
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafine PdAu nanoparticles immobilized on amine functionalized carbon black toward fast dehydrogenation of formic acid at room temperature†
Luming
Wu
a,
Baoxia
Ni
a,
Rui
Chen
a,
Chengxiang
Shi
a,
Pingchuan
Sun
 bc and
Tiehong
Chen
*ac
bc and
Tiehong
Chen
*ac
aInstitute of New Catalytic Materials Science, School of Materials Science and Engineering, Key Laboratory of Advanced Energy Materials Chemistry (MOE), Nankai University, Tianjin 300350, PR China. E-mail: chenth@nankai.edu.cn
bKey Laboratory of Functional Polymer Materials of Ministry of Education, College of Chemistry, Nankai University, Tianjin 300071, PR China
cCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300071, PR China
First published on 23rd September 2019
Abstract
Ultrafine and highly dispersed PdAu nanoparticles were immobilized on amine functionalized carbon black (VXC-72-NH2) for dehydrogenation of formic acid (FA). The introduction of amines is of vital importance for the formation of ultrafine PdAu nanoparticles (∼1.5 nm). Moreover, the presence of the amino groups also increased the electron density of PdAu nanoparticles, and this effect facilitated the formation of metal-formate, which further enhanced the rate of the catalytic dehydrogenation of FA. The as-prepared Pd0.6Au0.4/VXC-72-NH2 exhibited high catalytic activity and 100% H2 selectivity for dehydrogenation of formic acid without any additive, with turnover frequency (TOF) values of 7385 h−1 at 298 K and 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 724 h−1 at 333 K, which are the highest TOF values ever reported among heterogeneous catalysts for FA dehydrogenation.
724 h−1 at 333 K, which are the highest TOF values ever reported among heterogeneous catalysts for FA dehydrogenation.
1. Introduction
Supported noble metal nanoparticles (NPs) have drawn considerable attention due to their high activities in many heterogeneous catalytic reactions.1–3 However, owing to their high surface energy, metal NPs are generally thermodynamically unstable and prone to aggregate, resulting in loss of catalytic activity and recyclability.4 Therefore, support materials with a high surface area and suitable interaction with the metal NPs are crucial to achieve good dispersity of metal NPs and thus to improve the catalytic performance.Among all the available support materials, carbon-based materials with unique properties of high surface area, enhanced mobility of charge carriers and good stability have been widely used.5–7 However, for carbon-based materials, the surface hydrophobicity would result in poor dispersion in aqueous solution, and the lack of surface functional groups is to the disadvantage of interactions with supported metal NPs. Recently, nitrogen (N)-doped carbon has been prepared and used as the support for catalysts, and the electron-rich N dopant could bring about basic coordination sites to facilitate the anchoring of well-dispersed and ultrafine NPs for high activity.8–10 However, in most cases, the procedures presently used for the synthesis of N-functionalized carbon involve high temperature or long pyrolysis time, which would greatly hinder their practical applications.11,12 Hence, controllable surface modification of carbon materials by facile and low temperature methods to support metal NPs with high dispersion remains a critical challenge.
Hydrogen is a sustainable and clean energy carrier for the establishment of a fuel-cell-based hydrogen economy.13 Formic acid (FA, HCOOH) has emerged as one of the most promising hydrogen storage compounds due to its high hydrogen density (4.4 wt%), nontoxicity and excellent stability.14–17 Hydrogen stored in FA can be released via the dehydrogenation pathway (HCOOH → CO2 + H2) and the dehydration pathway (HCOOH → CO + H2O).18,19 The dehydration pathway is an undesired side reaction and should be strictly controlled because CO is highly toxic and capable of poisoning Pt-based fuel cell catalysts. In recent years, many heterogeneous catalysts have been developed for FA dehydrogenation,20–37 and among them amine functionalized graphene, MOFs and mesoporous silica are generally used to support gold (Au), palladium (Pd) and Pd-based alloy NPs. Depending on the nature of support materials and preparation methods, some catalysts exhibited efficient dehydrogenation activities. For instance, Jiang and coworkers prepared AuPd NPs supported on amine functionalized graphene, and the catalyst gave a TOF value of 4445.6 h−1 at 298 K.27 Masuda et al. reported PdAg NPs supported on mesoporous carbon functionalized with p-phenylenediamine, which exhibited a high TOF of 5638 h−1 at 348 K.25 Xu and coworkers prepared highly dispersed palladium nanoclusters immobilized on a nitrogen (N)-functionalized porous carbon support with a TOF of 8414 h−1 at 333 K.10
Compared with graphene, MOFs, mesoporous silica, mesoporous carbon, active carbons and carbon black are industrial-scale produced and cost-effective supports, which have widely been used as the support of noble metals for many industrial catalysts. Herein, we report a facile and efficient approach to synthesize well-dispersed PdAu NPs immobilized on amine functionalized carbon black (VXC-72). The introduction of amines is of vital importance for the synthesis of ultrafine PdAu nanoparticles (∼1.5 nm), and the as-prepared Pd0.6Au0.4/VXC-72-NH2 exhibited high catalytic activity and 100% H2 selectivity for dehydrogenation of formic acid without any additive, with TOF values of 7385 h−1 at 298 K and 17724 h−1 at 333 K, which are the highest TOF values ever reported among heterogeneous catalysts for FA dehydrogenation. This work may provide a general and applicable approach to improve the properties of carbon supported noble metal catalysts, which are widely employed in industrial applications such as hydrogenation, oxidation and electrocatalysis.
2. Experimental
2.1 Chemicals and materials
All the reagents were of analytical purity and used as received without further purification. VXC-72 was purchased from Ailian Electronic Technology Co., Ltd, Tianjin. Tetrachloroauric (III) acid was purchased from Shanghai Macklin Biochemical Co., Ltd. Sodium tetrachloropalladate, 3-aminopropyltriethoxysilane (APTES), formic acid (FA), and sodium borohydride (NaBH4) were purchased from Aladdin (China). Nitric acid (HNO3) and ethanol (CH3CH2OH) were purchased from Tianjin Chemical Reagents, China.2.2 Synthesis of the catalysts
:Pd in the PdAu/VXC-72-NH2 system was changed (0:1, 0.2:0.8, 0.6:0.4, 0.8:0.2, and 1:0). For comparison, Pd0.6Au0.4/VXC-72 was prepared using a similar method without the addition of APTES.
2.3 Characterization
Transmission electron microscopy (TEM) was performed on a JEM-2800 microscope with an energy dispersive X-ray spectroscopy (EDX) detector, working at 200 kV to analyze the surface morphology of the prepared samples. All the samples subjected to TEM measurements were ultrasonically dispersed in ethanol and dropped onto carbon films. Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku Smart Lab 3000 W powder diffractometer using Cu Kα radiation (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermofisher ESCALAB 250Xi spectrometer. The Raman spectra were obtained using a Raman spectrometer (SR-500I-A) using laser excitation at 532 nm. Nitrogen adsorption and desorption isotherms were measured on a BELSORP-mini II sorption analyzer at 77 K. The specific surface area was calculated by the BET (Brunauer–Emmett–Teller) method, the pore-size distribution was calculated from the adsorption branch using the BJH (Barett–Joyner–Halenda) method and the total pore volume was obtained at a P/P0 of 0.99. The contents of Pd and Au were determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES) on a Thermo Jarrell-Ash ICP-9000. The contact angle was measured on a JC2000D3M contact angle tester.2.4 FA dehydrogenation experiments
The hydrogen production from FA solution was carried out in a 25 mL flask. Typically, 0.1 g of the Pd0.6Au0.4/VXC-72-NH2 catalyst was kept in the flask, and then 5 mL of aqueous solution containing FA (1 M) was injected quickly. The catalytic reaction begun after the FA solution was added to the flask under continuous magnetic stirring (600 rpm) at 25 °C. The evolution of gas was monitored by using a gas burette. Concentrations of H2, CO2 and CO were measured on a GC (SP-2100A) with a thermal conductivity detector (TCD) and a flame ionization detector (FID)-methanator and the detection limit for CO was 10 ppm.2.5 Durability testing of the catalysts
For testing the durability of the as-prepared Pd0.6Au0.4/VXC-72-NH2 catalyst, 0.2 mL of pure FA was added to the reaction mixture after the complete conversion of the first-run composition of FA. Such recycle experiments for the synthesized catalyst for the decomposition of FA were carried out for 5 runs at 298 K by adding equivalent FA (5 mmol). After the durability testing, the catalyst was collected for XRD, XPS and TEM analyses.2.6 Calculation methods
The TOF value is based on the number of Au and Pd atoms in the catalyst, which is calculated from the initial TOF value, which is calculated when the conversion reaches 20%. The equation is as follows: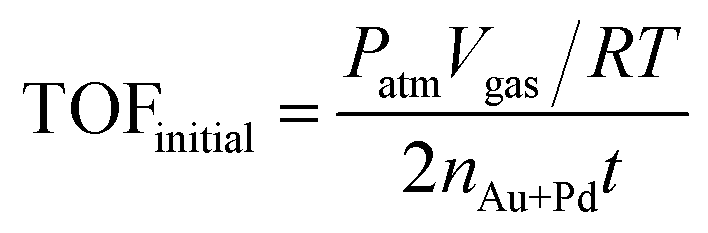 | (1) |
3. Results and discussion
The design and fabrication procedure of Pd0.6Au0.4/VXC-72-NH2 are summarized in Scheme 1. VXC-72 was first treated with highly concentrated nitric acid to generate grafting oxygen-containing functional groups (–COOH and –OH). According to the literature,26,38 3-aminopropyl-triethoxysilane (APTES) underwent hydrolysis in a water/ethanol mixture and the ethoxy groups were replaced by silanol groups, which would further react with the hydroxyl or carboxyl groups on the carbon black surface. Aqueous solutions of H2AuCl4 and Na2PdCl4 (with a Au:Pd molar ratio of 0.4:0.6) were added to the above APTES-treated VXC-72 solution and reduced with NaBH4 at −3 °C for 5 h. The product Pd0.6Au0.4/VXC-72-NH2 was obtained and used for catalytic H2 generation from FA solution.
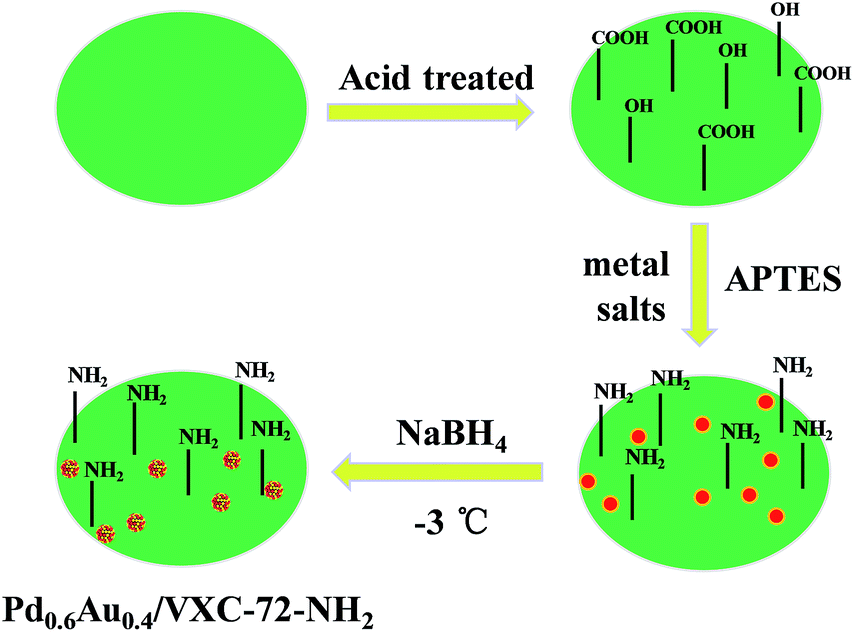 | ||
| Scheme 1 Schematic illustration of the preparation of Pd0.6Au0.4/VXC-72-NH2. | ||
The transmission electron microscopy (TEM) images of Pd0.6Au0.4/VXC-72-NH2 showed that PdAu NPs with an average particle size of 1.5 nm were homogenously dispersed on VXC-72-NH2 (Fig. 1a–c). In contrast, larger PdAu NPs with a mean size of 3.0 nm were observed for Pd0.6Au0.4/VXC-72 (Fig. 1d–f). In addition, VXC-72 is hard to disperse in water uniformly due to its hydrophobicity (the water contact angle is shown in Fig. 2a), while VXC-72-NH2 functionalized with amine groups exhibited excellent hydrophilicity. As shown in Fig. 2b, VXC-72-NH2 could be uniformly dispersed in aqueous solution, and the catalyst suspension remained stable even after 24 h. The hydrophilicity differences highlight the importance of amine functionalized VXC-72 as a support for mediating the ultrafine size and excellent dispersion of PdAu NPs. The energy-dispersive X-ray spectroscopy (EDX) result confirmed the existence of N, Au and Pd in Pd0.6Au0.4/VXC-72-NH2 (Fig. S1†). The accurate content of Au:Pd in Pd0.6Au0.4/VXC-72-NH2 was determined to be 0.42:0.58 by ICP-AES.
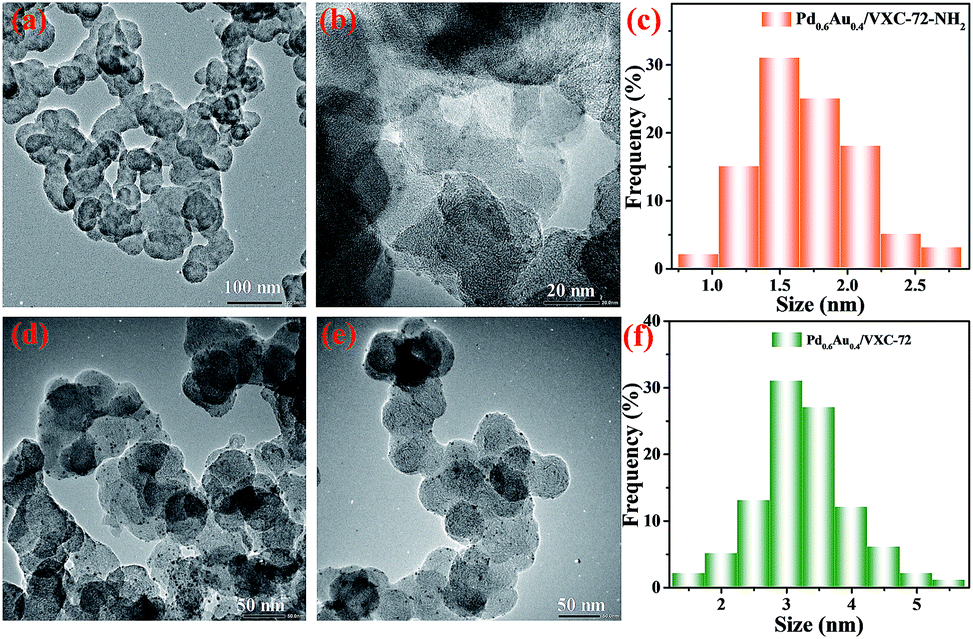 | ||
| Fig. 1 TEM images and size distributions of Pd0.6Au0.4/VXC-72-NH2 (a–c) and Pd0.6Au0.4/VXC-72 (d–f). | ||
 | ||
| Fig. 2 Water-contact angle of (a) VXC-72 and (b) VXC-72-NH2 and the corresponding photos of VXC-72 and VXC-72-NH2 suspension. | ||
Brunauer–Emmett–Teller (BET) surface areas were calculated from the N2 adsorption desorption isotherms of the samples (Fig. S2†). The detailed textural parameters are summarized in Table S1.† It can be seen that compared with VXC-72 (SBET = 120 m2 g−1), the surface area of amine functionalized carbon black was slightly reduced (SBET = 103 m2 g−1). No obvious change was observed after the deposition of Pd and Au onto VXC-72-NH2 (SBET = 108 m2 g−1).
The XRD patterns of the as-prepared catalysts are shown in Fig. 3a. Two broad peaks at around 24.9° and 43.3° were ascribed to the (002) and (200) diffractions of amorphous carbon, respectively.39,40 For the sample Pd0.6Au0.4/VXC-72, the diffraction peak at 38.7° was just between the standard (111) planes of cubic Au (JCPDS: 65-2870) and cubic Pd (JCPDS: 65-2867), indicating the formation of a PdAu alloy structure.41–43 For the sample Au/VXC-72-NH2 the diffraction peaks at 38.2° could be assigned to the (111) plane of metallic Au (JCPDS: 65-2870). However, Pd/VXC-72-NH2 and Pd0.6Au0.4/VXC-72-NH2 showed no obvious diffraction peaks, indicating that ultra-small Pd and PdAu particles were well dispersed on the support.24 Therefore, the presence of amines was the key factor for the formation of the ultrasmall PdAu NPs. Moreover, after annealing of Pd0.6Au0.4/VXC-72-NH2 in an Ar atmosphere (773 K, 3 h), there appeared a diffraction peak at about 38.7°, confirming that the PdAu NPs were in an alloy structure (Fig. S3†).42 In the Raman spectra (Fig. 3b) the wide bands at around 1344 and 1591 cm−1 were ascribed to the disordered graphite carbon (D-band) and sp2-carbon (G-band) within aromatic carbon rings, respectively.44 After acid treatment, the D/G ratio increased, indicating a decrease of the sp2-hybridized structure that generates defects and edge planes. The D/G ratio of VXC-72-NH2 remained almost the same as that of acid treated VXC-72, implying that amine functionalization induced no damage to the structure of the carbon matrix.
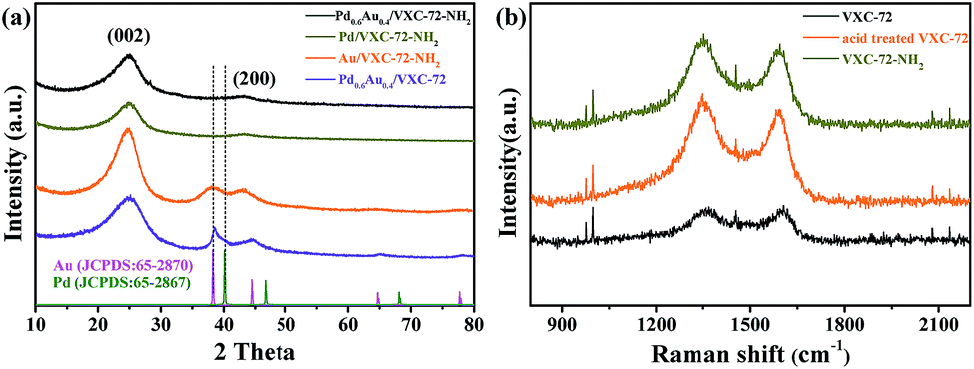 | ||
| Fig. 3 (a) XRD patterns of the as-synthesized catalysts and (b) Raman spectra of the as synthesized VXC-72 supports. | ||
X-ray photoelectron spectroscopy (XPS) investigation confirmed the existence of abundant O and N species after amine functionalization of VXC-72 (Fig. 4 and Table S2†). The binding energy of Au and Pd in Pd0.6Au0.4/VXC-72-NH2 indicated the valence of metallic Au0 and Pd0 (Fig. 5).45 Additionally, the XPS results showed that the binding energy of Pd 3d in Pd0.6Au0.4/VXC-72-NH2 was shifted to a higher value relative to that in Pd/VXC-72-NH2, while binding energies of Au 4f in Pd0.6Au0.4/VXC-72-NH2 were shifted to the lower value compared with that in Au/VXC-72-NH2. These shifts could be attributed to a partial electron-transfer from Pd to Au, which could be explained by the lower electronegativity of Pd than Au (Pd 2.2, Au 2.4) due to the alloy effect between Pd and Au. Moreover, the Pd 3d and Au 4f peaks of Pd0.6Au0.4/VXC-72-NH2 were both shifted to lower binding energies in comparison to those of Pd and Au on Pd0.6Au0.4/VXC-72, and the shifts indicated that some electrons were transferred from the VXC-72-NH2 substrate to the PdAu NPs, confirming the strong interaction between the VXC-72-NH2 support and PdAu NPs.41
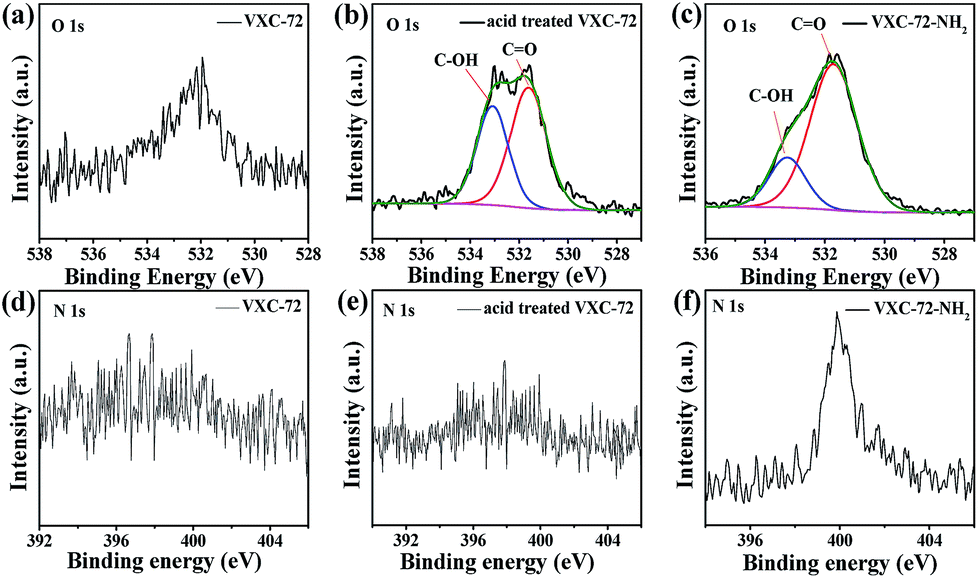 | ||
| Fig. 4 XPS spectra of O 1s for (a) VXC-72, (b) acid treated VXC-72 and (c) VXC-72-NH2 and N 1s for (d) VXC-72 (e) acid treated VXC-72 and (f) VXC-72-NH2. | ||
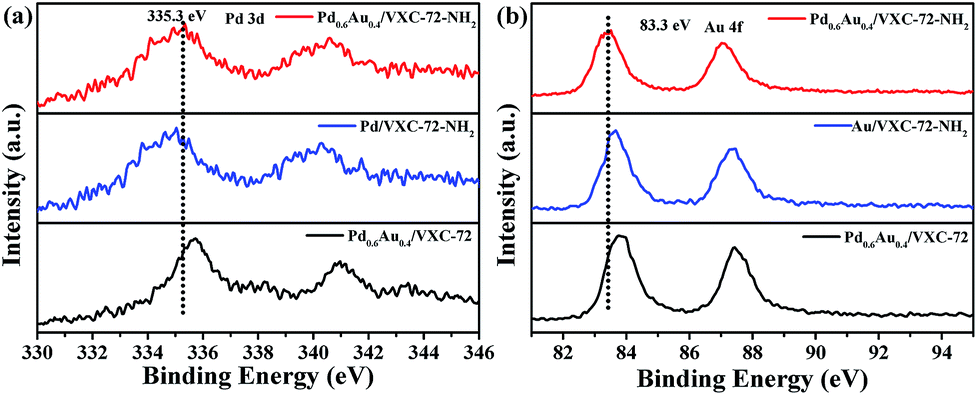 | ||
| Fig. 5 XPS spectra of (a) Pd 3d and (b) Au 4f in Pd0.6Au0.4/VXC-72-NH2, Pd0.6Au0.4/VXC-72, Pd/VXC-72-NH2 and Au/VXC-72-NH2. | ||
Fig. 6 shows the catalytic activity of the as-prepared catalysts for the dehydrogenation of FA at 298 K. It can be seen that without Pd addition, the Au/VXC-72-NH2 catalyst did not show any activity. The catalyst Pd0.6Au0.4/VXC-72-NH2 showed a high catalytic activity and 245 mL of gas was released within 8 min at 298 K, affording a turnover frequency (TOF) value of 7385 h−1, the highest value at room temperature reported so far. In contrast, the catalyst Pd0.6Au0.4/VXC-72 showed a much lower activity for the decomposition of FA under the same conditions, over which only 73 mL of gas was released in 20 min (Fig. 6c). The calculated TOF value of Pd0.6Au0.4/VXC-72-NH2 (7385 h−1) was greater than that on the Pd0.6Au0.4/VXC-72 catalyst (497 h−1) by a factor of 15.
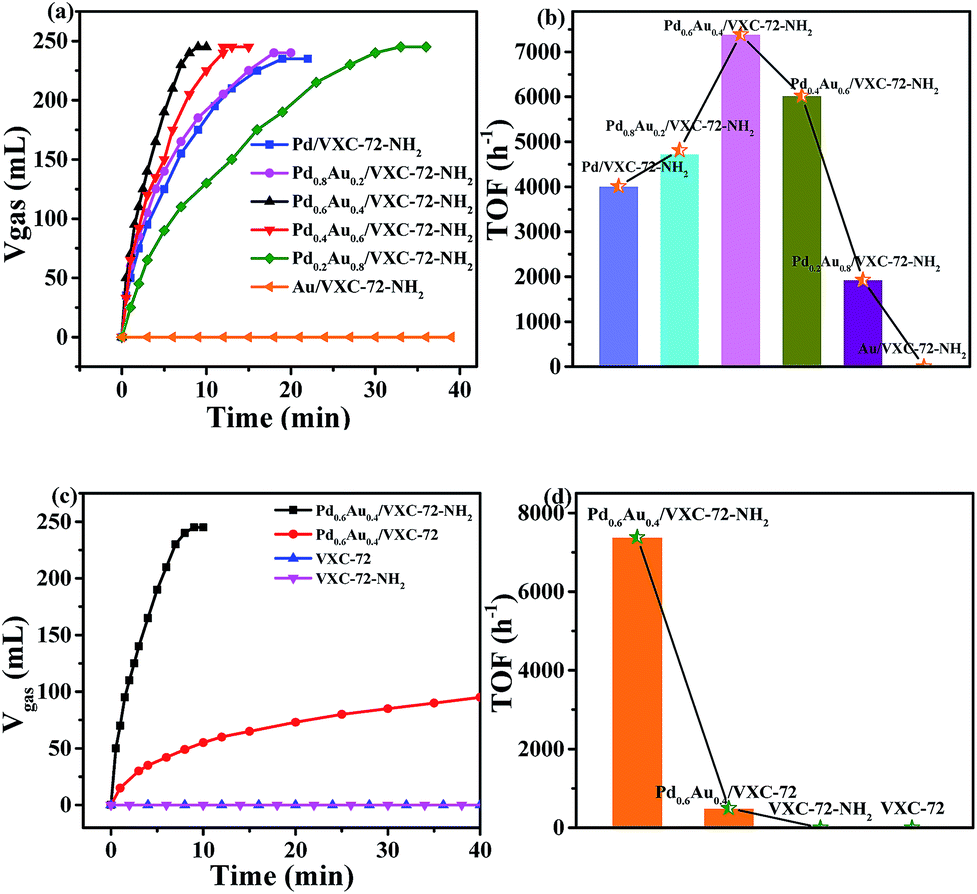 | ||
| Fig. 6 (a) Volume of the generated gas (CO2 + H2) from the decomposition of FA (1.0 M, 5.0 mL) versus time in the presence of PdxAu1−x/VXC-72-NH2 and (b) the corresponding TOF values; (c) volume of the generated gas (CO2 + H2) from the decomposition of FA versus time in the presence of Pd0.6Au0.4/VXC-72-NH2, Pd0.6Au0.4/VXC-72, VXC-72-NH2 and VXC-72 at 298 K ((nPd+Au/nFA) = 0.0034) and (d) the corresponding TOF values. The TOF value was calculated at a conversion of 20%. | ||
Obviously, the significant enhancement of the catalytic performance of Pd0.6Au0.4/VXC-72-NH2 could be ascribed to the hydrophilic VXC-72-NH2 support for the synthesis of ultrafine PdAu NPs. The catalytic activity is much higher than that of Pd or Pd-based alloy supported by amine functionalized rGO,21,24,26,27,37 mesoporous carbon,25 MOFs,29,35 and mesoporous silica,32–34,36 as listed in Table 1. As reported in the literature,15 for supported noble metal catalysts, the interaction between the metal and support is closely related to the catalytic activity. The functionalization of carbon black by amine converted the hydrophobic surface to a hydrophilic surface, which was beneficial to the formation of highly dispersed ultrafine PdAu NPs. Meanwhile the presence of amine groups modulated the electronic structure of PdAu NPs, and all these factors lead to greatly improved activity.
| Catalyst | Temp. (K) | Additive | TOF (h−1) | E a (kJ mol−1) | Ref. |
|---|---|---|---|---|---|
| a TOF values calculated for total metal atoms. b TOF values calculated on surface metal sites or active sites. c Initial TOF values calculated at the initial time or initial conversion of FA. d TOF values calculated at the complete time of gas release. | |||||
| Pd0.6Au0.4/VXC-72-NH2 | 298 | None | 7385a,c | 20.6 | This work |
| Pd0.6Au0.4/VXC-72-NH2 | 333 | None | 17724a,c |
20.6 | This work |
| (Co6)Ag0.1Pd0.9/rGO | 333 | HCOONa | 2739a,d | 43.1 | 1 |
| Pd/N-MSC-30 | 333 | HCOONa | 8414a,d | 43.7 | 10 |
| Pd@CN900 K | 298 | HCOONa | 5530b,c | 46.9 | 11 |
| Pd/PDA-rGO | 298 | HCOONa | 727a,d | 54.3 | 21 |
| Pd/S-1-in-K | 298 | HCOONa | 856a,d | 39.2 | 23 |
| PdAg/amine-mesoporous carbon | 348 | HCOONa | 5638a,c | — | 25 |
| NiPd/NH2–N-rGO | 298 | None | 954.3a,c | — | 26 |
| Au0.5Pd0.5/NH2–N-rGO | 298 | None | 4445.6a,c | — | 27 |
| Pd/SBA-15-PA | 298 | None | 355a,c | — | 28 |
| Pd–NH2-MIL-125 | 305 | None | 214a,c | — | 29 |
| Au@SiO2 | 403 | None | 958a,c | — | 31 |
| Pd–MnOx/SiO2–NH2 | 323 | None | 1300a,c | — | 32 |
| Pd60Au40/ZrSBA-15-AP | 298 | None | 1185a,c | 47 | 33 |
| SBA-15-amine/Pd | 298 | None | 293a,c | — | 34 |
| AuPd/MIL-101-NH2 | 298 | None | 526a,c | 32.5 | 35 |
| CrAuPdN–SiO2 | 298 | None | 730a,c | 49.8 | 36 |
| PdAuNi/f-GNS | 298 | None | 1090a,c | 55 | 37 |
| AuPd–MnOx/ZIF-8-rGO | 298 | None | 382.1a,c | — | 41 |
| Pd0.44Ag0.19–Mn0.37/N-SiO2 | 298 | None | 330a,c | 72.4 | 51 |
| Pd0.6Ag0.4@ZrO2/C/rGO | 333 | HCOONa | 4300a,d | 50.1 | 52 |
The gas mixture generated from FA catalyzed by Pd0.6Au0.4/VXC-72-NH2 was analyzed by gas chromatography (GC) to be CO2 and H2, and no CO could be detected from the evolved gas (Fig. 7), suggesting that the as-prepared Pd0.6Au0.4/VXC-72-NH2 has a 100% selectivity to H2 from FA. Gas generation over Pd0.6Au0.4/VXC-72-NH2 was completed in 9, 8, 7.5, 7 and 5.5 min at 298, 303, 313, 323 and 333 K, respectively (Fig. 8), corresponding to TOF values of 7385, 8491, 11037, 14179 and 17724 h−1. The apparent activation energy (Ea) was determined to be 20.6 kJ mol−1, which was lower than most of the reported values (Table 1).
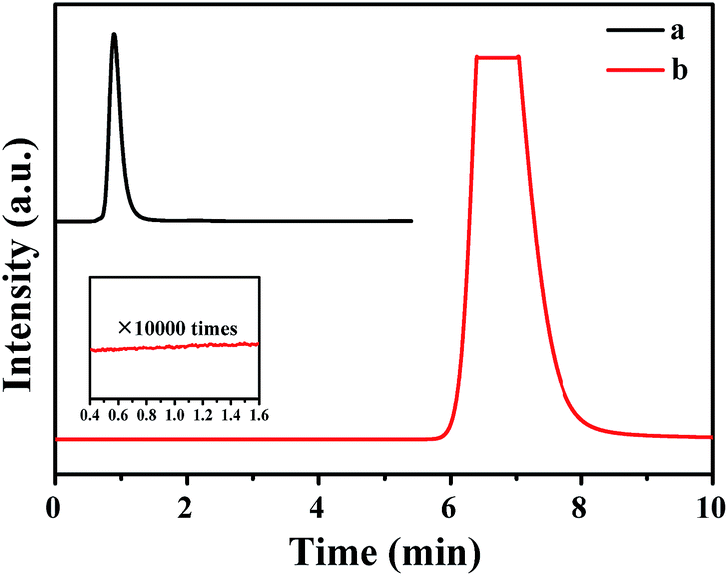 | ||
| Fig. 7 GC spectra collected using a FID-methanator for the (a) mixed gas of CO/Ar and (b) evolved gas from FA aqueous solution (1.0 M, 5.0 mL) over the Pd0.6Au0.4/VXC-72-NH2 catalyst at 298 K ((nPd+Au/nFA) = 0.0034). | ||
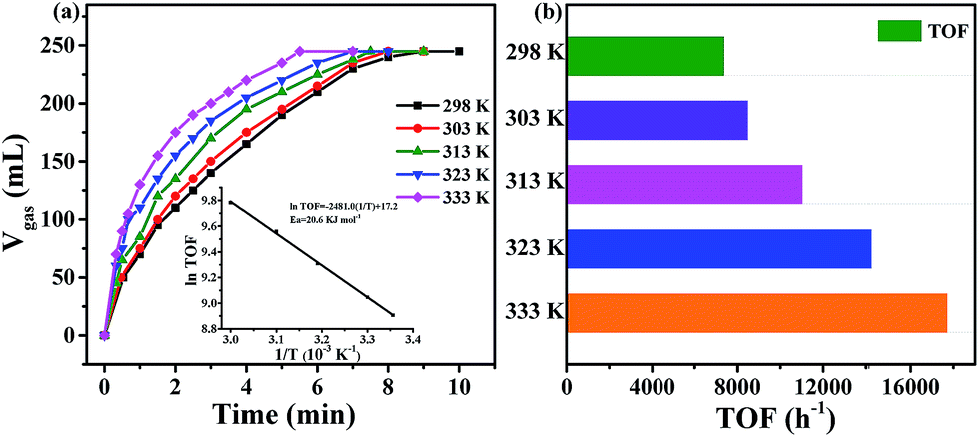 | ||
| Fig. 8 (a) Volume of generated gas (CO2+H2) versus the reaction time at 298 K ((nPd+Au/nFA) = 0.0034) and (b) TOF values of H2 generation for the dehydrogenation of FA over the Pd0.6Au0.4/VXC-72-NH2 catalyst at different reaction temperatures. Inset in (a): Arrhenius plot (ln (TOF) vs. 1/T). | ||
The recycling stability of Pd0.6Au0.4/VXC-72-NH2 was also evaluated by further addition of an equivalent of FA to the reaction mixture after the completion of the previous cycle. As shown in Fig. 9, there was no significant decrease in catalytic activity after the fifth run. In addition, the results of XRD (Fig. S4†) and TEM images (Fig. S5†) of Pd0.6Au0.4/VXC-72-NH2 after the fifth run showed no obvious change in the PdAu alloy size. The XPS result showed that after recycled use, the elements N, Au and Pd were stable on carbon black (Fig. S6†), and the atomic percentage is listed in Table S2.† The contents of Au and Pd in the samples determined by ICP-AES are listed in Table S3,† and the results showed that there was no obvious loss of N, Au and Pd in recycled Pd0.6Au0.4/VXC-72-NH2, indicating good catalytic durability and stability of the catalyst.
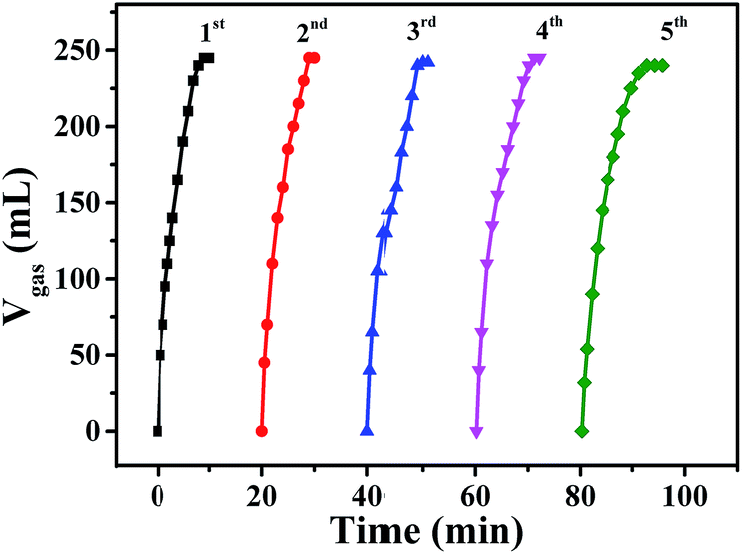 | ||
| Fig. 9 Durability test for the dehydrogenation of FA solution over Pd0.6Au0.4/VXC-72-NH2 at 298 K. | ||
The results above suggested that VXC-72-NH2 could efficiently anchor the PdAu NPs, preventing aggregation and overgrowth during the synthetic and catalytic processes. The amine groups on the support could serve the following roles. First, –NH2 would convert the surface of carbon black to be hydrophilic and give rise to ultrafine sizes and excellent dispersion of nanoparticles. Second, the alkaline –NH2 group could facilitate O–H bond dissociation of FA to produce –[H2NH]+ and metal-formate species. Moreover, the electron modification of VXC-72-NH2 toward PdAu NPs would increase the electron density of PdAu active centers to facilitate the formation of metal-formate, which enhanced the rate of the catalytic dehydrogenation of FA.46–48 As illustrated in Scheme 2, the alkaline –NH2 group could serve as a proton scavenger, which benefits the O–H bond dissociation in the FA molecule, resulting in the formation of a metal-formate intermediate along with the [H2NH]+ group during the initial step of the reaction. Subsequently, the PdAu–formate species undergo β-hydride elimination to produce CO2 and a palladium hydride species. Finally, H2 is produced from the palladium hydride species and [H]+.21,49,50
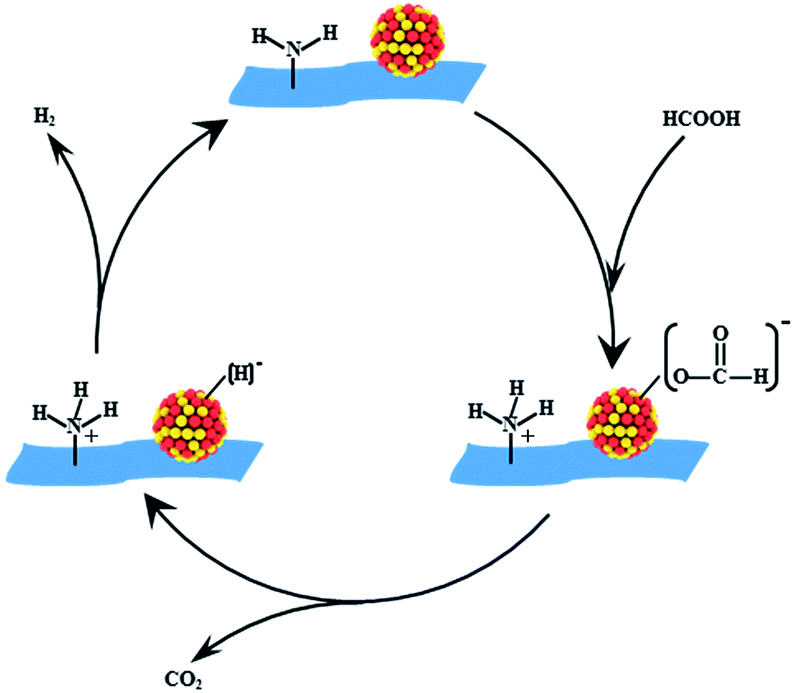 | ||
| Scheme 2 Possible reaction pathway of FA decomposition over Pd0.6Au0.4/VXC-72-NH2. | ||
4. Conclusions
In summary, ultrafine and highly dispersed PdAu NPs were immobilized on amine functionalized carbon black, and the catalyst Pd0.6Au0.4/VXC-72-NH2 exhibited excellent activity for FA dehydrogenation without any additives at room temperature. The functionalization of carbon black with amines converted the hydrophobic surface to a hydrophilic surface, which was beneficial to the formation of highly dispersed ultrafine PdAu NPs. Meanwhile the presence of amine groups could modulate the electronic structure of PdAu NPs, leading to greatly improved activity. As carbon supports (activated carbon and carbon black) are widely used for industrial supported noble metal catalysts, our study provides a general approach for the design of ultrafine and highly dispersed noble metal catalysts, not only for enhanced dehydrogenation of FA but also for more efficient industrial applications with carbon supported noble metal catalysts.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the NSFC (No. 21773128, 21534005, and 21421001).References
- Y. Chen, Q. L. Zhu, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2015, 137, 106–109 CrossRef CAS PubMed.
- B. Liu, H. Yao, W. Song, L. Jin, I. M. Mosa, J. F. Rusling, S. L. Suib and J. He, J. Am. Chem. Soc., 2016, 138, 4718–4721 CrossRef CAS PubMed.
- Q. L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- Q. Wang, N. Tsumori, M. Kitta and Q. Xu, ACS Catal., 2018, 8, 12041–12045 CrossRef CAS.
- B. Wu, Y. Kuang, X. Zhang and J. Chen, Nano Today, 2011, 6, 75–90 CrossRef CAS.
- Q. L. Zhu, D. C. Zhong, U. B. Demirci and Q. Xu, ACS Catal., 2014, 4, 4261–4268 CrossRef CAS.
- S. H. Ye, F. Y. Luo, Q. L. Zhang, P. Y. Zhang, T. T. Xu, Q. D. Wang, S. He, L. C. Guo, Y. Zhang, C. X. He, X. P. Ouyang, M. Gu, J. H. Liu and X. L. Sun, Energy Environ. Sci., 2019, 12, 1000 RSC.
- S. J. Jiang, Y. W. Ma, G. Q. Jian, H. S. Tao, X. Z. Wang, Y. N. Fan, Y. N. Lu, Z. Hu and Y. Chen, Adv. Mater., 2009, 21, 4953 CrossRef CAS.
- Z. Li, J. Liu, C. Xia and F. Li, ACS Catal., 2013, 3, 2440–2448 CrossRef CAS.
- Z. Li, X. Yang, N. Tsumori, Z. Liu, Y. Himeda, T. Autrey and Q. Xu, ACS Catal., 2017, 7, 2720–2724 CrossRef CAS.
- Q. Y. Bi, J. D. Lin, Y. M. Liu, H. Y. He, F. Q. Huang and Y. Cao, Angew. Chem., 2016, 55, 11849–11853 CrossRef CAS PubMed.
- J. Sun, H. Qiu, W. Cao, H. Fu, H. Wan, Z. Xu and S. Zheng, ACS Sustainable Chem. Eng., 2018, 7, 1963–1972 CrossRef.
- K. J. Jeon, H. R. Moon, A. M. Ruminski, B. Jiang, C. Kisielowski, R. Bardhan and J. J. Urban, Nat. Mater., 2011, 10, 286–290 CrossRef CAS PubMed.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- H. Zhong, M. Iguchi, M. Chatterjee, Y. Himeda, Q. Xu and H. Kawanami, Adv. Sustainable Syst., 2018, 2, 1700161 CrossRef.
- X. C. Yang, P. Pachfule, Y. Chen, N. Tsumoric and Q. Xu, Chem. Commun., 2016, 52, 4171 RSC.
- Z. P. Li and Q. Xu, Acc. Chem. Res., 2017, 50, 1449–1458 CrossRef CAS PubMed.
- Z. P. Li and Q. Xu, Nanoscale, 2015, 7, 8321 RSC.
- J. Cheng, X. J. Gu, X. Sheng, P. L. Liu and H. Q. Su, J. Mater. Chem. A, 2016, 4, 1887 RSC.
- K. Jiang, K. Xu, S. Zou and W. B. Cai, J. Am. Chem. Soc., 2014, 136, 4861–4864 CrossRef CAS PubMed.
- F. Z. Song, Q. L. Zhu, N. Tsumori and Q. Xu, ACS Catal., 2015, 5, 5141–5144 CrossRef CAS.
- D. A. Bulushev, M. Zacharska, E. V. Shlyakhova, A. L. Chuvilin, Y. Guo, S. Beloshapkin, A. V. Okotrub and L. G. Bulusheva, ACS Catal., 2015, 6, 681–691 CrossRef.
- N. Wang, Q. Sun, R. Bai, X. Li, G. Guo and J. Yu, J. Am. Chem. Soc., 2016, 138, 7484–7487 CrossRef CAS PubMed.
- H. Zhong, M. Iguchi, M. Chatter-jee, T. Ishizaka, M. Kitta, M. Q. Xu and H. Kawanami, ACS Catal., 2018, 8, 5355–5362 CrossRef CAS.
- S. Masuda, K. Mori, Y. Futamura and H. Yamashita, ACS Catal., 2018, 8, 2277–2285 CrossRef CAS.
- J. M. Yan, S. J. Li, S. S. Yi, B. R. Wulan, W. T. Zheng and Q. Jiang, Adv. Mater., 2018, 30, 1703038 CrossRef PubMed.
- S. J. Li, Y. T. Zhou, X. Kang, D. X. Liu, L. Gu, Q. H. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2019, 31, 1806781 CrossRef.
- K. Koh, M. Jeon, C. W. Yoon and T. Asefa, J. Mater. Chem. A, 2017, 5, 16150 RSC.
- M. Martis, K. Mori, K. Fujiwara, W. S. Ahn and H. Yamashita, J. Phys. Chem. C, 2013, 117, 22805–22810 CrossRef CAS.
- M. Yadav, T. Akita, N. Tsumori and Q. Xu, J. Mater. Chem., 2012, 22, 12582–12586 RSC.
- J. Mielby, A. J. Kunov-Kruse and S. Kegnæs, J. Catal., 2017, 345, 149–156 CrossRef CAS.
- A. Buluta, M. Yurderia, Y. Karatasa, M. Zahmakirana, H. Kivrakb, M. Gulcana and M. Kaya, Appl. Catal., B, 2015, 164, 324–333 CrossRef.
- Z. Z. Wang, X. F. Hao, D. W. Hu, L. Li, X. J. Song, W. X. Zhang and M. J. Jia, Catal. Sci. Technol., 2017, 7, 2213 RSC.
- K. Koh, J. E. Seo, J. H. Lee, A. Goswami, C. W. Yoo and T. Asefa, J. Mater. Chem. A, 2014, 2, 20444–20449 RSC.
- J. Cheng, X. J. Gu, P. L. Liu, H. Zhang, L. L. Ma and H. Q. Su, Appl. Catal., B, 2017, 218, 460–469 CrossRef CAS.
- M. Yurderi, A. Bulut, N. Caner, M. Celebi, M. Kayab and M. Zahmakiran, Chem. Commun., 2015, 51, 11417–11420 RSC.
- A. Bulut, M. Yurderi, M. Kaya, M. Aydemir, A. Baysal, F. Durapc and M. Zahmakiran, New J. Chem., 2018, 42, 16103 RSC.
- K. Kim, H. Oh, J. Kim, S. Ha, M. Kim, J. L. Yang and J. Kim, J. Nanosci. Nanotechnol., 2019, 19, 1525–1532 CrossRef CAS PubMed.
- Z. L. Wang, J. M. Yan, Y. Ping, H. L. Wang, W. T. Zheng and Q. Jiang, Angew. Chem., 2013, 52, 4406–4409 CrossRef CAS.
- Q. L. Zhu, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2015, 137, 11743–11748 CrossRef CAS.
- J. M. Yan, Z. L. Wang, L. Gu, S. J. Li, H. L. Wang, W. T. Zheng and Q. Jiang, Adv. Energy Mater., 2015, 5, 1500107 CrossRef.
- S. Wu, F. Yang, H. Wang, R. Chen, P. C. Sun and T. H. Chen, Chem. Commun., 2015, 51, 10887 RSC.
- Z. L. Wang, J. M. Yan, Y. F. Zhang, Y. Ping, H. L. Wang and Q. Jiang, Nanoscale, 2014, 6, 3073 RSC.
- K. A. Wepasnick, B. A. Smith, K. E. Schrote, H. K. Wilson, S. R. Diegelmann and D. H. Fairbrother, Carbon, 2011, 49, 24–36 CrossRef CAS.
- Z. L. Wang, H. L. Wang, J. M. Yan, Y. Ping, S. I. O, S. J. Li and Q. Jiang, Chem. Commun., 2014, 50, 2732–2734 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302 CrossRef CAS.
- S. Jones, J. Qu, K. Tedsree, X. Q. Gong and S. C. E. Tsang, Angew. Chem., Int. Ed., 2012, 51, 11275 CrossRef CAS.
- K. Tedsree, C. W. A. Chan, S. Jones, Q. Cuan, W. K. Li, X. Q. Gong and S. C. E. Tsang, Science, 2011, 332, 224 CrossRef CAS.
- K. Mori, M. Dojo and H. Yamashita, ACS Catal., 2013, 3, 1114–1119 CrossRef CAS.
- J. S. Yoo, F. Abild-Pedersen, J. K. Nørskov and F. Studt, ACS Catal., 2014, 4, 1226–1233 CrossRef CAS.
- A. Bulut, M. Yurderi, Y. Karatas, Z. Say, H. Kivrak, M. Kaya, M. Gulcan, E. Ozensoy and M. Zahmakiran, ACS Catal., 2015, 5, 6099–6110 CrossRef CAS.
- F. Z. Song, Q. L. Zhu, X. Yang, W. W. Zhan, P. Pachfule, N. Tsumori and Q. Xu, Adv. Energy Mater., 2018, 8, 1701416 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00462a |
| This journal is © The Royal Society of Chemistry 2019 |