
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multifunctional biodegradable brush polymer-drug conjugate for paclitaxel/gemcitabine co-delivery and tumor imaging†
Haotian
Sun
 a,
Lingyue
Yan
b,
Michael Yu Zarng
Chang
b,
Kevin A.
Carter
b,
Runsheng
Zhang
a,
Leigh
Slyker
a,
Jonathan F.
Lovell
b,
Yun
Wu
*b and
Chong
Cheng
*a
a,
Lingyue
Yan
b,
Michael Yu Zarng
Chang
b,
Kevin A.
Carter
b,
Runsheng
Zhang
a,
Leigh
Slyker
a,
Jonathan F.
Lovell
b,
Yun
Wu
*b and
Chong
Cheng
*a
aDepartment of Chemical and Biological Engineering, University at Buffalo, The State University of New York, Buffalo, New York 14260, USA. E-mail: ccheng8@buffalo.edu
bDepartment of Biomedical Engineering, University at Buffalo, The State University of New York, Buffalo, New York 14260, USA. E-mail: ywu32@buffalo.edu
First published on 27th May 2019
Abstract
A multifunctional biodegradable brush polymer-drug conjugate (BPDC) is developed for the co-delivery of hydrophobic paclitaxel (PTX) and hydrophilic gemcitabine (GEM) chemotherapeutics, as well as a tumor imaging agent. A novel ternary copolymer of conventional, acetylenyl-functionalized and allyl-functionalized lactides is prepared to serve as the backbone precursor of the BPDC. Acetylenyl groups of the copolymer are then reacted with azide-functionalized poly(ethylene glycol) (PEG) and cyanine5.5, a fluorescent probe, via azide–alkyne click reactions. Subsequently, the allyl groups of the yielded PEG-grafted brush polymer are used to covalently link PTX and GEM onto the backbone via thiol–ene click reactions. The resulting BPDC exhibits an average hydrodynamic diameter of 111 nm. Sustained and simultaneous release of PTX and GEM from the BPDC is observed in phosphate buffered saline, with the release of PTX showing sensitivity under mildly acidic conditions. In vitro studies using MIA PaCa-2 human pancreatic cancer cells illustrate the cellular uptake and cytotoxicity of the BPDC. In vivo, the BPDC undergoes long blood circulation and tumor accumulation, and enables optical tumor imaging. Further development and testing are warranted for multifunctional conjugated brush polymer systems that integrate combination chemotherapy and imaging.
1. Introduction
As one of the most commonly used combination therapies in clinical practice, combination chemotherapy using multiple anti-cancer drugs has attracted significant interest over the past decade.1–4 Synergistic drug combinations can reduce undesirable toxicity, suppress drug resistance, overcome side effects, and improve therapeutically relevant selectivity and the therapeutic index.5–11 In 2013, the U. S. Food and Drug Administration (FDA) approved the combination of paclitaxel (PTX) albumin-stabilized nanoparticle formulation with gemcitabine (GEM) for metastatic pancreatic cancer treatment. PTX and GEM have different mechanisms of action and there is no overlapping toxicity; therefore such a combination is appealing.12 The FDA recommended that PTX albumin-stabilized nanoparticle formulation is administered first on days 1, 8, and 15 of each 28 day cycle, immediately followed by separate administration of GEM on each day.13 In 2017, Vyxeos, as the first combination nanomedicine for cancer treatment (by using liposomes to encapsulate two drugs), was approved by the FDA, demonstrating the significant clinic applications of combination nanomedicines.14 Recent studies have also revealed the promising application potential of polymer-drug conjugates (PDCs) as combination nanomedicines.15–22Compared with separate administration of PTX and GEM, integrated single nanomedicine formulations for co-delivery of PTX and GEM can simplify the cancer treatment process.23 However, there is a vast difference in hydrophobicity between PTX and GEM. PTX is hydrophobic, with minimal solubility in aqueous solutions, while GEM is much more hydrophilic. Because similar water solubility of drugs is preferred in typical approaches for the fabrication of nanoparticle-based drug co-delivery systems,24 practically it is difficult to load drugs of very different hydrophilicity into the same nanoparticles.25–27 Besides the specific concern of drug loading, general issues on the size control, drug release behavior and side effects of scaffolds should also be addressed in the development of PTX/GEM-loaded co-delivery nanoparticles. To capitalize passive targeting via enhanced permeability and retention (EPR) effects, a hydrodynamic size range of approximately 10–100 nm is considered optimal.28 Relative to drug-encapsulated systems typically involving burst release of drugs, drug-conjugated systems allow for sustained drug release and may exhibit reduced systemic toxicity of drugs.28,29 Moreover, the co-delivery scaffolds need to be biocompatible and there should also be effective pathways to eliminate these scaffolds, in order to minimize their side effects.30
Several nanoparticle-based systems developed for the co-delivery of PTX and GEM have been reported.25,31,32 Zhang and co-workers developed an innovative approach to load a small molecule PTX–GEM conjugate, instead of two individual drugs, into lipid-coated biodegradable poly(lactic-co-glycolic acid) nanoparticles with an average hydrodynamic size of ∼70 nm.25 Although the reported loading amounts of the conjugate were relatively limited, this work demonstrated precise control of the molar ratio of the conjugated drugs, acid-sensitive sustained drug release, and remarkable in vitro therapeutic efficacy of conjugate-loaded nanoparticles. The co-delivery of PTX and GEM via PDCs has also been explored. Haam and co-workers reported multi-PDC nanocarriers consisting of two PDCs, i.e. poly(L-lysine)-carboxylate PTX and hyaluronic acid-conjugated GEM.31 PTX/GEM-containing nanoparticles (>200 nm) were formed from the two PDCs via electrostatic attractions, and showed a remarkable cellular uptake in target cells and synergistic cancer therapeutic efficacy. As demonstrated by Kopeček and co-workers,32 employing a PDC that integrates both PTX and GEM with the same base polymer is an appealing strategy for the co-delivery of the two drugs. Linear poly(N-(2-hydroxypropyl)methacrylamide) (PHPEA) was utilized as the base polymer in their investigation. While the specific PHPMA–PTX/GEM conjugate formulation showed moderate antagonism, further optimization of the structure and physicochemical properties of such a PDC with two types of conjugated drugs may be required to achieve highly promising therapeutic results.
Multifunctional systems integrating anticancer therapeutic delivery and cancer imaging hold great promise for the diagnosis and treatment of cancer.33–40 Because there are a variety of imaging technologies and a broad range of anticancer therapeutics in clinical use, in principle, there is a substantial research space for the development of such integrated systems. The integration of co-delivered anticancer drugs with cancer imaging agents is an approach that could be of interest.41–45 However, integrated systems demonstrating PTX/GEM co-delivery with cancer imaging agents have not yet been reported.
Herein, a brush polymer-drug conjugate (BPDC or densely grafted PDCs) with both hydrophobic PTX and hydrophilic GEM, together with a fluorophore, conjugated to the backbone of a brush-like polymer scaffold is reported for the co-delivery of two anticancer drugs and for cancer imaging. As a new class of PDCs, BPDCs not only possess the general merits of PDCs (such as explicit molecular structures and precise drug loading amounts), but also provide well-controlled nanoscopic dimensions and offer a protective chemical environment for the conjugated drugs.46–48 In recent years, with thoughtful consideration of concerns of long-term biocompatibility and side effects of polymer-based therapeutic systems, we have made significant research efforts to develop BPDCs with biodegradable architectures.49–51 Our previous studies have demonstrated the therapeutic efficacy of BPDCs in the delivery of individual drugs (i.e. PTX50 and doxorubicin51), whereas the objective of the current work is to further illustrate the feasibility of co-delivery of multiple drugs and cancer imaging via biodegradable BPDCs. PTX and GEM, with a mass feed ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 for conjugation reactions, were selected as anticancer drugs for combinational therapeutic effects in this work. This drug ratio was chosen because it is within the range of PTX/GEM mass ratios (from 1:1 to 1:25) studied in the literature on co-delivery of these two anticancer drugs.23,31,32,52–57
:2.5 for conjugation reactions, were selected as anticancer drugs for combinational therapeutic effects in this work. This drug ratio was chosen because it is within the range of PTX/GEM mass ratios (from 1:1 to 1:25) studied in the literature on co-delivery of these two anticancer drugs.23,31,32,52–57
The research design of this work is different from the original PTX–GEM combination polymer conjugate produced by Kopěcek and co-workers in both the base polymer and the conjugation linkages. In their work, PHPEA was used as the base polymer of the PDC, and enzyme-sensitive Gly–Phe–Leu–Gly (GFLG) conjugation linkers were employed. In this work, a polylactide-graft-poly(ethylene glycol) (PLA-g-PEG) brush polymer (BP) was designed as the base polymer, allowing the resulting BPDC to easily reach nanoscopic dimensions for capitalizing passive targeting via enhanced permeability and retention (EPR) effects.28 The PLA backbone was selected because of its biodegradability.58,59 PEG chains were chosen as the grafts of the scaffold because PEG is a commonly used solubilizing moiety for drug carriers.60 PEG can reduce cytotoxicity and increase the biocompatibility and blood circulation time of drugs.61 Because there are numerous PEG chains per BPDC molecule, PEG chains with a molecular weight (MW) of ∼900, instead of higher-MW ones, were selected in this work because they can lead to an appropriate hydrodynamic size and high wt% of drugs for the BPDC. Hydrolysable ester-thioether (–OCOCH2CH2S–) and amide-thioether (–NHCOCH2CH2S–) conjugation linkages were selected to link polymeric scaffolds with PTX and GEM, respectively. These conjugation linkages may facilitate drug release without enzyme assistance. The ester-thioether linkage for PTX conjugation was utilized previously in PDCs,62,63 with a chemical environment considerably different than that of BPDCs. The use of the amide-thioester linkage for GEM conjugation has not been reported.
Other specific design considerations of this work include the following aspects. While PTX and GEM were selected as anticancer drugs, a fluorescent probe, cyanine5.5 (Cy5.5), was chosen to provide near infrared imaging capabilities.64 Azide–alkyne and thiol–ene reactions were employed to conjugate PEG chains, Cy5.5, PTX and GEM in the synthetic design of the BPDC because of the chemoselectivity and orthogonality of these efficient click reactions.65–67 Besides drug release studies, both in vitro cytotoxicity and cellular uptake studies and in vivo imaging and biodistribution studies using a MIA PaCa-2 pancreatic cancer model were performed for the assessment of the therapeutic properties and tumor imaging function of the BPDC.57
2. Results and discussion
2.1. Synthesis
The synthesis of the BPDC carrying both PTX and GEM with a PLA-g-PEG BP scaffold is illustrated in Scheme 1. Acetylenyl/allyl-functionalized PLA 1 was prepared by ring-opening polymerization (ROP) of L-lactide (LA), acetylenyl-functionalized LA (ACLA), and allyl-functionalized LA (ALLA), using benzyl alcohol (BnOH) as the initiator and 4-dimethylaminopyridine (DMAP) as the organocatalyst in dichloromethane ([LA]0:[ALLA]0:[ACLA]0:[BnOH]0:[DMAP]0 = 40:30:30:1:4 at 35 °C for 7 days). According to gel permeation chromatography (GPC) analysis (Fig. 1a), it had a number average molecular weight (Mn) of 30.9 kDa and a narrow molecular weight dispersity (Đ) of 1.21, relative to linear polystyrenes. 1H NMR spectra of 1 were obtained in CDCl3 and DMSO-d6 (Fig. S1a and b†). Both spectra showed an area ratio of 1.00:9.55 for the resonance peak of terminal Ar–H at ∼7.3–7.4 ppm to that of –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) protons from the allyl group of ALLA at ∼5.8 ppm, indicating the presence of on average 49.8 ALLA units per molecule of 1. In Fig. S1a,† an area ratio of 9.55:8.93 for the resonance peak of –CH protons of ALLA to that of CH
protons from the allyl group of ALLA at ∼5.8 ppm, indicating the presence of on average 49.8 ALLA units per molecule of 1. In Fig. S1a,† an area ratio of 9.55:8.93 for the resonance peak of –CH protons of ALLA to that of CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) protons from the acetylenyl group of ACLA at 2.00–2.13 ppm further revealed the presence of on average 44.6 ACLA units per molecule of 1. Based on the area ratio of 9.55:84.2 for the resonance peak of –CH protons of ALLA and the overlapped resonance peak of CH2 protons of ALLA and –CH< protons of all monomer units, on average the presence of 68.4 LA units per molecule of 1 could be deduced. Therefore, the structural formula of 1 was poly(LA0.42-co-ALLA0.30-co-ACLA0.28)163, corresponding to a MNMRn of 26.0 kDa.
protons from the acetylenyl group of ACLA at 2.00–2.13 ppm further revealed the presence of on average 44.6 ACLA units per molecule of 1. Based on the area ratio of 9.55:84.2 for the resonance peak of –CH protons of ALLA and the overlapped resonance peak of CH2 protons of ALLA and –CH< protons of all monomer units, on average the presence of 68.4 LA units per molecule of 1 could be deduced. Therefore, the structural formula of 1 was poly(LA0.42-co-ALLA0.30-co-ACLA0.28)163, corresponding to a MNMRn of 26.0 kDa.
 | ||
| Scheme 1 Synthesis of the Cy5.5-labelled BPDC conjugated with PTX and GEM. | ||
 | ||
| Fig. 1 GPC curves obtained from the (a) RI detector and (b) UV/vis detector (685 nm). | ||
Subsequently, the azide–alkyne reaction of methoxyl PEG azide (CH3O-PEG-N3; MW = 897 Dalton, Fig. S2†) and Cy5.5 azide (Cy5.5-N3) with acetylenyl/allyl-functionalized PLA 1 was performed, using CuBr as the catalyst and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) as the ligand in DMF ([acetylenyl group of 1]0:[CH3O-PEG-N3]0:[Cy5.5-N3]0:[CuBr]0:[PMDETA]0 = 1:1.07:0.025:1:1; at room temperature for 45 h). As determined by GPC analysis using an RI detector, the resulting Cy5.5-labelled PLA-g-PEG, i.e. BP 2, had a Mn of 41.4 kDa and a Đ of 1.41, relative to linear polystyrenes (Fig. 1). When a UV-vis detector (685 nm) was used, the presence of the GPC curve of 2 indicated the successful conjugation of Cy5.5 moieties, and on average about one Cy5.5 moiety per molecule of 2 was estimated from the quantitative analysis of the UV-vis absorbance of 2. The 1H NMR spectrum of BP 2 was recorded using CDCl3 as the solvent (Fig. 2a). Along with the quantitative presence of resonances from protons of PEG grafts, the disappearance of resonances of characteristic protons of ACLA units (i.e., CHCH2CCH protons at ∼2.9 ppm and CHCH2CCH protons at ∼2.1 ppm) indicated the consumption of all acetylenyl groups on copolymer 1 and on average the presence of 43.6 PEG grafts per molecule of 2. Thus, BP 2 had a MNMRn of 65.7 kDa, with 59.5 wt% PEG. The quantitative presence of –CH protons from allyl groups at 5.68–5.89 ppm illustrated that the allyl groups of 1 were intact under the azide–alkyne reaction conditions.
 | ||
| Fig. 2
1H NMR spectra of (a) BP 2 in CDCl3; (b) BPDC 3 in CDCl3/CD3OD (v/v, 1:1). | ||
Finally, a UV-induced thiol–ene reaction of thiol-functionalized PTX (PTX-SH) and GEM (GEM-SH) with BP 2 was performed in a mixed solvent of CHCl3 and MeOH (v/v, 5:1), with 2,2-dimethoxy-2-phenylacetophenone (DMPA) as the photoinitiator. The thiol-functionalized drugs, PTX-SH and GEM-SH, were prepared according to the literature,62,68 and their 1H NMR spectra are shown in Fig. S3 and S4.† The feed ratio of reagents of the thiol–ene reaction was [allyl group of 2]0:[PTX-SH]0:[GEM-SH]0:[DMPA]0 = 1:0.12:0.91:0.50, with a mass ratio of PTX to GEM of 1:2.5.
The reaction system was irradiated under UV light (λmax = 365 nm) for 90 min, to yield the PTX/GEM-dully conjugated BPDC 3. GPC analysis showed that 3 had a Mn of 51.5 kDa, with a Đ of 1.35, relative to linear polystyrenes. 1H NMR analysis of 3 in CDCl3/CD3OD (v/v, 1:1) revealed its composition (Fig. 2b). According to the resonance intensities of –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) protons from the remaining allyl groups at ∼5.8 ppm, two aromatic protons from PTX at ∼8.1 ppm, >NCH protons from GEM at ∼8.3 ppm and 55% of allyl groups of BP 2 were consumed and the ratio of the number of conjugated units of PTX to GEM was 1.00:6.34 in the resulting BPDC 3. Thus, on average there were 3.8 PTX units and 23.8 GEM units per molecule of BPDC 3, with a mass ratio of PTX to GEM of 1:1.95. According to such 1H NMR results, BPDC 3 had a MNMRn of 77.6 kDa, with 4.1 wt% PTX and 8.1 wt% GEM.
protons from the remaining allyl groups at ∼5.8 ppm, two aromatic protons from PTX at ∼8.1 ppm, >NCH protons from GEM at ∼8.3 ppm and 55% of allyl groups of BP 2 were consumed and the ratio of the number of conjugated units of PTX to GEM was 1.00:6.34 in the resulting BPDC 3. Thus, on average there were 3.8 PTX units and 23.8 GEM units per molecule of BPDC 3, with a mass ratio of PTX to GEM of 1:1.95. According to such 1H NMR results, BPDC 3 had a MNMRn of 77.6 kDa, with 4.1 wt% PTX and 8.1 wt% GEM.
2.2. Characterization of nanostructures
BP 2 and BPDC 3 had amphiphilic structures, allowing them to form self-assembled nanoparticles (NPs) in aqueous solutions via self-assembly. These NPs of 2 and 3 were characterized by nanoparticle tracking analysis (NTA) and transmission electron microscopy (TEM). NTA showed that the NPs of 2 had an average hydrodynamic diameter (Dh) of 138 nm, with a peak Dh of 123 nm (Fig. S5a†). These NPs exhibited a number-average diameter of 45.1 ± 5.4 nm on the TEM grid (Fig. S5b and c†). TEM imaging revealed their core–shell micellar structures, with PLA-rich cores and PEG-rich shell domains indicated by RuO4 staining. As demonstrated by NTA assessment, the NPs of 3 had an average Dh of 111 nm, with a peak Dh of 70 nm (Fig. 3a). TEM images of the NPs of 3 showed a number-average diameter of 34.1 ± 3.9 nm with no core–shell structures (Fig. 3b–d). The fact that BPDC 3 with higher Mn formed smaller NPs as compared to BP 2 may be ascribed, at least partly, to the much more densely grafted and stiffened backbone of 3 that can limit the aggregation of 3 in aqueous solutions. It should be noted that drug loading may affect the aggregation and final size of 3, although the effect of drug loading was not studied in this work.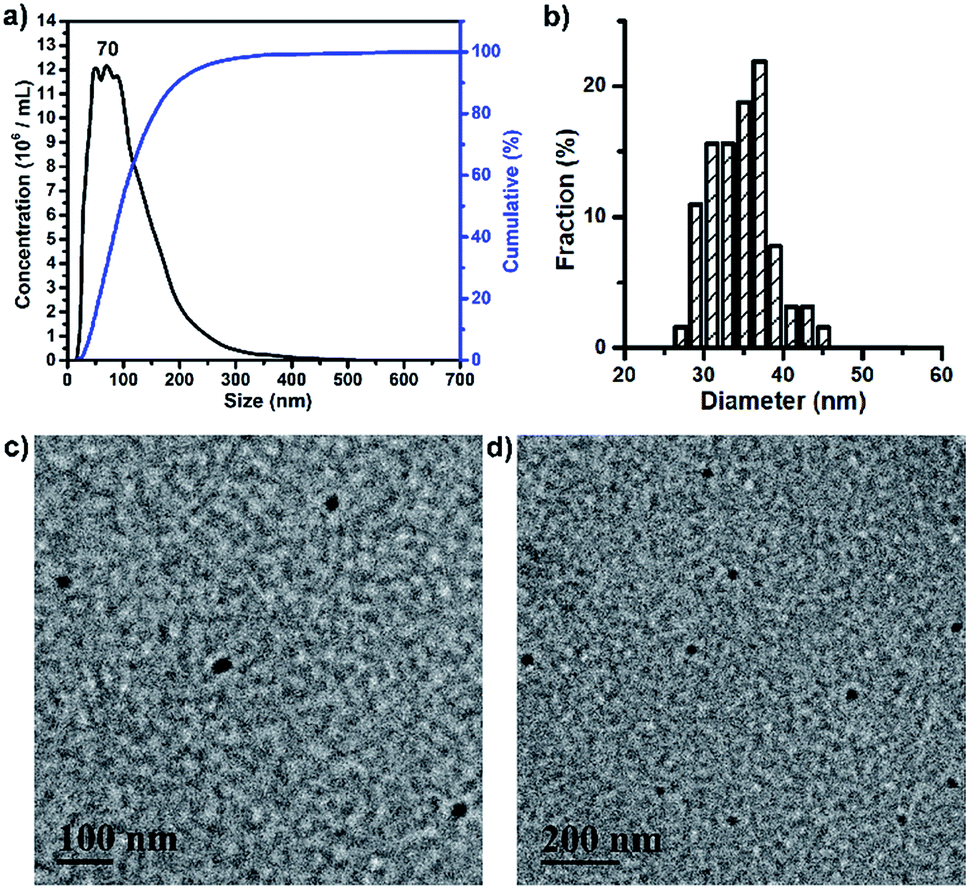 | ||
| Fig. 3 (a) Size distribution profile of BPDC 3 in H2O based on NTA measurements. (b) Histogram of the diameter distribution of BPDC 3 according to the TEM images. (c) and (d) TEM images of BPDC 3. TEM samples were stained with RuO4. | ||
2.3. Drug release
PTX and GEM were conjugated on BPDC 3via hydrolysable ester-thioether and amide-thioether linkages, respectively. Therefore, it is expected that aqueous solutions of BPDC 3 may enable sustained release of PTX and GEM. Accordingly, the drug release profiles of 3 were measured by analyzing the PBS buffer solutions of 3 at different pH (5.5 and 7.4) incubated at 37 °C, with high-performance liquid chromatography (HPLC) at different time intervals. Although PTX had very low solubility in aqueous solutions (<2.0 μg mL−1 in 0.01 M PBS buffer),62 HPLC analysis showed that PTX was fully dissolved in a mixed solvent of H2O and acetonitrile (ACN; v/v, 1:4) at a concentration of 5.1 μg mL−1, which was the highest concentration of PTX involved in this study. Therefore, all samples were diluted five times with ACN before measurements. The release profiles of PTX and GEM from BPDC 3 are shown in Fig. 4. In agreement with prior reports,62,63 the ester-thioether linkage between PTX and the PLA-based backbone of 3 was acid-labile. This result also suggests that the conjugation linkages should be quite exposed to aqueous media to facilitate acid-sensitive PTX release. On the other hand, the amide-thioester linkage between GEM and the PLA-based backbone of 3 did not exhibit acid-lability, presumably because amide hydrolysis requires higher activation energy at acidic pH due to the protonated amine leaving group.69 It should be noted that 90% PTX and 25% GEM were released from BPDC 3 within 120 h at pH 5.5, a similar pH to endolysosomes. Such results suggest that, after 3 is taken up by cancer cells via endocytosis, faster release of PTX relative to GEM might be achieved. This potentially can be favorable for cancer treatment because PTX is recommended to be administered before a separate administration of GEM in the FDA approved combination formulation.13
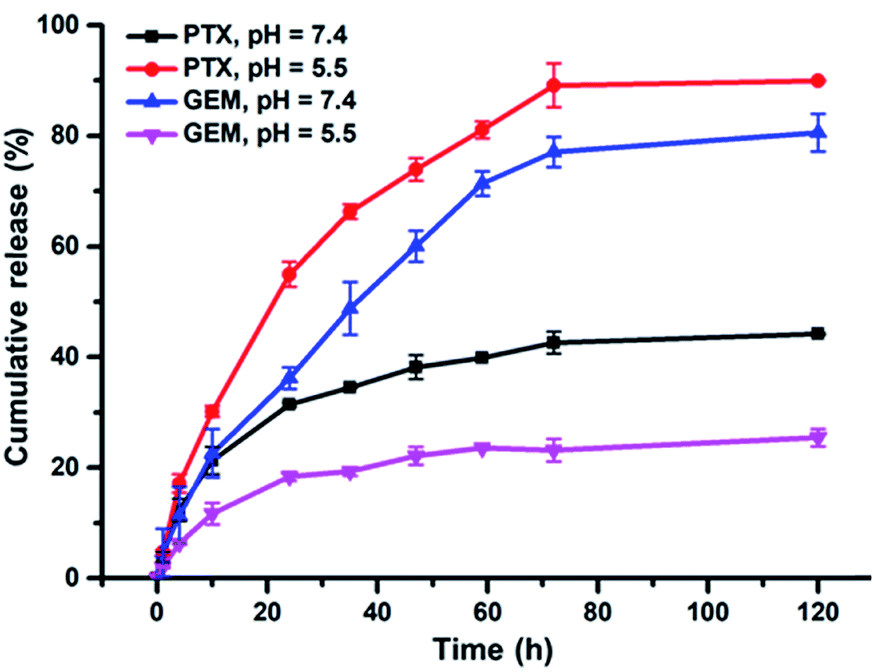 | ||
| Fig. 4 Drug release profiles of PTX and GEM from PBS solutions of BPDC 3 at pH of 5.5 and 7.4. | ||
2.4. In vitro cytotoxicity
In vitro cytotoxicity was examined to evaluate the therapeutic effects of BPDC 3. Human pancreatic MIA Paca-2 cells were treated with BPDC 3, a mixture of free PTX and GEM (PTX + GEM) or BP 2 (as the polymeric scaffold of 3). The same PTX/GEM concentrations ranging from 0.01/0.02 μg mL−1 to 5/10 μg mL−1 were used for BPDC 3 and PTX + GEM, and the same polymer concentrations were used for BPDC 3 and BP 2. At 72 h post treatment, the cell viability was measured using the alamarBlue assay, with vehicle treated cells as the control. As shown in Fig. 5, BP 2 did not exhibit noticeable cytotoxicity under the experimental conditions, demonstrating that the scaffold is highly biocompatible and thus it is a promising and potent carrier for various therapeutic and diagnostic agents. Although PTX and GEM had not been fully released at 72 h, BPDC 3 showed an overall better anti-cancer efficacy than PTX + GEM. When the PTX/GEM concentration was higher than 0.05/0.1 μg mL−1, BPDC 3 exhibited higher anti-cancer efficacy compared to PTX + GEM. At the highest PTX/GEM concentration of 5/10 μg mL−1 used in this study, BPDC 3 (40% cell viability) resulted in remarkably lower cell viability of MIA PaCa-2 cells than PTX + GEM (51% cell viability), demonstrating the higher therapeutic efficacy of BPDC 3 than the drug mixture.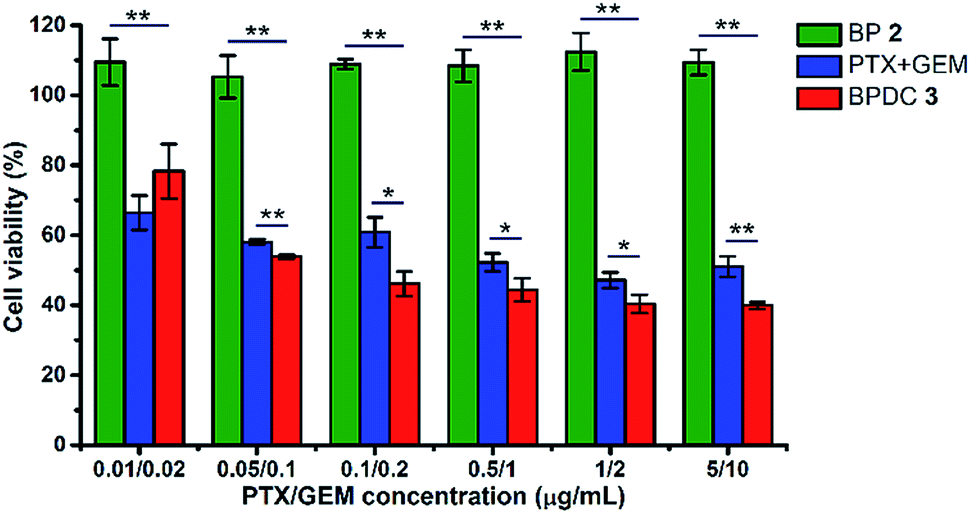 | ||
| Fig. 5 Cytotoxicity of BP 2, PTX + GEM, and BPDC 3 in MIA PaCa-2 cells. The cell viability was measured at 72 h post treatment. BP 2 had the same polymer concentrations as BPDC 3. Data were reported as the mean ± standard deviation (SD) of three independent experiments. One-way ANOVA was used to assess the statistical significance of the data. *p < 0.05 was considered as statistically significant. **p < 0.01 was considered as statistically highly significant. | ||
2.5. In vitro cellular uptake
Flow cytometry and confocal microscopy were used to assess the cellular uptake of BPDC 3. The conjugated Cy5.5 dye was used as the fluorescent probe to visualize the cellular uptake. MIA PaCa-2 cancer cells were treated with BPDC 3 at a Cy5.5 concentration of 0.2 μg mL−1. At 4 and 24 h post treatment, the cells were harvested and the uptake efficiency of BPDC 3 was quantitatively analyzed by flow cytometry. As shown in Fig. 6a and b, the cellular uptake started as early as 4 h post treatment and increased further during the following 20 h. From 4 to 24 h post treatment, the mean fluorescence intensity of the cells treated with BPDC 3 increased by 3.7 times, demonstrating the continuous cellular uptake. Confocal microscopy was used to confirm that BPDC 3 was accumulated inside the MIA PaCa-2 cells, instead of attaching on the cell surface (Fig. 6c). Images from confocal microscopy showed that BPDC 3 was taken up by the cells and remained in the cytoplasm of the cells. The fluorescence intensity of Cy5.5 in the cells increased with time. At 24 h post treatment, strong fluorescence from Cy5.5 was observed in all cells, indicating ∼100% cellular uptake efficiency of BPDC 3, which agreed with the results from flow cytometry analysis.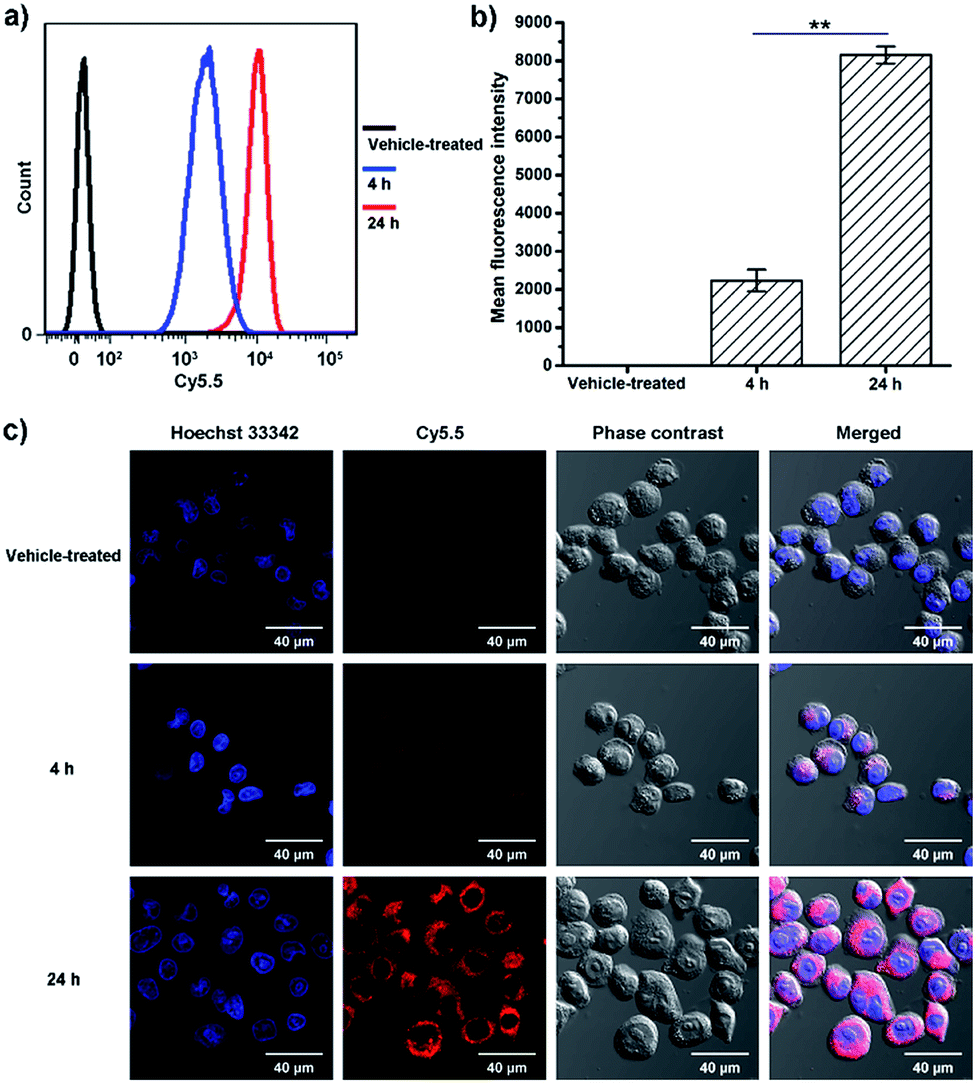 | ||
| Fig. 6 (a) Flow cytometry data showing the cellular uptake of BPDC 3 in MIA PaCa-2 cells at 4 and 24 h post treatment compared with vehicle treated cells. (b) Mean fluorescence intensity of Cy5.5 obtained from flow cytometry at 4 and 24 h post treatment. (c) Confocal microscopy images of MIA PaCa-2 cells at 4 and 24 h post treatment with BPDC 3, compared with vehicle treated cells. Cell nuclei were counterstained with Hoechst 33342. Data were reported as the mean ± standard deviation (SD) of three independent experiments. One-way ANOVA was used to assess the statistical significance of the data. **p < 0.01 was considered as statistically highly significant. | ||
We also investigated the cellular uptake efficiency of BP 2 to demonstrate its use as a versatile delivery platform. The absorbance spectra of BP 2 showed similar fluorescence intensity to BPDC 3 (Fig. S6†). The cellular uptake of BP 2 was also comparable to that of BPDC 3 and showed high delivery efficiency (Fig. S7†). Together with the high biocompatibility of BP 2, such cellular uptake results further supported the broad application potential of BP 2 as a delivery scaffold.
2.6. In vivo imaging and biodistribution
Vehicle control, Cy5.5-N3, and BPDC 3 solutions were intravenously injected into female athymic nude mice bearing subcutaneous MIA PaCa-2 pancreatic xenografts (tumor volume ∼ 100 mm3) at a Cy5.5 dose of 1.5 mg kg−1 body weight. At different time intervals post injection, in vivo fluorescence imaging of the mice was carried out (dorsal side and ventral side). As shown in Fig. 7, BPDC 3 was distributed across the mouse whole body immediately after injection and remained in circulation during the following 4 h. At 24 h post injection, weak fluorescence signals were still observed on the dorsal side of the mice. These results indicated that BPDC 3 had prolonged, up to 24 h, circulation time in vivo. Strong fluorescence signals were observed at tumor sites (dorsal side), showing the accumulation of BPDC 3 in the tumor tissue. On the other hand, Cy5.5-N3 quickly went to the liver and kidneys after the injection, and little was accumulated in tumors.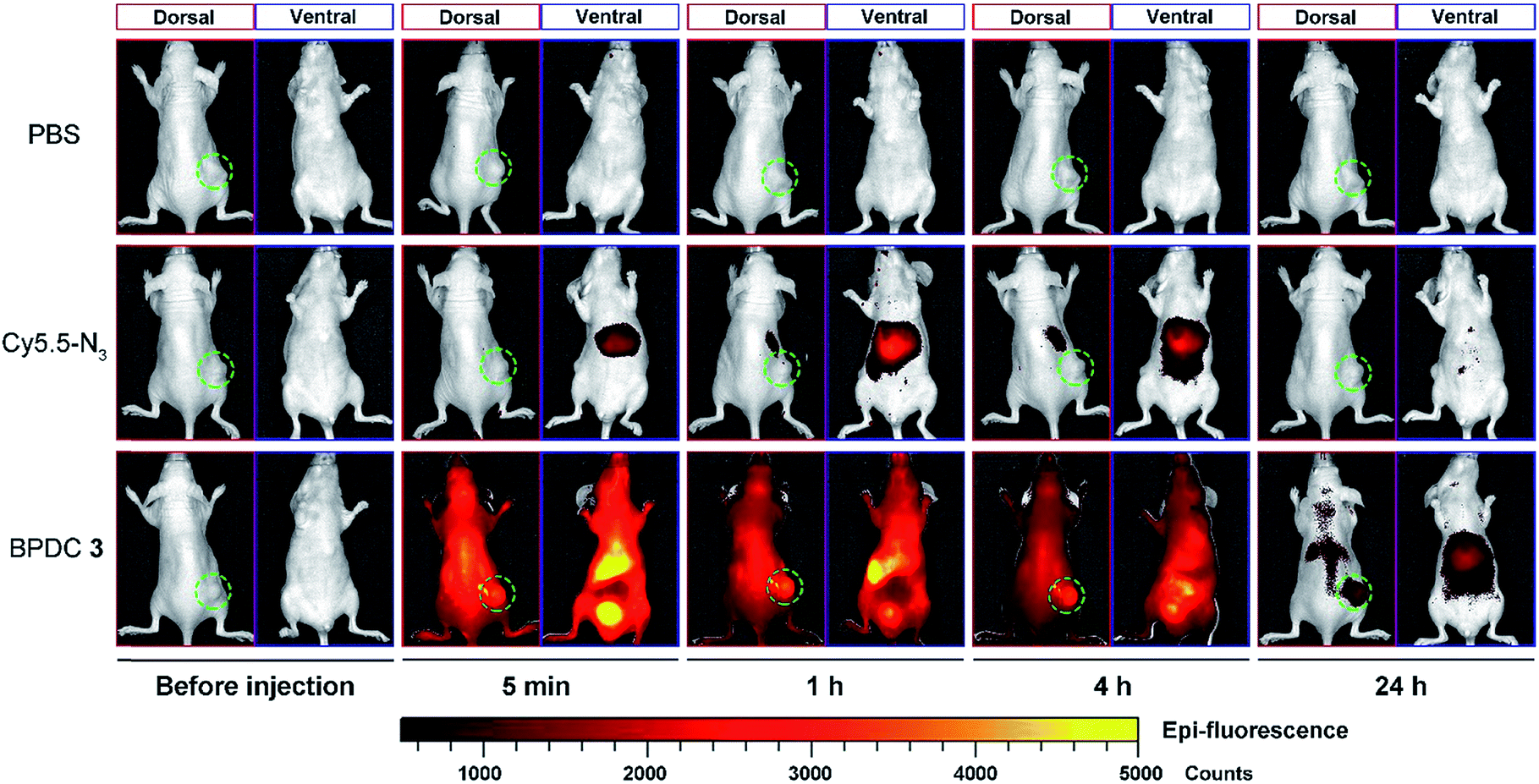 | ||
| Fig. 7 Representative whole body fluorescence imaging of mice (dorsal side and ventral side) before and at different time points post intravenous injection of the vehicle control, Cy5.5-N3, and BPDC 3 at a Cy5.5 dose of 1.5 mg kg−1 body weight. Tumors are circled in each image (dorsal side). | ||
Ex vivo evaluation of excised tumors and major organs at 24 h post injection is shown in Fig. 8. BPDC 3 treated mice showed significant fluorescence signals in tumors, while no accumulation of free Cy5.5-N3 was observed in the tumors, demonstrating that BPDC 3 was effectively taken up by the MIA PaCa-2 tumor cells in vivo presumably due to the EPR effect.28 We also observed fluorescence signals from the liver and kidneys in BPDC 3 treated mice, suggesting that BPDC 3 may be degraded and excreted through metabolism in the liver and kidneys.
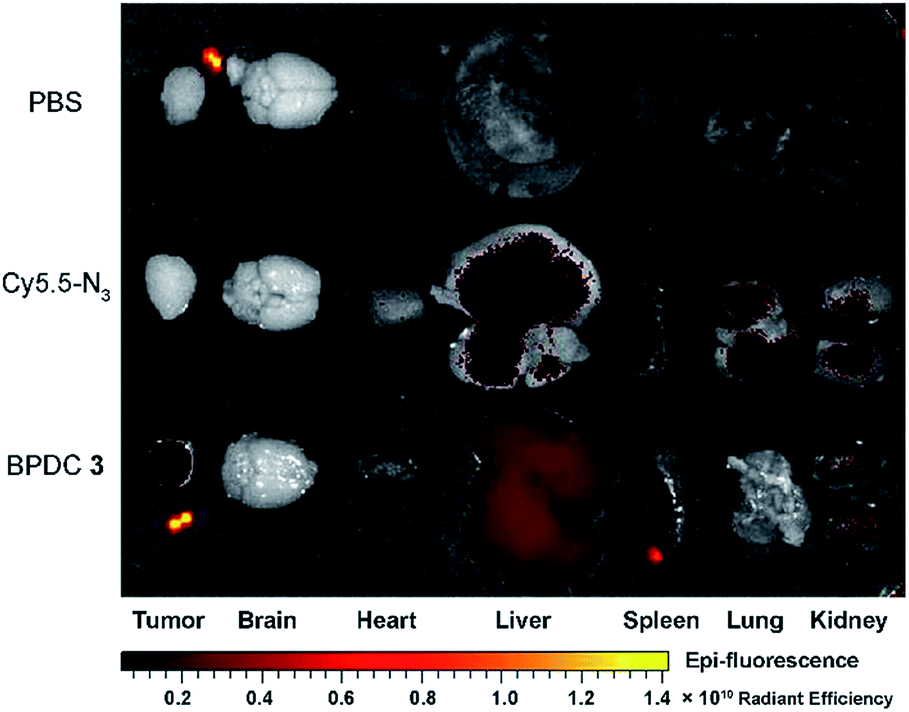 | ||
| Fig. 8 Ex vivo fluorescence imaging of excised tumors and major organs at 24 h post intravenous injection of the vehicle control, Cy5.5-N3, and BPDC 3. | ||
3. Experimental
3.1. Materials
(3S)-cis-3,6-Dimethyl-1,4-dioxane-2,5-dione (LA, 98%), DMAP (99%, prilled) and copper(I) bromide (CuBr, 99.999% trace metals basis) were purchased from Sigma-Aldrich. PMDETA (99+%), DMPA (99%) and sodium periodate (99%) were purchased from Acros Organics. BnOH was purchased from J. T. Baker. α-Methoxy-ω-azido polyethylene glycol (CH3O-PEG-N3, manufacturer suggested MW of PEG: 750 Dalton) was purchased from Rapp Polymere. Cy5.5-N3 was purchased from Lumiprobe Corporation. PTX (99%) was purchased from AvaChem Scientific. GEM (98+%) was purchased from Ark Pharm. Ruthenium dioxide (99.9%) was purchased from Pfaltz & Bauer. Acetone (Certified ACS grade), acetonitrile (HPLC grade), chloroform (CHCl3, HPLC grade), dichloromethane (DCM, HPLC grade), ethyl acetate (HPLC grade), diethyl ether (HPLC grade), methanol (MeOH, HPLC grade), and N,N′-dimethylformamide (DMF; HPLC grade) were purchased from Fisher Scientific. DMF and DCM were dried by distillation over CaH2. ACLA and ALLA were freshly prepared following our previous publications.49,70 PTX-SH and GEM-SH were prepared according to the approaches reported in the literature.62,683.2. Synthesis of poly(LA0.42-co-ALLA0.30-co-ACLA0.28)163 (1)
Living ring-opening polymerization (ROP) was performed using LA (572 mg, 3.97 mmol), ALLA (506 mg, 2.98 mmol), and ACLA (501 mg, 2.98 mmol) as monomers, BnOH (10.7 mg, 0.10 mmol) as the initiator, and DMAP (48.5 mg, 0.40 mmol) as the organocatalyst, in 2 mL dry DCM under a N2 atmosphere in a 10 mL sealed reaction flask ([LA]0:[ALLA]0:[ACLA]0:[BnOH]0:[DMAP]0 = 40:30:30:1:4). The reaction mixture was stirred for 7 days in an oil bath at 35 °C. The polymerization mixture was precipitated twice in a large amount of cold methanol, to obtain 1 (1.04 g, 65.7% yield) as a white solid after drying under vacuum. 1H NMR analysis indicated the formula of copolymer 1 as poly(LA0.42-co-ALLA0.30-co-ACLA0.28)163. 1H NMR (500 MHz, CDCl3, δ): 7.40–7.30 (m, Ar–H from BnOH), 5.87–5.69 (m, CH2CHCH2 of units from ALLA), 5.39–5.07 (br m, CHCH3 of units from LA; CHCH3 and CHCH2CHCH2 of units from ALLA; CHCH3 and CHCH2CCH of units from ACLA), 3.00–2.79 (br m, CHCH2CCH of units from ACLA), 2.79–2.56 (br m, CHCH2CHCH2 of units from ALLA), 2.13–2.00 (br s, CHCH2CCH of units from ACLA), and 1.68–1.41 (br m, CHCH3 of units from LA, ALLA, and ACLA). MNMRn = 26.0 kDa, MGPCn = 30.9 kDa, and ĐGPC = 1.21.
3.3. Synthesis of PLA-g-PEG/Cy5.5 (BP 2)
To a 10 mL reaction flask, 1 (138 mg), CH3O-PEG-N3 (229 mg), Cy5.5-N3 (4.10 mg), and CuBr (34.0 mg) were added with 4.0 mL of DMF. The flask was sealed and degassed with N2. Then PMDETA (41.1 mg) was added slowly using a syringe. The overall feed ratio was [acetylenyl group of 1]0:[CH3O-PEG-N3]0:[Cy5.5-N3]0:[CuBr]0:[PMDETA]0 = 1:1.07:0.025:1:1. Three freeze–pump–thaw cycles were conducted to further remove oxygen in the reaction system. The reaction mixture was stirred for 45 h in the dark at room temperature. The crude product was precipitated in a large amount of cold diethyl ether three times. Then residual CuBr was removed by passing through a short alumina column using DCM as an eluent to obtain 2 (216 mg, 62.3% yield) as a blue solid after drying under vacuum. 1H NMR analysis indicated that the acetylenyl groups of 1 were fully consumed. 1H NMR (500 MHz, CDCl3, δ): 7.69–7.55 (br m, CHCH2CCHN), 7.39–7.31 (m, Ar–H from BnOH), 5.89–5.68 (br m, CHCH2CHCH2 of units from ALLA), 5.53–5.34 (br m, CHCH2CCHN), 5.33–4.96 (br m, CHCH3 of units from LA; CHCH3 and CHCH2CHCH2 of units from ALLA; CHCH3 of units from ACLA), 4.64–4.42 (m, NCH2(CH2OCH2)18CH2OCH3), 3.95–3.82 (m, NCH2(CH2OCH2)18CH2OCH3), 3.81–3.43 (br m, NCH2(CH2OCH2)18CH2OCH3), 3.42–3.15 (br m, NCH2(CH2OCH2)18CH2OCH3 and CHCH2CCHN), 2.81–2.51 (br m, CHCH2CHCH2 of units from ALLA), and 1.69–1.38 (br m, CHCH3 of units from LA, ALLA, and ACLA). MNMRn = 65.7 kDa, MGPCn = 41.4 kDa, and ĐGPC = 1.41.
3.4. Synthesis of PLA-g-PEG/Cy5.5/PTX/GEM (BPDC 3)
In a 10 mL reaction flask, 2 (67.7 mg), PTX-SH (5.4 mg, containing 4.9 mg of PTX), GEM-SH (16.3 mg, containing 12.2 mg of GEM), and DMPA (6.56 mg) were dissolved in 3.6 mL of the mixed solvent of CHCl3 and MeOH (v/v, 5:1). The overall feed ratio was [allyl group of 2]0:[PTX-SH]0:[GEM-SH]0:[DMPA]0 = 1:0.12:0.91:0.50. Three freeze–pump–thaw cycles were conducted to remove oxygen from the reaction system. Then the mixture was irradiated under UV light (λmax = 365 nm) for 90 min at room temperature. The crude product was dialyzed against acetone for two days and then against a mixture of CHCl3 and MeOH (v/v, 5:1) for 1 day. The product was finally precipitated in 40 mL of cold diethyl ether to obtain 3 (84.9 mg, 95.9% yield) as a blue solid after drying under vacuum. The 1H NMR spectrum (500 MHz) of 3 in CDCl3/CD3OD (v/v, 1:1) is shown in Fig. 2b (assignments are not presented here due to numerous resonances from the conjugated drug moieties). MNMRn = 77.6 kDa, MGPCn = 51.5 kDa, and ĐGPC = 1.35.
3.5. Characterization methods
1H NMR spectra of all products were recorded using a 500 MHz Varian INOVA-500 spectrometer at 25 °C. The NMR solvents used in this study included CDCl3 (with tetramethylsilane as an internal standard), CD3OD and DMSO-d6. The Mn and Đ of functionalized PLA 1, BP 2, and BPDC 3 were determined by GPC. The employed Viscotek GPC system had a VE-1122 pump, two mixed-bed organic columns (PAS-103M-UL and PAS-105M-M), a VE-3580 refractive index (RI) detector, and a VE-3210 UV/vis detector. The GPC eluent was DMF containing 0.01 M LiBr (flow rate: 0.5 mL min−1, 55 °C). For each measurement, 0.1 mL of sample (∼3 mg mL−1 in DMF) was injected. The GPC instrument was calibrated by using linear polystyrene standards (Đ < 1.1, Varian). The morphology of BP 2 and BPDC 3 was analyzed by NTA and TEM. NTA was used to determine the Dh of the assembled NPs of BP 2 and BPDC 3 by tracking and recording the Brownian motions of the NPs. A NanoSight LM10 (Malvern Instruments, laser wavelength: 405 nm) was employed for NTA analysis. The samples had a concentration of ∼1 mg mL−1 in different aqueous environments. TEM images of the assembled NPs of BP 2 and BPDC 3 were recorded with a JEOL 2010 microscope. The samples were prepared by loading 7 μL of solution (∼0.1 mg mL−1 in water) on 400 mesh carbon-coated copper grids, followed by complete drying under vacuum, and positive staining for 3 h using 0.5% solution of ruthenium tetroxide (RuO4). The staining agent was freshly prepared by the reaction between sodium periodate and ruthenium dioxide in water for 1 h.3.6. Drug release study
HPLC was employed to study the drug release profiles of BPDC 3. Solutions of 3 in 1 × PBS (pH = 7.4) and 1 × PBS (pH = 5.5), at the same concentration of 0.62 mg mL−1 (containing 50.0 μg mL−1 conjugated GEM and 25.6 μg mL−1 conjugated PTX), were prepared. The solutions were placed on a shaking bed and incubated at 37 °C. At different time intervals, 0.2 mL aliquots of each solution were withdrawn and diluted with 0.8 mL of ACN. For each sample, 20 μL of the diluted solution was injected for HPLC measurement. A reversed phase C18-column (ZORBAX Eclipse XDB-C18, 4.6 × 150 mm, 5 μ) was used for chromatographic separation. The mobile phase consisted of Milli-Q water and ACN (HPLC grade), with linear gradients of water/ACN (9:1–6:4 v/v, 0–3 min; 6:4–3:7 v/v, 3–10 min; 3:7–6:4 v/v, 10–18 min; 6:4–9:1 v/v, 18–20 min). The eluent flow rate was 1.0 mL min−1, and the column compartment temperature was maintained at 30 °C. PTX and GEM molecules were detected with a diode array detector (DAD) (model: Agilent 1260 Infinity), according to their UV absorbance at 227 and 250 nm, respectively. Calibration curves of PTX and GEM were obtained in advance, based on the absorbance spectra of BP 2 and BPDC 3 recorded by using a VWR® Spectrophotometer (UV-1600PC).
3.7. Cell culture
MIA PaCa-2 cells were purchased from the American Type Culture Collection (CRL-1420, Manassas, VA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific; 11965-092) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific; 26140079), 1% penicillin–streptomycin and 1 mM sodium pyruvate (Thermo Fisher Scientific; 15140122). The cells were subcultured every 2–3 days and maintained in 5% CO2 with humidified air at 37 °C.3.8. In vitro cytotoxicity assay
The cytotoxicity of BPDC 3 was evaluated based on the viability of MIA PaCa-2 cells post treatment. MIA Paca-2 cells were seeded in a 96-well plate (Greiner Bio-one; 655180) at a density of 1 × 104 cells per well and allowed to grow overnight. The cells were treated with the vehicle control, a mixture of free PTX and GEM, BPDC 3, and BP 2. The PTX concentrations were 0.01, 0.05, 0.1, 0.5, 1, and 5 μg mL−1 and the GEM concentrations were 0.02, 0.1, 0.2, 1, 2, and 10 μg mL−1. BP 2 had the same molar concentrations as BPDC 3. For all groups, the total volume was 100 μL (the treatment: 10 μL; cell culture medium: 90 μL). At 72 h post treatment, the cell viability was measured by alamarBlue assay (Thermo Fisher Scientific, DAL1025) following the manufacturer's protocol. Briefly, one part of alamarBlue solution was diluted in ten parts of the culture medium, which was added to cells and incubated at 37 °C for 3 h in the dark. A TECAN microplate reader (San Jose, CA) was used to measure fluorescence intensity with the excitation and emission wavelengths at 560 nm and 590 nm, respectively. The cell viability was normalized to the vehicle control group.3.9. In vitro cellular uptake analysis
MIA PaCa-2 cells were seeded in 6-well plates (Greiner Bio-one, 655180) at a seeding density of 2 × 105 cells per well and cultured overnight. The cells were treated with BP 2 and BPDC 3 at a Cy5.5 concentration of 0.2 μg mL−1. Vehicle treated cells were the control. The cells were harvested at 4 and 24 h post treatment and fixed in 4% paraformaldehyde (Acros, 41678-5000). For flow cytometry analysis (BD Fortessa flow cytometer, BD bioscience, San Jose, CA), 10000 events were collected for each sample in an Alexa Fluor 700 channel (λex = 696 nm and λem = 719 nm). The fluorescence intensity of Cy5.5 was reported as mean ± SD. For confocal microscopy imaging, the cell nuclei were stained with 10 μg mL−1 Hoechst 33342 (Thermo Fisher Scientific H3570) at room temperature for 10 min. The cells were then mounted on glass slides and observed under an LSM 710 confocal microscope (ZEISS, Dublin, CA). The fluorescence signals from Hoechst 33342 were detected in the DAPI channel (excitation at 405 nm and emission at 453 nm). The fluorescence signals from BP 2 and BPDC 3 were observed in the Cy5.5 channel (excitation at 643 nm and emission at 707 nm).
3.10. In vivo imaging and biodistribution
MIA PaCa-2 cells (5 × 106) were inoculated at the flanks of female athymic nude mice (6 week old, Charles River Laboratories). When tumor volumes reached ∼100 mm3, the mice were randomly divided into three groups and treated with the vehicle control (n = 3), Cy5.5-N3 (n = 2) and BPDC 3 (n = 3) through intravenous injection at a Cy5.5 dose of 1.5 mg kg−1 body weight. Whole body imaging was performed with an IVIS Lumina II in vivo imaging system (PerkinElmer, Waltham, MA) before and 5 min, 1 h, 4 h, and 24 h after injection. At 24 h after injection, the mice were sacrificed. Tumors and major organs (brain, heart, liver, spleen, lung and kidneys) were harvested. The fluorescence intensities of Cy5.5 in tumors and organs were recorded by using the IVIS system to investigate the biodistribution of BPDC 3 and free Cy5.5-N3. All animal procedures were performed in accordance with the Animal Research Guidelines of University at Buffalo and approved by the Institutional Animal Care and Use Committee (IACUC) at the University at Buffalo.4. Conclusions
The design and synthesis of a novel multifunctional BPDC for co-delivery of hydrophobic PTX and hydrophilic GEM, as well as cancer imaging, has been demonstrated. The BPDC with well-defined structures was prepared by successive azide–alkyne and thiol–ene click functionalization reactions, using an acetylenyl/alkyl-functionalized PLA as the backbone precursor. With both hydrophobic and hydrophilic components, the BPDC formed self-assembled NPs in aqueous solutions. Sustained release of PTX and GEM from the PBS solutions of the BPDC was observed, and specifically the release of PTX demonstrated acid-labile behavior. Both the BPDC and the corresponding scaffold can be readily taken up by MIA PaCa-2 cells. While the scaffold is non-cytotoxic, the BPDC exhibited higher therapeutic efficacy towards MIA PaCa-2 cells than the mixture of PTX and GEM at most of the tested drug concentrations. Moreover, the Cy5.5-labelled BPDC showed a much longer circulation time than a small molecule Cy5.5 reagent, and effectively accumulated at tumor sites for fluorescence imaging. Overall, this work illustrates that the multifunctional BPDC represents a promising design of integrated systems enabling co-delivery of multiple anticancer drugs of different hydrophilicity and tumor imaging.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U. S. National Science Foundation [DMR-1609914; CBET-1337860] and the U. S. National Institutes of Health [R21 EB024095-01]. The authors thank Prof. Mark T. Swihart and Mr Zheng Fu for kind support during NTA measurements, Dr Yueling Qin for technical support during TEM measurements, and the UB North Campus Confocal Imaging Facility in the Department of Biological Sciences along with their NSF grant [National Science Foundation Major Research Instrumentation Grant # DBI 0923133].Notes and references
- P. Parhi, C. Mohanty and S. K. Sahoo, Drug Discovery Today, 2012, 17, 1044–1052 CrossRef CAS PubMed.
- R. Pushpalatha, S. Selvamuthukumar and D. Kilimozhi, J. Drug Delivery Sci. Technol., 2017, 39, 362–371 CrossRef CAS.
- X. Xu, W. Ho, X. Zhang, N. Bertrand and O. Farokhzad, Trends Mol. Med., 2015, 21, 223–232 CrossRef CAS PubMed.
- C. M. J. Hu, S. Aryal and L. Zhang, Ther. Delivery, 2010, 1, 323–334 CrossRef CAS.
- Y. Sun, C. Kang, A. Zhang, F. Liu, J. Hu, X. Zhong and J. Xie, European Journal of BioMedical Research, 2016, 2, 12–18 CrossRef.
- J. Lehar, A. S. Krueger, W. Avery, A. M. Heilbut, L. M. Johansen, E. R. Price, R. J. Rickles, G. F. Short, J. E. Staunton, X. W. Jin, M. S. Lee, G. R. Zimmermann and A. A. Borisy, Nat. Biotechnol., 2009, 27, 659–666 CrossRef CAS PubMed.
- L. F. Zhang, A. F. Radovic-Moreno, F. Alexis, F. X. Gu, P. A. Basto, V. Bagalkot, S. Y. Jon, R. S. Langer and O. C. Farokhzad, ChemMedChem, 2007, 2, 1268–1271 CrossRef CAS PubMed.
- Y. Ma, D. Liu, D. Wang, Y. J. Wang, Q. Fu, J. K. Fallon, X. G. Yang, Z. G. He and F. Liu, Mol. Pharmaceutics, 2014, 11, 2623–2630 CrossRef CAS PubMed.
- L. Liao, J. Liu, E. C. Dreaden, S. W. Morton, K. E. Shopsowitz, P. T. Hammond and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 5896–5899 CrossRef CAS PubMed.
- D. Goldstein, R. H. El-Maraghi, P. Hammel, V. Heinemann, V. Kunzmann, J. Sastre, W. Scheithauer, S. Siena, J. Tabernero, L. Teixeira, G. Tortora, J. L. Van Laethem, R. Young, D. N. Penenberg, B. Lu, A. Romano and D. D. Von Hoff, J. Natl. Cancer Inst., 2015, 107, dju413 CrossRef PubMed.
- D. R. Vogus, A. Pusuluri, R. Chen and S. Mitragotri, Bioeng. Transl. Med., 2018, 3, 49–57 CrossRef CAS PubMed.
- R. Colomer, Oncology, 2004, 18, 8–12 Search PubMed.
- F. De Vita, J. Ventriglia, A. Febbraro, M. M. Laterza, A. Fabozzi, B. Savastano, A. Petrillo, A. Diana, G. Giordano, T. Troiani, G. Conzo, G. Galizia, F. Ciardiello and M. Orditura, BMC Cancer, 2016, 16, 709 CrossRef.
- M. L. Crain, Oncol. Times, 2018, 40, 30 CrossRef.
- D. R. Vogus, V. Krishnan and S. Mitragotri, Curr. Opin. Colloid Interface Sci., 2017, 31, 75–85 CrossRef CAS.
- D. R. Vogus, M. A. Evans, A. Pusuluri, A. Barajas, M. Zhang, V. Krishnan, M. Nowak, S. Menegatti, M. E. Helgeson, T. M. Squires and S. Mitragotri, J. Controlled Release, 2017, 267, 191–202 CrossRef CAS PubMed.
- A. Pusuluri, V. Krishnan, V. Lensch, A. Sarode, E. Bunyan, D. R. Vogus, S. Menegatti, H. T. Soh and S. Mitragotri, Angew. Chem., Int. Ed., 2019, 58, 1437–1441 CrossRef CAS PubMed.
- J. J. Arroyo-Crespo, C. Deladriere, V. J. Nebot, D. Charbonnier, E. Masiá, A. Paul, C. James, A. Armiñán and M. J. Vicent, Adv. Funct. Mater., 2018, 22, 1800931 CrossRef.
- T. Plyduang, A. Armiñán, J. Movellan, R. M. England, R. Wiwattanapatapee and M. J. Vicent, Macromol. Rapid Commun., 2018, 39, 1800265 CrossRef PubMed.
- H. Baabur-Cohen, L. I. Vossen, H. R. Krüger, A. Eldar-Boock, E. Yeini, N. Landa-Rouben, G. Tiram, S. Wedepohl, E. Markovsky, J. Leor, M. Calderón and R. Satchi-Fainaro, J. Controlled Release, 2017, 257, 118–131 CrossRef CAS PubMed.
- D. Kasala, S.-H. Lee, J. W. Hong, J.-W. Choi, K. Nam, Y. H. Chung, S. W. Kim and C.-O. Yun, Biomaterials, 2017, 145, 207–222 CrossRef CAS PubMed.
- K. Nam, H. Y. Nam, P.-H. Kim and S. W. Kim, Biomaterials, 2012, 33, 8122–8130 CrossRef CAS PubMed.
- H. Meng, M. Wang, H. Liu, X. Liu, A. Situ, B. Wu, Z. Ji, C. H. Chang and A. E. Nel, ACS Nano, 2015, 9, 3540–3557 CrossRef CAS PubMed.
- C.-M. J. Hu and L. F. Zhang, Biochem. Pharmacol., 2012, 83, 1104–1111 CrossRef CAS PubMed.
- S. Aryal, C. M. J. Hu and L. F. Zhang, Small, 2010, 6, 1442–1448 CrossRef CAS PubMed.
- T. Lammers, V. Subr, K. Ulbrich, P. Peschke, P. E. Huber, W. E. Hennink and G. Storm, Biomaterials, 2009, 30, 3466–3475 CrossRef CAS PubMed.
- S.-H. Hu, S.-Y. Chen and X. Hu, ACS Nano, 2012, 6, 2558–2565 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS PubMed.
- R. Tong and J. Cheng, Polym. Rev., 2007, 47, 345–381 CrossRef CAS.
- Y. Zhang, H. F. Chan and K. W. Leong, Adv. Drug Delivery Rev., 2013, 65, 104–120 CrossRef CAS PubMed.
- I. Noh, H.-O. Kim, J. Choi, Y. Choi, D. K. Lee, Y.-M. Huh and S. Haam, Biomaterials, 2015, 53, 763–774 CrossRef CAS PubMed.
- N. Larson, J. Yang, A. Ray, D. L. Cheney, H. Ghandehari and J. Kopeček, Int. J. Pharm., 2013, 454, 435–443 CrossRef CAS PubMed.
- W. Du, Z. Xu, A. M. Nystrom, K. Zhang, J. R. Leonard and K. L. Wooley, Bioconjugate Chem., 2008, 19, 2492–2498 CrossRef CAS PubMed.
- J. Xie, G. Liu, H. S. Eden, H. Ai and X. Chen, Acc. Chem. Res., 2011, 44, 883–892 CrossRef CAS PubMed.
- Y. Xiao, H. Hong, A. Javadi, J. W. Engle, W. Xu, Y. Yang, Y. Zhang, T. E. Barnhart, W. Cai and S. Gong, Biomaterials, 2012, 33, 3071–3082 CrossRef CAS PubMed.
- H. K. Sajja, M. P. East, H. Mao, Y. A. Wang, S. Nie and L. Yang, Curr. Drug Discovery Technol., 2009, 6, 43–51 CrossRef CAS.
- L. L. Lock, Y. Li, X. Mao, H. Chen, V. Staedtke, R. Bai, W. Ma, R. Lin, Y. Li, G. Liu and H. Cui, ACS Nano, 2017, 11, 797–805 CrossRef CAS PubMed.
- J. Kim, J. E. Lee, S. H. Lee, J. H. Yu, J. H. Lee, T. G. Park and T. Hyeon, Adv. Mater., 2008, 20, 478–483 CrossRef CAS.
- V. P. Torchilin, AAPS J., 2007, 9, E128–E147 CrossRef CAS PubMed.
- K. K. Ng, J. F. Lovell and G. Zheng, Acc. Chem. Res., 2011, 44, 1105–1113 CrossRef CAS PubMed.
- J. G. Croissant, D. Zhang, S. Alsaiari, J. Lu, L. Deng, F. Tamanoi, A. M. Al Malik, J. I. Zink and N. M. Khashab, J. Controlled Release, 2016, 229, 183–191 CrossRef CAS PubMed.
- M. Chen, X. Liang, C. Gao, R. Zhao, N. Zhang, S. Wang, W. Chen, B. Zhao, J. Wang and Z. Dai, ACS Nano, 2018, 12, 7312–7326 CrossRef CAS PubMed.
- J. Sun, L. Sun, J. Li, J. Xu, Z. Wan, Z. Ouyang, L. Liang, S. Li and D. Zeng, Acta Biomater., 2018, 75, 312–322 CrossRef CAS PubMed.
- A. J. Mieszawska, Y. Kim, A. Gianella, I. van Rooy, B. Priem, M. P. Labarre, C. Ozcan, D. P. Cormode, A. Petrov, R. Langer, O. C. Farokhzad, Z. A. Fayad and W. J. M. Mulder, Bioconjugate Chem., 2013, 24, 1429–1434 CrossRef CAS PubMed.
- C. Gao, X. Liang, S. Mo, N. Zhang, D. Sun and Z. Dai, ACS Appl. Mater. Interfaces, 2018, 10, 3219–3228 CrossRef CAS PubMed.
- J. Zou, G. Jafr, E. Themistou, Y. Yap, Z. A. P. Wintrob, P. Alexandridis, A. C. Ceacareanu and C. Cheng, Chem. Commun., 2011, 47, 4493–4495 RSC.
- J. A. Johnson, Y.-Y. Lu, A. O. Burts, Y.-H. Lim, M. G. Finn, J. T. Koberstein, N. J. Turro, D. A. Tirrell and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 559–566 CrossRef CAS PubMed.
- B. Parrish and T. Emrick, Bioconjugate Chem., 2007, 18, 263–267 CrossRef CAS PubMed.
- Y. Yu, J. Zou, L. Yu, W. Jo, Y. K. Li, W. C. Law and C. Cheng, Macromolecules, 2011, 44, 4793–4800 CrossRef CAS.
- Y. Yu, C.-K. Chen, W.-C. Law, J. Mok, J. Zou, P. N. Prasad and C. Cheng, Mol. Pharmaceutics, 2013, 10, 867–874 CrossRef CAS PubMed.
- Y. Yu, C. K. Chen, W. C. Law, H. Sun, P. N. Prasad and C. Cheng, Polym. Chem., 2015, 6, 953–961 RSC.
- M. Lei, S. Sha, X. Wang, J. Wang, X. Du, H. Miao, H. Zhou, E. Bai, J. Shi and Y. Zhu, RSC Adv., 2019, 9, 5512–5520 RSC.
- Y. Di, Y. Gao, X. Gai, D. Wang, Y. Wang, X. Yang, D. Zhang, W. Pan and X. Yang, RSC Adv., 2017, 7, 24030–24039 RSC.
- D. Yun, H. O. Kim, H. Y. Son, Y. Choi, I. Noh, J. W. Lim, J. Kim, H. Chun, G. Park, D. K. Lee and S. I. Jang, J. Mater. Chem. B, 2017, 5, 6317–6324 RSC.
- W. Yang, Q. Hu, Y. Xu, H. Liu and L. Zhong, Mater. Sci. Eng., C, 2018, 89, 328–335 CrossRef CAS PubMed.
- K. K. Frese, A. Neesse, N. Cook, T. E. Bapiro, M. P. Lolkema, D. I. Jodrell and D. A. Tuveson, Cancer Discovery, 2012, 2, 260–269 CrossRef CAS PubMed.
- N. Awasthi, C. H. Zhang, A. M. Schwarz, S. Hinz, C. G. Wang, N. S. Williams, M. A. Schwarz and R. E. Schwarz, Carcinogenesis, 2013, 34, 2361–2369 CrossRef CAS PubMed.
- Y. Yu, J. Zou and C. Cheng, Polym. Chem., 2014, 5, 5854–5872 RSC.
- R. Tong, N. P. Gabrielson, T. M. Fan and J. Cheng, Curr. Opin. Solid State Mater. Sci., 2012, 16, 323–332 CrossRef CAS PubMed.
- P. Bailon and C. Y. Won, Expert Opin. Drug Delivery, 2009, 6, 1–16 CrossRef CAS PubMed.
- J. M. Harris, N. E. Martin and M. Modi, Clin. Pharmacokinet., 2001, 40, 539–551 CrossRef CAS PubMed.
- J. Zou, F. W. Zhang, S. Y. Zhang, S. F. Pollack, M. Elsabahy, J. W. Fan and K. L. Wooley, Adv. Healthcare Mater., 2014, 3, 441–448 CrossRef CAS PubMed.
- H. Sun, M. Y. Z. Chang, W.-I. Cheng, Q. Wang, A. Commisso, M. Capeling, Y. Wu and C. Cheng, Acta Biomater., 2017, 64, 290–300 CrossRef CAS PubMed.
- V. Ntziachristos, E. A. Schellenberger, J. Ripoll, D. Yessayan, E. Graves, A. Bogdanov Jr, L. Josephson and R. Weissleder, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12294–12299 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nystrom, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef CAS PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- M. Dasari, A. P. Acharya, D. Kim, S. Lee, S. Lee, J. Rhea, R. Molinaro and N. Murthy, Bioconjugate Chem., 2013, 24, 4–8 CrossRef CAS PubMed.
- M. A. G. Loudon and J. N. Jacob, J. Am. Chem. Soc., 1981, 103, 4508–4515 CrossRef.
- J. Zou, C. C. Hew, E. Themistou, Y. K. Li, C. K. Chen, P. Alexandridis and C. Cheng, Adv. Mater., 2011, 23, 4274–4277 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00282k |
| This journal is © The Royal Society of Chemistry 2019 |