
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescence dynamics and enhancement of the UV and visible emissions of Tm3+ in LiYF4:Yb3+,Tm3+ upconverting nanoparticles†
Steven L.
Maurizio
 ,
Gabriella
Tessitore
,
Gabrielle A.
Mandl
and
John A.
Capobianco
*
,
Gabriella
Tessitore
,
Gabrielle A.
Mandl
and
John A.
Capobianco
*
Department of Chemistry and Biochemistry, Centre for NanoScience Research, Concordia University, 7141 Sherbrooke St. W. Montreal, Quebec, H4B 1R6, Canada. E-mail: John.Capobianco@Concordia.ca
First published on 18th October 2019
Abstract
To maximize the intrinsic luminescence efficiency of the higher energy emissions of Tm3+ in LiYF4:Yb3+,Tm3+ upconverting nanoparticles, we investigated a specific range of Tm3+ dopant concentrations. Reported to be optimized at 25% Yb3+, 0.5% Tm3+, due to the multitude of Tm3+-to-Tm3+ interactions, the Tm3+ concentration commonly used may not be suitable for strong UV and visible emissions. Thus, we varied the concentration of Tm3+ in LiYF4 nanoparticles between 0.08 and 0.55% to elucidate the effect of moderate changes of the dopant concentration on the UV, visible and NIR emissions. We determined a new optimized concentration of 0.24% Tm3+ for maximal UV and visible emissions (nominally 0.2%). An extensive analysis of the luminescence spectra in the UV, visible and NIR regions and decay time measurements provides evidence for new luminescence mechanisms involving cross-relaxation pathways from the UV-emitting states of Tm3+. Furthermore, we performed studies on an azobenzene derivative to demonstrate the substantial enhancement of the UV emissions by the newly optimized composition as evidenced by an increase in the degree of trans–cis photoisomerization.
1. Introduction
Lanthanide-doped upconverting nanoparticles (UCNPs) have garnered interest in the past few decades owing to their versatile optical properties.1 Fluoride hosts, specifically NaREF4 (RE = Y, La, Gd or Lu) and LiYF4 doped with Tm3+ and Yb3+ have been used to produce emissions in the ultraviolet (UV), visible and near-infrared (NIR) regions of light upon excitation with NIR irradiation via upconversion.2 The upconversion process is affected by the properties of the material, which include the phonon energies of the host lattice, the presence of defects, the crystal field and the doping levels of the lanthanide ions.3 These properties affect the overall optical characteristics of the materials, such as the absorption and emission bandwidth, the absorption cross-section, the probability of energy transfer and the population mechanisms of the different excited states.4–8Mahalingam et al. first described the synthesis of LiYF4:Yb3+,Tm3+ UCNPs and reported the upconverted emissions from the UV to the NIR upon 980 nm excitation.9 Taking advantage of the high depth penetration, reduced photodamage and minimal autofluorescence of the NIR excitation light, Tm3+-doped upconverting nanoparticles have found applications in drug delivery systems and in photodynamic therapy.10–12 These applications are viable because LiYF4:Yb3+,Tm3+ UCNPs show a higher UV-to-NIR emission ratio than the sodium ternary hosts. The UV emission bands centered at 288, 345 and 360 nm are assigned to the 1I6 → 3H6, 3F4 and 1D2 → 3H6 transitions, respectively. These emissions have been used as an in situ source of UV light to facilitate photomediated reactions that require UV excitation. In addition, UV light produced by LiYF4:Yb3+,Tm3+ UCNPs can induce reversible interconversion between states in organic photochromic compounds such as spiropyrans, azobenzenes, fulgides, etc. that may be exploited for light-induced drug delivery.13,14 However, the relatively low UV emission intensity produced is a major limitation of this approach for in vivo applications. This implies that the NIR-to-UV upconversion efficiency of UCNPs is the limiting factor for their use in nanomedicine, whether it is for photocleavable drug release, photodynamic therapy, or inducing photoresponse of photochromic molecules. Therefore, there is a pressing need to optimize the UV emissions in order to increase the viability of these systems for clinical use.15–18
Two solutions can be adopted: tuning of the concentration of the Tm3+ ions or excite using a higher power density. An increased Tm3+ concentration would provide additional pathways for concentration quenching via non-radiative processes such as cross-relaxation, and a higher excitation power would induce heating which is detrimental in biological applications. A number of studies using LiYF4:Yb3+,Tm3+ UCNPs with a nominal Tm3+ concentration of 0.5% have been exploited, but optimization of Tm3+ concentrations to achieve more intense UV emissions have not been reported in these systems.10–13,19
In this study, we investigate the effects of slight changes in the Tm3+ concentration on the UV, visible and NIR transitions. We also evaluate the sensitization mechanisms and the cross-relaxation processes that are involved in the higher energy emitting states of Tm3+ (specifically the 1I6 and 1D2 levels). Due to the complexity of the Tm-to-Tm interactions, the reported cross-relaxation mechanisms in the literature do not address the 1I6 level. Our investigation reveals a new cross-relaxation process that involves the higher emitting states of Tm3+ and corroborates the choice of the new optimized concentration for Tm3+ of 0.24% to limit the non-radiative depopulation of the 1I6 emitting state. As a proof-of-concept, the newly optimized LiYF4:Yb3+,Tm3+ UCNPs were employed to demonstrate the improved efficiency of the trans–cis photoisomerization of trans-3-(4-phenylazophenoxy)propanol.
2. Experimental
2.1 Materials
Lanthanide oxides (Ln2O3) were purchased of highest available purity from Alfa Aesar (Y2O3: 99.99%, Yb2O3: 99.998% and Tm2O3: 99.997%). Trifluoroacetic acid (99.5+%) and lithium trifluoroacetate (97%) were purchased from Alfa Aesar. Technical grade oleic acid (90%) and 1-octadecene (90%) were used without further purification.2.2 LiYF4:Yb3+,Tm3+ upconverting nanoparticle synthesis
The synthesis of LiYF4 UCNPs doped with Tm3+ and Yb3+ was carried out using a previously established one-pot thermal decomposition synthesis method.9 1.25 mmol of oxide reagents (75 − x% Y2O3, 25% Yb2O3 and x% Tm2O3, where x = 0.1, 0.2, 0.4 and 0.5) were added to 10 mL of 50% v/v aqueous trifluoroacetic acid and refluxed for 16 hours, producing the trifluoroacetate precursors. The resulting precursors were then mixed with 2.5 mmol CF3COOLi in 20 mL oleic acid and 20 mL of 1-octadecene. This mixture was degassed at 120 °C for 30 minutes under constant stirring and vacuum, followed by an increase at a rate of 10 °C min−1 to 315 °C under constant stirring and an Ar-flow. This mixture was allowed to react for 1 hour and then cooled to room temperature in ambient conditions. The produced nanoparticles were then precipitated in ethanol, collected by centrifugation and washed by dispersing in hexanes. Samples were stored as a solid pellet in ethanol.2.3 Transmission electron microscopy
10 μL of a 1 mg mL−1 solution of UCNPs in toluene was evaporated on a formvar/carbon film supported on a 300 mesh copper grid (3 mm in diameter). Transmission electron microscopy (TEM) images were taken using a Jeol-JEM-2100F microscope operating at 200 kV equipped with a charge coupled device camera (Gatan).2.4 Powder X-ray diffraction
Powder X-ray diffraction (PXRD) patterns were recorded on solid sample UCNPs using a Bruker D2 Phaser equipped with a LynxEye detector, Cu source at a generation power of 30 kV and 10 mA. Scan range was set from 10–90° 2θ with a step size of 0.01° and an integration time of 2 s.2.5 Inductively coupled plasma-mass spectrometry
UCNP samples were digested in preparation for inductively coupled plasma-mass spectrometry (ICP-MS) analysis. 100 μL of a 1 mg mL−1 UCNP solution in toluene was added to 1 mL of concentrated HCl and 100 μL of H2O2. Test tubes were covered and heated to 115 °C for 16 hours to digest the nanoparticles. The solvent was then evaporated, and the resulting precipitate was dissolved in 5% aqueous HNO3, producing 5 ppm solutions to analyze. The analysis was completed using an Agilent 7500ce ICP-MS equipped with a quartz Scott-type spray chamber, an off-axis Omega lens ion focus and an octopole reaction system with a quadrupole mass spectrometer analyzer (operating at a frequency of 3 MHz).2.6 Upconversion luminescence spectroscopy
Upconversion emission spectra were obtained using 976 nm excitation from a Coherent 6-pin 15 fiber-coupled F6 series laser diode (operating at a power density of 42.9 W cm−2), controlled using an ILX Lightwave LDC-3744B. For comparative measurements, 1 mg mL−1 solutions of UCNPs in toluene were prepared. Visible emissions were recorded at a right angle to the incident beam, dispersed using a Jarrell-Ash Czerny-Turner 25-102 1 m double monochromator (1180 grooves per mm) coupled to a cooled Hamamatsu R943-02 photomultiplier tube. UV emissions were recorded at a right angle to the incident beam, dispersed using an Oriel 77250 1/8 m monochromator (2400 grooves per mm) and detected with a Hamamatsu R4632 photomultiplier tube. Output signals were processed through an SR440 Stanford Research Systems preamplifier and converted using an SR400 Stanford Research Systems gated photon counter. NIR emissions were recorded at a right angle to the incident beam, dispersed using a Spectral Products DK-240 monochromator (600 grooves per mm) and detected with a Spectral Products AD-131 InGaAs detector, with output signals processed using the provided software.2.7 Synthesis and characterization of 3-(4-phenylazophenoxy) propanol
3-(4-phenylazophenoxy)propanol was synthesized according to a previously reported protocol.12 The 1H-NMR spectrum was recorded on a 500 MHz Bruker Scientific NMR spectrometer at 25 °C with DMSO-d6 as a solvent. MALDI-TOF-MS was performed using a 2,5-dihydroxybenzoic acid matrix prepared as a saturated solution in a solvent mixture of acetonitrile and water with 0.1% trifluoroacetic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). The MALDI spectrum was recorded on an UltrafleXtreme time-of-flight spectrometer operated in positive ion, reflectron mode with a Bruker Daltonics Smartbeam II laser operating at a 2 kHz repetition rate. The spectrum was acquired using an accelerating voltage of +20 kV.
:1 v/v). The MALDI spectrum was recorded on an UltrafleXtreme time-of-flight spectrometer operated in positive ion, reflectron mode with a Bruker Daltonics Smartbeam II laser operating at a 2 kHz repetition rate. The spectrum was acquired using an accelerating voltage of +20 kV.
2.8 Photoisomerization measurements
Photoisomerization measurements were performed on 3-(4-phenylazophenoxy)propanol using a previously established procedure.12 A sample of 1 mg mL−1 UCNPs and 4.3 × 10−6 M 3-(4-phenylazophenoxy)propanol in hexanes, was irradiated at 30 min increments using a 976 nm laser (Coherent 6-pin 15 fiber-coupled F6 series laser diode operating at a power density of 42.9 W cm−2, controlled using an ILX Lightwave LDC-3744B). The recording of a UV-Vis absorption spectrum was obtained using a Cary 5000 Series UV-Vis-NIR Spectrophotometer (Agilent Technologies), operating with a scan speed of 600 nm s−1 and a resolution of 1 nm.2.9 Excited state decay times
Tm3+ decay times for powder UCNP samples were collected using a pulsed 976 nm laser diode (Coherent 6-pin 15 fiber-coupled F6 series, operating at 0.7 W cm−2, controlled using an ILX Lightwave LDP-3811), with a 200 μs pulse width. Emissions were recorded using the same experimental setup as the upconversion emission spectra, varying the acquisition time at a single observable wavelength. Instrument response curves for both detector setups are present in Fig. S1.†Yb3+ decay times for powder UCNP samples were collected using a chopped 976 nm laser diode (Sky Lasers PL-980-2000B, operating at 13 W cm−2, chopped with a Stanford Research Systems SR540 Optical Chopper operating at 45 Hz). Emissions were dispersed using a Jarrell-Ash Czerny-Turner 25-102 1 m double monochromator (1180 grooves per mm), collected using a North Coast Scientific EO-817P Ge-detector, and recorded using a Tektronix TDS 520A digital oscilloscope.
3. Results and discussion
Synthesized LiYF4:25% Yb3+,x% Tm3+ UCNPs were characterized by transmission electron microscopy (TEM), as presented in Fig. 1A–D. Inductively coupled plasma-mass spectrometry (ICP-MS) results shown in Table S1† provide the measured ionic concentration of Y3+, Tm3+, and Yb3+ in the samples studied. The concentrations reported in Table S1† were used to determine the sample composition, rather than their nominal values, to take into account the possible discrepancies between them due to the narrow range of concentrations studied. The LiYF4:25% Yb3+,x% Tm3+ UCNPs possess a diamond-like morphology with an aspect ratio of approximately 2:1, in agreement with previously reported results.9,12 The powder X-ray diffraction (PXRD) patterns (Fig. S2†) obtained from the nanocrystals confirmed their highly crystalline nature and their tetragonal structure (space group I41/a).20 From the particle size distribution (Fig. S3†) for the four samples synthesized, one can observe a substantial overlap in the particle size distribution and the average size ranges from 87.5 to 95.7 nm. Due to the overlapping size distributions, size-related discrepancies in the spectroscopic properties of the compositions studied may be ruled out.
 | ||
| Fig. 1 TEM images for LiYF4:25% Yb3+,x% Tm3+ UCNPs, with x = (A) 0.55%, (B) 0.46%, (C) 0.24% and (D) 0.08% Tm3+. | ||
The upconversion luminescence spectra of the 1 mg mL−1 toluene dispersion of the UCNPs upon irradiation with 976 nm light are shown in Fig. 2A. The observed bands in the UV, visible and NIR correspond to the 1I6 → 3H6, 3F4 and the 1D2 → 3H6 transitions (at 288, 345 and 360 nm, respectively), the 1D2 → 3F4 and the 1G4 → 3H6, 3F4 transitions (at 450, 475 and 650 nm, respectively) and the 3H4 → 3H6 transition (at 800 nm). An energy level diagram depicting the energy transfer upconversion (ETU) processes is shown in Fig. S4.† From the comparison of the four upconversion emission spectra in Fig. 2A, there is a substantial change in the emission intensities in the UV, visible and NIR regions as a function of Tm3+ concentration. Interestingly, the intensity of the bands in the UV/visible and those in the NIR do not follow the same trend as a function of Tm3+ concentration (Fig. 2B). We observed a maximized intensity for the emissions from the 1I6, 1D2, and 1G4 excited states upon decreasing the Tm3+ concentration to 0.24%, whereas the emission intensity from the 3H4 → 3H6 transition is maximized at 0.55%. Recent comprehensive investigations on the Tm3+ dopant concentration in NaYF4 UCNPs have observed maximized UV and visible emissions with 0.5% Tm3+.18 It should be noted that the weighted average lanthanide-to-lanthanide distance in a unit cell is further apart in NaYF4 (6.15 Å) compared to LiYF4 (5.16 Å), therefore it is expected that a lower dopant concentration is necessary to avoid cross-relaxation mechanisms that quench the higher energy excited states in LiYF4, and the observed 0.24% Tm3+ is consistent with this statement.21,22 These initial results provide evidence of possible non-radiative processes that quench the UV and visible emissions at concentrations higher than 0.24% Tm3+, which consequently increase the emission intensity of the NIR transition.
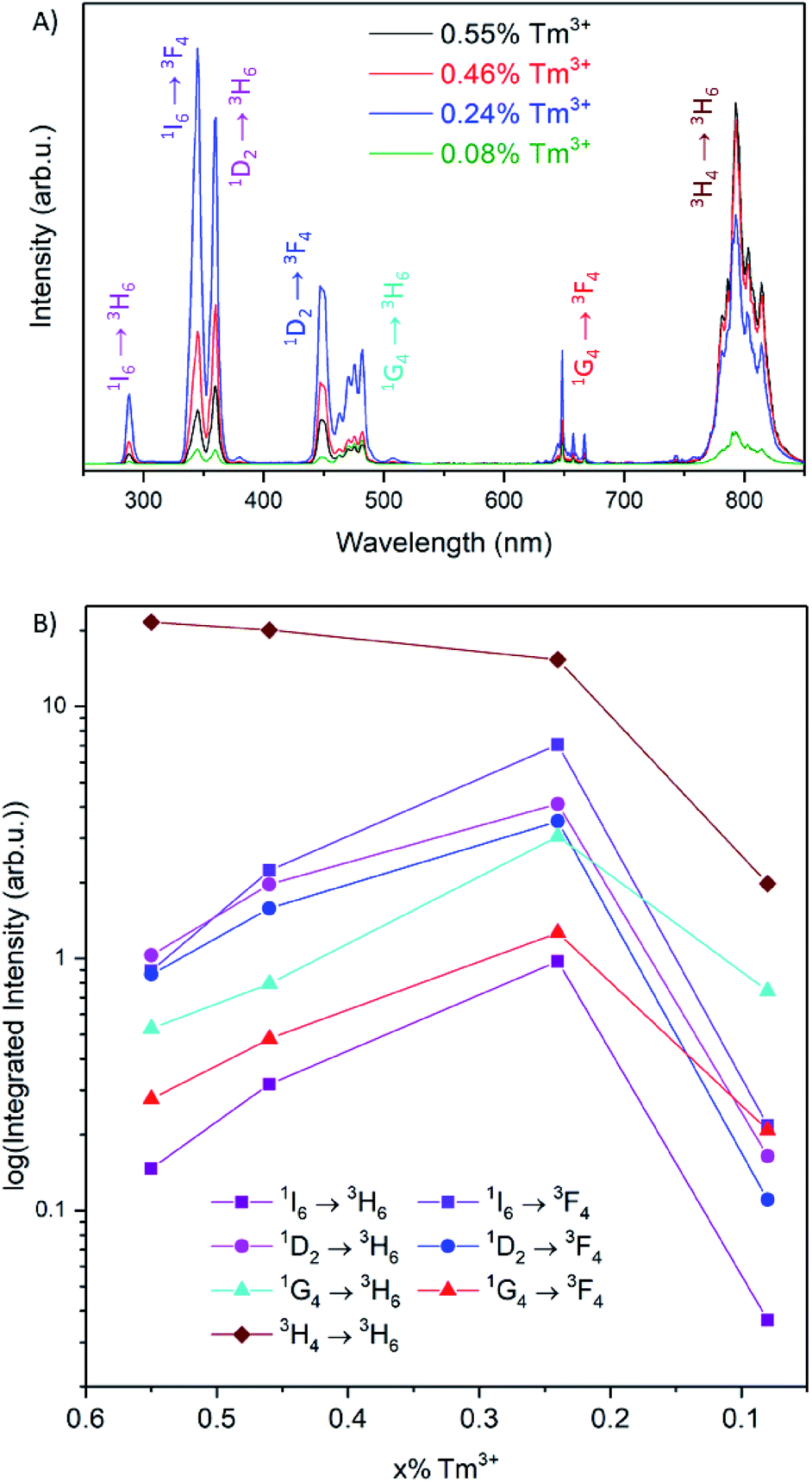 | ||
| Fig. 2 (A) Emission spectra for LiYF4:25% Yb3+,x% Tm3+ UCNPs after 976 nm excitation, measured in colloidal solutions of 1 mg mL−1 in toluene. (B) Integrated emission intensities for each transition of Tm3+ as a function of dopant concentration. | ||
To understand the dependence of the UV, visible and NIR emission intensities as a function of the concentration of Tm3+, the excited state population pathways were investigated. Typically, decreasing luminescence intensities are attributed to concentration quenching via cross-relaxation or energy migration.23,24 Since the optimal Tm3+ concentration for maximum NIR emission intensity were found to differ from the optimal concentration for the UV and visible emissions, the population mechanisms for each emitting state were evaluated in detail. The emission spectra were normalized to the 1D2 or the 1I6 transitions to examine the relative emission probability from each transition (Fig. 3 and S5,† respectively). These spectra show that the emissions from the 1G4 excited state have a much greater relative intensity at the lowest Tm3+ concentration studied, at 0.08%, in comparison to 0.24% Tm3+. The 3H4 → 3H6 transition at 800 nm exhibits a similar change for these two compositions. It is well-documented that the 1G4 and 3H4 excited states are involved in cross-relaxation mechanisms that populate the 1D2 state, and this result supports these findings.25–28 As the concentration of Tm3+ increases from 0.08 to 0.24%, the cross-relaxation mechanisms have an increased probability due to the decrease in interionic distance. This manifests itself as a decrease in the emission intensities from the 1G4 and 3H4 states, and an increase in the emission intensities from the 1D2 and consequently the 1I6 states.20,25–27
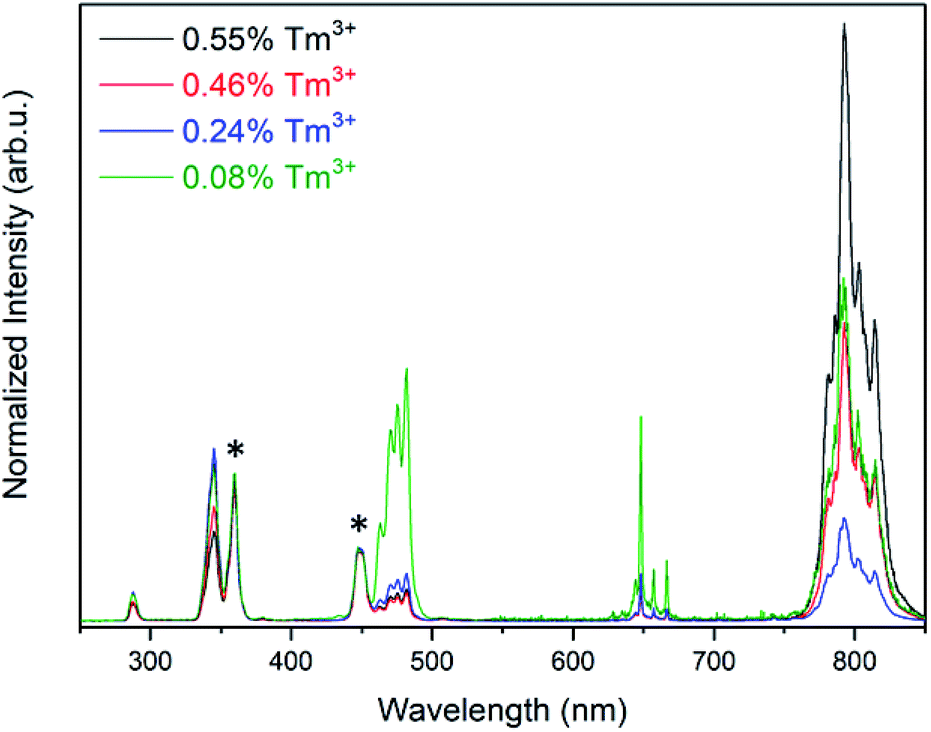 | ||
| Fig. 3 Emission spectra for LiYF4:25% Yb3+,x% Tm3+ after 976 nm excitation, normalized to the 1D2 transitions indicated by the asterisks. | ||
It is well established that the 1I6 state is solely populated via energy transfer upconversion between an excited Yb3+ ion and an excited Tm3+ ion in the 1D2 state.25,29,30 Thus, the population of the 1I6 state should be dependent on the population of the 1D2 state, regardless of the Tm3+ concentration. However, the observed results contradict this reasoning, as the emission intensities from these states were not found to be dependent when normalized to either transition. The emission intensities from the 1I6 transitions were found to decrease in relative intensity with respect to the 1D2 transitions as a function of Tm3+ concentration (Fig. S6†).
Furthermore, at concentrations greater than 0.24% Tm3+, the observed increase in relative intensity of the 3H4 → 3H6 transition indicates an increase in the population of the 3H4 state as a function of dopant concentration. Combined with the observations with respect to the 1I6 and 1D2 states, this suggests the presence of a possible cross-relaxation pathway involving the 1I6, 1D2 and 3H4 states. This cross-relaxation mechanism becomes prominent at concentrations above 0.24% Tm3+, and only becomes apparent when investigating such a low and narrow concentration range (0.08–0.55% Tm3+).
To elucidate the mechanism involved between these states, the luminescence decay curves (Fig. 4) were recorded from each excited state of Tm3+ in the concentration range between 0.55% and 0.24%, and their respective rise and decay times are reported in Table 1.
 | ||
| Fig. 4 Luminescence decay curves for LiYF4:25% Yb3+,x% Tm3+ UCNPs with x= (A) 0.55%, (B) 0.46% and (C) 0.24%, for the reported emissions from the 1I6, 1D2, 1G4, and 3H4 states after a 200 μs pulse of 976 nm excitation. | ||
| Transition | 0.55% Tm3+ | 0.46% Tm3+ | 0.24% Tm3+ |
|---|---|---|---|
| Rise times (μs) | |||
| 1I6 → 3F4 | 181 | 209 | 217 |
| 1D2 → 3H6 | 183 | 222 | 369 |
| 1G4 → 3H6 | 284 | 297 | 435 |
| 3H4 → 3H6 | 301 | 332 | 367 |
|
|||
| Decay times (μs) | |||
| 1I6 → 3F4 | 318 | 387 | 619 |
| 1D2 → 3H6 | 383 | 462 | 689 |
| 1G4 → 3H6 | 700 | 851 | 1136 |
| 3H4 → 3H6 | 1116 | 1223 | 1665 |
The luminescence decay curves show that there is a shortening in rise and decay times as the concentration of Tm3+ increases. Faster rise dynamics are indicative of more efficient energy transfer from the Yb3+ ions to the Tm3+ ions.31 Increasing the Tm3+ concentration corresponds to shortening of the Yb3+-to-Tm3+ distance, which results in more efficient energy transfer, as evidenced by the decreasing decay times of the Yb3+ emission at 1030 nm (Fig. S7†). Consequently, lower Tm3+ concentrations result in differing rise times for each excited state, due to the weaker Yb3+-to-Tm3+ interactions. Further, given the multitude of different interactions involving the 1G4 state, the increased rise time for this level at the lowest Tm3+ concentration indicates a reduced probability for cross-relaxation pathways to occur.25–28
The decrease in decay time between the samples containing 0.24% and 0.55% Tm3+ is 48.6% for the 1I6 → 3F4 transition, 44.4% for the 1D2 → 3H6 transition, 38.6% for the 1G4 → 3H6 transition and 32.9% for the 3H4 → 3H6 transition. A decrease in decay time is associated with the occurrence of non-radiative depopulation of a level via energy migration to traps, energy transfer to solvent or ligand molecules, and cross-relaxation processes.31 Analyzing each of these non-radiative contributions to the decay times, the first two processes are dependent on the particle size and dopant concentrations. Smaller particles have a greater surface area-to-volume ratio, which results in an increased number of traps and higher probability of a lanthanide ion to reside on the particle surface and interact with the ligand or solvent molecules.32,33 Moreover, it has been demonstrated that these two quenching mechanisms depend on the concentration of Yb3+.34 Considering that the size of the particle and concentration of Yb3+ does not change between the samples studied and the change in Tm3+ concentration is modest, quenching of luminescence due to energy migration to traps or energy transfer to the ligand is not expected to change significantly between the samples. Cross-relaxation processes have been demonstrated to extensively influence the luminescence dynamics in Tm3+-doped UCNPs, and can explain the change in the decay times as a function of the Tm3+ concentration.32
In the absence of cross-relaxation pathways, the population of the 1I6 level should depend only on the ETU efficiency and the 1D2 population dynamics.30 The increased ETU probability demonstrated by the Yb3+ decay curves should result in a shorter decay time of the 1I6 level.31 This is evidenced by the results in Fig. 4; the rise and decay times of the 1I6 and 1D2 excited states are relatively short and are influenced to a greater extent by the increasing Tm3+ dopant concentration, in comparison to the 1G4 and 3H4 excited states. The relative shortening of the decay time for the 1I6 level is greater than the other excited states, indicating there is a stronger influence of the cross-relaxation processes on the luminescence dynamics of the 1I6 transitions. Moreover, it is worth noting that the shortening in decay time of the 1I6 level is greater than that of the 1D2 level, which is involved in the population of the 1I6 level via ETU.22,29 This can be attributed to greater interactions between Tm3+ ions, which results in the increased probability for cross-relaxation mechanisms to occur between these two states.
In summary, the 1G4 → 3H6, 3F4 transitions do not change in relative intensity at concentrations above 0.24% Tm3+, demonstrating that this level is not involved in other mechanisms aside from those discussed previously with respect to Fig. 3. The emission intensities and dynamics of the 1I6 and 1D2 levels are not correlated, as would be expected by the excitation mechanism of the 1I6 state via ETU (Fig. S4†), suggesting there is a possible sensitization of the 1D2 excited state from the 1I6 state at higher Tm3+ concentrations. Furthermore, the 3H4 → 3H6 transition continues to increase in intensity with increasing concentrations of Tm3+, as shown in Fig. 2B, which is in contrast with the trend of all the other emissions. Therefore, a cross-relaxation mechanism involving the 1I6, 1D2 and 3H4 levels would account for the observed phenomena. This mechanism involves the depopulation of the 1I6 level and the population of the 1D2 and 3H4 levels, as demonstrated from the analysis of the emission spectra and luminescence decay times. The proposed mechanism, 1I6 + 3F4 → 1D2 + 3H4, is presented in Fig. 5. For Tm3+ concentrations above 0.24%, the luminescence spectra and decay curves support this mechanism and explain the disproportional quenching of the 1I6 and 1D2 emissions. The excited states involved in this mechanism are resonant, differing by less than one phonon of LiYF4, which further enforces the proposed mechanism.35,36
 | ||
| Fig. 5 Energy level diagram depicting the 1I6 + 3F4 → 1D2 + 3H4 cross-relaxation mechanism. | ||
The proposed mechanism can be corroborated upon investigation of the NIR emission spectrum, shown in Fig. 6. Upon 976 nm irradiation, the emissions from the 3H4 → 3F4 (1485 nm) and 3F4 → 3H6 (1730 nm) transitions are observed. With increasing Tm3+ dopant concentrations, there is an increase in these emission intensities, in contrast to the decrease in visible and UV emission intensities discussed previously (Fig. 2B). This suggests that the population of the NIR-emitting states occurs via the depopulation of the higher energy states. A correlation between the emission intensities of the NIR-emitting transitions was found, which can be rationalized by a previously reported cross-relaxation mechanism, 3H4 + 3H6 → 3F4 + 3F4, which results in an increase in the relative emission intensity at 1730 nm with increasing Tm3+ concentrations, shown in Fig. S8.†20 This serves to explain why the 3H4 → 3H6 transition at 800 nm increases to a lesser extent as the Tm3+ concentration is increased, as both cross-relaxation mechanisms (1I6 + 3F4 → 1D2 + 3H4 and 3H4 + 3H6 → 3F4 + 3F4) involve the 3H4 level. Thus, the depopulation of the 1I6 level ultimately results in the population of and emission from the 3F4 level. The importance of investigating the entire emission spectrum, including the NIR region, to understand the dynamics of the upconversion luminescence in nanoparticles is further demonstrated by these results.
 | ||
| Fig. 6 NIR emission spectrum for LiYF4:25% Yb3+,x% Tm3+ UCNPs after 976 nm excitation, normalized to the 2F5/2 → 2F7/2 emission of Yb3+, indicated by the asterisk. | ||
As a proof of concept to further validate the improvements of the UV and visible luminescence from the sample with 0.24% Tm3+, we performed experiments on a UV-photoswitchable azobenzene derivative. Azobenzene undergoes trans–cis isomerization upon excitation with UV irradiation, which has been successfully demonstrated using the UV emissions from Tm3+ in UCNPs.11,12 Azobenzenes are particularly attractive for light-mediated drug delivery systems, due to their significant change in shape, polarity, and absorption spectra upon isomerization.37,38 The trans isomer of azobenzene has a strong π → π* transition centered in the UV, and a weak n → π* in the blue region, whereas the cis isomer has a weaker, blue-shifted π → π* transition, with a slightly more intense n → π* transition (Fig. 7A).
 | ||
| Fig. 7 (A) UCNP emission spectrum with absorption spectra of azopropOH, depicting spectral overlap to induce a photoisomerization. (B) AzopropOH photoisomerization reaction scheme. (C) Absorption spectra of azopropOH in solution with LiYF4:25% Yb3+,x% Tm3+ UCNPs of 0.55% and (D) 0.24% Tm3+ after 210 minutes of 976 nm irradiation. | ||
Notwithstanding the successful demonstration of trans–cis isomerization of azobenzenes using Tm3+-doped nanoparticles, the dopant concentrations in LiYF4:Yb3+,Tm3+ UCNPs have not been optimized for their UV emissions, and thus the isomerization has been demonstrated at relatively high nanoparticle concentrations with high NIR power densities.
A comparison of the photoswitching capabilities of the UCNPs with 0.55% Tm3+ and 0.24% Tm3+ was performed on 3-(4-phenylazophenoxy)propanol (azopropOH) with the UCNPs under 976 nm irradiation. 1H-NMR and mass spectrometry results for the synthesized azopropOH are presented in Fig. S9 and S10,† respectively. This azobenzene derivative was used in this study because the absorption maximum for the π → π* transition is centered at 342 nm and overlaps well with the UV emissions from the UCNPs, presented in Fig. 7A and B.38 Isomerization of the azopropOH was studied by evaluating the decrease in the π → π* transition by UV-Vis absorption spectroscopy as a function of NIR irradiation time. As shown in Fig. 7C and D, after 210 minutes of irradiation, no significant isomerization of the azopropOH can be detected using the UCNPs containing 0.55% Tm3+ as evidenced by the lack of change in the π → π* absorption band. In contrast, a significant decrease in the π → π* transition was observed after 30 minutes of irradiation using the optimized 0.24% Tm3+ UCNPs, from which a photostationary state was achieved after 210 minutes of NIR irradiation, resulting in approximately 20% decrease in the intensity of the π → π* transition. This represents a significant improvement in the capability of Tm3+-doped UCNPs to isomerize azobenzene, as other reports in the literature demonstrate the isomerization at higher UCNP concentrations and excitation powers, and some achieve photostationary states after much longer irradiation times.12,39,40 Additionally, the interactions of the azopropOH molecules and UCNPs in this experiment were limited to interactions through Brownian motion, which further demonstrates the increased efficiency of the system.
4. Conclusions
In order to investigate the influence of Tm3+ concentration on the UV and visible emissions in LiYF4:Yb3+,Tm3+ upconverting nanoparticles, emission spectroscopy and decay time measurements were performed. The Tm3+ concentration was varied between 0.08 and 0.55%, and a new optimized concentration of 0.24% Tm3+ was established (nominally 0.2%) with respect to the improvement of the UV and visible emissions. The small concentration range investigated corresponded to a dramatic change in luminescence intensities for the UV, visible and NIR emissions, emphasizing the multitude of Tm3+-to-Tm3+ interactions. The performed measurements were used to determine the depopulation mechanisms of the higher-energy levels in Tm3+, and a new cross-relaxation mechanism of 1I6 + 3F4 → 1D2 + 3H4 was proposed. To demonstrate the significant enhancement in the UV emissions, photoswitching experiments involving 3-(4-phenylazophenoxy)propanol were performed. Trans–cis isomerization of the azobenzene derivative was observed to be far more efficient with the optimized Tm3+ concentration of 0.24%. This new composition provides a number of new opportunities in any application exploiting NIR-induced photochemistry from the UV and visible emissions of UCNPs, where lower powers and reduced UCNP concentrations are desired.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. A. C. is a Concordia University Research Chair in Nanoscience and is grateful to Concordia University for financial support. J. A. C. is grateful to the Natural Science and Engineering Research Council (NSERC) of Canada for the sustained support of his research. S. L. M. is grateful to Concordia University for financial support from the Triskelion Fellowship. G. T. is grateful to Concordia University for financial support from the Horizon Post-Doctoral Program. G. A. M. is grateful for the Concordia Merit Scholarship and NSERC graduate scholarship for financial support.Notes and references
- G. Tessitore, G. A. Mandl, M. G. Brik, W. Park and J. A. Capobianco, Nanoscale, 2019, 11, 12015–12029 RSC.
- F. Auzel, J. Lumin., 1990, 45, 341–345 CrossRef CAS.
- S. Hüfner, Optical Spectra of Transparent Rare Earth Compounds, Academic Press, 1978 Search PubMed.
- H.-X. Mai, Y.-W. Zhang, R. Si, Z.-G. Yan, L.-D. Sun, L.-P. You and C.-H. Yan, J. Am. Chem. Soc., 2006, 128, 6426–6436 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- B. Zhou, B. Shi, D. Jin and X. Liu, Nat. Nanotechnol., 2015, 10, 924–936 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- G. Tessitore, S. L. Maurizio, T. Sabri and J. A. Capobianco, Angew. Chem., Int. Ed., 2019, 58, 9742–9751 CrossRef CAS PubMed.
- V. Mahalingam, F. Vetrone, R. Naccache, A. Speghini and J. A. Capobianco, Adv. Mater., 2009, 21, 4025–4028 CrossRef CAS.
- M. M. Dcona, Q. Yu, J. A. Capobianco and M. C. T. Hartman, Chem. Commun., 2015, 51, 8477–8479 RSC.
- P. A. Rojas-Gutierrez, S. Bhuckory, C. Mingoes, N. Hildebrandt, C. DeWolf and J. A. Capobianco, ACS Appl. Nano Mater., 2018, 1, 5345–5354 CrossRef CAS.
- G. A. Mandl, P. A. Rojas-Gutierrez and J. A. Capobianco, Chem. Commun., 2018, 54, 5847–5850 RSC.
- B. F. Zhang, M. Frigoli, F. Angiuli, F. Vetrone and J. A. Capobianco, Chem. Commun., 2012, 48, 7244–7246 RSC.
- H. K. Bisoyi and Q. Li, Angew. Chem., Int. Ed., 2016, 55, 2994–3010 CrossRef CAS PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- C. Zhao, X. Kong, X. Liu, L. Tu, F. Wu, Y. Zhang, K. Liu, Q. Zeng and H. Zhang, Nanoscale, 2013, 5, 8084 RSC.
- Y. Cui, S. Zhao, Z. Liang, M. Han and Z. Xu, J. Alloys Compd., 2014, 593, 30–33 CrossRef CAS.
- M. Kraft, C. Würth, E. Palo, T. Soukka and U. Resch-Genger, Methods Appl. Fluoresc., 2019, 7, 024001 CrossRef CAS PubMed.
- Q. Yu, E. M. Rodriguez, R. Naccache, P. Forgione, G. Lamoureux, F. Sanz-Rodriguez, D. Scheglmann and J. A. Capobianco, Chem. Commun., 2014, 50, 12150–12153 RSC.
- A. Braud, S. Girard, J. L. Doualan, M. Thuau, R. Moncorgé and A. M. Tkachuk, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 5280–5292 CrossRef CAS.
- A. Grzechnik, P. Bouvier, M. Mezouar, M. D. Mathews, A. K. Tyagi and J. Köhler, J. Solid State Chem., 2002, 165, 159–164 CrossRef CAS.
- A. Grzechnik, K. Syassen, I. Loa, M. Hanfland and J. Y. Gesland, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 104102 CrossRef.
- B. Tian, J. Zhao, Y. Tian, X. Li, J. Zhang, J. Sun, R. Hua and B. Chen, J. Nanosci. Nanotechnol., 2015, 15, 281–289 CrossRef CAS PubMed.
- J. Martincik, M. Nikl, S. Ishizu, K. Fukuda, T. Suyama, A. Beitlerova, K. Polak, V. Babin and A. Yoshikawa, Radiat. Meas., 2010, 45, 403–405 CrossRef CAS.
- P. Villanueva-Delgado, K. W. Krämer, R. Valiente, M. de Jong and A. Meijerink, Phys. Chem. Chem. Phys., 2016, 18, 27396–27404 RSC.
- F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5642–5643 CrossRef CAS PubMed.
- M. Misiak, K. Prorok, B. Cichy, A. Bednarkiewicz and W. Stręk, Opt. Mater., 2013, 35, 1124–1128 CrossRef CAS.
- B. S. Cao, J. L. Wu, Z. Q. Feng and B. Dong, Mater. Chem. Phys., 2013, 142, 333–338 CrossRef CAS.
- G. Wang, W. Qin, J. Zhang, J. Zhang, Wangyan, C. Cao, L. Wang, G. Wei, P. Zhu and R. Kim, J. Phys. Chem. C, 2008, 112, 12161–12167 CrossRef CAS.
- C. Cao, W. Qin, J. Zhang, Y. Wang, P. Zhu, G. Wang, G. Wei, L. Wang and L. Jin, J. Fluorine Chem., 2008, 129, 204–209 CrossRef CAS.
- J. Bergstrand, Q. Liu, B. Huang, X. Peng, C. Würth, U. Resch-Genger, Q. Zhan, J. Widengren, H. Ågren and H. Liu, Nanoscale, 2019, 11, 4959–4969 RSC.
- B. M. Walsh, N. P. Barnes, M. Petros, J. Yu and U. N. Singh, J. Appl. Phys., 2004, 95, 3255–3271 CrossRef CAS.
- L. Cademartiri and G. A. Ozin, Concepts of Nanochemistry, Wiley-VCH, 2009 Search PubMed.
- S. Fischer, N. D. Bronstein, J. K. Swabeck, E. M. Chan and A. P. Alivisatos, Nano Lett., 2016, 16, 7241–7247 CrossRef CAS PubMed.
- S. A. Miller, H. E. Rast and H. H. Caspers, J. Chem. Phys., 1970, 52, 4172–4175 CrossRef CAS.
- M. Dulick, G. E. Faulkner, N. J. Cockroft and D. C. Nguyen, J. Lumin., 1991, 48–49, 517–521 CrossRef CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- J. Liu, W. Bu, L. Pan and J. Shi, Angew. Chem., Int. Ed., 2013, 52, 4375–4379 CrossRef CAS PubMed.
- L. Wang, H. Dong, Y. Li, C. Xue, L.-D. Sun, C.-H. Yan and Q. Li, J. Am. Chem. Soc., 2014, 136, 4480–4483 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00556k |
| This journal is © The Royal Society of Chemistry 2019 |