Teflon cylinder and placed it above the level of HF solution, in order to facilitate the gas–solid process to get a better application, making powder wrapping more full. After sealing with stainless steel, the coated powder cylinder evaporates with HF solution. For comparison, they prepared another sample in an argon-filled glove box. After it had been firmly sealed, the autoclave was heated. Soon thereafter, they also synthesized improved desirable products based on normal cathode materials synthesized by the vapor–solid growth mechanism, as shown by Fig. 3g.16 The SEM image of the as-synthesized Fe3O4/G nanocomposites demonstrates that the 50 nm Fe3O4 nanoparticles were homogeneously anchored upon the graphene sheets. After reacting with HF vapor and post-heat-treating, the Fe3O4/G nanoparticles transformed entirely to FeF3/G, as revealed in Fig. 3h, during which the morphology underwent a transformation from 50 nm nanoparticles to rectangular rods with a length ranging from 100 nm to 500 nm and a width of approximately 150 nm. As revealed in Fig. 3i, the TEM image demonstrates the distinct structure of the generated FeF3/G, namely, the rectangular rods became completely enwrapped with multi-sheet graphene. The image of one rod marked in Fig. 3i was magnified and is revealed in Fig. 3k, expressly representing FeF3, showing a length of 300 nm and a width of 150 nm randomly in the graphene matrix. Because the graphene matrix can hold the volume change and serve as a current collector, it is considered that the electrochemical properties of iron-based fluoride nanocomposites can be polished by the preparation of FeF3 composites based on graphene.2.3 Hydrothermal method
The biggest advantage of the hydrothermal method is that the crystalline powder can be obtained directly without sintering at high temperature, which avoids the possibility of forming a hard agglomerates of particles, and also saves the grinding and the resulting impurities. In the hydrothermal process, the crystal structure of the nanoparticles can be controlled by adjusting the reaction conditions, the crystal morphology and the grain purity can be controlled, which is beneficial for crystals with few growth defects and good orientation, and the crystallinity of the synthesized product is high and the particle size of the product is easy to control. The obtained powders have high purity, good dispersion, uniform distribution, no agglomeration, good crystal shape, controllable shape, and favorablity for environmental purification. A hydrothermal method can be used to prepare monocrystalline and special compound powders with two or more components. Powder materials such as metals, oxides and composite oxides can be prepared. The particle size ranges from 0.1 μm to several microns, some of which can reach tens of nanometers. Since 3D technologies are being continuously developed, here, Zhang et al.48 fabricated a novel MnO2/Ni/PVDF in-line fiber membrane through the hydrothermal method. Ni, which serves as a conductive 3D framework for the manageable deposition of a MnO2 nanosheet network, was attached suitably to the porous PVDF fibers. Since it served as an anode in LIBs, the 3D MnO2/Ni/PVDF in-line fiber membrane demonstrated the following strong points: (1) the electrospun PVDF nanofiber membranes possessed high porosity (75%), and the micropores that were created in the smooth fibers provided easier access for the electrolyte in the electrodes; (2) the PVDF fibers attached to a layer of nickel had high electrical conductivity; (3) the mesoporous nanosheet MnO2 network was firmly fixed on the highly conductive Ni/PVDF fiber to fabricate the MnO2/Ni/PVDF in-line fiber membrane. Additionally, the 3D Ni/PVDF frame and the outer MnO2 network coat could prevent structural collapse and destruction. The properties of these composites cause the in-line MnO2/Ni/PVDF fiber-combined film to possess a very high charge and discharge capacity and enhanced performance.To improve the cycling ability and rate performance, Song et al. also made a lot of effort. They built up a close interface by the hydrothermal method, including iron oxide nanoparticles in mesoporous carbon (CMK-3) in an order manner;52 the coupling connection then remains rather than being topologically converted to ferric fluoride hydrate by the iron oxide precursor; finally, the mesoporous nanostructured FeF2 is constructed from the hydrate. The detailed procedure is distinctly shown in Fig. 4a as well. The electrochemical properties of FeF3 hydrated nanocrystalline compounds were improved by the thin layer morphology and porous structure of FeF3 nanoparticles. The thin layer morphology of the nanostructure can improve the specific capacity by providing sufficient reaction area for electrochemical reaction, while a nanoporous structure can improve the polarization in the process of discharge–charge for the sake of improving the rate of the electrode capacity and capacity retention.
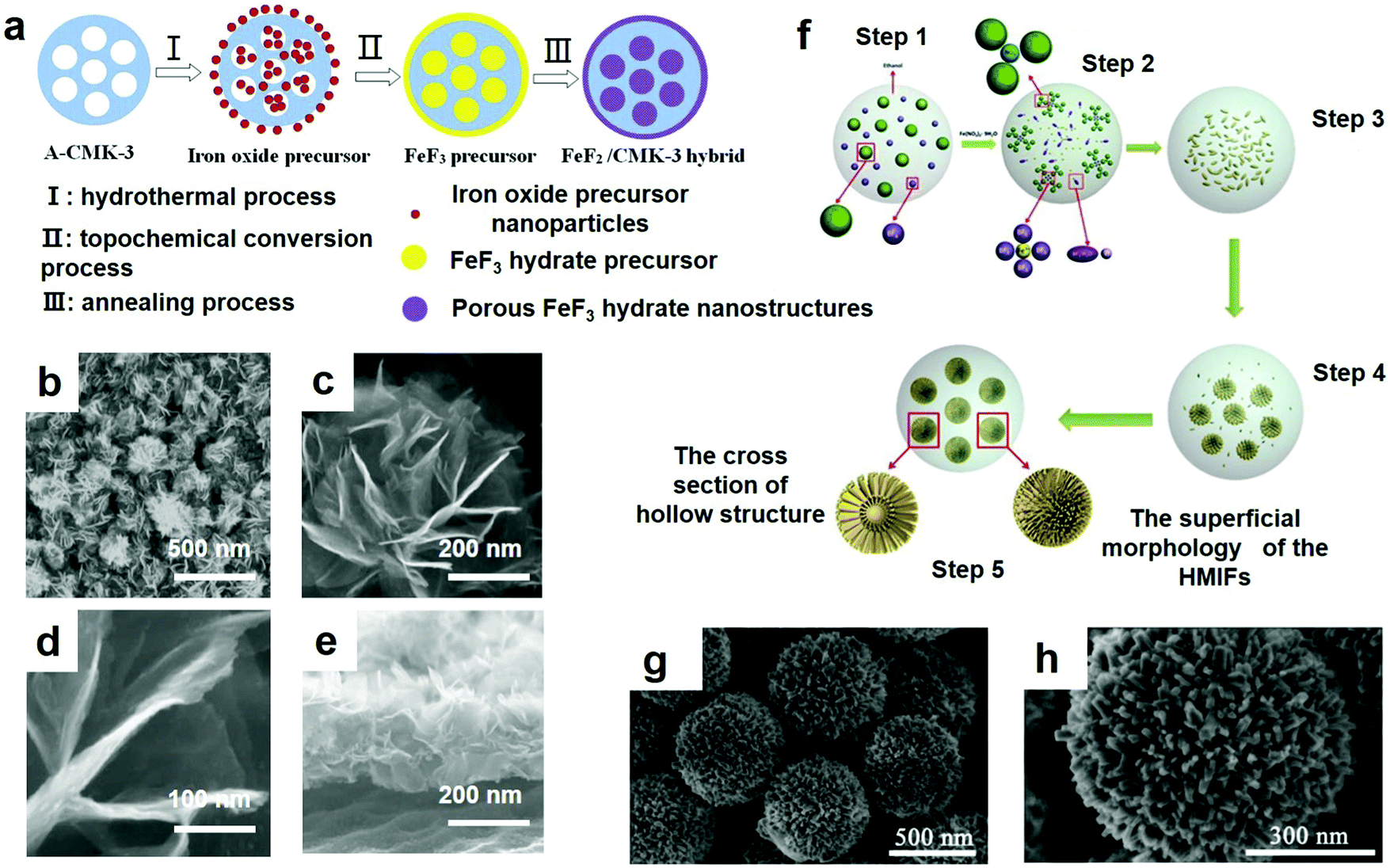 | ||
| Fig. 4 (a) Synthesis of porous FeF2 graded nanostructured CMK-3 hybrid composites; (b) the low magnification SEM image of the FeF3·0.33H2O flower-like array; (c) the locally enlarged SEM image of the array; (d) the SEM of nanopetals of the FeF3·0.33H2O microflower; (e) the SEM of the side view of the FeF3·0.33H2O flower-like array; (f) scheme of the shape mechanism of hierarchical mesoporous structured iron based fluorides (HMIFs) with partially hollow structure obtained at 90 °C, 36 h with 1 mL fluorine source; (g and h) SEM images. (a) Reproduced with permission.53 Copyright 2015, The Royal Society of Chemistry; (b–e) reproduced with permission.54 Copyright 2014, Elsevier-Ltd; (f–h) reproduced with permission.42 Copyright 2014, The Royal Society of Chemistry. | ||
2.4 Solvothermal method
A solvothermal method was developed based on the hydrothermal method, but the employed solvent is an organic solvent rather than water. When a solvothermal reaction occurs in a non-aqueous solvent, one or more of the precursors is dissolved under liquid or supercritical circumstances, is dispersed in the solution and becomes more active; as the reaction occurs, the products slowly build up. Therefore, it has the advantages of faster reaction kinetics, shorter processing times, a higher crystallinity and yield, and uniform particle size distributions. Most importantly, it is cost-effective, environmentally friendly, and easy to scale. The materials obtained from this method usually show great enhancements in discharge/charge, cycling ability, rate capability, and inexpensive synthesis in comparison with conventional methods. Relatively, the method is very ideal to synthesize iron-based fluorides that possess optimal morphology by using such an appropriate chemical method and the solvothermal method has been widely applied in preparing nanomaterials, porous materials, and hollow materials in recent years.55–57Recently, 3D hierarchical structures equipped with lower dimensional nanoscale building blocks have attracted increasing attention.58–61 An ordered 3D hierarchy not only inherited the better features of the original building blocks but also exhibited the new physicochemical properties of the secondary architectures. The 3D hierarchically structured electrodes provide a short electron transport path and Li+ diffusion determined by the small thickness of the nanounits, strong connection among building blocks and excellent porosity for the effective transport of Li+. However, the fabrication of self-supported 3D hierarchical nanostructure arrays with desirable architectures continues to be an enormous obstacle. Li et al. reported the synthesis of a porous 3D hierarchical iron fluoride flower-like array directly grown on Ti foil by a solvothermal approach and studied the morphology of the product. Compared with other corrosive fluoride sources (such as HF), 1-butyl-3-methylimidazolium tetrafluoroborate ILs (BmimBFC4) serve as the fluoride source and are shown to be more environmentally friendly and exhibit better operational security.62–64 To the best of our knowledge, there is no report on nanostructures iron fluoride arrays grown directly on a conducting substrate.
The SEM image of a representative sample consisting of uniform, flower-like architectures approximately 1 μm in diameter is illustrated in Fig. 4b. Fig. 4c and d show the morphology of the flower-like nanostructures in detail, which reveals that the entire structure of the 3D hierarchical architecture is constructed by dozens of nanopetals. These nanopetals are approximately 10 nm thick and 500 nm wide, and the centers are connected to each other to form a 3D layered structure. Fig. 4e shows a side view image of the array, showing that these micro-flowers developed from the Ti foil and exhibited good contact with the substrate.
There is thereby a critical need but it is still a significant challenge to seek a simple approach to synthesizing iron fluoride with excellent properties. Soon after, Lu et al. successfully synthesized HMIFs by an easy solvothermal method.42 The ionic liquid (IL) BmimBFC4 was used as a fluoride source, while Fe(NO3)3·9H2O served as the iron source. Its synthesis route is shown in Fig. 4f. The sample was annealed at 90 °C for 36 h with 1 mL of the fluorine source, and the SEM images of the obtained products are revealed in Fig. 4g and h. As clearly exhibited in Fig. 4g, the products have formed emblematical hierarchical nanostructures with a relatively uniform size distribution with diameters of approximately 600 nm. In addition, Fig. 4h clearly shows many ordered and radial nanorods formed during the fabrication of the HMIFs.
2.5 Microwave synthesis method
Compared with other methods for the synthesis of fluoride materials, it is generally accepted that microwave-assisted synthesis is a promising method. In the fields of synthetic chemistry and material preparation, the microwave-assisted method has been a rapidly growing area of research since it was reported.65,66 Different from traditional heating methods, the advantages of microwave synthesis methods depend on the rapid volume heating process, which provides reagents and solvents. Furthermore, intermediates and products can be rapidly and uniformly heated, thereby gaining a higher reaction rate and efficiency, lower energy consumption and decreased time, which is favorable for the formation of small monodisperse nanostructures.67 In particular, when the IL participates in the reaction system, the high polarity generated by a large amount of organic cations becomes an eminent medium for absorbing microwaves, resulting in a very high heating rate. In recent years, due to the combined advantages of room temperature ILs and microwave heating, nanostructured materials with tailorable morphologies have been easily prepared.68To achieve more insight, many researchers have carried out a lot of research and many great breakthroughs have been reported for this method. Jiang et al. stand out in this field. They synthesized a product by a microwave-assisted synthesis method,23 as shown in Fig. 5a. BMMimBF4 served as a co-solvent, a source of fluorine and the structure of the indicator. Fe(NO3)3·9H2O served as the source of iron and water to promote IL hydrolysis. To prepare Fe2F5·H2O, Fe(NO3)3·9H2O was gradually added to the IL under rapid stirring, and the solution remained until the IL medium formed a milky white precipitate. They washed the resulting product with acetone and centrifuged it so that they could remove the residual IL and other organic substances and get more pure products. The Fe2F5·H2O/G compounds were synthesized under the same conditions, except that more G was added before the other steps. Since the Fe(NO3)3·9H2O powder was added into the BMMimBF4 medium, the water would first be separated from the Fe(NO3)3·9H2O and then enter into the hydrophilic IL medium. Fe3+ and NO3− could be respectively dissolved by the BF4− anions and larger imidazolium cations. Weakly coordinating BF4 anions were readily hydrolyzed to form BF3·H2O and F− in the presence of water. In the last step, the solvated Fe3+ combined with the F− to form small monodisperse nanostructures. Because of the interfacial tension, a well-separated spherical morphology was formed. To prepare Fe2F5·H2O/G compounds, this procedure started with the interaction between the imidazolium cation groups of the IL and the p-electrons of graphene with extra graphene added. Therefore, the prepared fluoride particles were firmly fixed on the surface of graphene.
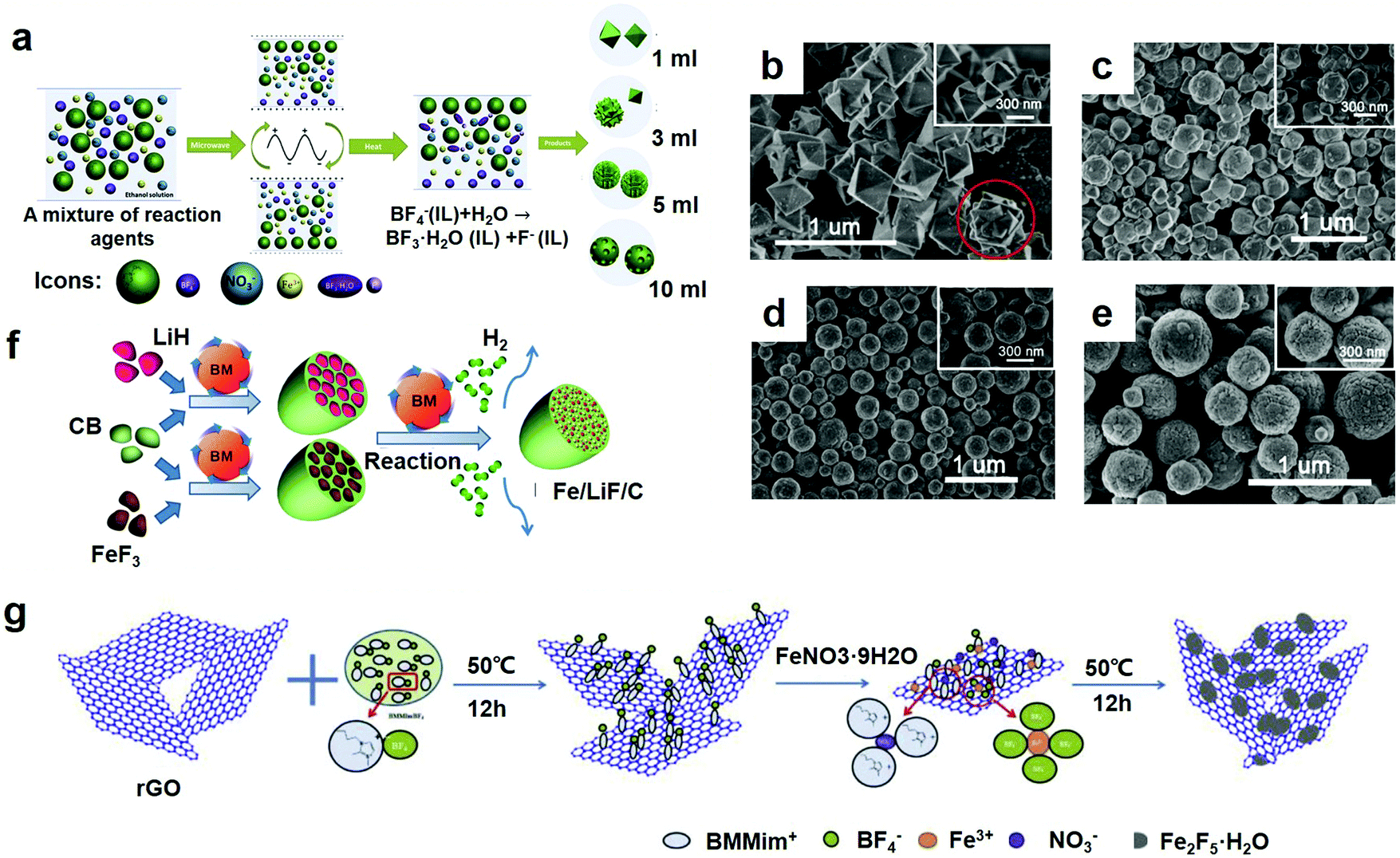 | ||
| Fig. 5 (a) Schematic of the synthesis of different morphologies for Fe1.9F4.75·0.95H2O materials; SEM images of the F-1 sample (b), F-3 sample (c), F-5 sample (d) and F-10 sample (e); (f) preparation of Fe/LiF/C nanocomposites by mechanical ball milling and chemical ball milling. BM stands for ball milling and CB represents carbon black; (g) scheme of the synthesis mechanism of Fe2F5·H2O/reduction of graphene oxide (rGO) cathode materials. (a–e) Reproduced with permission.32 Copyright 2013, Elsevier-Ltd; (f) reproduced with permission.30 Copyright 2016, Elsevier-Ltd; (g) reproduced with permission.69 Copyright 2016, Elsevier-Ltd. | ||
As for this method, others have performed a number of corresponding studies. The product was synthesized by Lu et al. through the microwave method.42 Through optimization of the synthesis temperature, reaction time, and amount of IL, a series of nanostructured iron-based fluoride materials with different morphologies and phases were fabricated. In particular, for the sample HMIFs obtained at a relatively high temperature for 36 h with 1 mL of fluorine, first, the metal precursor nitrate salts were dissolved in ethanol solvent and stirred at room temperature to form solution A. Then, at room temperature, BmimBF4, which served as a fluorine source, was added to solution A. They then transferred the mixtures to a Teflon autoclave, sealed and heated it, and kept it at this temperature. After the reaction was complete, they washed and centrifuged the product to remove the ILs and other organic substances. They dried the products under vacuum and saved them for future investigation. The products were defined as F-X (F stands for iron-based fluorides, and X stands for the amount of IL used in the preparation) for the sake of distinguishing the different morphologies obtained for the materials. Therefore, the samples F-1, F-3, F-5, and F-10 were respectively prepared with 1 mL, 3 mL, 5 mL and 10 mL of BmimBF4 during the preparation process. The SEM images further confirm the details of the morphological changes, as shown in Fig. 5b–e. Fig. 5b–e show that as the amount of IL increased from 1 mL to 10 mL, the morphology of the products exhibits a continuous transformation from nanostructured octahedrons (F-1) to a mixture of nanostructured octahedrons and nanostructured spheres (F-3), to nanostructured spheres (F-5), and finally, to nanostructured spheres with worm-like nanopores on the surface (F-10). Interestingly, the nanostructured octahedrons exhibited in sample F-1 developed into the nanostructured spheres shown in samples F-3, F-5 and F-10. In Fig. 5b and most of the nanostructured spheres in Fig. 5c, semi-formed nanostructured spheres can clearly confirm this. In Fig. 5d and e, however, the complete growth of the nanospheres generates a uniform morphology of the nanostructured spheres with excess IL. By adjusting the amount of IL in the reaction system, it is easy to control the morphology of the Fe1.9F4.75·0.95H2O fabricated via a microwave-assisted heating method. The surface area of spherical nanoparticles is proportional to the square of the diameter, and the volume is proportional to the cube of the diameter, so the specific surface area/volume is inversely proportional to the diameter. As the particle diameter becomes smaller, the specific surface area will increase significantly, indicating that the percentage of surface atoms will increase significantly. At the same time, the surface atoms are highly active and unstable. They are easy to combine with foreign atoms to form stable structures and have better properties.
2.6 Ion synthesis method
ILs are salts of cations and anions that are liquid at or near room temperature. They do not burn, have good electrical conductivity, have high thermal stability, are liquid at a wide range of temperatures, and can dissolve many organic and inorganic substances. They can also be recycled, and are a new solvent system. ILs do not volatilize, their vapor pressure is basically zero, they are known as environmentally friendly green solvents and can replace the volatile organic compounds used in many chemical reactions. The synthesis Fe2F5·H2O/rGO by an ionic-liquid-assisted method will be covered in this section when it comes to the characteristics of graphene and the advantages of iron-based fluoride. They chose a green route compared to hydrazine or dimethyl hydrazine as a reducing agent to reduce GO. The rGO suspension was quickly obtained under strongly basic conditions and was then developed into paper-like rGO using a rotary evaporation instrument.70 In the final step, on the surface of the paper-like rGO, they obtained uniform loading of the Fe2F5·H2O/rGO composite through in situ crystallization of Fe2F5·H2O. In addition, the physicochemical and electrochemical properties of the resulting paper-like Fe2F5·H2O/rGO sample will be determined in detail. Fig. 5g shows a schematic of the possible reaction. Under basic conditions, the prepared rGO showed abundant surface defects, which favored the nucleation and growth of the objective product on the surface of rGO. Additionally, a small number of residual functional groups on the prepared composite rGO surface facilitates the hydrophobic dispersion of rGO in the hydrolysis of BF4− to form BF3·H2O as well as F− iron. At the end, the solvated proton Fe3+ reacts with F−, contributing to the in situ crystallization of Fe2F5·H2O on the as-prepared rGO to obtain the Fe2F5·H2O/rGO mixtures.During the process, the IL BMMimBF4 not only serves as a source of green fluoride rather than the corrosive HF, but also as a dispersant for the reduction of rGO. Furthermore, the use of GO rather than natural flake graphite as a starting material to obtain rGO avoids additional oxidation treatment, and the assisted cooperation between the IL and rGO results in the excellent dispersion of rGO and, finally, the anchoring of the Fe2F5·H2O particles in the rGO surface.
2.7 Chemical ball-milling method
Chemical ball-milling is a method of crushing the powder into nanocrystalline particles by making use of the rotation or vibration of the ball mill to make the hard ball impact, grind, and stir the raw material. The chemical ball-milling method can obviously reduce the activation energy of the reaction, refine the grain size, enhance the activity of the powder, improve the sintering ability, induce the chemical reaction at low temperature, and finally comminute the metal or alloy powder into nanocrystalline particles.In the current work, Fan et al. successfully solved the challenges presented in the conversion reaction of FeF3.30 In chemical ball-milling, to prepare the lithiated precursor of FeF3, LiF and Fe nanoparticles were simultaneously dispersed in the carbon matrix. This lithiated inverse reaction nanocomposite (Fe/LiF/C) differs from most of the currently used inverse reaction materials but is similar to the cathode materials used in commercial LIBs. Improvements in performance, and an understanding of the electrochemical reaction mechanism of the lithiated conversion reaction composite represent important progress in the design of practical cathode materials for next-generation batteries.
This production process is schematically illustrated in Fig. 5f and is described in detail in the Experimental section. Nanocomposites of FeF3/C and the lithiating agent LiH/C were prepared by ball-milling FeF3 first and then LiH separately from carbon black. In the process of “mechanical” ball-milling, FeF3 and LiH were uniformly dispersed on the nanoscale, leading to the excellent dispersion of the FeF3/C and LiH/C nanocomposites. The “chemical” ball-milling method was also adopted by ball-milling the FeF3/C and LiH/C nanocomposites to produce a Fe/LiF/C nanocomposite in situ by reaction (1):
| 3LiH + FeF3 → 3LiF + Fe + 1.5H2 ΔGf = −588 kJ mol−1 | (1) |
The chemical reactions between nano-LiH and nano-FeF3 materials formed the LiF/Fe/C nanocomposite. The resulting products have a similar structure and particle size, and they formed electrochemically by the lithiation of FeF3. Furthermore, Fe and LiF are smaller in size and much narrower in distribution than in the mixture of Fe and LiF fabricated through conventional mechanical ball-milling. These differences generate the high strength and resistance to crushing due to the nanometer-sized iron. In the conventional mechanical process of ball-milling, the flimsy and vulnerable nature of both FeF3 and LiH41 accelerate the pulverization step to fabricate nano-Fe and nano-LiF during the processes of subsequent mechanical and chemical ball-milling.
2.8 Sol–gel method
The sol–gel method is to use compounds with high chemical activity components as precursors, mix these raw materials uniformly in the liquid phase, perform hydrolysis and condensation chemical reactions, and form a stable transparent sol system in solution. The gel was formed by the slow polymerization of the colloidal particles, and the gel network was filled with the solvent of losing fluidity to form the gel. The gel was dried and sintered to prepare molecular and even nano-substructure materials. The sol–gel method has many unique advantages over other methods: (1) because the raw materials used in the sol–gel method are first dispersed into the solvent to form a low-viscosity solution, homogeneity at the molecular level can be obtained in a very short time, and when the gel is formed, the reactants may be homogeneously mixed at the molecular level. (2) Due to the reaction of the solution, it is easy to add some trace elements uniformly and quantitatively, so as to realize homogeneous doping at the molecular level. (3) Compared with a solid state reaction, the chemical reaction will be carried out easily and only at a lower synthesis temperature. It is generally considered that the diffusion of the components in the sol–gel system is in the nanometer range, while in the solid state reaction, the diffusion of the components is within the micron range. As a result, the reaction is easy and the temperature is low. (4) Various new materials can be prepared under suitable conditions.Previous research on FeF3/C showed that the ex situ paint-coated layer of the conductive additive is crucial for the fabrication of FeF3/C compounds possessing excellent electrochemical properties. Is the fabrication of FeF3 conductive additive in situ coatings possible yet? How does the coating process influence the materials? Obviously, it is worthwhile to explore the growth and properties of the cathode materials. Zhang et al. synthesized an Fe2O3 precursor via a sol–gel process in 2012.71 This paper reports the fabrication and characterization of an anhydrous core–shell-structured FeF3@Fe2O3 compound obtained by in situ Fe2O3 coating. The results show that the coated Fe2O3 plays an effective role in improving the electrochemical properties. Fig. 6a–h show the colors of the FeF3@Fe2O3 composites at different heating times. This may be the first academic report on the use of a metal fluorinated coating as a cathode material. Since Fe2O3 is a semiconductor possessing a lower band interval (2.0–2.1 eV) and greater electronic conductivity than FeF3, it is hoped that the coating will increase the electron conductivity of the FeF3@Fe2O3 mixed materials. For convenience, the FeF3@Fe2O3 compounds acquired through heating for 15 s, 30 s, 60 s, 120 s, 240 s and 480 s are separately labeled C15, C30, C60, C120, C240 and C480. Fig. 6i–n illustrates the morphology of the pure FeF3.
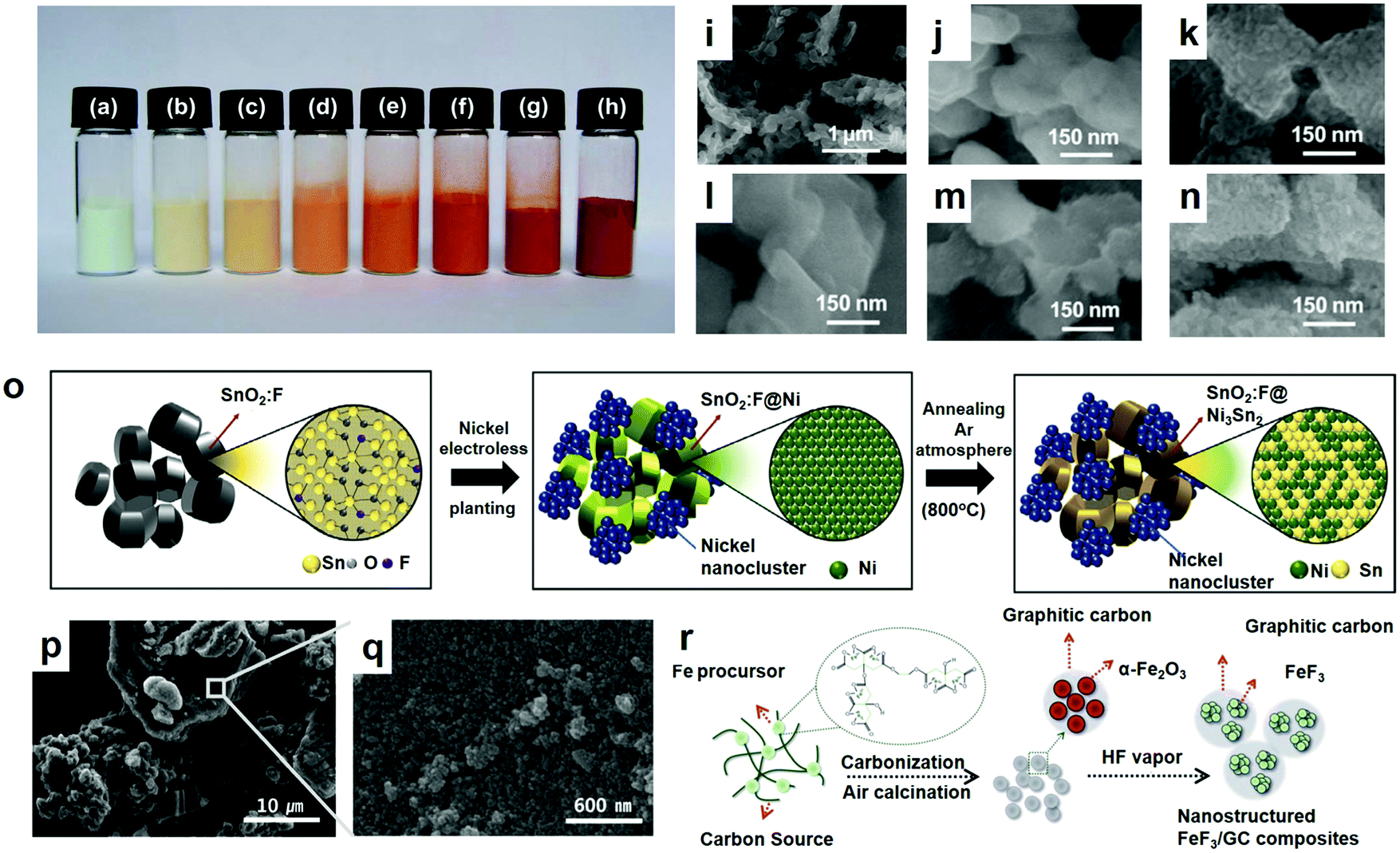 | ||
| Fig. 6 Coloration for the FeF3@Fe2O3 composites at different heating times: (a) 0 s (FeF3), (b) 15 s, (c) 30 s, (d) 60 s, (e) 120 s, (f) 240 s, (g) 480 s and (h) 5 h; SEM images of the FeF3@Fe2O3 composites: (i and j) FeF3, (k) C30, (l) C60, (m) C120 and (n) C240; (o) preparation of FeF3/graphitic carbon (GC) compounds material schematic; (p and q) SEM images of SnO2:F@Ni3Sn2/Ni-nc; (r) schematic diagram of the formation of SnO2:F@Ni3Sn2/Ni-nc through electroless plating and annealing. (a–n) Reproduced with permission.18 Copyright 2016, The Royal Society of Chemistry; (o) reproduced with permission.72 Copyright 2016, The Royal Society of Chemistry; (p–r) reproduced with permission.73 Copyright 2016, Elsevier-Ltd. | ||
2.9 Others
The steps of the synthetic electrode materials described in this part are mostly the comprehensive use of various methods. The suitable methods are used in each step to optimize the performance of the synthesized electrode materials. As shown in Fig. 6p, the Ni nanoclusters remained, and the surface of the annealed SnO2:F@Ni3Sn2/Ni-nanocluster (nc) was relatively rough and composed of ultra-small nanocrystals (<5 nm). Herein, Poizot et al.73 used graphite as a normal working anode for LIBs with a comparatively low specific capacity and studied the successful synthesis of Ni3Sn2-covered fluorine-containing nanoclustered tin oxide (SnO2:F@Ni3Sn2/Ni-nc), a new hybrid anode material. SnO2:F@Ni3Sn2/Ni-nc was synthesized via an easy Ni electroless plating and annealing approach (Fig. 6o). As a result, SnO2 has aroused the attention of many people because of its low cost, rich abundance, environmental friendliness and good theoretical capacity.74–77 Nevertheless, the tremendous volume change (>300%) occurring in the course of successive alloying/dealloying results in the loss of large amounts of capacity during cycling, resulting in the comminution and cracking of the electrodes.77,78 Currently, the best way to improve the electrical conductivity of an active substrate is through fluorine doping.79–81 Despite the superior electrical properties of SnO2:F, the volumetric expansion as well as side reactions between the electrolyte and electrode are still problematic. In order to solve these problems, Ni–Sn intermetallic composites have aroused a wide range of interest. Nevertheless, because the Ni (1728 K) has a much higher melting point than Sn (505 K), Ni–Sn alloys which have a well-defined substance and controllable morphology are difficult to fabricate. However, Ni–Sn intermetallic composites may not be able to hold back forming the Li2O, resulting in low initial columbic efficiency (CE). Recently, Liu et al.82 reported a highly porous Ni–Sn intermetallic micro cage made using a solvothermal method, with a regular quasi-spherical morphology as well as high dispensability. The Ni–Sn alloys prepared using the chemical reduction method had a high crystallinity, which has been reported by Dhanapal et al.83 And they proposed a new convenient and cost-efficient synthetic method of graphitized carbon-encapsulated FeF3 nanoparticles via mixing FeCl3 and citric acid during the original period of the synthesis. Here, they use the FeCl3 and ethylene glycol (EG) as an iron precursor and a cross-linker respectively, and citric acid (C6H8O7) as the source of carbon and a chelating agent. Briefly, FeCl3 chelated with citric acid firstly, forming a citric acid infiltration of the Fe3+–citrate complexes, which was then cross-linked with EG.84 Subsequent annealing and hydrothermal treatment of the FeF3 nanoparticles caused HF vapor to be encapsulated in the graphitic carbon array (Fig. 6q). In the process of synthesis, Fe3+ ions were reduced in situ to Fe(0) in order to catalyze the formation of graphitic carbons and FeF3 through the following reaction with HF.85 The synthesized FeF3 compound with graphitic carbon layers exhibited evidently modified electrochemical properties, such as improved reversible capacity, capacity retention, and rate capability, when compared with bare FeF3. Next, we will discuss their performance with some related examples (Table 1).| Electrode materials | Morphology | Special capacity (mA h g−1) | Cycle number | Ref. |
|---|---|---|---|---|
| a LNMO (LiNi0.5Mn1.5O4−δ). b 3DOM (three-dimensionally ordered macroporous). | ||||
| Fe/LiF/C | Nanoparticles | <300 | 50th | 30 |
| CoF2-coated LNMOa | Nanoparticles | 264.4 | 100th | 86 |
| Fe3O4–FeF2@CFx | Nanoparticles | 718 | 100th | 87 |
| FeF2–CMK-3 | Nanoparticles | 571 | 1000th | 63 |
| FeF2 | Film | 571 | 10th | 88 |
| FeF2.5·0.5H2O–MWCNTs | Nanoparticles | 324.7 | 100th | 47 |
| Li[Li0.2Mn0.54Ni0.13Co0.13]O2 | Powders | 190 | 100th | 89 |
| FeF3/GC | Particles | 421 | 50th | 72 |
| FeF3/rGO1.7 | Particles | 196 | 50th | 90 |
| FeF3 | Powders | 230 | 60th | 91 |
| FeF3 | — | 146.5 | 10th | 92 |
| FeF3·0.33H2O/C | Particles | 233.9 | 40th | 93 |
| FeF3·3H2O | Nanoparticles | 476 | 50th | 39 |
| FeF3/LiMn2O4 | Powders | 122.7 | 200th | 94 |
| FeF3·0.33H2O | Nanoparticles | 712 | 35th | 35 |
| FeF3·3H2O | Nano spheres | 222 | 50th | 40 |
| FeF3 | Powders | 237 | 30th | 95 |
| Fe/Fe2O3/C | Nanoparticles | 196.3 | 50th | 33 |
| FeF3/G | Nanocrystals | 185.6 | 100th | 16 |
| 3DOMb FeF3 | Nanoparticles | 210 | 100th | 62 |
| FeF3/C | Nanocrystals | 710 | 100th | 96 |
| FeF3, FeF3/Fe2O3 | Particles | 201 | 12th | 18 |
| Fe1−xCoxF3 (x = 0.05) | Nanoparticles | 151.7 | 100th | 63 |
FeF3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetylene black = 70:25 :acetylene black = 70:25 |
Nanoparticles | 200 | 50th | 97 |
| FeF3/V2O5 | Particles | 209 | 30th | 53 |
| FeF3, FeF3/MoS2 | Particles | 169.6 | 30th | 98 |
| FeF3/C | Nanoparticles | 390–410 | 20th | 30 |
| FeF3·0.33H2O | Nanoparticles | 1000 | 120th | 99 |
| FeF3:acetylene black | Nanoparticles | 676 | 25th | 100 |
| β-FeF3·3H2O | Nanoparticles | 146.5 | 10th | 92 |
| FeF3·0.33H2O | Nanoparticles | 106.7 | 100th | 31 |
| FeF3·0.33H2O | Nanopetal | 123 | 50th | 54 |
| Fe1.9F4.75·0.95H2O | Nanopores | 145 | 30th | 101 |
| FeF3 | Films | 571.2 | 30th | 102 |
| FeF3 | Nanoparticles | ∼200 | 80th | 103 |
| Iron fluoride–graphene | Nanoparticles | 205 | 40th | 104 |
| FeF3 | Particles | >200 | 50th | 105 |
| Fe1.9F4.75·0.95H2O | Nanorods | 148 | 100th | 42 |
3. Electrochemical energy storage
3.1 Lithium-ion batteries
As a common battery in our life, LIBs have the following advantages: (1) high voltage, the working voltage of a single battery is as high as 3.6–3.9 V, which is three times higher than those of Ni–Cd and Ni–H batteries. (2) Specific energy is high, since the actual specific energy available at present is 100–125 kg−1 and 240–300 L−1, and in the future, with the development of technology, the specific energy can be as high as 150 W h kg−1 and 400 W h L−1. (3) Cycle life is long and it can up to 500 times, or even more than 1000 times. For small current discharge appliances, battery life will multiply the competitiveness of appliances. (4) LIBs have the quality of good safety, no pollution, and no memory effect. One of the major drawbacks of Ni–CD batteries in some processes (such as sintered type) is the “memory effect”, which severely restrains the use of batteries. But LIBs have no such problem. (5) The self-discharge rate of Li-ions stored at room temperature is about 10%, which is much lower than those of Ni–CD (25–30%) and NiMH (30–35%).Notably, these cathode materials of iron fluoride nanocomposites exhibit an excellent rate capability, with a discharge capacity of 176, 145 and 113 mA h g−1 at 0.1, 0.2 and 0.5C, respectively. This provides LIBs with a high specific capacity as well as high magnification. The first synthesis of iron fluoride nanoparticles immobilized with two-dimension (2D) graphene sheets will be introduced, which have a small uniform size for rapid Li-ion diffusion. As schematically shown in Fig. 7a, the chemical circuit diagram is distinct. Representative CV curves of the as-prepared SnO2 anode at a scan rate of 0.1 mV s−1 from 3.00 to 0.05 V are exhibited in Fig. 7b. There are three peaks at approximately 1.21, 0.48 and 0.17 V during the first cathodic scan. The peak at 1.21 V, which is irreversible, is legitimately related to the decomposition of the electrolyte due to the generation of a solid electrolyte interface (SEI) on the electrode surface. As exhibited in Fig. 7c, the galvanostatic discharge/charge properties for the independent SnO2 nanosheets/nickel/PVDF ternary composite anode possess the voltage range of 3.00–0.05 V versus Li/Li+. The discharge/charge capacities of the 3D architecture SnO2 anode are evaluated to be 1533.0 and 1099.9 mA h g−1 during the first cycle at a current density of 200 mA g−1, respectively, illustrating an initial coulombic efficiency of 71.8%.
 | ||
| Fig. 7 (a) Chemical synthesis diagram of iron fluoride–graphene nanocomposites for Li-ion battery cathode materials; (b) CV curves of the freestanding SnO2/Ni/PVDF ternary composite at a scan rate of 0.1 mV s−1 between 0.05 V and 3.00 V; (c) the charge/discharge profiles of the freestanding SnO2/Ni/PVDF ternary composite at different cycles at a current density of 200 mA g−1; (d) cycling profiles of different iron fluoride samples at 1C in the voltage range of 2.0–4.5 V (1C = 237 mA h g−1); (e) rate capacity profiles of different iron fluoride samples; (f) chemical bonding between Fe and F. The schematic drawings of the structure of (g) FeF3·0.33H2O and (h) FeF3, respectively. (a) Reproduced with permission.104 Copyright 2013, The Royal Society of Chemistry; (b and c) reproduced with permission.29 Copyright 2015, Elsevier-Ltd; (d–i) reproduced with permission.49 Copyright 2017, Elsevier-Ltd. | ||
The electrochemical properties of different iron fluoride samples were studied in coin cells employing lithium foil as a counter/reference electrode and S200/SP, S300/SP, S400/SP, and S500/SP as the working electrodes. The specific capacities are counted based only on the quality of iron fluorides. S200/SP, S300/SP, S400/SP and S500/SP delivered initial discharge specific capacities of 163.4, 187.1, 198.1 and 170 mA h g−1, respectively (Fig. 7d). To test the rate performances of the different samples, the cells are discharged–charged at a variety of current densities from 0.1 to 1C and finally back to 0.1C (Fig. 7e). S300/SP shows superior rate performances, demonstrating a high initial discharge capacity of 193 mA h g−1, as well as achieving reversible capacities of 173, 161, 150, and 146 mA h g−1 at 0.1C, 0.2C, 0.5C and 1.0C, respectively. The discharge capacity of S300/SP at 1C in the voltage range of 2.0–4.5 V was higher than that of FeF3·0.33H2O/C and FeF3·0.33H2O/CMK-3 (approximately 130 mA h g−1).
However, the connection mechanisms are completely different in FeF3·0.33H2O and FeF3. For FeF3·0.33H2O, six octahedra are connected through corner-sharing, forming many distinct hexagonal cavities in FeF3·0.33H2O (Fig. 7f). The water molecules are located in the center of the cavities to stabilize the structure. The growth of the (002) plane forms a large inner tunnel structure in FeF3·0.33H2O (Fig. 7g), which can facilitate the insertion and extraction for Li+. As a result, Li+ can be more easily intercalated and extracted during the charge–discharge process in FeF3·0.33H2O, which results in the excellent cycle and rate performances compared with other samples. FeF3 shows a distorted ReO3 structure without water (Fig. 7h and i). The octahedra are closer to each other to form a narrow tunnel, making Li+ accommodation more difficult. Therefore, as a kind of a stable structure medium, water can improve the electrochemical performance of the battery more effectively and safely.
3.2 Sodium-ion batteries
The principle of SIBs is similar to that of lithium-ion batteries. Compared with the LIBs, the SIBs have the advantages of rich reserves of raw materials and low price of a sodium salt. Compared with the ternary cathode material of LIBs, the cost of the raw material is reduced by half by using iron, manganese and nickel base cathode materials. Because of the sodium salt characteristics, it is possible to use a low concentration electrolyte (the same concentration electrolyte, the conductivity of which is about 20% higher than the lithium electrolyte) to reduce the cost. Sodium ions do not form alloys with aluminum, and the negative electrode can be used as a collector. The cost can be further reduced by about 8% and the weight by about 10% the SIBs are allowed to discharge to zero volts because it has no overdischarge characteristics. The energy density of SIBs is more than 100 W h kg−1, which is comparable to that of lithium iron phosphate batteries, but its cost advantage is obvious, which is expected to replace the traditional lead–acid battery in large-scale energy storage.The detailed synthesis route of the FeF3/C composites is illustrated in Fig. 8a and b shows the circular charge/discharge voltage profiles, which confirm that the sample provides a very high sodium storage capacity of 280 mA h g−1, at a current density of 75 mA g−1. An abnormal coulombic efficiency, which is generated by irreversible capacity loss was observed in the experiment, including the necessity of SEI and other side reactions.
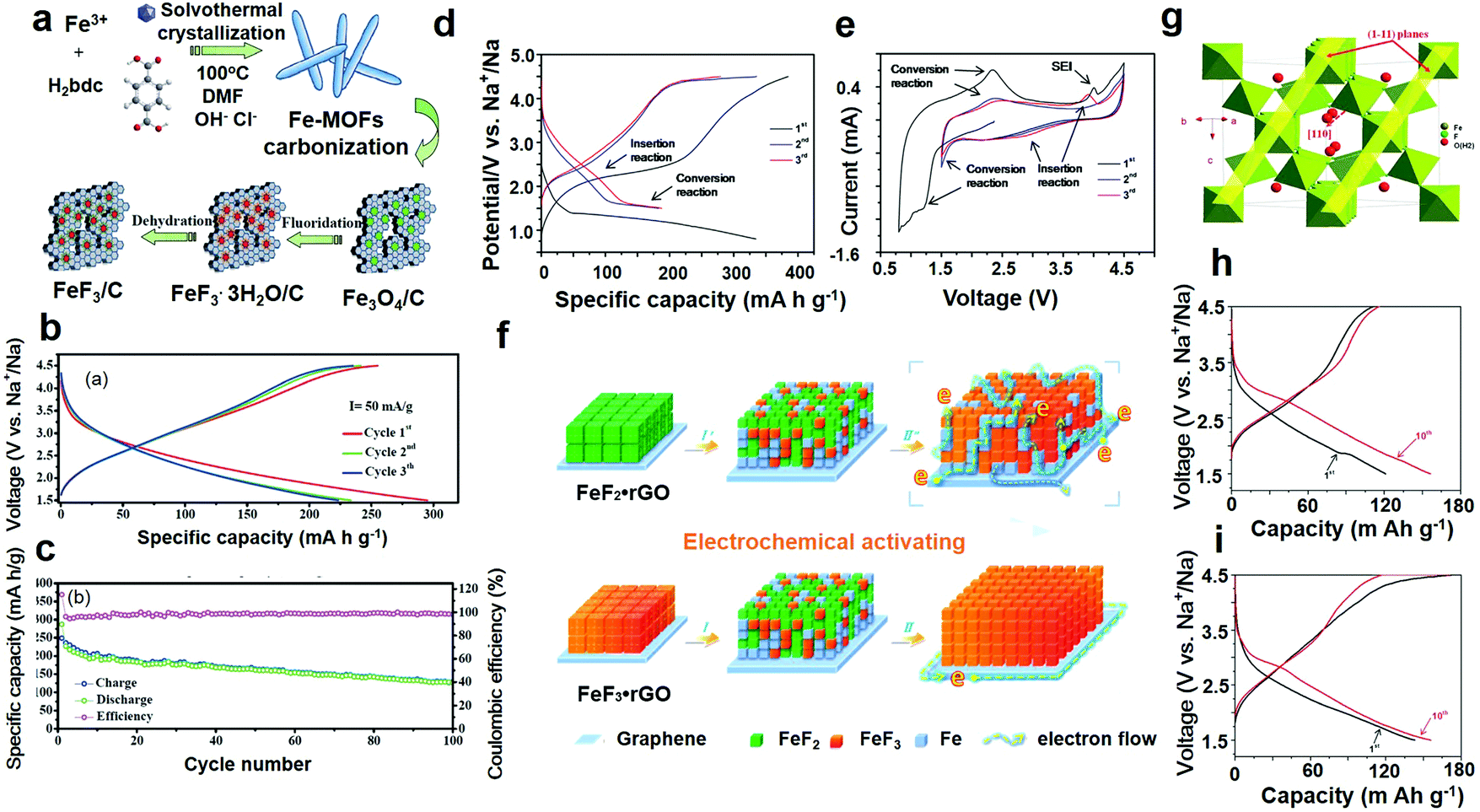 | ||
| Fig. 8 (a) Schematic diagram of the fabrication of FeF3/C nanocomposites, MOF represents metal–organic frameworks; (b) charge and discharge voltage profile of the 1st, 2nd and 3rd cycles; (c) cycle performance at a continuous current density of 75 mA g−1; (d) electrochemical activation of the FeF2–rGO compound for in situ production of the FeF3–Fe–rGO compound charge–discharge curves; (e) cyclic voltammetry profiles, electrochemical performance of the above in situ produced FeF3–Fe–rGO composite; (f) schematic summarization and comparison of the electrochemical reaction mechanisms of the FeF3–rGO and FeF2–rGO; (g) schematic representation of FeF3·0.5H2O; (h) galvanostatic charge–discharge voltage profile of the 1st, 2nd and 10th cycle of bare FeF3·0.5H2O; (i) charge–discharge curves of the 1st, 2nd and 10th cycles of the FeF3·0.5H2O–rGO composite. (a–c) Reproduced with permission.106 Copyright 2017, The Royal Society of Chemistry; (d–f) reproduced with permission.107 Copyright 2014 Elsevier-Ltd; (g–i) reproduced with permission.108 Copyright 2015, The Royal Society of Chemistry. | ||
This characteristic is also in accordance with the CV result, in other words, the initial section of the curve differs from the latter. To further highlight the superiority of the FeF3/C-700-3 h for the cathode materials of SIBs, the cycle performance of the specimen was measured in the range of 1.5–4.5 V (C = 75 mA g−1), as shown in Fig. 8b. The FeF3/C-700-3 h had a great performance, and the initial discharge capacity was 227 mA h g−1, respectively. In addition, the composites had a high discharge capacity of 163 mA h g−1 after 50 cycles, with a fading rate of approximately 0.45% per cycle after 100 cycles, which shows that the sodium insertion/extraction process is reversible. Fig. 8e shows the first CV curve of FeF2–rGO in the range of 0.8–4.5 V; among them, the reduction peak of the capacity of 1.2 V can be attributed to the conversion reaction (2).
| FeF2 + 2Na+ + 2e− → Fe + 2NaF | (2) |
| 2NaF + Fe → xFeF2 + yFeF3 + (1 − x − y)Fe + 2Na+ + 2e−(3y/2 + x = 1) | (3) |
Interestingly, the CV profile of FeF2–rGO changes gradually in the continuous cycle (1.5–4.5 V). It is explained in detail that the conversion reaction of the redox peak to the FeF2 capacity of 1.5 and 2.3 V, respectively, shows that the conversion of FeF2 to FeF3 is gradual. These CV results are consistent with the gradual change in the charge/discharge profiles (Fig. 8d). It is important to note that most of the discharge capacity is above 1.7 V, indicating that the Na+ implantation reaction occurs mainly at high capacity. When Fe2+ cannot be reduced to Fe+, Na+ embedding in the FeF2 framework is not possible. Further confirmation of the successful generation of the FeF2 material agrees with the abovementioned reaction process and mechanism. To facilitate understanding, the schematic diagram summarizes the above discussion points (Fig. 8f).
As shown in Fig. 8g, H2O is located at the center of the FeF6 eight-sided cage. The unit cell volume of FeF3·0.5H2O (1127.4 Å3) was larger than that of the FeF3 pyrochlore (1100.7 Å3) probably due to the existence of water molecules in the pyrochlore structure. Fig. 8h shows the charge and discharge curves of the composite materials in the range of 1.5–4.5 V. In the first cycle, the discharge capacity was as high as 239 mA h g−1 at a current density of 0.05C. In the sixth cycle, the discharge capacity reached a maximum of 266 mA g−1 and reached an obvious platform at 2.8 V, indicating that the Na+ inserted in the active material was more than the theoretical capacity (220 mA g−1, containing Na+). The electrochemical properties of the composite were better, which is also due to the unique growth of the active material. The nanoparticles adhere to the high surface area particles of the rGO layer at a loading of 155 m2 g−1, which is 3.7 times the value of the pure material. The graphs of the discharge capacities at different current rates (Fig. 8i) show that, when the current density is 0.1 and 0.25C, the shape of the initial charge–discharge curve is basically unchanged; the discharge capacity of the composite is 190 and 153 mA h g−1, respectively, indicating that the composite has a good performance for multiple ratios. However, the discharge capacity decreased to 90 mA h g−1 and 64 mA h g−1 at a high current density of 0.5 and 1C, respectively, showing a relatively rapid decline in capacity. What is worth mentioning here is that the cells were galvanostatically cycled five times in the range 1.0–4.5 V at a current density of 0.05C preferential to any measurement.
3.3 Zn–air batteries
Rechargeable metal–air batteries (Zn–air and Li–air) are drawing increasing attention in energy storage and charging applications because of their countless strong points. (1) The specific energy of Zn–air batteries is twice as much as that of LIBs. The maximum stroke of an electric vehicle powered by it can reach 400 km, while that of the same car with a lead–acid battery of the same mass is no more than 100 km. (2) The manufacturing process is simple and the cost is low. (3) Renewable utilization. After the zinc electrode is used, it can be reused by regeneration and reduction. In addition, the zinc electrode can also be mechanically charged. The zinc electrode will be removed from the battery and charged in a special tank. A zinc air electrode can be reused many times, and can be a directly charged Zn–air battery, referred to as secondary Zn–air batteries. (4) Because the charge of Zn–air batteries is mainly to replace the plate, the regeneration of the plate can be concentrated. The plate can be distributed like a store without having to set up a dedicated charging station. This can not only save a lot of forwarding investment, but also bring a lot of convenience to users. Next, we will introduce the application of iron fluoride in Zn–air batteries.16,109–112 The catalytic activity of these catalysts is strongly dependent upon two crucial factors: the electronic structure of the surface graphitic layer, which can be adjusted by electronic modulation of suitable metal cores, determines the turnover frequency (TOF) per active site (intrinsic activity); on the other hand, the density of the active sites, the effective area of the modulated surface carbon, is a function of the metal core size, loading, and dispersion. Consequently, tremendous work can be done to controllably tune the activity with respect to these two aspects. MOF constructed by organic linkers and metal ions can act to perfectly encapsulate metal ions due to their well-defined coordination environment and ordered arrangement in the framework. However, high-temperature pyrolysis often leads to structural collapse, thus making further growth of the metal particles uncontrollable. In this section, they have designed a unique two-stage encapsulation technique to acquire carbon-encapsulated Ni, Fe, or NiFe2 alloy nanocrystals (denoted as Ni@NCx, Fe@NCx, and NiFe@NCx, respectively) with an ultra-small core size, high metal loading, and high dispersion.As illustrated in Fig. 9a, MOF-based NiFe (or single Ni, Fe metal)–MILs are employed as metal ion encapsulators at low temperature. A homemade single-cell Zn–air battery was fabricated and tested to reveal the in situ cell performance of our bifunctional catalyst under real battery operating conditions (Fig. 9e). Fig. 9f presents the polarity as well as the specific power curves for Zn–air batteries based on NiFe@NCx and Pt/C + IrO2 cathodes, respectively. The NiFe@NCx catalyst showed larger current density and maximal power density compared with those of the Pt/C + IrO2 catalyst. The specific capacity of the NiFe@NCx based battery was estimated to be 583.7 mA h g−1, consistent with a gravimetric energy density of 732.3 W h kgZn−1, which is higher than that of the Pt/C + IrO2-based battery (specific capacity 499.1 mA h g−1, energy density 543.2 W h kgZn−1). The discharge/charge polarity curves for the Zn–air batteries equipped with NiFe@NCx and a commercial catalyst (Pt/C and IrO2) cathode is shown in Fig. 9g. An obviously lower charge and discharge voltage interval was observed with the NiFe@NCx cathode compared to the commercial one, indicating a better recharge ability. Furthermore, the total of the charge–discharge overpotential for the NiFe@NCx cathode-based Zn–air battery was 0.78 V at a current density of 50 mA cm−2, which is significantly lower than that of the Pt/C + IrO2 cathode (1.1 V). The overpotential is the most crucial benchmark in estimating the performance of a bifunctional electrocatalyst. The NiFe@NCx cathode-based battery showed an initial voltage interval of 0.39 V as well as a high full-cycle efficiency of 76.7% at 10 mA cm−2. After 205 cycles, a slight loss in performance was found for the NiFe@NCx cathode (0.29 V increase in the voltage gap), while the commercial cathode illustrated a greater increase in the voltage interval (0.48 V, Fig. 9h).
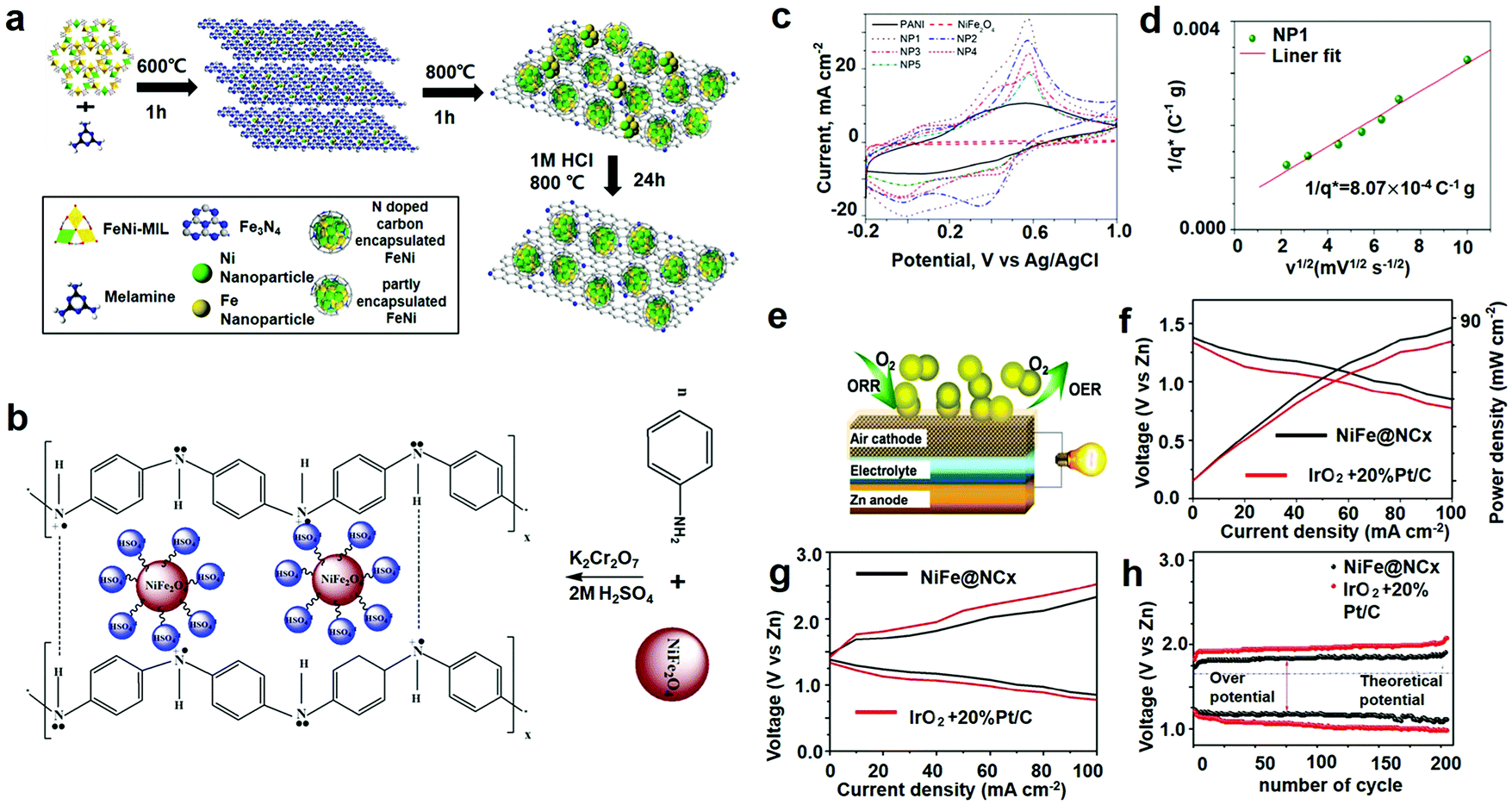 | ||
| Fig. 9 (a) Schematic illustration of the synthetic strategy of the transition-metals (TMs)@NCx composite; (b) preparation and charge compensation in a polyaniline (PANI)–NiFe2O4 composite; (c) CV curves of PANI, NiFe2O4 and PANI–NiFe2O4 composites; (d) Trasatti plot; (e) schematic depiction of the Zn–air battery structure; (f) discharge curves and power density of Zn–air batteries; (g) charge and discharge polarization curves for the batteries; (h) the cycle stability of rechargeable Zn–air battery, the galvanostatic discharge–charge cycling curves were performed at 10 mA cm−2 and a duration of 600 s per cycle. (a and e–h) Reproduced with permission.6 Copyright 2016, American Chemical Society; (b–d) reproduced with permission.113 Copyright 2012, Elsevier-Ltd. | ||
3.4 Supercapacitors
SCs can store energy by separating or storing charges. The process of charge storage includes not only the storage on the double layer, but also the redox reaction between the electrolyte ions and the electrode active material.114 When ions in electrolyte (such as H− OH− K+ or Li+) diffuse from solution to the electrode/solution interface under the action of an external electric field, they will enter the bulk phase of the active oxide on the electrode surface through the redox reaction on the interface. In this way, a large number of charges are stored in the electrode. When discharge occurs, the ions entering the oxide will return to the electrolyte through the reversion of the above redox reaction. At the same time, the stored charge is released through the external circuit.115,116 In addition, they exhibit an extended cycle life and are useful for a wide range of applications. The use of a suitable electrode material allows SCs to achieve a high power or energy density, a long cycle life and high flexibility. Therefore, the conversion reaction compounds such as MFx (M: Fe, Co, Ni) have the advantage of transferring multiple electrons in each formula unit, thus showing a high theoretical capacity. Among them, iron fluoride is one of the most excellent cathode materials. Due to its outstanding theoretical capacity as well as good thermal stability, it has attracted widespread interest. Meanwhile, the rate performance of SCs has been greatly improved because of the coupling of nanoparticle materials with conductive carbon. And the influence of in situ Fe2O3 which coated on the electrochemical properties was studied. It was pointed out that a little amount of Fe2O3 had an electrochemical performance though it was not as good as the performance acquired by mixing with carbon-based materials, which also can contribute a little to the performance of SCs.In addition to graphene, other materials involved in the preparation of the electrode can also make a great contribution to the performance of the electrode. Fig. 9b represents the conductivity and charge compensation of a PANI–NiFe2O4 composite. Fig. 9c illustrates a typical cyclic voltammogram of PANI, in which NiFe2O4 and PANI–NiFe2O4 composites at 20 mV s−1 exhibit a voltage window of −0.2 and 1.0 V vs. Ag/AgCl. Every CV curve contains two pairs of redox peaks in addition to NiFe2O4, demonstrating the pseudocapacitive reaction of the different materials. The observation of the first pair of redox peaks is a result of the redox transition from the leucoemeraldine to platonic emeraldine phase. Similarly, the second pair of redox peaks results from a transition between platonic emeraldine and the bipolaronic penigraniline form.117–119 The CV curve of NiFe2O4 also indicates oxidation and a decrease of the peaks; the observed peak current is extremely low compared with those of PANI and PANI/NiFe2O4 composites. Compared to other systems, the area under the CV curve of NiFe2O4 is relatively small. As is known to all, the specific capacity of the electroactive material is consistent with the CV curve coverage area.120,121 As shown in Fig. 9c, the CV curve coverage area varies with the loading concentration of NiFe2O4 in the PANI. The CV curve shows that the area covered by the NP1(PANI–NiFe2O4 composites) sample is higher than that of the PANI and other composites, probably because of the large availability of active sites of PANI122 as well as the increased conductivity of the network due to the formation of hydrogen bonds.123–125 To determine the source of the enhanced properties of NP1, the diffusion coefficients for all samples were calculated using the Randles–Sevcik equation (eqn (4)).126
| Ip = (2.687 × 105)n3/2ACD1/2v1/2 | (4) |
In the redox reaction, n represents the number of transferred electrons, A stands for the area of the electrode in cm2, D is the diffusion coefficient in cm2 s−1, and C is the concentration in mol cm−3. The Trasatti plot of NP1, which is shown in Fig. 9d, symbolically represents the sum of the charge reserved in the inner and outer surfaces of the fabricated electrode in the electrochemical reaction process. The curve of 1/q* vs. v1/2 facilitates the calculation of the general charge reserved in the NP1. Similarly, the 1/q* vs. v1/2 provides a convenient way to calculate the sum of the charge reserved in the outer surfaces of the NP1 electrode. The curve illustrates that the general sum of the charge reserved in the fabricated NP1 electrodes is 1.85 × 103 C g−1, while the charge reserved in the outer surfaces was 238.26 C g−1. Therefore, the general charge reserved in the material differs from that in the outer surface, and the charge reserved in the inner surface of the electrode is 1.61 × 103 C g−1. The results show that, even at a high scan rate, the inner surfaces of the fabricated electrode contribute more to the electrochemical reaction than the outer surfaces.127–130 This further confirms that the NP1 electrodes have excellent capacitance retention as well as reversibility even at high scan rates.
It is found that metal fluoride can improve the electrochemical performance of LIBs to some extent, but the metal fluoride of nanomaterials shows better performance. This is due to the fact that nanostructures can effectively alleviate various strains resulting from volume changes and increased specific capacity. In addition, nanostructures can also provide a larger area for contact between electrodes and electrolytes, thus shortening the length of lithium ion diffusion. Better performance has been achieved. However, nanostructured electrodes cannot be amplified because of their complex process, so they can only be studied in the laboratory. And it is not difficult to find out from the above studies that nanometal fluorides, which are based on carbon materials such as G, GO, rGO and so on, can accommodate changes in volume and act as a current collector. Therefore, the focus of current work is to improve the nanostructure of the electrode. Many layered porous nanostructures and nanorods have been developed and used. These materials require complex processes, including synthetic spin templates and impregnation of precursors, so we still need to explore the development of simple and scalable hierarchical nanostructures.
4. Conclusions and outlook
In conclusion, this review mainly introduces the electrochemical applications of transition metal (Fe, Co, Ni) fluoride materials in LIBs, SIBs, Zn–air batteries and SCs. Their synthesis methods, morphology and electrochemical properties were extensively introduced. Although LIBs and SIBs are now the mainstay of energy storage devices. It is undeniable that Zn–air batteries will become the mainstream of the future with their greater capacity, lower prices and more environmentally friendly use of electrode materials. But in the aspect of service life, they need to be further improved in order to obtain more far-reaching development prospect.Transition metal (Fe, Co, Ni) fluoride materials have shown exciting achievements in previous studies, and their unique advantages, including high surface area, high porosity, adjustable pore size, and controllable structure, make great contributions to their electrochemical performance. Although numerous impressive advancements have been made, research should be continued to achieve more enhanced products. The following primary shortcomings have made metal fluoride electrodes unsuitable for practical applications: (1) LiF is a product of the conversion reaction but also highly insulated. Consequently, based on metal electrodes in the reaction, there are often many problems. (2) During cycling, the volume changes can lead to contact between the electrodes and the collector and can cause an irreversible and obvious decomposition due to the SEI film. To address the above problems, some effective strategies have been suggested. (1) The formation of highly reactive carbon–metal–fluoride nanocomposites (CMFNCs) or transition metal–LiF nanocomposites through high energy mechanical ball-milling. Nanocrystals which have abundant active interfaces include many defects, which can result in the electronic and ionic activities. Moreover, the particles connect to each other electrically due to the positive effect of conductive carbon, which compensates for the high resistance of the fluoride. (2) Characterizing metal fluoride materials is important to gain a better understanding of the electrochemical potential behavior, such as the transition from electron to ion transport and the mechanism governing the charge transfer reaction at the electrode/electrolyte interface to optimize and enhance their performance for their realization as another type of electrode. It is important to take an overview approach towards improving the performances. In particular, a high energy density means high security risks, and it is imperative to satisfy all the safety requirements in developing efficient, sustainable ‘green’ power sources.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC – 21671170, 21673203, and 21201010), the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP), the Program for New Century Excellent Talents of the University in China (NCET-13-0645), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (XKYCX17-038), the Six Talent Plan (2015-XCL-030), and the Qinglan Project. We also acknowledge the Priority Academic Program Development of Jiangsu Higher Education Institutions and the technical support we received at the Testing Center of Yangzhou University.References
- J. Jiang, L. Li, M. Xu, J. Zhu and C. M. Li, ACS Appl. Mater. Interfaces, 2016, 8, 16240–16247 CrossRef CAS PubMed.
- R. Marom, S. F. Amalraj, N. Leifer, D. Jacob and D. Aurbach, J. Mater. Chem., 2011, 21, 9938–9954 RSC.
- C. Li, L. Gu, X. Guo, D. Samuelis, K. Tang and J. Maier, Nano Lett., 2012, 12, 1241–1246 CrossRef CAS PubMed.
- C. Li, L. Gu, J. Tong and J. Maier, ACS Nano, 2011, 5, 2930–2938 CrossRef CAS PubMed.
- C. Li, C. Yin, L. Gu, R. E. Dinnebier, X. Mu, P. A. Van Aken and J. Maier, J. Am. Chem. Soc., 2013, 135, 11425–11428 CrossRef CAS PubMed.
- J. Zhu, M. Xiao, Y. Zhang, Z. Jin, Z. Peng, C. Liu, S. Chen, J. Ge and W. Xing, ACS Catal., 2016, 6, 6335–6342 CrossRef CAS.
- X. Xiao, S. Zheng, X. Li, G. Zhang, X. Guo, H. Xue and H. Pang, J. Mater. Chem. B, 2017, 5, 5234–5239 RSC.
- S. Zheng, H. Xue and H. Pang, Coord. Chem. Rev., 2018, 373, 2–21 CrossRef CAS.
- J. Chen, Z. Yu, P. Zhu, J. Wang, Z. Gan, J. Wei, Y. Zhao and S. Wei, J. Mater. Chem. B, 2015, 3, 34–38 RSC.
- Q. Wu, M. Chen, K. Chen, S. Wang, C. Wang and G. Diao, J. Mater. Sci., 2016, 51, 1572–1580 CrossRef CAS.
- M. Wang, Y. Hu, J. Han, R. Guo, H. Xiong and Y. Yin, J. Mater. Chem. A, 2015, 3, 20727–20735 RSC.
- L. Fan, L. Tang, H. Gong, Z. Yao and R. Guo, J. Mater. Chem., 2012, 22, 16376–16381 RSC.
- J. Han, S. Lu, C. Jin, M. Wang and R. Guo, J. Mater. Chem. A, 2014, 2, 13016–13023 RSC.
- M. Zhu, C. Wang, D. Meng and G. Diao, J. Mater. Chem. A, 2013, 1, 2118–2125 RSC.
- M. Chen, W. Li, X. Shen and G. W. Diao, ACS Appl. Mater. Interfaces, 2014, 6, 4514–4523 CrossRef CAS PubMed.
- R. Ma, Z. Lu, C. Wang, H.-E. Wang, S. Yang, L. Xi and J. C. Y. Chung, Nanoscale, 2013, 5, 6338–6343 RSC.
- J. Hu, Y. Zhang, D. Cao and C. Li, J. Mater. Chem. A, 2016, 4, 16166–16174 RSC.
- W. Zhang, L. Ma, H. Yue and Y. Yang, J. Mater. Chem., 2012, 22, 24769–24775 RSC.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 RSC.
- J. Jiang, L. Li, M. Xu, J. Zhu and C. M. Li, ACS Appl. Mater. Interfaces, 2016, 8, 16240–16247 CrossRef CAS PubMed.
- H. Pan, Y. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 RSC.
- L. Wu, X. H. Hu, J. F. Qian, F. Pei, F. Y. Wu, R. J. Mao, X. P. Ai, H. X. Yang and Y. L. Cao, J. Mater. Chem. A, 2013, 1, 7181–7184 RSC.
- S. Chen, M. Xue, Y. Li, Y. Pan, L. Zhu and S. Qiu, J. Mater. Chem. A, 2015, 3, 20145–20152 RSC.
- L. Ji, M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. Zhang, Energy Environ. Sci., 2011, 4, 5053–5059 RSC.
- N. Yamakawa, M. Jiang, B. Key and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 10525–10536 CrossRef CAS PubMed.
- C. Yuan, J. Li, L. Hou, X. Zhang, L. Shen and X. W. Lou, Adv. Funct. Mater., 2012, 11, 4592–4597 CrossRef.
- M. Zhou, L. Zhao, T. Doi, S. Okada and J. Yamaki, J. Power Sources, 2010, 195, 4952–4957 CrossRef CAS.
- T. Bao, H. Zhong, H. Zheng, H. Zhan and Y. Zhou, Mater. Lett., 2015, 158, 21–24 CrossRef CAS.
- T. Zhao, L. Li, R. Chen, H. Wu, X. Zhang, S. Chen, M. Xie, F. Wu, J. Lu and K. Amine, Nano Energy, 2015, 15, 164–176 CrossRef CAS.
- X. Fan, Y. Zhu, C. Luo, L. Suo, Y. Lin, T. Gao, K. Xu and C. Wang, ACS Nano, 2016, 10, 5567–5577 CrossRef CAS PubMed.
- J. Tan, L. Liu, H. Hu, Z. Yang, H. Guo, Q. Wei, X. Yi, Z. Yan, Q. Zhou, Z. Huang, H. Shu, X. Yang and X. Wang, J. Power Sources, 2014, 251, 75–84 CrossRef CAS.
- R. R. Gaddam, D. Yang, R. Narayan, K. Raju, N. A. Kumar and X. S. Zhao, Nano Energy, 2016, 26, 346–352 CrossRef CAS.
- R. Ma, M. Wang, P. Tao, Y. Wang, C. Cao, G. Shan, S. Yang, L. Xi, J. C. Y. Chung and Z. Lu, J. Mater. Chem. A, 2013, 1, 15060–15067 RSC.
- R. G. Cao, W. Xu, D. P. Lv, J. Xiao and J. G. Zhang, Adv. Energy Mater., 2015, 5, 1402273 CrossRef.
- C. L. Li, L. Gu, S. Tsukimoto, P. A. van Aken and J. Maier, Adv. Mater., 2010, 22, 3650–3654 CrossRef CAS PubMed.
- L. Di Carlo, D. E. Conte, E. Kemnitz and N. Pinna, Chem. Commun., 2014, 50, 460–462 RSC.
- S.-T. Myung, S. Sakurada, H. Yashiro and Y.-K. Sun, J. Power Sources, 2013, 223, 1–8 CrossRef CAS.
- Y.-L. Shi, N. Wu, M.-F. Shen, Y.-L. Cui, L. Jiang, Y.-H. Qiang and Q.-C. Zhuang, ChemElectroChem, 2014, 1, 645–654 CrossRef.
- C. L. Li, X. K. Mu, P. A. van Aken and J. Maier, Adv. Energy Mater., 2013, 3, 113–119 CrossRef CAS.
- Q. Chu, Z. Xing, J. Tian, X. Ren, A. M. Asiri, A. O. Al-Youbi, K. A. Alamry and X. Sun, J. Power Sources, 2013, 236, 188–191 CrossRef CAS.
- S.-W. Kim, D.-H. Seo, H. Gwon, J. Kim and K. Kang, Adv. Mater., 2010, 22, 5260–5624 CrossRef CAS PubMed.
- Y. Lu, Z. Wen, J. Jin, K. Rui and X. Wu, Phys. Chem. Chem. Phys., 2014, 16, 8556–8562 RSC.
- Y. Lu, Z. Wen, J. Jin, X. Wu and K. Rui, Chem. Commun., 2014, 50, 6487–6490 RSC.
- C. Li, L. Gu, S. Tsukimoto, P. A. van Aken and J. Maier, Adv. Mater., 2010, 22, 3650–3654 CrossRef CAS PubMed.
- Y. Shen, X. Wang, H. Hu, M. Jiang, X. Yang and H. Shu, J. Power Sources, 2015, 283, 204–210 CrossRef CAS.
- J. Liu, W. Liu, S. Ji, Y. Wan, M. Gu, H. Yin and Y. Zhou, Chem. – Eur. J., 2014, 20, 5815–5820 CrossRef CAS PubMed.
- S. Y. Wei, X. Y. Wang, R. Zhang, H. Hu, Y. Q. Shen and J. Liu, RSC Adv., 2016, 6, 97759–97769 RSC.
- Y. Zhang, Z. P. Luo, Q. Z. Xiao, T. L. Sun, G. T. Lei, Z. H. Li and X. J. Li, J. Power Sources, 2015, 297, 442–448 CrossRef CAS.
- Y. Bai, X. Zhou, C. Zhan, L. Ma, Y. Yuan, C. Wu, M. Chen, G. Chen, Q. Ni, F. Wu, R. Shahbazian-Yassar, T. Wu, J. Lu and K. Amine, Nano Energy, 2017, 32, 10–18 CrossRef CAS.
- Y. Xu, W. Shen, A. Zhang, H. Liu and Z. Ma, J. Mater. Chem. A, 2014, 2, 12982–12990 RSC.
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- H. Song, H. Cui and C. Wang, J. Mater. Chem. A, 2015, 3, 22377–22384 RSC.
- Z. S. Li, B. Z. Wang, C. L. Li, J. J. Liu and W. Q. Zhang, J. Mater. Chem. A, 2015, 3, 16222–16228 RSC.
- B. Li, Z. Cheng, N. Zhang and K. Sun, Nano Energy, 2014, 4, 7–13 CrossRef CAS.
- J. Yang, X. Duan, W. Guo, D. Li, H. Zhang and W. Zheng, Nano Energy, 2014, 5, 74–81 CrossRef CAS.
- H. Zhang, X. Yu, D. Guo, B. Qu, M. Zhang, Q. Li and T. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 7335–7340 CrossRef CAS PubMed.
- M. K. Devaraju and I. Honma, Adv. Energy Mater., 2012, 2, 284–297 CrossRef CAS.
- Z. Zhu, D. Zhang, H. Yan, W. Li and Q. Lu, J. Mater. Chem. A, 2013, 1, 5492–5496 RSC.
- G. Zhao, Y. Lin, T. Zhou, Y. Lin, Y. Huang and Z. Huang, J. Power Sources, 2012, 215, 63–68 CrossRef CAS.
- Y. Tang, X. Rui, Y. Zhang, T. M. Lim, Z. Dong, H. H. Hng, X. Chen, Q. Yan and Z. Chen, J. Mater. Chem. A, 2013, 1, 82–88 RSC.
- A. Q. Pan, H. Bin Wu, L. Zhang and X. W. (David) Lou, Energy Environ. Sci., 2013, 6, 1476–1479 RSC.
- D. Ma, Z. Cao, H. Wang, X. Huang, L. Wang and X. Zhang, Energy Environ. Sci., 2012, 5, 8538–8542 RSC.
- L. Liu, M. Zhou, L. Yi, H. Guo, J. Tan, H. Shu, X. Yang, Z. Yang and X. Wang, J. Mater. Chem., 2012, 22, 17539–17550 RSC.
- L. Li, F. Meng and S. Jin, Nano Lett., 2012, 12, 6030–6037 CrossRef CAS PubMed.
- D. L. Ma, H. G. Wang, Y. Li, D. Xu, S. Yuan, X. L. Huang, X. B. Zhang and Y. Zhang, Nano Energy, 2014, 10, 295–304 CrossRef CAS.
- Z. H. Yang, S. Zhao, Y. J. Pan, X. Y. Wang, H. H. Liu, Q. Wang, Z. J. Zhang, B. Deng, C. S. Guo and X. Q. Shi, ACS Appl. Mater. Interfaces, 2017, 12, 3142–3151 Search PubMed.
- M. Tsuji, M. Hashimoto, Y. Nishizawa, M. Kubokawa and T. Tsuji, Chem. – Eur. J., 2005, 11, 440–452 CrossRef CAS PubMed.
- K. Ding, Z. Miao, Z. Liu, Z. Zhang, B. Han, G. An, S. Miao and Y. Xie, J. Am. Chem. Soc., 2007, 129, 6362–6363 CrossRef CAS PubMed.
- X. Bian, Q. Fu, Q. Pang, Y. Gao, Y. Wei, B. Zou, F. Du and G. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 3308–3318 CrossRef CAS PubMed.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- W. Zhang, P. N. Duchesne, Z. L. Gong, S. Q. Wu, L. Ma, Z. Jiang, S. Zhang, P. Zhang, J. X. Mi and Y. Yang, J. Phys. Chem. C, 2013, 117, 11498–11505 CrossRef CAS.
- T. Kim, W. J. Jae, H. Kim, M. Park, J.-M. Han and J. Kim, J. Mater. Chem. A, 2016, 4, 14857–14864 RSC.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- H.-X. Zhang, C. Feng, Y.-C. Zhai, K.-L. Jiang, Q.-Q. Li and S.-S. Fan, Adv. Mater., 2009, 21, 2299–2304 CrossRef CAS.
- Y. Yu, C.-H. Chen and Y. Shi, Adv. Mater., 2007, 19, 993–997 CrossRef CAS.
- J. Ming, Y. Wu, J. B. Park, J. K. Lee, F. Zhao and Y. K. Sun, Nanoscale, 2013, 5, 10390–10396 RSC.
- A.-Y. Kim, J. S. Kim, C. Hudaya, D. Xiao, D. Byun, L. Gu, X. Wei, Y. Yao, R. Yu and J. K. Lee, Carbon, 2015, 94, 539–547 CrossRef CAS.
- C. Zhu, X. Xia, J. Liu, Z. Fan, D. Chao, H. Zhang and H. J. Fan, Nano Energy, 2014, 4, 105–112 CrossRef CAS.
- G. L. Xu, Y. F. Xu, J. C. Fang, F. Fu, H. Sun, L. Huang, S. Yang and S. G. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6316–6323 CrossRef CAS PubMed.
- J. Sun, L. Xiao, S. Jiang, G. Li, Y. Huang and J. Geng, Chem. Mater., 2015, 27, 4594–4603 CrossRef CAS.
- L. Cheng, X. Li, H. Liu, H. Xiong, P. Zhang and Y.-Y. Xia, J. Electrochem. Soc., 2007, 154, A692–A697 CrossRef CAS.
- J. Liu, Y. Wen, P. A. Van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 6387–6392 CrossRef CAS PubMed.
- K. Dhanapal, V. N. Narayanan and A. Stephen, J. Magn. Magn. Mater., 2016, 406, 103–109 CrossRef CAS.
- D. Lv, W. Wen, X. Huang, J. Bai, J. Mi, S. Wu and Y. Yang, J. Mater. Chem., 2011, 21, 9506–9512 RSC.
- H. B. Peng, T. G. Ristroph, G. M. Schurmann, G. M. King, J. Yoon, V. Narayanamurti and J. A. Golovchenko, Appl. Phys. Lett., 2003, 83, 4238–4239 CrossRef CAS.
- S. Chong, Y. Chen, W. Yan, S. Guo, Q. Tan, Y. Wu, T. Jiang and Y. Liu, J. Power Sources, 2016, 332, 230–239 CrossRef CAS.
- H. Ming, J. Ming, W.-J. Kwak, W. Yang, Q. Zhou, J. Zheng and Y.-K. Sun, Electrochim. Acta, 2015, 169, 291–299 CrossRef CAS.
- M. F. Parkinson, J. K. Ko, A. Halajko, S. Sanghvi and G. G. Amatucci, Electrochim. Acta, 2014, 125, 71–82 CrossRef CAS.
- C. Li, J. Xu, J. Xia, W. Liu, X. Xiong and Z. Zheng, Solid State Ionics, 2016, 292, 75–82 CrossRef CAS.
- H. Jung, H. Song, T. Kim, J. K. Lee and J. Kim, J. Alloys Compd., 2015, 647, 750–755 CrossRef CAS.
- T. Bao, H. Zhong, H. Zheng, H. Zhan and Y. Zhou, Electrochim. Acta, 2015, 176, 215–221 CrossRef CAS.
- C. Chen, X. Xu, S. Chen, B. Zheng, M. Shui, L. Xu, W. Zheng, J. Shu, L. Cheng, L. Feng and Y. Ren, Mater. Res. Bull., 2015, 64, 187–193 CrossRef CAS.
- X. Xu, S. Chen, M. Shui, L. Xu, W. Zheng, J. Shu, L. Cheng, L. Feng and Y. Ren, Ceram. Int., 2014, 40, 3145–3148 CrossRef CAS.
- S. Zhao, Y. Bai, Q. Chang, Y. Yang and W. Zhang, Electrochim. Acta, 2013, 108, 727–735 CrossRef CAS.
- Y. Bai, X. Zhou, Z. Jia, C. Wu, L. Yang, M. Chen, H. Zhao, F. Wu and G. Liu, Nano Energy, 2015, 17, 140–151 CrossRef CAS.
- M. Burbano, M. Duttine, O. Borkiewicz, A. Wattiaux, A. Demourgues, M. Salanne, H. Groult and D. Dambournet, Inorg. Chem., 2015, 54, 9619–9625 CrossRef CAS PubMed.
- N. Yabuuchi, M. Sugano, Y. Yamakawa, I. Nakai, K. Sakamoto, H. Muramatsu and S. Komaba, J. Mater. Chem., 2011, 21, 10035–10041 RSC.
- W. Wu, X. Wang, X. Wang, S. Yang, X. Liu and Q. Chen, Mater. Lett., 2009, 63, 1788–1790 CrossRef CAS.
- J. Yang, Z. Xu, H. Sun and X. Zhou, Electrochim. Acta, 2016, 220, 75–82 CrossRef CAS.
- S. Tawa, T. Yamamoto, K. Matsumoto and R. Hagiwara, J. Fluorine Chem., 2016, 184, 75–81 CrossRef CAS.
- Y. Lu, Z. Wen, K. Rui, X. Wu and Y. Cui, J. Power Sources, 2013, 244, 306–311 CrossRef CAS.
- Y. Makimura, A. Rougier and J.-M. Tarascon, Appl. Surf. Sci., 2006, 252, 4587–4592 CrossRef CAS.
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, Chem. Commun., 2012, 48, 9909–9911 RSC.
- J. Liu, Y. Wan, W. Liu, Z. Ma, S. Ji, J. Wang, Y. Zhou, P. Hodgson and Y. Li, J. Mater. Chem. A, 2013, 1, 1969–1975 RSC.
- Y. Lu, Z. Wen, J. Jin, X. Wu and K. Rui, Chem. Commun., 2014, 50, 6487–6490 RSC.
- L. Zhang, S. Ji, L. Yu, X. Xu and J. Liu, RSC Adv., 2017, 7, 24004–24010 RSC.
- D. Ma, H. Wang, Y. Li, D. Xu, S. Yuan, X. Huang, X. Zhang and Y. Zhang, Nano Energy, 2014, 10, 295–304 CrossRef CAS.
- G. Ali, S. H. Oh, S. Y. Kim, J. Y. Kim, B. W. Cho and K. Y. Chung, J. Mater. Chem. A, 2015, 3, 10258–10266 RSC.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y. J. Zhu, X. G. Han, Y. H. Xu, Y. H. Liu, S. Y. Zheng, K. Xu, L. B. Hu and C. S. Wang, ACS Nano, 2013, 7, 6378–6386 CrossRef CAS PubMed.
- A. Eftekhari and M. Mohamedi, Energy Storage Mater., 2017, 6, 211–229 Search PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- L. Liu, H. Guo, M. Zhou, Q. Wei, Z. Yang, H. Shu, X. Yang, J. Tan, Z. Yan and X. Wang, J. Power Sources, 2013, 238, 501–515 CrossRef CAS.
- Y. Li, X. Zhao, Q. Xu, Q. Zhang and D. Chen, Langmuir, 2011, 27, 6458–6463 CrossRef CAS PubMed.
- C. L. Li, L. Gu, J. Tong, S. Tsukimoto and J. Maier, Adv. Funct. Mater., 2011, 21, 1391–1397 CrossRef CAS.
- C. Hu, E. Chen and J. Lin, Electrochim. Acta, 2002, 47, 2741–2749 CrossRef CAS.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- L. Chen, L.-J. Sun, F. Luan, Y. Liang, Y. Li and X.-X. Liu, J. Power Sources, 2010, 195, 3742–3747 CrossRef CAS.
- D. P. Lv, M. L. Gordin, R. Yi, T. Xu, J. X. Song, Y. B. Jiang, D. Choi and D. H. Wang, Adv. Funct. Mater., 2014, 24, 1059–1066 CrossRef CAS.
- C. Li, L. Gu, J. Tong, S. Tsukimoto and J. Maier, Adv. Funct. Mater., 2011, 21, 1391–1397 CrossRef CAS.
- A. M. Cao, J. S. Hu, H. P. Liang and L. J. Wan, Angew. Chem., Int. Ed., 2005, 44, 4391–4395 CrossRef CAS PubMed.
- P. Sen and A. De, Electrochim. Acta, 2010, 55, 4677–4684 CrossRef CAS.
- J. Yan, W. Sun, T. Wei, Q. Zhang, Z. Fan and F. Wei, J. Mater. Chem., 2012, 22, 11494–11502 RSC.
- S. Zheng, X. Li, B. Yan, Q. Hu, Y. Xu, X. Xiao, H. Xue and H. Pang, Adv. Energy Mater., 2017, 7, 1602733 CrossRef.
- Q. Zhu, H. Hu, G. Li, C. Zhu and Y. Yu, Electrochim. Acta, 2015, 156, 252–260 CrossRef CAS.
- B. Li, P. Gu, Y. Feng, G. Zhang, K. Huang, H. Xue and H. Pang, Adv. Funct. Mater., 2017, 27, 1605784 CrossRef.
- X. Guo, S. Zheng, G. Zhang, X. Xiao, X. Li, Y. Xu, H. Xue and H. Pang, Energy Storage Mater., 2017, 9, 150–169 CrossRef.
Footnote |
| † Nannan Zhang and Xiao Xiao contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |