
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceClean rhodium nanoparticles prepared by laser ablation in liquid for high performance electrocatalysis of the hydrogen evolution reaction†
Giulia Alice
Volpato‡
 a,
David
Muneton Arboleda‡
b,
Riccardo
Brandiele‡
a,
Francesco
Carraro
a,
Giovanni Battista
Sartori
a,
Andrea
Cardelli
a,
Denis
Badocco
a,
Paolo
Pastore
a,
Stefano
Agnoli
a,
Christian
Durante
*a,
Vincenzo
Amendola
*a and
Andrea
Sartorel
*a
a,
David
Muneton Arboleda‡
b,
Riccardo
Brandiele‡
a,
Francesco
Carraro
a,
Giovanni Battista
Sartori
a,
Andrea
Cardelli
a,
Denis
Badocco
a,
Paolo
Pastore
a,
Stefano
Agnoli
a,
Christian
Durante
*a,
Vincenzo
Amendola
*a and
Andrea
Sartorel
*a
aDepartment of Chemical Sciences, University of Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: christian.durante@unipd.it; vincenzo.amendola@unipd.it; andrea.sartorel@unipd.it
bCentro de Investigaciones Ópticas CIOp (CONICET-CIC-UNLP), Facultad de Ingeniería UNLP, La Plata, Argentina
First published on 16th October 2019
Abstract
Rhodium nanoparticles (NPs) were prepared by a one-step, green and facile procedure consisting in laser ablation of a bulk Rh target immersed in pure water (W–Rh-NPs) or ethanol (E–Rh-NPs). When embedded in mesoporous carbon based inks, both W–Rh-NPs and E–Rh-NPs show excellent activity towards the hydrogen evolution reaction in acidic media, operating close to the thermodynamic potential with 85–97% faradaic yields and low Tafel slopes of 50–54 mV per decade in the low overpotential region (η < 20 mV). A superior activity of W–Rh-NPs with respect to E–Rh-NPs is ascribed to the absence of surface carbon reducible species derived from the synthesis in organic solvent, and thus confirms the importance of the use of water as the preferred medium for laser synthesis of clean nanocrystals in liquid environment. These results provide an important contribution to the impelling need for the preparation of nano-catalysts based on energy critical materials by clean, sustainable and low cost routes.
Rh-based materials are desired catalysts for diverse industrial reactions such as petroleum refining and fine chemical production.1 Besides the resistance to high temperature, oxidation, and corrosion, Rh possesses partially filled 4d electron levels which enable the moderate adsorption of reactants and thus promote the formation of intermediates.1–4 The development of Rh-based nano-catalysts has thus received increasing interest, also by virtue of their compelling properties towards electrocatalysis for energy applications, in particular for the hydrogen evolution reaction (HER).5–9 To this purpose, Rh nanocrystals are often obtained by solution-based routes, either one-pot or multi-step, in environments containing salts, carbohydrates, oil, hydrofluoric acid or organic solvent,6–8,10,11 by various procedures such as microwave heating,1 solvothermal,8 sonochemical7 or electrochemical synthesis.1 These methods allow the achievement of the conditions required for the conversion of Rh precursor (e.g., RhCl3, Na3RhCl6 or Rh acetate) into metal nanocrystals, typically in the presence of stabilizing agents (oleylamine), strong reducing agents (NaBH4) or high temperature.1
Despite its catalytic value, there are no reports about the synthesis or Rh catalysts for HER by laser ablation is liquid (LAL), which has the potential of being a fast, easy, reproducible, robust, clean, green and cost-effective approach.12–15 In LAL, a bulk metal plate is directly converted into a colloid of the same metal by a pulsed laser beam, under ambient conditions and without the need for additional chemicals dissolved in the liquid phase.12,13 Therefore, when the appropriate conditions are selected, LAL provides the remarkable advantage of producing nanocrystals with a clean surface, with no undesired chemical species blocking the catalytic active atoms, as often observed with the synthesis routes using organic compounds or salts either as reagents or capping agents. In general, any residual species in solution can adsorb on the catalyst surface when NPs are embedded in a functional substrate, which in turn drastically diminishes the catalytic performance.16
We present herein the preparation of Rh NPs by laser ablation of a bulk Rh target in pure liquids (water or ethanol), and the evaluation of their performances for HER, with particular emphasis on the role of the medium where the synthesis is conducted.
The Rh colloids were generated according to the typical LAL procedure (Fig. 1A),17 by focusing a 1064 nm pulsed laser beam with fluence of 7.6 J cm−2 on a bulk rhodium plate immersed in water or ethanol. After LAL, Rh NPs were collected by centrifugation for further characterization and catalytic studies.
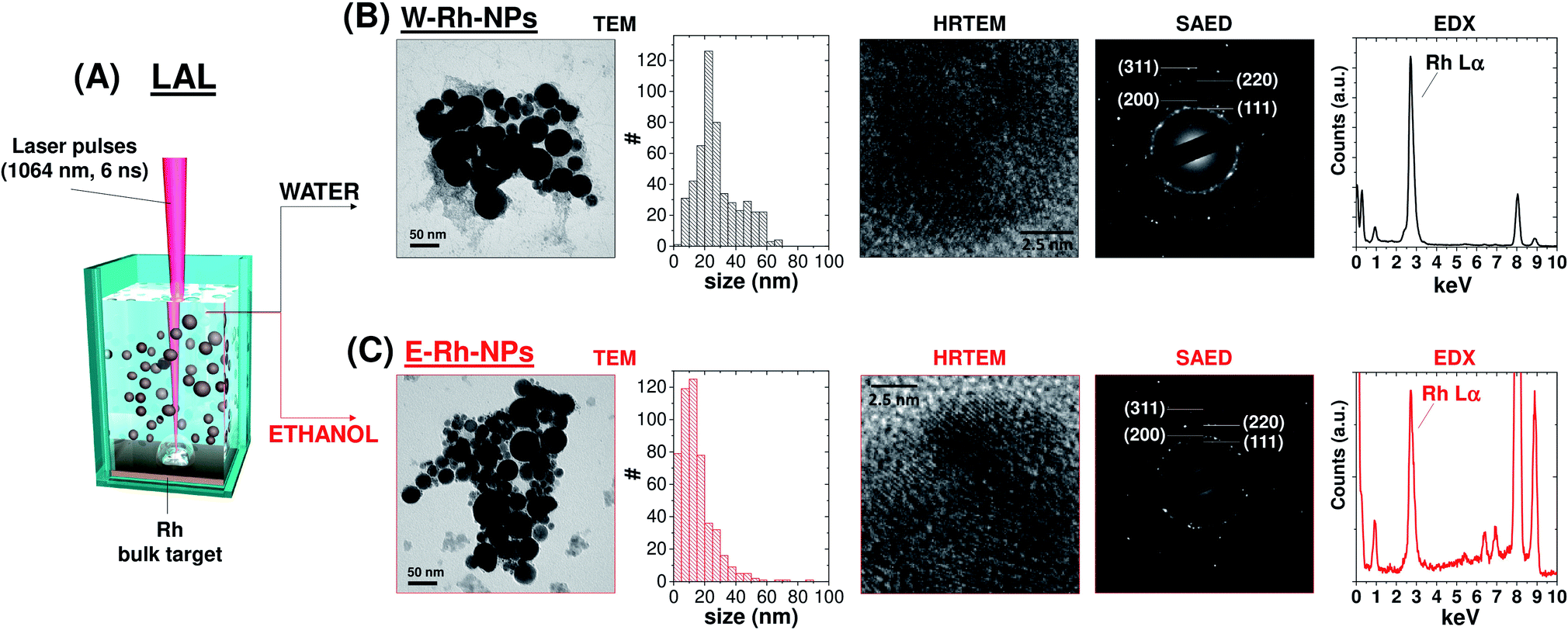 | ||
| Fig. 1 (A) Sketch of LAL methodology: ns pulses (1064 nm, 6 ns) are focused on a bulk Rh target dipped in either water or ethanol, and Rh NPs are collected as a colloid. (B and C) Representative TEM, HRTEM, SAED, EDX results and the size histograms of the W–Rh-NPs (B) and E–Rh-NPs (C) samples. | ||
The morphology and crystal structure of Rh NPs obtained in water (W–Rh-NPs, Fig. 1B and S1 in ESI†) and ethanol (E–Rh-NPs, Fig. 1C and S1 in ESI†) were assessed by transmission electron microscopy (TEM) analysis. It can be seen that NPs possess a regular spherical shape in both cases. In fact, metal nanoparticles generated by laser ablation in pure liquids are generally spherical for surface free-energy minimization, and the surface energy of Rh is notably larger than other noble metals such as Pt, Au and Pd, due to the large cohesive energy of Rh atoms in their face centered cubic (fcc) lattice.18
The statistical analysis performed on 510 NPs for each sample (see size histograms in Fig. 1B and C) showed average diameters of 28 ± 14 nm for W–Rh-NPs and 14 ± 11 nm for E–Rh-NPs, consistent with the typical range found for metal nanoparticles obtained with near infrared laser pulses in the absence of any interfering chemical compound or stabilizer.19 In fact, the presence of chemical stabilizers in the liquid during the synthesis is associated to a smaller average size, because the stabilizing molecules can readily coat the surface of the nanoparticles, preventing their further growth and coalescence.12 Smaller average size has been observed also when running LAL with 532 nm pulses.20 At this wavelength, the same laser beam used for ablation of the target can be also absorbed by the previously generated NPs that are dispersed in the liquid layer above the target, which are photo-fragmented, resulting in a final product with smaller average size than using near infrared pulses.
The lattice planes of Rh NPs can be clearly observed from the HRTEM images of the two samples (see Fig. 1B and C and S1 in ESI†), and the measured crystal lattice spacing is 0.22 nm, in agreement with the (111) plane of fcc rhodium. Despite the surface area of Rh NPs is dominated by low index facets, HRTEM also evidenced that each NPs is polycrystalline and endowed at the grain boundaries with some atomic steps and kink sites typically associated with coordinatively unsaturated atoms. Generally, this type of structural defects is desired for optimal performance in catalysis.18
The selected area electron diffraction (SAED) analysis on clusters of W–Rh-NPs or E–Rh-NPs clearly showed concentric rings of spots, thus confirming particles crystallinity. In particular, the first four spot rings of both the NPs obtained in water or ethanol correspond to diffractions from the (111), (200), (220) and (311) lattice planes, and match with the reference fcc rhodium structure (JCPDS 05-0685). Notably, no rhodium oxide diffraction peaks were detected in the SAED patterns. However, Rh has a renowned thermodynamic tendency to oxidation and surface passivation.21 Therefore, to assess the surface chemical composition and electronic state of Rh NPs, Raman spectroscopy was performed on both samples. In fact, a broad Raman peak, centred at 540 cm−1 (Fig. 2A), was clearly detected in both samples, matching with the Raman fingerprint of Rh2O3.21 Only in case of the Rh NPs obtained in ethanol, some surface amorphous carbon contamination was detected by Raman spectroscopy, as apparent from the double broad band at 1300–1500 cm−1. This is likely due to adsorption of solvent pyrolysis by-products, as previously observed during LAL of noble metal targets in ethanol and other organic solvents.19 Since no amorphous carbon bands are detected in the W–Rh-NPs, one can infer that pure water is a more suitable environment for the synthesis, in addition to the obvious advantages brought from water in comparison to organic solvents in terms of sustainability, costs and safety (i.e. lack of flammability).
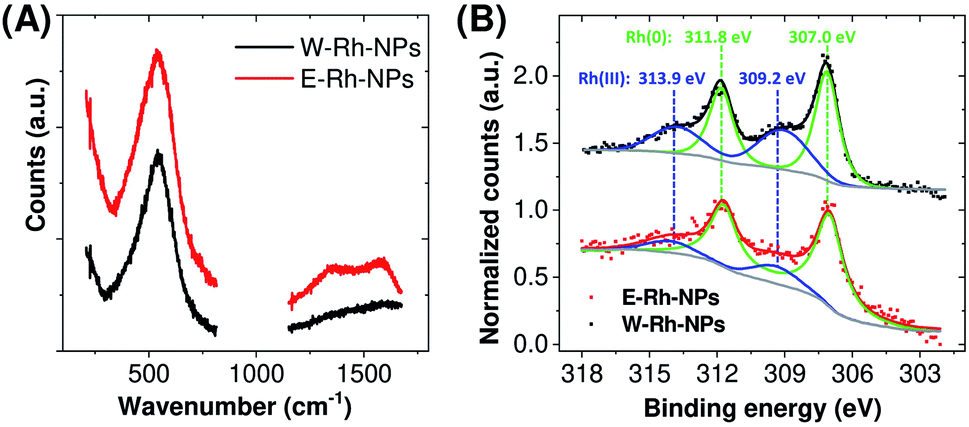 | ||
| Fig. 2 (A) Raman spectra of Rh NPs obtained in water or ethanol. (B) X-ray photoelectron spectroscopy (XPS) spectra of Rh NPs E–Rh/C and W–Rh/C inks obtained in different liquid environments. | ||
For the electrocatalytic characterization, inks were prepared by loading the E–Rh-NPs and W–Rh-NPs onto finely dispersed mesoporous carbon particles (Vulcan XC-72; 30/70 w/w Rh/C ratio) suspended in ethanol/water mixture containing 0.02% Nafion. The resulting E–Rh/C and W–Rh/C inks were then homogeneously drop casted onto glassy carbon electrodes (GCE, 0.071 or 0.196 cm2 geometric surface area), keeping a fixed metal loading of 20 μgRh cm−1;2 this procedure allowed a proper dispersion of the nanoparticles onto the substrate, as evidenced by scanning electron microscopy (Fig. S2 in ESI†). The X-ray photoemission spectra of the inks deposited on carbon paper are reported in Fig. 2B. The Rh 3d region is characterized by the 3d3/2 and 3d5/2 doublet. In agreement with Raman analysis, the core level Rh 3d5/2 photoemission peak indicated two chemically shifted components, namely Rh(0) and Rh(III) at binding energies of 307.0 eV and 309.2 eV, respectively. This confirms the presence of both oxidation states on the surface of the Rh NPs.7 After deconvolution, the percentage of the surface Rh(0) component was estimated to be 73% for W–Rh-NPs and 84% for E–Rh-NPs. The slightly higher fraction of the oxide in the W–Rh-NPs sample is consistent with the higher oxidizing properties of water with respect to ethanol. It is worth to highlight that the presence of the oxide was proposed to have a key role in efficient hydrogen evolution reaction by Rh/Rh2O3 nanostructures bifunctional catalyst in alkaline environment, due to the favoured balancing of the surface properties coming from the weakening of hydrogen binding energy and the increase of oxophilicity and ability of OH− binding.7
The electrochemical features of E–Rh/C and W–Rh/C (0.5 M H2SO4, 20 mV s−1) under argon saturated solution are similar, with an anodic wave at Epa = 0.70 V vs. reversible hydrogen electrode (RHE), attributed to surface oxidation of metallic Rh to higher oxidation states, i.e. Rh(I)–Rh(III).7,22 In the backward scan, two cathodic peaks are observed at Epc = 0.60 and 0.32 V vs. RHE, and attributed to stepwise reduction of higher oxidized states to Rh(0).22
The electrochemical surface area (ESA) of the electrodes was evaluated by anodic CO stripping,23–25 by comparing the cyclic voltammetries (0.5 M H2SO4, 20 mV s−1) under argon saturated solution of the electrodes before and after exposure treatment with CO (Fig. 3A).§ The resulting ESA were 0.67 ± 0.10 and 0.91 ± 0.10 cm2 for E–Rh/C and W–Rh/C inks, respectively. The electrochemical active surface area (ECSA) was then obtained by normalization of the ESA with respect to the metal surface loading (1.4 μg, by considering the metal loading of 20 μgRh cm−2 on the 0.071 cm2 geometric surface area). The resulting ECSA were 48 ± 9 and 65 ± 7 m2 gRh−1 for E–Rh/C and W–Rh/C inks, respectively, and are in the range reported for Rh/C systems investigated in literature for HER (see Tables S1 and S2 in ESI†).7
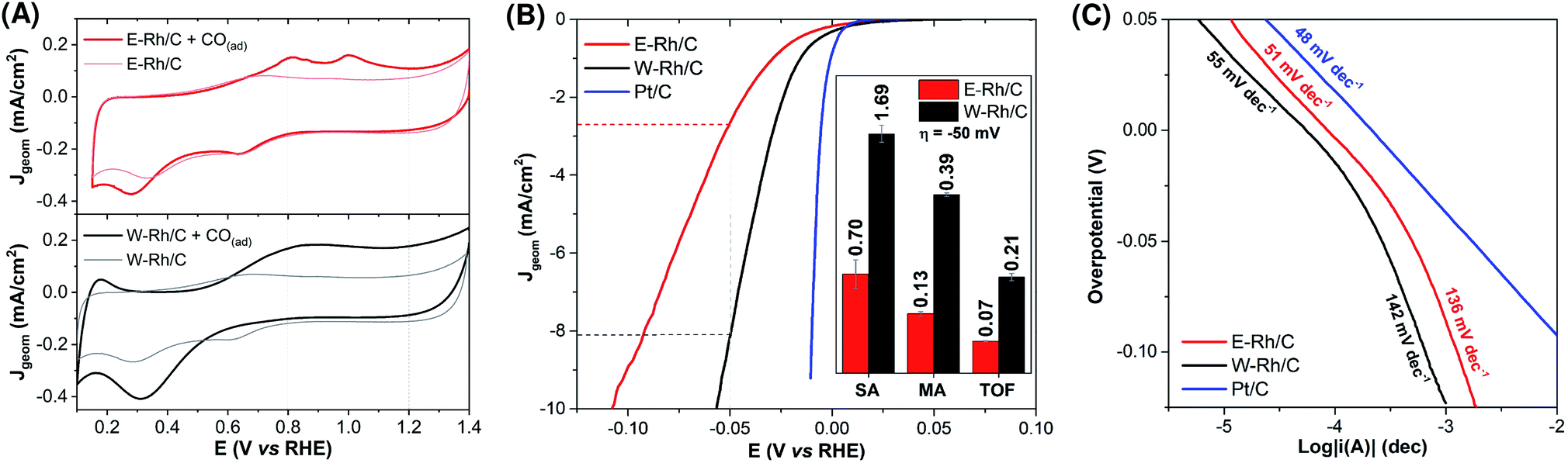 | ||
| Fig. 3 Electrochemical characterization of Rh/C inks at fixed metal loading (20 μgRh cm−2). (A) CO ad-layer stripping voltammograms (0.5 M H2SO4, 20 mV s−1) for the determination of the electrochemical surface area (ESA) of E–Rh/C and W–Rh/C inks deposited on GCE (0.071 cm2): voltammograms of the inks before (lighter lines) and after (darker lines) CO adsorption; ESA is derived from the integrated area of CO-stripping peak (integration range: 0.8 ÷ 1.2 V). (B) Linear sweep voltammograms (2 mV s−1 iR compensated) in 0.1 M H2SO4 of E–Rh/C (red), W–Rh/C (black) and commercial Pt/C (blue) deposited on GCE (0.196 cm2) with equal metal loading (20 μgM cm−2). Inset: comparison of specific activities (SA, expressed in mA cmM−2), mass activities (MA, mA μgM−1) and turnover frequencies (TOF, s−1) at −50 mV of overpotential for W–Rh/C and E–Rh/C. (C) Tafel plots for E–Rh/C, W–Rh/C and commercial Pt/C on GCE (0.071 cm2) in 0.1 M H2SO4, obtained from LSV at 0.5 mV s−1 with RDE at 1600 rpm. | ||
The electrocatalytic activity of the E–Rh/C and W–Rh/C materials towards the hydrogen evolution reaction (HER) was then investigated by linear sweep voltammetry (LSV) in 0.1 M H2SO4 (Fig. 3B; Jgeom is the current density normalized with respect to geometric area; see also Fig. S3 in ESI† for LSV where current density normalized with respect to ESA, JESA, is reported). For both Rh based materials, the LSV scans show the rising of catalytic cathodic currents attributed to proton reduction to hydrogen at potentials close to the thermodynamic one. A relevant parameter is the overpotential η required to reach Jgeom of −1 and −10 mA cm−2: W–Rh/C required only −16 ± 1 mV and −57 ± 1 mV of overpotential to achieve Jgeom of −1 mA cm−2 and −10 mA cm−2, respectively, whereas E–Rh/C needed η = −32 ± 4 mV and −110 ± 10 mV, respectively (Table S1†). Concerning performance evaluation with respect to the literature benchmarks, Table S1† collects some figures of merit. Higher overpotentials were observed for Rh nanoparticles onto silicon wires (η comprised between −84 and −129 mV at Jgeom = −10 mA cm−2),11 while η up to −36 mV (Jgeom = −10 mA cm−2) were obtained with Rh/silicon quantum dot/carbon quantum dot composites.6 The current benchmark for Rh HER electrocatalysis is constituted by Rh–Rh2O3 nanoparticles supported onto nitrogen doped carbon, showing performance comparable to Pt/C (η = −13 mV and −24 mV for Rh–Rh2O3 and for Pt/C, respectively, at Jgeom = −10 mA cm−2, see Table S1† and Fig. 3).7¶ More in general, the high performance of Rh materials for the HER is ascribed to the hydrogen adsorption free energy ΔGH close to the ideal value of 0,26 and to low kinetic barriers for the hydrogen evolution steps, that position Rh materials close to the top of the volcano plot for HER.26
Focusing on the comparison of reactivity between the nanoparticles under investigation in this work, the higher activity of W–Rh/C with respect to E–Rh/C in terms of lower overpotential was supported also by a higher specific activity (SA of 1.69 and 0.70 mA cmRh−2 for W–Rh/C and E–Rh/C, respectively, determined at η = −50 mV), mass activity (MA of 0.39 and 0.13 mA μgRh−1 for W–Rh/C and E–Rh/C, respectively, determined at η = −50 mV), and turnover frequency (TOF, of 0.21 and 0.07 s−1 for W–Rh/C and E–Rh/C, respectively, determined at η = −50 mV), see histograms in inset of Fig. 3B.
Electrochemical impedance spectroscopy (EIS) is frequently exploited to obtain useful insights about the activity of nanocatalysts.27–29 In our case, EIS further confirmed the better activity of W–Rh/C with respect to E–Rh/C also in terms of lower charge transfer resistance (RCT) and lower time constant (τCT) for the electron transfer process (Fig. S4 and Table S3 in ESI†); for the sake of comparison, at E = −40 mV vs. RHE, RCT and τCT are 11 Ω and 0.13 ms for W–Rh/C, and 64 Ω and 0.90 ms for E–Rh/C.
Chronopotentiometries at Jgeom = −10 mA cm−2 were performed in order to confirm hydrogen evolution and to check the stability of the material and possible modifications or sintering of the nanoparticles. Different tests were run by supporting the inks onto carbon paper or onto GCE (Fig. S5 in ESI†).|| Hydrogen evolution was confirmed in all cases, while the faradaic yield was shown to depend only on the ink and not on the carbon support, reaching values of 97 ± 4 and 85 ± 3 for W–Rh/C and E–Rh/C, respectively. More stable performances were achieved for inks casted onto carbon paper with respect to glassy carbon, likely ascribed to a better adhesion of the Rh/C materials on this porous medium. During 2 h chronopotentiometry at Jgeom = −10 mA cm−2, the W–Rh/C showed a constant operating potential around −60 mV, while the E–Rh/C required a progressive cathodic shift up to −165 mV in order to maintain the current density (Fig. S5 in ESI†). For both W–Rh/C and E–Rh/C, ex situ SEM and XPS analyses comparing fresh with stressed samples did not show any significant structural or chemical modification of the nanoparticles in the carbon inks (Fig. S7 and S8 in ESI†), in line with previous examples showing high robustness of Rh materials towards corrosion.6,7
Tafel analysis on E–Rh/C and W–Rh/C materials was performed by LSV at low scan rate with rotating disk electrode (Fig. 3C). Similar Tafel slopes of 50 and 54 mV dec−1 were obtained for W–Rh/C and E–Rh/C, respectively, in the low overpotential region (η < −20 mV),** while higher slopes of 130 and 133 mV dec−1 were registered for W–Rh/C and E–Rh/C, respectively, at higher overpotentials (η > −20 mV). These results suggest a Volmer/Heyrovsky HER mechanism, with the Heyrovsky step being rate determining for both these catalysts.4,5,20,30
Interestingly, the W–Rh/C and E–Rh/C materials maintained electrocatalytic activity for HER also under alkaline conditions (0.1 M KOH), with W–Rh/C still showing superior performance with respect to the E–Rh/C. Indeed, overpotentials of −1 mV and −20 mV were needed for W–Rh/C and E–Rh/C, respectively, in order to reach a Jgeom of −1 mA cm−2, and of −113 ± 4 mV and −157 ± 8 mV for W–Rh/C and E–Rh/C, respectively, to reach Jgeom of −10 mA cm−2 (Fig. S10 and Table S2 in ESI†). The higher performance of W–Rh/C with respect to E–Rh/C was also confirmed by higher surface activity (SA, 1.89 and 1.49 mA cmRh−2 for W–Rh/C and E–Rh/C, respectively), mass activity (MA, 0.44 and 0.26 mA μgRh−1 for W–Rh/C and E–Rh/C, respectively), and turnover frequency (TOF, 0.23 and 0.14 s−1 for W–Rh/C and E–Rh/C, respectively), all determined at η = −0.1 V. The Tafel slope analysis obtained by LSV experiments indicate similar slopes of 41 and 44 mV per decade. Typically under alkaline conditions a Volmer/Tafel mechanism is envisaged, where the Tafel is the rate determining step.7 These key performance indicators suggest a comparable or even better performance of the Rh nanoparticles with respect to commercial Pt/C (η = −103 mV to reach Jgeom = −10 mA cm−2, SA = 0.87 mA cm−2Pt, MA = 0.33 mA μgPt−1, TOF = 0.33 s−1 at η = −0.1 V, Tafel slope = 113 mV per decade, see Table S2†). The origin of such excellent performance in alkaline media was recently attributed to the presence of oxophilic surface rhodium oxide sites, favouring adsorptive dissociation of water, and thus forming reactive hydrogen adsorbed intermediates on the neighbouring Rh metallic sites.7 However, in our case, a decrease of performances was observed during consecutive scans, indicating a fast damaging of the material, and thus requiring a further engineering of the ink in order to maintain the electrocatalytic stability.
In summary, we reported the first study about laser generated Rh nanoparticles for HER. Two pure liquids (water and ethanol) were compared as the synthetic environments, allowing the production of Rh nanoparticles with comparable structure but markedly different electrocatalytic activity. In particular, the performances of the W–Rh/C nano-catalysts are comparable to those typically reported in the literature for Rh nanoparticles obtained by more complex or not environmentally friendly synthetic routes (Table S1†). Under alkaline conditions, the HER activity of the W–Rh/C nano-catalysts is comparable or even superior to commercial Pt/C catalyst.
The better performance of W–Rh/C with respect to E–Rh/C, in terms of higher SA, MA, TOF, low charge transfer resistance and FY, can be explained by the residual presence in the E–Rh/C sample of amorphous carbon due to solvent pyrolysis during LAL, inhibiting the electrocatalytic HER process.23
These results thus highlight the importance of conducting the synthesis in water, to achieve clean nanocrystals with high catalytic activity. In this way, laser synthesis is capable to satisfy the impelling need for the preparation of nano-catalysts based on energy critical materials by sustainable and low-cost routes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was performed with the support of the University of Padova STARS grant “4NANOMED” and “Iniziative di Cooperazione Universitaria 2017” and the Ministry of Foreign Affairs and International Cooperation “Great relevance project” with Protocol No. 0191594. AS thanks the Department of Chemical Sciences at the University of Padova for funding (Project PHOETRY, P-DiSC #10BIRD2018-UNIPD). GAV acknowledges Fondazione Cariparo for funding her PhD grant.Notes and references
- L. Luo, H. Li, Y. Peng, C. Feng and J. Zeng, ChemNanoMat, 2018, 4, 451–466 CrossRef CAS.
- K. Kani, J. Kim, B. Jiang, M. S. A. Hossain, Y. Bando, J. Henzie and Y. Yamauchi, Nanoscale, 2019, 11, 10581–10588 RSC.
- S. Y. Kim, A. Yu, Y. Lee, H. Y. Kim, Y. J. Kim, N.-S. Lee, C. Lee, Y. Lee and M. H. Kim, Nanoscale, 2019, 11, 9287–9295 RSC.
- C. Lin, G. Wu, H. Li, Y. Geng, G. Xie, J. Yang, B. Liu and J. Jin, Nanoscale, 2017, 9, 1834–1839 RSC.
- L. Zhang, L. Liu, H. Wang, H. Shen, Q. Cheng, C. Yan and S. Park, Nanomaterials, 2017, 7, 103 CrossRef PubMed.
- Q. Dan, F. Liao, Y. Sun, S. Zhang, H. Huang, W. Shen, Z. Kang, Y. Shi and M. Shao, Electrochim. Acta, 2019, 299, 828–834 CrossRef.
- M. K. Kundu, R. Mishra, T. Bhowmik and S. Barman, J. Mater. Chem. A, 2018, 6, 23531–23541 RSC.
- H. Duan, D. Li, Y. Tang, Y. He, S. Ji, R. Wang, H. Lv, P. P. Lopes, A. P. Paulikas, H. Li, S. X. Mao, C. Wang, N. M. Markovic, J. Li, V. R. Stamenkovic and Y. Li, J. Am. Chem. Soc., 2017, 139, 5494–5502 CrossRef CAS PubMed.
- Z. Pu, I. S. Amiinu, D. He, M. Wang, G. Li and S. Mu, Nanoscale, 2018, 10, 12407–12412 RSC.
- N. Zhang, Q. Shao, Y. Pi, J. Guo and X. Huang, Chem. Mater., 2017, 29, 5009–5015 CrossRef CAS.
- L. Zhu, H. Lin, Y. Li, F. Liao, Y. Lifshitz, M. Sheng, S. T. Lee and M. Shao, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- J. Zhang, M. Chaker and D. Ma, J. Colloid Interface Sci., 2017, 489, 138–149 CrossRef CAS PubMed.
- S. Reichenberger, G. Marzun, M. Muhler and S. Barcikowski, ChemCatChem, 2019, 11, 4489–4518 CrossRef CAS.
- Z. Lin, J. Li, L. Li, L. Yu, W. Li and G. Yang, J. Mater. Chem. A, 2017, 5, 773–781 RSC.
- D. Liang, S. Wu, J. Liu, Z. Tian and C. Liang, J. Mater. Chem. A, 2016, 4, 10609–10617 RSC.
- Y. Jiang, J. Su, Y. Yang, Y. Jia, Q. Chen, Z. Xie and L. Zheng, Nano Res., 2016, 9, 849–856 CrossRef CAS.
- I. Vassalini, L. Borgese, M. Mariz, S. Polizzi, G. Aquilanti, P. Ghigna, A. Sartorel, V. Amendola and I. Alessandri, Angew. Chem., Int. Ed., 2017, 56, 6589–6593 CrossRef CAS PubMed.
- B. Jiang, C. Li, Ö. Dag, H. Abe, T. Takei, T. Imai, M. S. A. Hossain, M. T. Islam, K. Wood, J. Henzie and Y. Yamauchi, Nat. Commun., 2017, 8, 3–4 CrossRef PubMed.
- V. Amendola and M. Meneghetti, Phys. Chem. Chem. Phys., 2013, 15, 3027–3046 RSC.
- X. Liu, Q. Han, Y. Zhang, X. Wang, S. Cai, C. Wang and R. Yang, Appl. Surf. Sci., 2019, 471, 929–934 CrossRef CAS.
- A. A. Tolia, R. J. Smiley, W. N. Delgass, C. G. Tokoudis and M. J. Weaver, J. Catal., 1994, 150, 56–70 CrossRef CAS.
- H. Gouveia, R. Landers and J. F. C. Boodts, Electrochim. Acta, 2007, 52, 2359–2369 CrossRef CAS.
- T. H. M. Housmans, J. M. Feliu and M. T. M. Koper, J. Electroanal. Chem., 2004, 572, 79–91 CrossRef CAS.
- N. F. Yu, N. Tian, Z. Y. Zhou, L. Huang, J. Xiao, Y. H. Wen and S. G. Sun, Angew. Chem., Int. Ed., 2014, 53, 5097–5101 CAS.
- C. A. Campos-Roldán, R. G. González-Huerta and N. Alonso-Vante, Electrochim. Acta, 2018, 283, 1829–1834 CrossRef.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- X. Yang, Z. Zhao, X. Yu and L. Feng, Chem. Commun., 2019, 55, 1490–1493 RSC.
- Y. Wang, Z. Liu, H. Liu, N. T. Suen, X. Yu and L. Feng, ChemSusChem, 2018, 11, 2724–2729 CrossRef CAS PubMed.
- F. Wang, X. Yang, B. Dong, X. Yu, H. Xue and L. Feng, Electrochem. Commun., 2018, 92, 33–38 CrossRef CAS.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods; SEM images, additional LSV, chronopotentiometry, additional XPS analysis, Tafel plots, benchmarking of Rh NPs. See DOI: 10.1039/c9na00510b |
| ‡ Authors with equal contributions. |
| § The electrode carrying the E–Rh/C ink shows two CO-stripping peaks at ca. 0.8 and 1.0 V; the presence of CO-stripping peak multiplicity was observed with Pt nanoparticles, and its origin is still under debate, see also (ref. 19). |
| ¶ For a commercial Pt/C sample (Pt/C), having 4 ÷ 7 nm size Pt nanoparticles, and the same metal and carbon loading of W–Rh/C and E–Rh/C, η = −11 ± 1 mV at Jgeom = −10 mA cm−2, see Table S1†. |
| || The stability of the two catalysts was also tested by repeated cycling: for both, 30% of the residual current density was maintained after 1000 potential cycles (Fig. S6 in ESI†), with partial detachment of the ink coating from GCE. Therefore, the poor stability under this specific test are ascribed to mechanical stress and to low surface adhesion of the inks onto the flat GCE surface, thus highlighting the importance of the nature of the substrate and of ink optimization. |
| ** Tafel slopes obtained with controlled potential electrolysis (Fig. S9 in ESI†) showed similar values of 50 mV per decade for W–Rh/C and 42 mV per decade for E–Rh/C respectively, in the low overpotential region (η < −20 mV); a deviation to higher slopes (280–320 mV per decade) was observed at higher overpotential due to damaging of the ink coating along the chronoamperometries. |
| This journal is © The Royal Society of Chemistry 2019 |