
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCorrelation of near-unity quantum yields with photogenerated excitons in X-type ligand passivated CsPbBr3 perovskite quantum dots†
Sunjoong
Park‡
,
Hyunjin
Cho‡
,
Wonseok
Choi
,
Hanfeng
Zou
and
Duk Young
Jeon
 *
*
Display Materials Laboratory, Department of Materials Science and Engineering, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 305-338, Republic of Korea. E-mail: dyj@kaist.ac.kr
First published on 13th May 2019
Abstract
We investigated the exciton decay dynamics of CsPbBr3 perovskite quantum dots (PQDs) through an X-type ligand passivation process. 1-Dodecanethiol (DDT), as an X-type ligand, covers Br vacancies of PQDs and then the photoluminescence quantum yield (PLQY) sharply improved from 76.1% to 99.8%. Impressively, after passivation, the photoluminescence (PL) lifetime of PQDs decreased from 3.16 ns to 2.42 ns, contrary to the PLQY increase. To clarify this phenomenon, we observed exciton decay dynamics by varying the temperature. Thereby, we found that shallow traps from Br vacancies not only reduce the PLQY but also induce a longer lifetime related to the nonradiative exciton decay leading to an increase in the lifetime. Our results are novel and important in a way that we provide a systematic understanding of the exciton decay dynamics by combining two key concepts together: (1) near unity PLQY via ligand engineering and (2) temperature-dependent photogenerated excitons.
Lead-halide perovskite quantum dots (QDs) such as all-inorganic perovskites (CsPbX3, X = Cl, Br, or I) and organic–inorganic hybrid perovskites (CH3NH3PbX3) have attracted significant attention due to their superior optical properties.1–4 Especially, CsPbBr3 perovskite QDs (PQDs) have a high photoluminescence quantum yield (PLQY), narrow emission bandwidth and an emission wavelength easily tunable by changing halide atoms or controlling the nanocrystal size.5–9 All these excellent features make PQDs promising candidate materials for optoelectronic applications including applications in lasers,10–12 photodetectors,13,14 solar cells15,16 and light-emitting diodes (LEDs).17,18 Prior to being applied in various fields, the correlation between the chemical surface state and the high efficiency of the PQDs should be studied. In general, conventional inorganic QDs such as CdSe have required surface treatments to obtain a high PLQY. (1) The outer shell (CdS) which has a larger band gap can passivate the deep trap sites of the core (CdSe) and localize excitons. (2) Capping ligands cover the surface defects of QDs. Interestingly, PQDs can achieve a high PLQY with only ligand passivation except for the outer shell. The reason is that Br− vacancies on the surface of PQDs create shallow trap sites19–21 whereas Cd based QDs form surface defects in deep midgap states, which lead to the loss of their PL characteristics.22–24
Since semiconductor QDs have large surface/volume ratios, the optical properties change with surface conditions. Since PQDs are covered with only ligands, we should consider the chemical interaction between ligands and surface defects. In particular, Br vacancies result in an imperfect Pb octahedral [PbBr6] structure, which is essential for the formation of the electronic structure around the band edge. As a result, PQDs could not achieve unity PLQY, and exciton decay dynamics was also affected. For this reason, we should gain deep insight into ligand passivation on PQDs, especially the relationship between exciton decay dynamics and surface traps. Fortunately, the Peng group gave some hints on the correlation between exciton decay dynamics and surface traps of CdSe/CdS heterostructure QDs.25 Shallow electron traps of surface defects can be fatal for QDs and may result in a loss of their characteristic radiative decay and they create a relatively short photoluminescence (PL) lifetime. In addition, the A. Paul Alivisatos group reported the surface mechanism of PQDs with X-type ligands.19 Oleate as a hard Lewis base has a negligible effect on the PLQY, whereas thiol ligands as a soft Lewis base passivate the surface Br vacancies and significantly enhance the PLQY. Furthermore, L. Ruan et al. showed that 1-octanethiol ligands fill the surface defects of PQDs and raise the stability.26 Thus, ligand engineering in PQDs can be a milestone for engineering optoelectronic properties.
In this work, we have studied the mechanism of exciton decay by passivating the Br vacancies of PQDs with thiol ligands. Impressively, the PLQY of PQDs passivated with thiol ligands improved from 76.1% to 99.8%, whereas the PL lifetime decreased from 3.16 ns to 2.36 ns. To clarify this phenomenon, the relationship between exciton decay dynamics and surface traps was investigated by varying the temperature from low temperature (80 K) to room temperature (300 K). We have found that shallow electron traps from halide vacancies show a relatively short lifetime and can be filled by thiol ligands. Our results provide deep insight into the exciton decay dynamics and will help to design ideal PQDs for further application.
Results and discussion
Perovskite quantum dots (PQDs) were synthesized by reported methods using oleic acid and oleylamine as surface capping ligands.7 However, oleylamine as a hard X-type Lewis base cannot completely cover the Br vacancies and the absolute PLQY of PQDs do not approach unity (absolute PLQY of PQDs is ∼76.1%). In order to passivate the Br vacancies, 1-dodecanethiol (DDT) with a soft X-type Lewis base was introduced.Fig. 1 shows the schematic illustration of the detailed DDT passivation mechanism of PQDs. In the case of PQDs covered with oleic acid and oleylamine, there are some Br vacancies. Br atoms can be detached from the perovskite surface in the form of oleylammonum bromide ligand pairs or volatile HBr.19 These Br vacancies result in an imperfect Pb octahedral [PbBr6] structure, which is essential for the formation of the electronic structure around the band edge.27 In this situation, we introduced an X-type ligand DDT to fill the Br vacancies. According to following eqn (1) and (2), oleate and thiol groups are used to cover the vacancies caused by the dissociated Br atom. However, oleate as a hard Lewis base hardly binds to the surface well because the trap sites act as a soft Lewis acid. In contrast, thiol ligands as a soft Lewis base can passivate the surface Br vacancies and then significantly enhance the PLQY. As a result, PQDs passivated using DDT (DDT-PQDs) achieved a near unity PLQY (∼99.8%). Fig. S1† shows the photograph of PQDs and DDT-PQDs with/without UV irradiation.
| Pb–VBr + R–COO/Pb–COO–R | (1) |
| Pb–VBr + R–S/Pb–S–R | (2) |
 | ||
| Fig. 1 Scheme of the dodecanethiol passivation process. Cs, Pb, and Br are depicted by orange, black and green spheres respectively. For PQDs, there are Br atom vacancies which result in PbBr6 octahedral destruction (the upper right site in PQDs). Dodecanethiol is an appropriate soft Lewis base to bind with Pb atoms, and then Br vacancies are filled by the thiol group (yellow sphere) in dodecanethiol. | ||
In order to compare the microstructure of PQDs via DDT passivation, transmission electron microscopy (TEM) and X-ray diffraction (XRD) analysis were performed. Fig. 2a and b show that morphologies of both PQDs and DDT-PQDs are identical and their XRD patterns present peaks at the same diffraction angles, which implies that the crystal structure of PQDs is orthorhombic (Fig. 2c).28–30 The XRD results suggest that the DDT ligands do not induce structural transformation of PQDs. However, the particle size of PQDs slightly increases from 7.31 ± 1.12 nm to 7.78 ± 1.28 nm with DDT passivation (Fig. S2†). It can be constructed by dissociation of pristine ligands with oleic acid during the DDT ligand passivation process. These results are also supported by the PL and absorbance spectral results as shown in Fig. 2d. Due to the quantum confinement effect,31 the increase of the QD size induces a slight red-shifted peak of the first exciton absorption from 486 nm to 490 nm and also a red-shifted PL peak from 514 nm to 515 nm with a full width half-maximum (FWHM) of 22 nm (Fig. 2d).
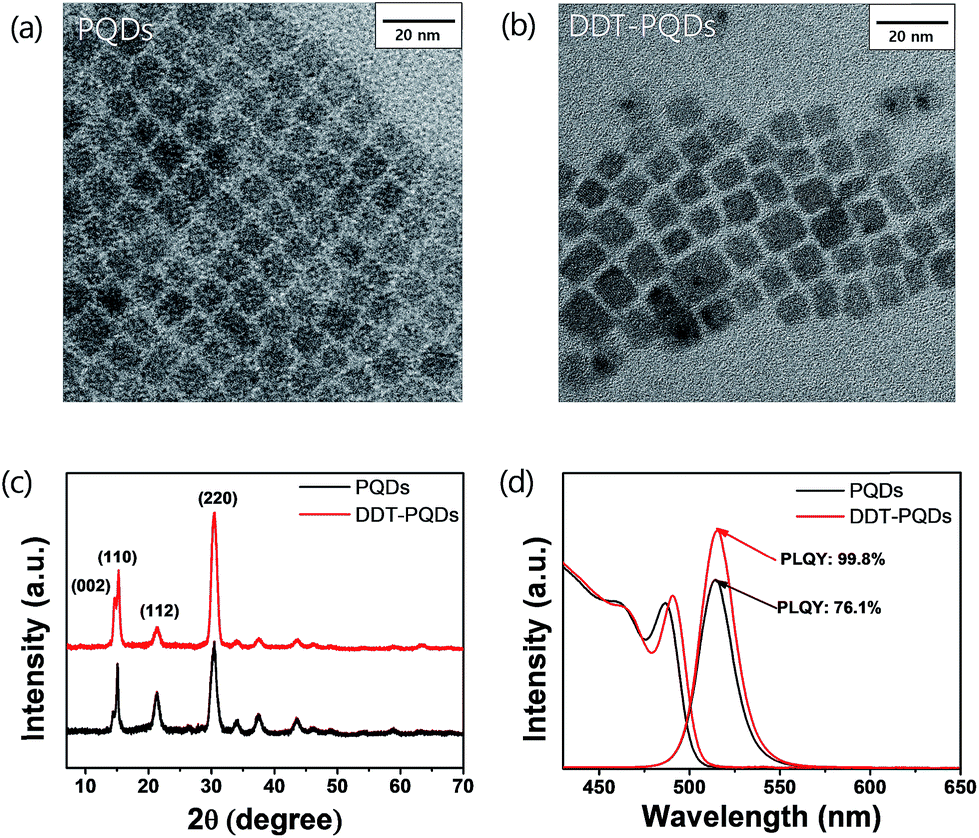 | ||
| Fig. 2 TEM images of (a) PQDs and (b) DDT-PQDs. (c) XRD patterns of PQDs and DDT-PQDs belonging to the orthorhombic crystal system (d) PL spectra (400 nm excitation) and UV-vis spectra of PQDs and DDT-PQDs. | ||
To gain better insight into the chemical interaction between PQDs and DDT ligands, we carried out X-ray photoelectron spectroscopy (XPS) analysis. All XPS spectra were calibrated with respect to the C 1s peak at 284.8 eV, the internal standard for the carbon–carbon bond (Fig. S3†).
Fig. 3a and b exhibit the XPS spectra of Pb 4f with respect to PQDs and DDT-PQDs. It has been reported that the Pb 4f core level consists of two distinct peaks; the higher and lower bind energy regions correspond to Pb–Br and Pb–oleate, respectively.32–34 The concentration of Pb–oleate in the PQDs is calculated to be 3.14%. However, the concentration is dramatically reduced to 0.99% after passivation. These results indicate that DDT is closely related to the exchanging ligand and covers the defect sites.
 | ||
| Fig. 3 Pb 4f XPS spectra of (a) PQDs and (b) DDT-PQDs; blue and red dashed lines represent the Pb–Br and Pb–oleate region respectively. (c) Br 3d XPS spectra of PQDs and DDT-PQDs. (d) Br/Pb atomic ratio of PQDs and DDT-PQDs from XPS survey data. (e) 1H NMR spectra for PQDs, DDT-PQDs, and DDT in toluene-d8. The methylene and –SH resonance signals are indicated in the 0.5–3.0 ppm region. | ||
Note that PQDs have a lattice structure with imperfect Pb octahedral [PbBr6] structures due to Br vacancies. After DDT passivation, DDT-PQDs form a rigid [PbBr6] octahedral structure, and they achieved a near unity PLQY. Br 3d spectra of DDT-PQDs also slightly shifted in the main peak towards the high binding energy state (Fig. 3c). These results indicate that DDT-PQDs have a stronger Pb–Br bond and a modified chemical environment for the [PbBr6] octahedral structure after DDT passivation. Our findings are consistent with the tendency of previously reported XPS spectra.32,33,35 Additionally, in the case of the PQDs with a perfect crystal structure, they have the chemical formula CsPbBr3. Furthermore, the atomic ratio of [Br]/[Pb] is fixed at 3.0. However, the calculated ratios with and without passivation using DDT ligands were measured to be 2.40 and 2.49. These values denote the existence of imperfections in the PQDs (illustrated in Fig. 3(d)). Imperfections of the CsPbBr3 perovskite are mainly caused by the dissociation of Br atoms in the PQDs.
To further study the chemical interaction between DDT ligands and Br vacancies on the surface of PQD nanocrystals, nuclear magnetic resonance (NMR) spectroscopy was performed. After the purification process, the precipitates were dispersed in a toluene-d8 solvent for NMR analysis. Fig. 3e shows the 1H NMR spectra of PQDs, DDT-PQDs and free DDT ligands, respectively. There are some distinct peaks attributed to oleic acid, oleylamine, DDT and impurity resonance such as residual ODE and the toluene solvent.36 DDT-PQDs still have resonance signals 5 of oleylamine and oleic acid at around 2 ppm attributed to hydrogen resonance from methylene near the double bonded carbon.19,37 This result reveals that introducing DDT ligands into PQDs induces an additional defect passivation effect as well as a ligand exchange process. In addition, we can find some evidence of DDT passivation. First, the –SH functional group peak of the free DDT ligand is located at around 1.3 ppm. However, after DDT ligand treatment, there are no more signals from H resonance of the –SH terminal functional group because the thiol group is tightly bound to Br vacancies.38,39 This can be identified from the magnification spectra of DDT in Fig. S4.† Second, another proof of Br vacancy passivation is that there are broadening effects and downfield chemical shift of the H resonance signals 1, 2 and 3 of the DDT ligand. The resonances with blue highlighted numbers in Fig. 3e are distinguishing peaks of the DDT ligand related to thiol, α-methylene (1), β-methylene (2), and γ-methylene (3).40,41 Therefore, Br vacancies can be covered by introducing DDT ligands and these findings are consistent with the improved PLQY.
To gain deep insight into the relationship between exciton decay dynamics and Br vacancies, the PL lifetime was calculated by using time-correlated single photon counting (TCSPC). Fig. 4 exhibits the PL decay curves of perovskite QD thin films at various temperatures from 80 K to 300 K. The PL lifetime of both PQDs and DDT-PQDs increases as the temperature increases, but the exciton radiative decay channels are different in a certain temperature region. (1) At low temperatures (80–180 K), the average lifetimes of all samples are similar (the inset of Fig. 4a and b). Since the electron–phonon interaction is minimized,42 most excitons are emitted as radiative decay in a single channel. Tables 1 and S1† show the optimized parameters (τ1, f1, τ2, and f2) for fitting the PL decay curves. We observed that excitons are mainly released in fast decay components (τ1 and f1), which may imply a radiative decay term. (2) At high temperatures (180–300 K), the average lifetimes of all perovskite QDs increase with temperature due to free carriers generated by the exciton fission43 (Fig. 4a and b). PQDs show a slow decay component and a trend with linearly increasing average lifetimes. Due to the mechanism of non-radiative recombination, introduction of the slow component indicates the existence of non-radiative recombination. The portion of the slow component increases from 5.3% to 35.17% with increasing temperature (Table 1 and Fig. 4c). Surprisingly, this component is rarely detected after passivation using DDT ligands because the thiol group can lead to the inhibition of non-radiative recombination through elimination of Br vacancies (Fig. 4d). Therefore, a lot of PQDs, which are covered by the DDT ligands, show a mono-exponential decay curve (Fig. S5†). Moreover, a component in the decay curve is not dramatically increased from the lifetime at 80 K compared to PQDs with pristine ligands such as oleic acid. As a result, the average lifetime of DDT-PQDs (2.42 ns) is shorter than that of PQDs (3.16 ns) at 300 K. In particular, slow lifetime components (τ2, and f2) of PQDs sharply increased with rising temperatures (Table 1). However, DDT-PQDs with a PLQY of ∼99.8% predominantly have exciton radiative decay in a single fast channel regardless of the temperature.
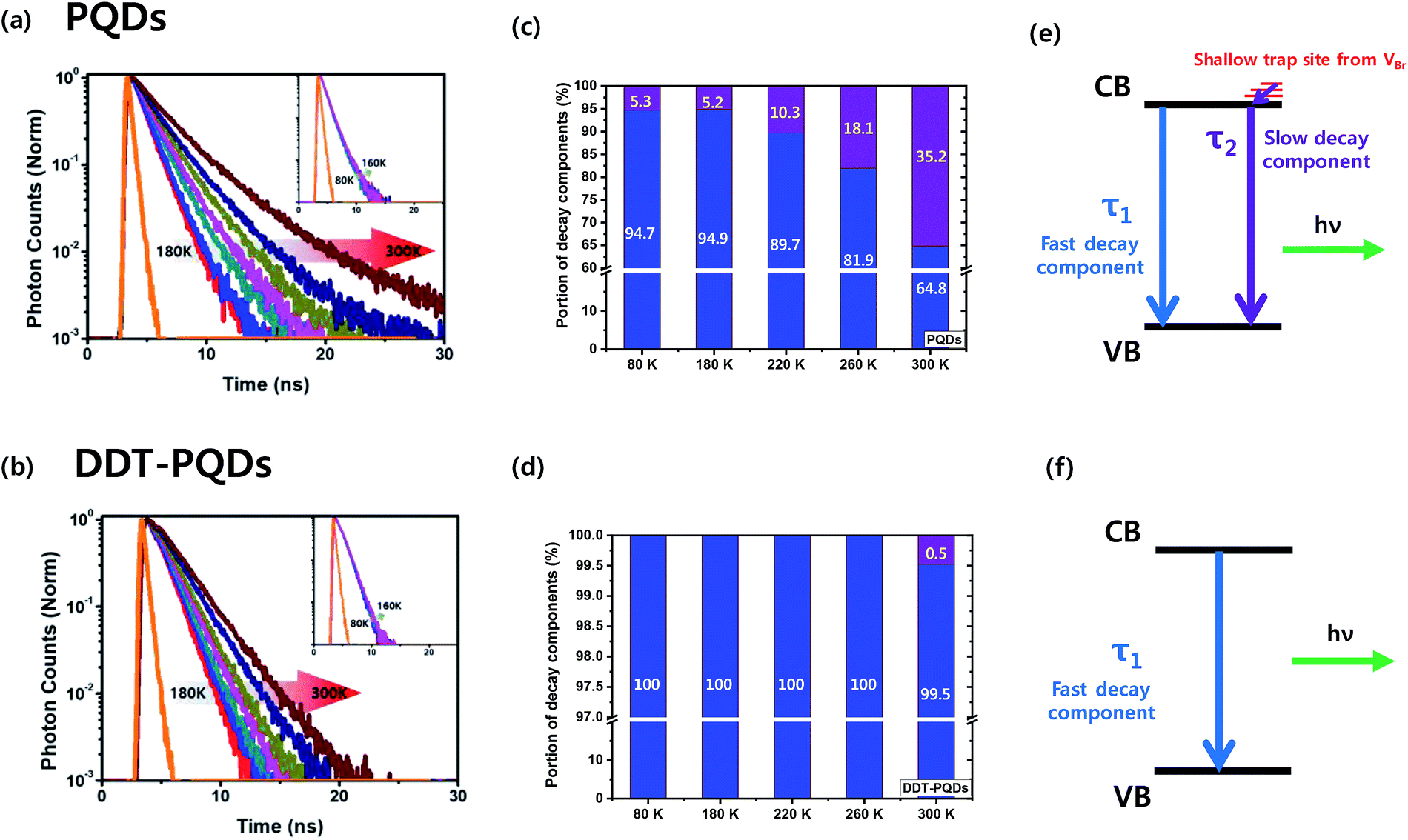 | ||
| Fig. 4 PL decay curves of (a) PQDs and (b) DDT-PQDs at various temperatures from 180 K to 300 K. The insets are obtained at a temperature below 160 K (orange: IRF curve, red: 180 K, blue: 200 K, dark cyan: 220 K, pink: 240 K, dark yellow: 260 K, royal: 280 K, and wine: 300 K). A portion of decay components of (c) PQDS and (d) DDT-PQDS at various temperatures (80–300 K, blue: τ1 and purple: τ2). The mechanism of the recombination process in (e) PQDs and (f) DDT-PQDs. CB: conduction band and VB: valence band. | ||
| Temperature [K] | τ 1 [ns] | f 1 [%] | τ 2 [ns] | f 2 [%] | τ avg [ns] | χ 2 | |
|---|---|---|---|---|---|---|---|
| PQDs | 80 | 1.04 | 94.7 | 3.17 | 5.3 | 1.15 | 1.09 |
| 180 | 1.16 | 94.85 | 3.02 | 5.15 | 1.25 | 0.93 | |
| 220 | 1.44 | 89.7 | 3.21 | 10.3 | 1.62 | 0.99 | |
| 260 | 1.83 | 81.94 | 3.65 | 18.06 | 2.16 | 1.26 | |
| 300 | 2.32 | 64.83 | 4.7 | 35.17 | 3.16 | 1.09 | |
| DDT-PQDs | 80 | 1.22 | 100 | — | — | 1.22 | 0.80 |
| 180 | 1.19 | 100 | — | — | 1.19 | 0.89 | |
| 220 | 1.40 | 100 | — | — | 1.40 | 0.83 | |
| 260 | 1.71 | 100 | — | — | 1.71 | 0.74 | |
| 300 | 2.36 | 99.52 | 15.82 | 0.48 | 2.42 | 1.15 |
The TCSPC data imply that the slow decay component comes from shallow trap sites of Br vacancies. Density functional theory (DFT) calculations from previous research suggest that the location of the shallow trap level is 0.16–0.23 eV above the conduction band edge.44,45 These results can be described with a simple recombination model as shown in Fig. 4(e) and (f). The slow component is caused by trapping and relaxation of electrons in shallow traps and it appears in all temperature ranges (80–300 K) in PQDs. But in DDT-PQDs, most of the shallow traps are passivated and then the slow component is only identified at 300 K.
The DDT-PQDs have better stability because of the mono-exponential system and have higher applicability in down-converted light emitting diodes (LEDs) than PQDs. To verify the working stability of LEDs, green PQD LEDs were fabricated by introducing perovskite QDs into a blue light emitting chip (450 nm). The blue light emitting chip was driven by 50 mA current density at 3.8 V and encapsulation was not performed. Fig. 5a and S6† show the PL/PLinitial intensity ratio for LED working time. In the case of DDT-PQDs, the PL intensity is maintained, but that of the PQDs sharply drops to 24% over the exposure time. As shown in the inset of Fig. 5a, PQDs show a significant decrease in the PL intensity after one hour compared to DDT-PQDs. This result implies that the PQDs, which have Br vacancies, show weaker LEDs working stability than the DDT-PQDs. Fig. 5b shows the CIE value of DDT-PQDs and PQDs. As shown in the PL data in Fig. 2d, because of excellent optical properties, both PQDs (0.111, 0.771) and DDT-PQDs (0.103, 0.769) have high CIE values. However, when DDT-PQDs are applied to various display devices, they are more advantageous in terms of stability and higher color purity than PQDs.
 | ||
| Fig. 5 (a) Change of the PL emissions and photographs (inset) of the PQDs (down) and DDT-PQDs (up) with respect to LED working time. (b) CIE color coordinates of LEDs. | ||
Our findings suggest that shallow traps related to Br vacancies are associated closely with the reduction of the PLQY and the introduction of the slow decay channel related to the non-radiative recombination. Additionally, the DDT ligand was proved to suppress non-radiative recombination. Further PL lifetime data are provided in the ESI.†
Conclusion
In conclusion, we explored the exciton decay dynamics of CsPbBr3 QDs. Through the DDT passivation process, the Br vacancies of PQDs were covered and the PLQY of DDT-PQDs improved from 76.1% to 99.8%. XPS and NMR spectrum analysis corroborated that DDT passivates Br vacancies resulting in the formation of a rigid PbBr6 octahedral structure. Interestingly, the PL lifetime of DDT-PQDs was found to decrease (from 3.16 ns to 2.42 ns at 300 K) as opposed to the PLQY enhancement. The TCSPC analysis revealed that DDT-PQDs mainly underwent a single radiative decay, whereas PQDs show additional slow nonradiative decay in a longer lifetime channel with rising temperature. These results indicate that the Br vacancies of PQDs affect the exciton decay channel as well as reduce the PLQY. Our findings not only provide a deeper insight into the exciton decay dynamics of PQDs but also present an informative guide to design further applications.Experimental
Materials
Cesium carbonate (99.9%, Sigma-Aldrich), lead bromide (99.999%, Sigma-Aldrich), oleylamine (70%, Sigma-Aldrich), oleic acid (90%, Sigma-Aldrich), 1-octadecene (90%, Sigma-Aldrich), ethyl acetate (90%, Alfa Aesar), n-hexane (95.0%, Samchun), and 1-dodecanethiol (≥98%, Sigma-Aldrich).Characterization
PL spectra were recorded using a Hitachi F-7000 luminescence spectrometer, and UV-vis absorbance spectra were recorded using a Hamamatsu Quantaurus-QY C11347 absolute PL quantum yield spectrometer. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-3010 transmission electron microscope. X-ray diffraction patterns (XRD) were recorded with a RIGAKU high-resolution powder X-ray diffractometer. Nuclear magnetic resonance (NMR) spectra were recorded using an Agilent 400 MHz 54 mm NMR DD2 spectrometer with toluene-d8 as the solvent. X-ray photoelectron spectroscopy (XPS) spectra were recorded using a Thermo VG Scientific K-alpha photoelectron spectrometer system. Fourier-transform infrared spectroscopy (FTIR) spectra were recorded with a JASCO FT/IR 680 plus spectrometer. Time correlated single photon counting (TCSPC) was performed using an Edinburgh FL920 fluorescence lifetime spectrometer with an excitation source of 375 nm (picosecond pulsed diode laser, EPL-375) and calculated using the F900 program with an instrument response function (IRF). The LED results were obtained on a DARSA PRO-5000.Synthesis of CsPbBr3 quantum dots
Cs oleate was fabricated using 0.204 g Cs2CO3, 0.625 mL oleic acid and 10 mL ODE in a three-neck flask. The flask was connected to a Schlenk line and the mixture was stirred vigorously under vacuum at 120 °C for 1 h, and then the temperature was raised to 150 °C in an argon atmosphere to keep Cs oleate clear until injection.0.690 g PbBr2, 5 mL oleic acid and 5 mL oleylamine were added to 50 mL ODE in a three-neck flask connected to a Schlenk line. To degas and dry, the mixture was stirred vigorously under vacuum at 120 °C for 30 min, and then the temperature was raised to 180 °C under argon flux.
Then 4 mL Cs oleate was injected into the PbBr2 ODE solution and the flask was transferred to an ice-water bath after 5 seconds of reaction. When the temperature decreased to 50 °C, the crude product was centrifuged at 7800 rpm for 5 min. The precipitates were re-dispersed in hexane and ethyl acetate was added to the QD hexane solution at a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and then centrifuged at 7800 rpm for 5 min. The precipitate was dissolved in hexane, and then centrifuged at 15000 rpm for 5 min. The supernatant was filtered into a vial. Thus, the purified CsPbBr3 quantum dots were dispersed in hexane.
:2 and then centrifuged at 7800 rpm for 5 min. The precipitate was dissolved in hexane, and then centrifuged at 15000 rpm for 5 min. The supernatant was filtered into a vial. Thus, the purified CsPbBr3 quantum dots were dispersed in hexane.
DDT passivation process
16 μL of 1-dodecanethiol was added to 2 mL of the CsPbBr3 quantum dot solution (10 mg mL−1). The mixture was stirred vigorously for 3 days. Large particles are precipitated by centrifugation at 15000 rpm for 5 min and supernatant DDT-PQDs were collected in a vial.
Conflicts of interest
There are no conflicts to declare.Notes and references
- I. Levchuk, P. Herre, M. Brandl, A. Osvet, R. Hock, W. Peukert, P. Schweizer, E. Spiecker, M. Batentschuk and C. J. Brabec, Chem. Commun., 2017, 53, 244–247 RSC.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- F. Zhang, H. Zhong, C. Chen, X.-g. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- P. Wang, X. Bai, C. Sun, X. Zhang, T. Zhang and Y. Zhang, Appl. Phys. Lett., 2016, 109, 063106 CrossRef.
- Y. Yang, Y. Zheng, W. Cao, A. Titov, J. Hyvonen, J. R. Manders, J. Xue, P. H. Holloway and L. Qian, Nat. Photonics, 2015, 9, 259 CrossRef CAS.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- H. Huang, A. S. Susha, S. V. Kershaw, T. F. Hung and A. L. Rogach, Adv. Sci., 2015, 2, 1500194 CrossRef PubMed.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- C.-Y. Huang, C. Zou, C. Mao, K. L. Corp, Y.-C. Yao, Y.-J. Lee, C. W. Schlenker, A. K. Jen and L. Y. Lin, ACS Photonics, 2017, 4, 2281–2289 CrossRef CAS.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- G. C. Xing, N. Mathews, S. S. Lim, N. Yantara, X. F. Liu, D. Sabba, M. Gratzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- L. T. Dou, Y. Yang, J. B. You, Z. R. Hong, W. H. Chang, G. Li and Y. Yang, Nat. Commun., 2014, 5, 5404 CrossRef CAS PubMed.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS PubMed.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- J. S. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- J. H. Li, L. M. Xu, T. Wang, J. Z. Song, J. W. Chen, J. Xue, Y. H. Dong, B. Cai, Q. S. Shan, B. N. Han and H. B. Zeng, Adv. Mater., 2017, 29, 1603885 CrossRef PubMed.
- J. Pan, L. N. Quan, Y. B. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. J. Yuan, L. Sinatra, N. M. Alyami, J. K. Liu, E. Yassitepe, Z. Y. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS PubMed.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS PubMed.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- J. Kang and L.-W. Wang, J. Phys. Chem. Lett., 2017, 8, 489–493 CrossRef CAS PubMed.
- W. Deng, H. Fang, X. Jin, X. Zhang, X. Zhang and J. Jie, J. Mater. Chem. C, 2018, 6, 4831–4841 RSC.
- A. J. Houtepen, Z. Hens, J. S. Owen and I. Infante, Chem. Mater., 2017, 29, 752–761 CrossRef CAS.
- W. Choi, D. Kim, H. Cho, M. Kim, J. Choi and D. Y. Jeon, Nanoscale, 2019 10.1039/c9nr01044k.
- Y. Gao and X. Peng, J. Am. Chem. Soc., 2015, 137, 4230–4235 CrossRef CAS PubMed.
- L. Ruan, W. Shen, A. Wang, Q. Zhou, H. Zhang and Z. Deng, Nanoscale, 2017, 9, 7252–7259 RSC.
- M. I. Bodnarchuk, S. C. Boehme, S. Ten Brinck, C. Bernasconi, Y. Shynkarenko, F. Krieg, R. Widmer, B. Aeschlimann, D. Günther and M. V. Kovalenko, ACS Energy Lett., 2018, 4, 63–74 CrossRef PubMed.
- S. W. Eaton, M. Lai, N. A. Gibson, A. B. Wong, L. Dou, J. Ma, L.-W. Wang, S. R. Leone and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1993–1998 CrossRef CAS PubMed.
- P. Cottingham and R. L. Brutchey, Chem. Commun., 2016, 52, 5246–5249 RSC.
- A. Swarnkar, R. Chulliyil, V. K. Ravi, M. Irfanullah, A. Chowdhury and A. Nag, Angew. Chem., Int. Ed., 2015, 54, 15424–15428 CrossRef CAS PubMed.
- Y.-H. Kim, C. Wolf, Y.-T. Kim, H. Cho, W. Kwon, S. Do, A. Sadhanala, C. G. Park, S.-W. Rhee and S. H. Im, ACS Nano, 2017, 11, 6586–6593 CrossRef CAS PubMed.
- M. Li, X. Zhang, K. Matras-Postolek, H. S. Chen and P. Yang, J. Mater. Chem. C, 2018, 6, 5506–5513 RSC.
- J. Y. Woo, Y. Kim, J. Bae, T. G. Kim, J. W. Kim, D. C. Lee and S. Jeong, Chem. Mater., 2017, 29, 7088–7092 CrossRef CAS.
- L. R. Pederson, J. Electron Spectrosc. Relat. Phenom., 1982, 28, 203–209 CrossRef CAS.
- M. Liu, G. H. Zhong, Y. M. Yin, J. S. Miao, K. Li, C. Q. Wang, X. R. Xu, C. Shen and H. Meng, Adv. Sci., 2017, 4, 1700335 CrossRef PubMed.
- J. De Roo, M. Ibanez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Koyalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- C. C. Reinhart and E. Johansson, Chem. Mater., 2015, 27, 7313–7320 CrossRef CAS.
- X. Zhou, J. M. El Khoury, L. Qu, L. Dai and Q. Li, J. Colloid Interface Sci., 2007, 308, 381–384 CrossRef CAS PubMed.
- B. A. Korgel, S. Fullam, S. Connolly and D. Fitzmaurice, J. Phys. Chem. B, 1998, 102, 8379–8388 CrossRef CAS.
- M. Gromova, A. Lefrançois, L. Vaure, F. Agnese, D. Aldakov, A. Maurice, D. Djurado, C. Lebrun, A. de Geyer, T. U. Schülli, S. Pouget and P. Reiss, J. Am. Chem. Soc., 2017, 139, 15748–15759 CrossRef CAS PubMed.
- M. Hasan, D. Bethell and M. Brust, J. Am. Chem. Soc., 2002, 124, 1132–1133 CrossRef CAS PubMed.
- K. Wei, Z. J. Xu, R. Z. Chen, X. Zheng, X. G. Cheng and T. Jiang, Opt. Lett., 2016, 41, 3821–3824 CrossRef CAS PubMed.
- B. T. Diroll, H. Zhou and R. D. Schaller, Adv. Funct. Mater., 2018, 28, 1800945 CrossRef.
- M. Sebastian, J. A. Peters, C. C. Stoumpos, J. Im, S. S. Kostina, Z. Liu, M. G. Kanatzidis, A. J. Freeman and B. W. Wessels, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 235210 CrossRef.
- H. Shi and M.-H. Du, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 174103 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Further information on the size of QDs, XPS, NMR and lifetime. See DOI: 10.1039/c9na00292h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |