
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInstantaneous, room-temperature, open-air atmosphere, solution-phase synthesis of perovskite quantum dots through halide exchange employing non-metal based inexpensive HCl/HI: ensemble and single particle spectroscopy†
Saptarshi
Mandal
 a,
Debjit
Roy
a,
Chayan K.
De
a,
Swarnali
Ghosh
a,
Mrinal
Mandal
a,
Ananya
Das
a and
Prasun K.
Mandal
*ab
a,
Debjit
Roy
a,
Chayan K.
De
a,
Swarnali
Ghosh
a,
Mrinal
Mandal
a,
Ananya
Das
a and
Prasun K.
Mandal
*ab
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, West Bengal 741246, India. E-mail: prasunchem@iiserkol.ac.in
bCentre of Advanced Functional Materials, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur, West Bengal 741246, India
First published on 9th July 2019
Abstract
Herein, the instantaneous synthesis of highly crystalline, uniform-sized (ca. 11.3 ± 0.1 nm), blue-to-green to yellow to red-emitting all-inorganic perovskite quantum dots (CsPbBr3 and mixed halide PQDs) was achieved at room temperature under an open-air atmosphere (no glove box) through halide exchange in the solution phase employing easily available, inexpensive non-metal-based halide sources such as HCl and HI. No complicated pre-treatment of the halide source was required. Moreover, these PQDs were stable for a few weeks under an open-air atmosphere. The PL emission spectra are quite narrow, and the PLQYs are quite high (80% for even Br/I mixed PQDs). At the single particle level, the 〈ON fraction〉 has been noted to vary from 75% to 85% for different PQDs, the mON values are close to 1.0, and the mOFF values are >1.5. The latter indicates that long ON durations are more probable. The increase in the ON event truncation time (from 2.7 to 4.0 s) and the concomitant decrease in the OFF event truncation time (from 6.6 to 4.3 s) could be correlated with the increase in the PLQY (from 0.55 to 0.75). In addition, an interesting memory effect could be observed in both the ON and the OFF event durations.
Introduction
During the last decade, research on perovskite quantum dot (PQD)-based solar cells,1–3 lasers,4–6 light-emitting diodes,7–9 and bioimaging10 has reached its pinnacle. Among the different kinds of PQDs, all-inorganic PQDs are most preferred due to their high stability;11–13 however, a few concerns have been raised regarding the application of all-inorganic PQDs, e.g., their synthesis is conducted (a) at high temperatures, (b) under an inert atmosphere, and (c) takes a long time (i.e. several hours), and (d) the metal halide sources are expensive and not easily available.14–17Halide exchange reactions in all-inorganic perovskites have been considered as an efficient method to synthesize mixed halide PQDs.18–22 However, in addition to the abovementioned concerns, (i) the complicated pre-treatment of the halide precursors (oleylamine-X, tetrabutylamine-X, octadecylamine-X, and PbX2, X = Cl, Br, and I)18–21 and/or poor solubility of metal salts (MX2/MX, M = Zn, Mg, Cu, Ca, and K; X = Cl, Br, and I) in toluene/DMF at RT are the matters of concern.22–26 The mixing of the previously synthesized CsPbX3 (X = Cl, Br, and I) (through the hot-injection technique) at room temperature (RT) is another way to solve this problem.27 However, the precursors of PQDs (CsPbX3, X = Cl/Br/I) are constructed using the hot-injection technique under an inert atmosphere; this is again a matter of concern.
Therefore, it would be interesting if all these reactions could be performed at RT under an open atmosphere, the halide exchange reactions could be performed using easily available and inexpensive non-metal-based halides such as HCl/HI, and all the reactions could be instantaneous in nature. To the best of our knowledge, there is no study in the literature in which both the synthesis of CsPbBr3 as well as the halide exchange reactions have been performed in the solution phase at RT under an open-air atmosphere using non-metal-based, easily available and inexpensive halide sources such as HCl/HI. Therefore, it would be highly advantageous if all the abovementioned propositions could be achieved; however, clearly, the achievement of these propositions is very challenging; thus, in this study, we report the instantaneous synthesis (i.e., the overall synthesis takes less time) of both CsPbBr3 and halide-exchanged PQDs at RT (i.e., without heating) under an open-air atmosphere (i.e., without using any glove box). Importantly, the halide exchange reaction was carried out in the liquid phase using inexpensive, easily available, non-metal-based halide sources such as HCl and HI. Moreover, no complicated pre-treatment of the halide precursors, as reported in the literature, was necessary for our successful solution-phase halide exchange reaction as we could directly use HCl and HI. In short, we could achieve (Scheme 1) all the abovementioned challenging propositions. To the best of our knowledge, to date, no such complete study has been reported in the literature.
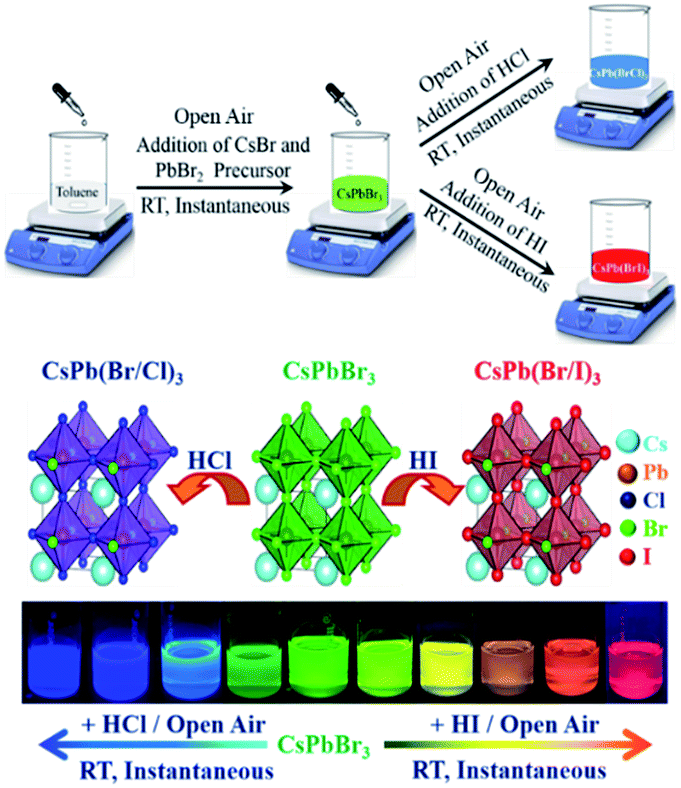 | ||
| Scheme 1 Room-temperature, open-air atmosphere, instantaneous synthesis of CsPbBr3 and mixed halide perovskites using non-metal and inexpensive halide sources such as HCl/HI. | ||
Results and discussion
At first, CsPbBr3 was synthesized adopting the room-temperature supersaturated recrystallization method with a modified procedure.24,28 In a typical synthesis, CsPbBr3 was instantaneously formed at RT under an open-air atmosphere (Video S1†). Moreover, mixed halide PQDs were synthesized in the solution phase instantaneously at room temperature under an open-air atmosphere (henceforth, termed as IRTOA) using the non-metal based, easily available, and inexpensive HCl/HI as the halide exchange material. The instantaneous synthesis of both CsPb(Br/Cl)3 and CsPb(Br/I)3 mixed halide PQDs could be visualized (Videos S1† and S2); the instantaneous nature of the halide exchange reaction could be controlled by choosing a proper solvent pair (see S1†).After the formation of the mixed CsPb(Br/Cl)3 PQD (λem = 430 nm), Cl-exchanged mixed CsPb(Br/Cl)3 PQDs with increasing emission maxima were formed through the IRTOA method with the addition of HBr. Similarly, after the formation of the mixed CsPb(Br/I)3 PQD (λem = 630 nm), the I-exchanged mixed CsPb(Br/I)3 PQDs were formed following the IRTOA process with the addition of HBr (Fig. S3†). Thus, the reversibility of this halide exchange process through the IRTOA method could be evidenced. Herein, note that in our case, even the CsPbBr3 PQD was synthesized through the IRTOA method. Thus, all steps of the reactions were instantaneous and carried out at RT under an open-air atmosphere. The instantaneous reaction saves a lot of time, the RT synthesis saves a lot of heat/electrical energy, the open-atmosphere synthesis reduces the difficulties experienced during an inert-atmosphere synthesis procedure, and the usage of easily available and inexpensive HCl/HBr/HI as the halide exchange source materials reduces the cost of the reaction. All these parameters will be extremely useful for the large-scale synthesis of the all-inorganic blue to green to red-emitting PQDs necessary for various applications.
Regarding stability under an ambient air condition, the PQD 1–5 (the mixed halide CsPb(Br/Cl)3 and CsPbBr3 PQDs) were stable in the solution (toluene) phase for a month (Fig. S4(a)†). However, the PQD 6–10 (the mixed halide CsPb(Br/I)3 PQDs) were not very stable in a toluene medium, and the intensity dropped down to 50% in less than two weeks (Fig. S4(a)†). The stability of these PQDs was slightly better in n-octane as compared to that in the toluene medium (Fig. S4(b)†). Thus, the blue to red-emitting PQDs synthesized through the IRTOA process are as stable (if not more stable) as the PQDs synthesized through the hot injection, inert-atmosphere synthesis using expensive halide exchange materials.
Herein, since all the PQDs were synthesized via a completely different IRTOA procedure in the solution phase using inexpensive HX (X = Cl/I) as compared to those synthesized using the methods (requiring several hours, high temperatures, an inert atmosphere inside glove box, and the use of expensive metal halide sources) reported in the literature, it was necessary to determine the quality of the PQDs. The successful halide exchange reaction and gradual change in halide concentration could be confirmed using the EDAX peak positions (Fig. S5†), HRTEM and PXRD (Fig. 1). As can be observed from Fig. 1(a), the sizes of the mixed halide PQDs were not significantly altered because of the halide exchange reaction. The average size of the Cl-exchanged CsPb(Br/Cl)3 PQD was 11.31 ± 0.96 nm (λem = 430 nm), that of CsPbBr3 was 11.26 ± 0.84 nm (λem = 513 nm) and that of the I-exchanged CsPb(Br/I)3 PQD was 11.39 ± 0.80 nm (λem = 630 nm) (Fig. 1(a)). The excellent HRTEM images (Fig. 1(a)) as well as the bright spots in the FFT pattern (Fig. 1(a)) confirm the very high crystallinity of the PQDs synthesized using the abovementioned ingenious approach. The constancy of the quality and the size/shape of the halide-exchanged PQDs as compared to those of CsPbBr3 confirmed that no strong detrimental effect was caused by the H+ ion of HX (X = Cl/I). The inter-planar spacings obtained from the HRTEM images have been calculated to be 0.57 nm for the Cl-exchanged CsPb(Br/Cl)3 PQD (λem = 430 nm), 0.58 nm for the CsPbBr3 PQD (λem = 513 nm) and 0.61 nm for the I-exchanged CsPb(Br/I)3 PQD (λem = 630 nm). These inter-planar spacings correspond to the {100} plane.27 The inter-planar spacings corresponding to the {100} plane have also been calculated from the PXRD patterns (Fig. 1(b)). The inter-planar spacing obtained via HRTEM and PXRD matches quite well with that of the {100} plane; thus, because of the exchange of the smaller sized Cl− (1.81 Å) in place of the larger sized Br− (1.96 Å), the inter-planar spacing decreases from 0.58 nm to 0.57 nm. Similarly, because of the exchange of the larger sized I− (2.20 Å) in place of the smaller sized Br−, the inter-planar spacing increases from 0.58 nm to 0.61 nm.18 The shifting of the peaks corresponding to the (100), (110), (200), (210), (211), and (202) planes in the PXRD pattern (Fig. 1(b)) confirms the successful halide exchange of CsPbBr3 by both Cl− and I−. The shifting of the PXRD peak positions and changes in the inter-planar spacing in the HRTEM images are expected because of the size difference of halides.18,27 However, this should not have a very significant effect on the overall size of the PQD, and the same has been observed herein, i.e., the overall sizes of all PQDs are in the range from 11.26 to 11.39 nm. It has been reported in the literature that if the peaks corresponding to the 2θ values of ∼15° and 32° do not exhibit any significant splitting, the PQD crystal is cubic in nature.22 As can be observed from Fig. 1(b), in our case, the peaks corresponding to the 2θ values of ∼15° and 32° do not exhibit any significant splitting. Hence, based on the literature studies, we would like to conclude that our PQDs are cubic in nature.14,18,22,27 Moreover, note that none of the peaks in the PXRD pattern shows splitting in our case. The single peak positions without splitting confirm the uniform homogeneous nature of the PQDs formed because of halide exchange.22
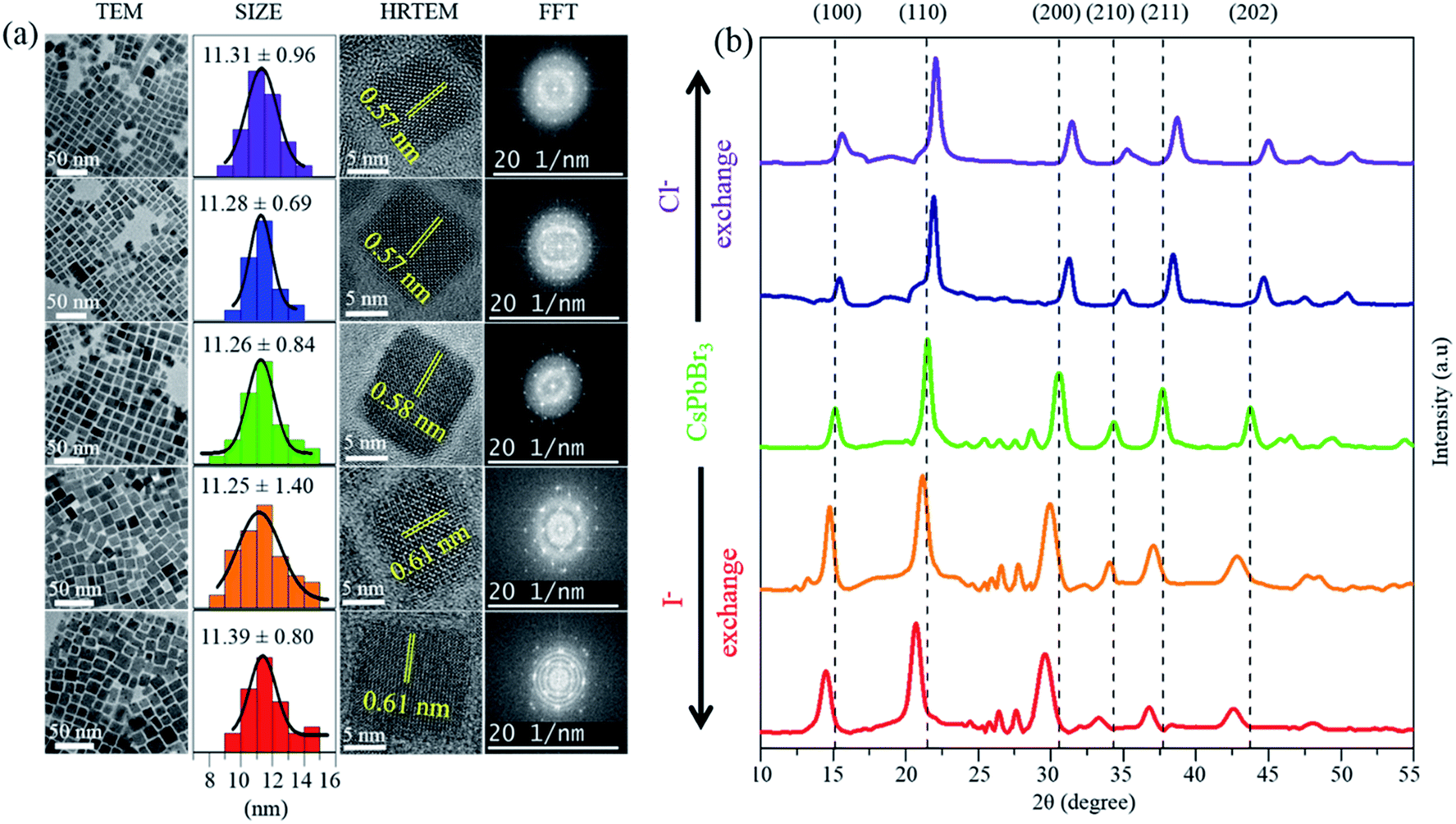 | ||
| Fig. 1 Structural characterization of PQDs. (a) TEM image analysis of PQDs. (b) XRD pattern of PQDs. | ||
Thus, we can conclude that the IRTOA method of the solution-phase halide exchange reaction does not alter the inner cationic sub-lattice.22 Preservation of the size and shape and the cubic nature of the high-quality PQD crystals are indicative of the ingenuity of the halide exchange process adopted in our study.
Moreover, we performed the IRTOA synthesis of several PQDs using HCl and HI as halide sources. Herein, note that the instantaneous halide exchange reactions were nearly quantitative as we never observed any PL peak corresponding to themother PQD CsPbBr3. As can be seen from Fig. 2(a) with a series of these pure and mixed halide all-inorganic PQDs we could cover emission wavelength ranged from blue (430 nm) to red (650 nm) (Fig. 2(b)).
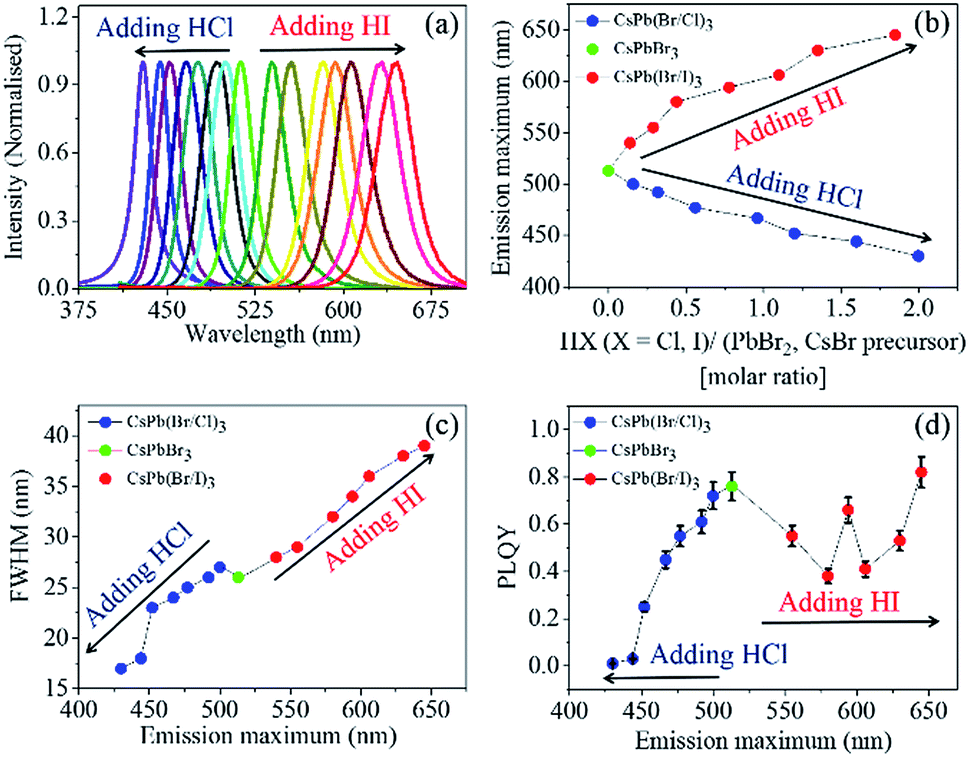 | ||
| Fig. 2 (a) PL emission spectra of the PQDs prepared by halide exchange from CsPbBr3. (b) Evolution of the emission maximum with the addition of HCl and HI to a fixed amount of the CsPbBr3 solution. (c) Evolution of FWHM with halide exchange. (d) Evolution of PLQY with halide exchange. | ||
The PL emission spectra are quite narrow (FWHM varying from 17 to 39 nm) (Fig. 2(c)). As the size/atomic number of the halide increases, the FWHM of the PL emission band also increases.27 Quite interestingly, the mixed halide Br/I PQDs exhibit PL emission spectra that are narrower (FWHM ∼ 35 nm) than those reported in the literature (FWHM ∼ 50 nm)18,19,27 for the PQDs synthesized at high temperatures.
In literature, it is generally shown that the PLQY of CsPbBr3 is highest, and it decreases with the formation of the mixed halide PQDs CsPb(Br/Cl)3 and CsPb(Br/I)3.19 The PLQY values reported herein for the CsPb(Br/Cl)3 and CsPb(Br/I)3 PQDs synthesized through the IRTOA process are either similar to or the same as those reported for the PQDs synthesized following several hours syntheses under an inert atmosphere through the hot injection procedure.18,19,27,29 In the case of CsPb(Br/Cl)3, the magnitude of PLQY decreases monotonically as the relative percentage of chlorine increases. However, in the case of CsPb(Br/I)3, the PLQY value initially decreases with a decrease in the concentration of CsPbBr3, whereas it increases beyond the PL maximum of 600 nm; this may perhaps be because of the lowering of the conduction band energy level with respect to the low-lying trap states with an increase in the relative percentage of iodine.30
For example, for CsPb(Br/I)3 PQDs (synthesized through the hot-injection inert atmosphere method), the PLQY values reported in most of the studies are ∼70%.27 Herein, we have reported a similar PLQY value for the Br/I mixed halide PQDs (a maximum of 82%) (Fig. 2(d)). The PLQY values of our CsPb(Br/Cl)3 PQDs are lower than that of CsPbBr3, and a similar trend has been observed for the CsPb(Br/Cl)3 PQDs synthesized through the inert-atmosphere, hot-injection technique.19 Thus, the optical quality of the PQDs synthesized through the IRTOA method employing non-metal-based halide sources, such as HCl/HI, is similar to that of the PQDs synthesized through the high-temperature, inert-atmosphere synthesis for several hours. As a next step, we probed the PL behaviour of these PQDs using pico-nano second-based time-correlated single photon counting (TCSPC) and femtosecond-based transient absorption techniques. As can be observed from Fig. 3(a) (TCSPC-based PL decay monitored at PL emission maximum), as the halide changes from Cl to Br to I, the PL decay becomes slower in nature. In other words, as the size/atomic number of the halide increases, the PL decay becomes much slower (Table S1†). In literature, for the PQDs synthesized through the hot-injection, inert-atmosphere synthesis, a similar pattern of PL decay has been reported.14,27 The higher excited-state lifetime and broader PL emission spectra for the I-exchanged PQDs possibly indicate that low lying triplet states are involved in the PL emission process for the I-exchanged PQDs. Very recently, similar theoretical results have been reported in the literature.31 Detailed investigations in this direction are currently underway. To investigate whether our PQDs exhibit similar ultrafast dynamics as that reported in the literature at the femtosecond time scale, we have explored CsPbBr3 as a case study material with the excitation wavelength of 400 nm as this particular wavelength has been used in most of the studies reported in the literature on PQDs. As can be observed from Fig. 3(b), the bleach dynamics of the CsPbBr3 PQD monitored at 501 nm yielded a rise followed by a decay phenomenon. The rising part of the plot could be fitted with the time constant of 590 (±20) fs, and the decay part of the dynamics could be fitted with 50 (±10) and 250 (±50) ps. These values are quite similar to those reported in the literature for PQDs.32–34 Thus, we can conclude that the PQDs synthesized herein by the IRTOA method employing halide exchange with inexpensive HX (X = Cl/I) exhibit similar ultrafast dynamics as that exhibited by the PQDs synthesized via the time-consuming, inert-atmosphere, hot-injection procedure. Detailed ultrafast dynamical analyses of other PQDs are currently under investigation.
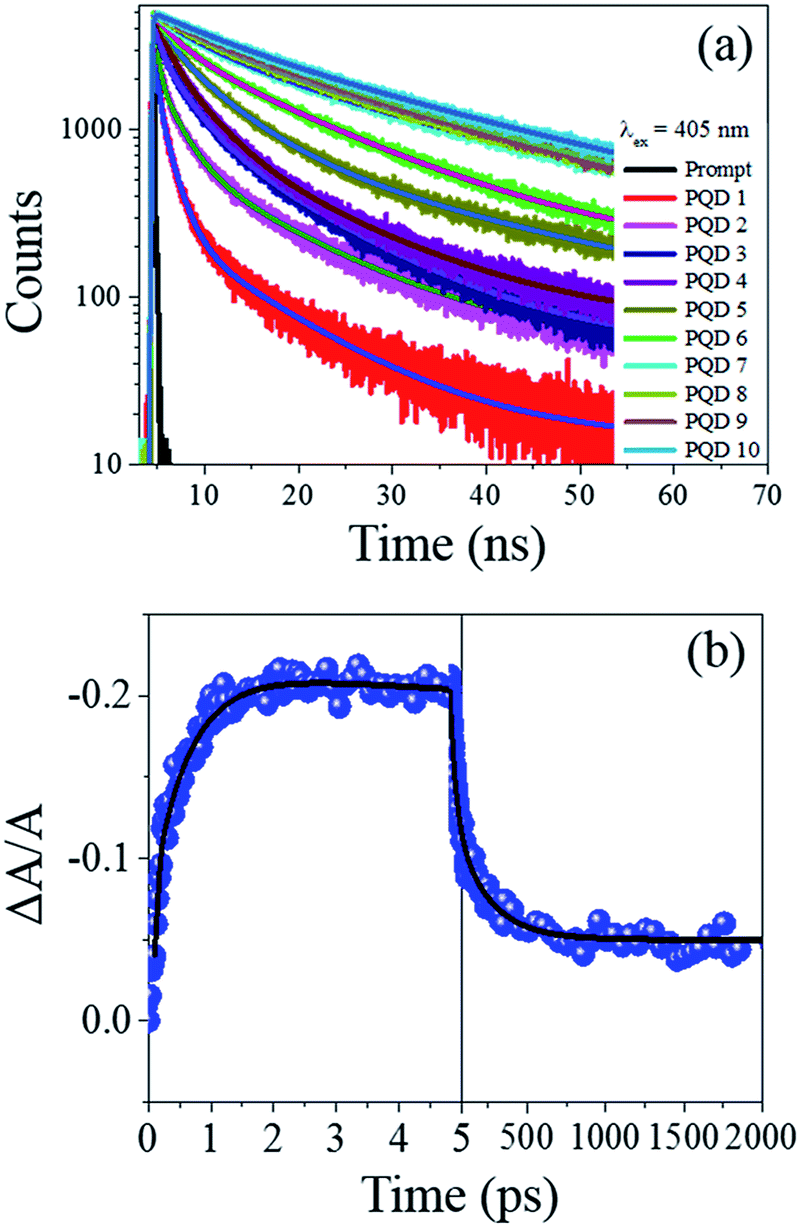 | ||
| Fig. 3 Fast PL decay of different PQDs (a). Ultrafast (b) optical behaviour of CsPbBr3 PQD. Pump wavelength = 400 nm. | ||
As a next step, we probed how these PQDs would behave at the extremely sensitive single particle level. As the PLQY of these PQDs is quite high, these PQDs can be easily observed using our home-built total internal reflection (TIRF)-based single molecule spectroscopic setup (Fig. 4(a)).35 We carried out single particle measurements for the PQD 3 (Br/Cl mixed halide PQD, λem = 477 nm), PQD 5 (CsPbBr3, λem = 513 nm) and PQD 6 (Br/I mixed halide PQD, λem = 555 nm) with 405 nm CW laser excitation and 100 ms binning time. Note that to date, the single particle spectroscopic results for three different PQDs ((Br/Cl)3, Br3, and (Br/I)3) under the same experimental conditions have not been reported in the literature. Typical single particle time traces are shown in Fig. 4(b). Blinking of these PQD single particles can be visualized clearly (Movie S7†). As observed from Fig. 4(c), for the PQD 3, i.e., PQD with the mixed Br/Cl halide, a well separated bimodal distribution of the PL intensities was obtained, and the threshold was maintained at 3σ above the peak intensity of the lower intensity distribution (where σ is the standard deviation of the lower intensity distribution). Thus, a clear blinking behaviour with distinct ON and OFF events could be noticed (for other PQDs, see Fig. S8†). The horizontal dashed line shown in Fig. 4(b) indicates the threshold value adopted herein; above this threshold value, the PL intensity is considered to be ON, and below this threshold value, the PL intensity is considered to be OFF.
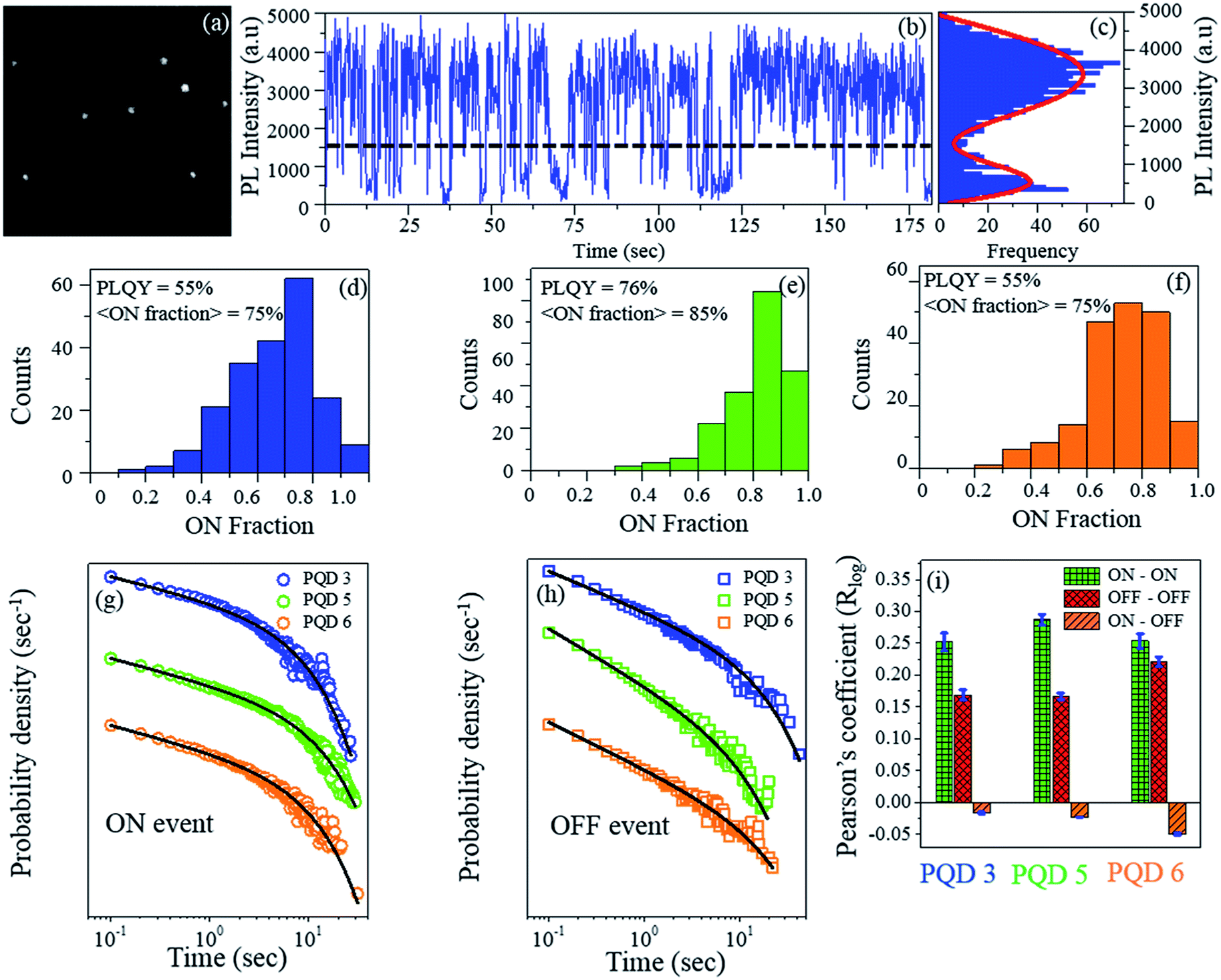 | ||
| Fig. 4 (a) Single particle image with the PQD 3. (b) Single particle time trace with PQD 3. (c) PL intensity histogram. Percentage ON fraction for three PQDs: ((d) PQD 3, (e) PQD 5, and (f) PQD 6). Ensemble level PLQY values have been mentioned in the inset. Probability density distribution of the (g) ON event duration and (h) OFF event duration of all three PQDs. (i) Pearson's logarithm correlation coefficient (Rlog) of subsequent event durations (ON–ON, OFF–OFF, and ON–OFF) for all three PQDs. | ||
From the PQD 3 (λem = 477 nm) to PQD 5 (λem = 513 nm) to PQD 6 (λem = 555 nm), the percentage of the ON fraction initially increases from 75% to 85% and then decreases to 75% (Fig. 4(d), (e) and to Fig. 4(f), respectively). The ensemble level PLQY values for the respective PQDs have been mentioned in the inset of Fig. 4(d)–(f). The single particle level average % ON fraction values of the respective PQDs seem to be correlated with the ensemble level PLQY values. The percentage of PQDs having the ON fraction higher than 70% is ∼50 for the PQD 3, ∼85 for the PQD 5, and ∼60 for the PQD 6. Hence, at the single particle level, a large % of the PQD population has high ON fraction value even after halide exchange with both Cl− and I−. Herein, note that the higher the ON fraction, the higher the probability of visualization of PQDs for the single particle tracking; moreover, the higher the percentage ON fraction, the better the quality of the PQD. Thus, we can conclude that at the single particle level, the CsPbBr3 PQD and the mixed halide PQDs (PQD 3 and PQD 6) remain mostly in the ON-state. These high ON fraction values are mostly similar to the values obtained for the PQDs synthesized through the inert atmosphere, hot-injection synthesis.36–38 This, in turn, demonstrates the ingenuity of the halide exchange process adopted herein. Thus, the IRTOA halide exchange reaction with inexpensive HX (X = Cl/I) yields PQDs with equally good (if not better) optical properties.
To investigate the blinking dynamics in detail, we calculated the probability density of the ON and OFF event durations (Fig. 4(g) (ON event durations) and Fig. 4(h) (OFF event durations)). For all three PQDs, the probability density distributions of both the ON and the OFF event durations truncate from the power law behaviour at longer event durations and are best fitted with a truncated power law equation (Fig. S9†).38,39 The power law exponent (m) is <1.0 for the ON events (mON) and >1.50 for the OFF events (mOFF) for all three PQDs (i.e., for all the samples, the mOFF magnitude is higher than mON (Table 1)). As can be observed from Table 1, the ‘m’ value for a particular event (ON or OFF) does not change significantly from PQD 3 to PQD 5 to PQD 6. However, for a particular sample, the difference between the ‘m’ values for the ON and OFF events is quite significant (Table 1). For a particular sample, the relative magnitude of ‘m’ will dictate which event (ON or OFF) will be more probable for a particular event duration. Hence, the ‘m’ values become important while comparing the probability density distribution of the ON and OFF events for a particular sample. A relatively higher value of mOFF than that of mON suggests (1.5 as compared to 0.9) that the slope of the ON event probability density distribution is less steeper than that of the OFF event probability density distribution. This, in turn, signifies that the probability of the ON event durations will be higher than that of the OFF event durations.40,41 After halide exchanges with the Cl− and I− ions, the PLQY decreases from 76% (PQD 5) to 55% for both PQD 3 and PQD 6 (Table 1). A similar trend was also observed for the ON event truncation time, which decreased from 4.0 s (PQD 5) to 2.7 s for the Cl− exchange (PQD 3) and to 2.9 s for the I− exchange (PQD 6) (Table 1). However, an opposite trend was observed for the OFF event truncation times, which increased from 4.3 s (PQD 5) to 5.6 s (PQD 3) and 6.6 s (PQD 6) (Table 1). A decrease in the ON event truncation time (from PQD 5 to PQD 3/PQD 6) suggests that the ON event distributions truncate earlier (in PQD 3/PQD 6 as compared to those in PQD 5); this would make the longer ON events less probable after halide exchange.
| PQD | PLQY | ON | OFF | ||
|---|---|---|---|---|---|
| m ON | ON truncation time (s) | m OFF | OFF truncation time (s) | ||
| PQD 3 | 0.55 ± 0.02 | 0.90 ± 0.04 | 2.7 ± 0.1 | 1.55 ± 0.06 | 5.6 ± 0.2 |
| PQD 5 | 0.76 ± 0.02 | 0.94 ± 0.05 | 4.0 ± 0.2 | 1.85 ± 0.10 | 4.3 ± 0.2 |
| PQD 6 | 0.55 ± 0.03 | 0.95 ± 0.05 | 2.9 ± 0.1 | 1.80 ± 0.09 | 6.6 ± 0.3 |
Similarly, the longer OFF events become more probable (in PQD 3/PQD 6 as compared to those in PQD 5) as the OFF event duration distribution truncates later in the mixed halide PQDs (PQD 3/PQD 6). Thus, for different PQD samples, an increase in the ON event truncation time and concomitant decrease in the OFF event truncation time could be correlated with the increase in the PLQY values. Thus, we could successfully correlate the physical parameters obtained from the ensemble as well as the single particle level.
We also studied the presence of the memory effect in all three PQD samples. Although studies have been reported on the memory effects in QDs,41–44 the same has not been explored for the all-inorganic PQDs to date. The two-dimensional (2D) joint probability distribution of the adjacent event durations (logarithms of event duration) for both ON and OFF events has been plotted with colour coding (Fig. S11†). A 2D joint probability distribution of the adjacent ON events (ON event to next ON event) for all three PQDs has been plotted in Fig. S11(a, g, and m),† and Fig. S11(b, h, and n)† represent the same for the ON events that are sufficiently separated (e.g., 1st ON event to 50th ON event). Then, the latter has been subtracted from the former. The subtracted 2D joint probability distribution (Fig. S11(c, i, and o)†) in the matrix plot shows a significant diagonalization, indicating the presence of residual memory in the ON event durations.41,42 Similar analyses on the OFF event durations also show the presence of memory among the OFF event durations (Fig. S11(f, l, and r)†).
To quantify the residual memory, Pearson's logarithm correlation coefficient for the adjacent event durations was calculated. For all three PQDs, both ON–ON and OFF–OFF events are positively correlated; however, the ON-OFF events are negatively correlated (Fig. 4(i)). The magnitude of the Pearson's logarithm correlation coefficient for the ON–ON events is higher than that for the OFF–OFF events. Similar results have been reported for the CdSe and InP-based QDs.41–46 As can be observed from Fig. 4(i), the ON–OFF events are anti-correlated. Thus, the existence of a residual memory in the all-inorganic pure CsPbBr3 and mixed halide PQDs could be evidenced.
We have compared our result with those of two of the recently reported studies.21,47 First, in these two studies, the authors have synthesized CsPbBr3via the hot-injection method under an inert atmosphere condition. This is different from the way we have synthesized CsPbBr3, i.e., via the IRTOA method. Second, in both the studies,21,47 the chloride/bromide exchange has been achieved using HCl/HBr in the vapour phase. Moreover, in one study,21 the Br → I anion exchange has not been performed by HI. Instead, the authors have used an OLAM-I precursor, which has been synthesized by the reaction of oleylamine (OAM) with I2 overnight.
In the other study,46 the I exchange has been achieved by HI vapour towards a sensing purpose. In our process, all halide exchanges were performed using HX (X = Cl/Br/I) through the IRTOA method in the solution phase and not in the vapour phase for sensing purpose as abovementioned in this study. Third, neither of the reported21,47 vapour phase halide replacements was instantaneous, and it took 20–60 minutes for the halide exchange. However, in our case, all halide exchanges took place instantaneously. Fourth, in none of these reports,21,47 quantitative procedures of mixed halide PQD synthesis covering a large range of PL emission maxima have been reported. However, in our case, we have not only synthesized mixed halide PQD with a range of PL emission maxima, but also characterized them all using TEM, EDAX, PXRD, among other techniques. Thus, in these two literature studies,21,47 (i) CsPbBr3 has been synthesized via the hot-injection inert atmosphere method, and then, (ii) vapour phase halide exchange was conducted, (iii) which took 20–60 minutes, and (iv) by this procedure, only one mixed halide PQD with a fixed PL emission maximum can be synthesized. However, in this study, we report (i) the synthesis of CsPbBr3via the RT open-air atmosphere method. Then, the mixed halide PQDs were synthesized in (ii) the solution phase, (iii) instantaneously, and (iv) a wide range of PL emission maxima (from 430 nm to 650 nm) could be covered. Thus, our study is significantly different from the abovementioned two literature studies.
Conclusions
In conclusion, herein, the instantaneous synthesis of blue to green to yellow to red-emitting all-inorganic perovskite quantum dots (PQDs) was achieved at room temperature under an open-air atmosphere (no glove box) through halide exchange from CsPbBr3 employing non-metal based inexpensive and easily available halide sources such as HCl and HI. All PQDs were highly crystalline and cubic-shaped with the size uniformity of ca. 11.3 ± 0.1 nm. These PQDs were stable for a few weeks under an open-air atmosphere. The PL emission spectra are quite narrow, and the PLQY values are quite high (80% for even Br/I mixed PQDs). A very regular increase in the PL lifetime has been observed as the halide changes from Cl to Br to I. Ultrafast dynamics revealed the excited-state time constants at the femtosecond to nanosecond timescales, similar to those of PQDs synthesized through the hot injection, inert atmosphere procedure. This kind of room-temperature (no use of heat energy/electrical energy), open-air atmosphere (no use of cumbersome inert atmosphere), instantaneous (less time consuming), nearly quantitative synthesis of PQDs using inexpensive and easily available HCl/HI is extremely important for the large-scale synthesis of PQDs for various applications. At the single particle level, the 〈ON fraction〉 has been noted to vary from 75% to 85% for different PQDs; this indicates that the blue-green-yellow-red-emitting PQDs are quite bright and mostly in the ON state even at the single particle level. The magnitudes of the mON values are close to 1.0, and the mOFF values are >1.5, indicating that long ON durations are more probable. The increase in the ON event truncation time (from 2.7 to 4.0 s) and a concomitant decrease in the OFF event truncation time (from 6.6 to 4.3 s) could be correlated with an increase in the PLQY (from 0.55 to 0.75). An interesting memory effect could be observed in both the ON and the OFF event durations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. K. M. thanks IISER Kolkata for financial help and instrumental facilities. Support received from the CSIR India, Project No. 01(2848)/16/EMR-II is gratefully acknowledged. S. M. thanks IISER Kolkata; D. R., M. M., and A. D. thank CSIR; C. K. D. and S. G. thank DST-INSPIRE for the fellowship. The authors thank Dr Prakriti Ranjan Bangal (IICT, India) for the femtosecond measurement.References
- J. Liang, C. Wang, Y. Wang, Z. Xu, Z. Lu, Y. Ma, H. Zhu, Y. Hu, C. Xiao, X. Yi, G. Zhu, H. Lv, L. Ma, T. Chen, Z. Tie, Z. Jin and J. Liu, J. Am. Chem. Soc., 2016, 138, 15829 CrossRef CAS PubMed.
- Q. A. Akkerman, M. Gandini, F. Di Stasio, P. Rastogi, F. Palazon, G. Bertoni, J. M. Ball, M. Prato, A. Petrozza and L. Manna, Nat. Energy, 2016, 2, 16194 CrossRef.
- S. Christodoulou, F. Di Stasio, S. Pradhan, A. Stavrinadis and G. Konstantatos, J. Phys. Chem. C, 2018, 122, 7621 CrossRef CAS.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- Y. Fu, H. Zhu, C. C. Stoumpos, Q. Ding, J. Wang, M. G. Kanatzidis, X. Zhu and S. Jin, ACS Nano, 2016, 10, 7963 CrossRef CAS PubMed.
- Y. Xu, Q. Chen, C. Zhang, R. Wang, H. Wu, X. Zhang, G. Xing, W. W. Yu, X. Wang, Y. Zhang and M. Xiao, J. Am. Chem. Soc., 2016, 138, 3761 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162 CrossRef CAS PubMed.
- Q. Shan, J. Li, J. Song, Y. Zou, L. Xu, J. Xue, Y. Dong, C. Huo, J. Chen, B. Han and H. Zeng, J. Mater. Chem. C, 2017, 5, 4565 RSC.
- J. S. Yao, J. Ge, B. N. Han, K. H. Wang, H. B. Yao, H. L. Yu, J. H. Li, B. S. Zhu, J. Z. Song, C. Chen, Q. Zhang, H. B. Zeng, Y. Luo and S. H. Yu, J. Am. Chem. Soc., 2018, 140, 3626 CrossRef CAS PubMed.
- H. Zhang, X. Wang, Q. Liao, Z. Xu, H. Li, L. Zheng and H. Fu, Adv. Funct. Mater., 2017, 27, 1604382 CrossRef.
- M. Kulbak, D. Cahen and G. Hodes, J. Phys. Chem. Lett., 2015, 6, 2452 CrossRef CAS PubMed.
- M. Kulbak, S. Gupta, N. Kedem, I. Levine, T. Bendikov, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2016, 7, 167 CrossRef CAS PubMed.
- J. Song, L. Xu, J. Li, J. Xue, Y. Dong, X. Li and H. Zeng, Adv. Mater., 2016, 28, 4861 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692 CrossRef CAS PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008 CrossRef CAS PubMed.
- D. Zhang, S. W. Eaton, Y. Yu, L. Dou and P. Yang, J. Am. Chem. Soc., 2015, 137, 9230 CrossRef CAS PubMed.
- J. Shamsi, Z. Dang, P. Bianchini, C. Canale, F. D. Stasio, R. Brescia, M. Prato and L. Manna, J. Am. Chem. Soc., 2016, 138, 7240 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635 CrossRef CAS PubMed.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276 CrossRef CAS PubMed.
- A. Haque, V. K. Ravi, G. S. Shanker, I. Sarkar, A. Nag and P. K. Santra, J. Phys. Chem. C, 2018, 122, 13399 CrossRef CAS.
- F. Palazon, Q. A. Akkerman, M. Prato and L. Manna, ACS Nano, 2016, 10, 1224 CrossRef CAS PubMed.
- S. Fang, G. Li, Y. Lu and L. Li, Chem.–Eur. J., 2018, 24, 1898 CrossRef CAS PubMed.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648 CrossRef CAS PubMed.
- S. Seth and A. Samanta, Sci. Rep., 2016, 6, 37693 CrossRef CAS PubMed.
- C. Guhrenz, A. Benad, C. Ziegler, D. Haubold, N. Gaponik and A. Eychmüller, Chem. Mater., 2016, 28, 9033 CrossRef CAS.
- S. K. Balakrishnan and P. V. Kamat, ACS Energy Lett., 2016, 2, 88 CrossRef.
- C. Bi, S. Wang, W. Wen, J. Yuan, G. Cao and J. Tian, J. Phys. Chem. C, 2018, 122, 5151 CrossRef CAS.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435 CrossRef CAS.
- Y. Su, X. Chen, W. Ji, Q. Zeng, Z. Ren, Z. Su and L. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 33020 CrossRef CAS PubMed.
- A. Swarnkar, V. K. Ravi and A. Nag, ACS Energy Lett., 2017, 2, 1089 CrossRef.
- M. A. Becker, R. Vaxenburg, G. Nedelcu, P. C. Sercel, A. Shabaev, M. J. Mehl, J. G. Michopoulos, S. G. Lambrakos, N. Bernstein, J. L. Lyons, T. Stoferle, R. F. Mahrt, M. V. Kovalenko, D. J. Norris, G. Raino and A. L. Efros, Nature, 2018, 553, 189 CrossRef CAS PubMed.
- P. Maity, J. Dana and H. N. Ghosh, J. Phys. Chem. C, 2016, 120, 18348 CrossRef CAS.
- N. Mondal and A. Samanta, Nanoscale, 2017, 9, 1878 RSC.
- N. Soetan, A. Puretzky, K. Reid, A. Boulesbaa, H. F. Zarick, A. Hunt, O. Rose, S. Rosenthal, D. B. Geohegan and R. Bardhan, ACS Photonics, 2018, 5, 3575 CrossRef CAS.
- D. Roy, T. Routh, A. V. Asaithambi, S. Mandal and P. K. Mandal, J. Phys. Chem. C, 2016, 120, 3483–3491 CrossRef CAS.
- A. Swarnkar, R. Chulliyil, V. K. Ravi, M. Irfanullah, A. Chowdhury and A. Nag, Angew. Chem., Int. Ed., 2015, 54, 15424 CrossRef CAS PubMed.
- A. Zhang, C. Dong and J. Ren, J. Phys. Chem. C, 2017, 121, 13314 CrossRef CAS.
- N. A. Gibson, B. A. Koscher, A. P. Alivisatos and S. R. Leone, J. Phys. Chem. C, 2018, 122, 12106–12113 CrossRef CAS.
- G. Yuan, C. Ritchie, M. Ritter, S. Murphy, D. E. Gómez and P. Mulvaney, J. Phys. Chem. C, 2018, 122, 13407–13415 CrossRef CAS.
- A. A. Cordones and S. R. Leone, Chem. Soc. Rev., 2013, 42, 3209–3221 RSC.
- D. Roy, S. Mandal, C. K. De, K. Kumar and P. K. Mandal, Phys. Chem. Chem. Phys., 2018, 20, 10332–10344 RSC.
- F. D. Stefani, X. Zhong, W. Knoll, M. Han and M. Kreiter, New J. Phys., 2005, 7, 197 CrossRef.
- J. P. Hoogenboom, J. Hernando, M. F. García-Parajó and N. F. van Hulst, J. Phys. Chem. C, 2008, 112, 3417–3422 CrossRef CAS.
- S. Volkán-Kacsó, P. A. Frantsuzov and B. Jankó, Nano Lett., 2010, 10, 2761–2765 CrossRef PubMed.
- C. K. De, T. Routh, D. Roy, S. Mandal and P. K. Mandal, J. Phys. Chem. C, 2018, 122, 964–973 CrossRef CAS.
- D. Roy, A. Das, C. K. De, S. Mandal, P. R. Bangal and P. K. Mandal, J. Phys. Chem. C, 2019, 123, 6922–6933 CrossRef CAS.
- X. Chen, H. Hu, Z. Xia, W. Gao, W. Gou, Y. Qu and Y. Ma, J. Mater. Chem. C, 2017, 5, 309–313 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00406h |
| This journal is © The Royal Society of Chemistry 2019 |