
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupercritical water assisted preparation of recyclable gold nanoparticles and their catalytic utility in cross-coupling reactions under sustainable conditions†
Abbasriyaludeen Abdul
Raheem‡
ab,
Pitchai
Thangasamy‡
 ab,
Marappan
Sathish
*ab and
Chandrasekar
Praveen
*ab
ab,
Marappan
Sathish
*ab and
Chandrasekar
Praveen
*ab
aMaterials Electrochemistry Division, Central Electrochemical Research Institute (CSIR Laboratory), Alagappapuram, Karaikudi-630003, Tamil Nadu, India. E-mail: chandrasekar.praveen@gmail.com; msathish@cecri.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, Uttar Pradesh, India
First published on 27th June 2019
Abstract
Preparation of gold nanoparticles (AuNPs) in environmentally friendly water without using any reducing agents under supercritical conditions is demonstrated. PXRD, XPS, FE-SEM and HR-TEM analysis confirmed the formation of phase-pure and crystalline AuNPs of the size of ∼10–30 nm. The catalytic potential of AuNPs was manifested through a generalized green procedure that could accommodate both Sonogashira as well as Suzuki coupling under aqueous conditions at low catalytic loading (0.1 mol%). The AuNP catalyst was found to be recuperated after the reaction and reused for up to six catalytic cycles with no leaching out of gold species as confirmed through ICP-OES analysis. With no confinement of AuNP catalysis to cross-coupling reaction, synthetic extension to one-flask preparation of π-conjugated semiconductors (4 examples) and their optoelectronic properties were also investigated. Other significant features of the present work include short reaction time, site-selectivity, wide substrate scope, high conversion, good chemical yields and applicability in gram-scale synthesis. Overall, the results of this paper signify an operationally sustainable supercritical fluid processing method for the synthesis of AuNPs and their catalytic application towards cross-coupling reactions in green media.
1. Introduction
Noble metal nanoparticles (viz. Ag, Au, Pt and Pd) have received considerable attention in recent years, due to their unique morphology-dependent electronic, optical and catalytic properties.1,2 Because of the robust and green nature of gold catalysts, organic transformations based on gold nanoparticles (AuNPs) have received a special place in the area of heterogeneous gold catalysis.3–10 The main advantage that AuNPs possess compared to their bulk form is the high surface area to volume ratio (sa/vol) and modifiable shapes which lead to high chemical reactivity. Their easy recyclability, reuse and environmentally friendly appeal established their catalytic potential for a myriad of organic reactions. Based on the preparation method, the size of AuNPs or their distribution on the support can be controlled, which would directly influence the reactivity as well as selectivity of the given chemical transformation. Therefore, preparation of AuNPs with controlled size and shape is of utmost significance not only to catalytic but also towards other applications such as sensors, biomedical devices and energy storage/conversion.11–13 It is appropriate to mention that most of the chemical methods require citric acid, hydrazine or sodium borohydride as reducing agents for the synthesis of AuNPs14,15 and thus possess significant environmental impacts.16 Reportedly, this issue was addressed through the rapid reduction of [AuCl4]− species in sonicated water. However, it leads to the formation of triangular and hexagonal Au nanoplates coexisting with spherical AuNPs with a diameter of ∼40 nm.17 To circumvent this limitation, a preparative method for the controlled growth of AuNPs without using reducing agents/additional immobilizing stabilizers/rare earth oxide support in an environmentally benign route is of paramount importance. With no confinement to gold, recent years have witnessed many greener synthetic routes for the preparation of an array of transition metal nanoparticles.18 Especially, the synthesis, characterization and application of different metallic nanoparticles constitute an important class of catalytic systems in cross-coupling reactions.19–24 On the other hand, it is pertinent to mention that the catalytic potential of AuNPs is exemplified in oxidative esterification and hydrogen transfer reactions.25 As part of our research interest in new reaction development26–30 based on gold catalysis,31–37 we became interested in the sustainable preparation of AuNPs and subsequent catalytic application in cross-coupling reactions. In particular, we desired to develop a simple and practical procedure for AuNP catalyzed Sonogashira and Suzuki reactions. Differing from previous gold catalysis disclosures on these reactions (Fig. 1), our working hypothesis comprises a generalized procedure that could accommodate both Sonogashira38–45 as well as Suzuki46–49 reactions. Keeping in mind the principles of green chemistry, our focus is also to execute the abovementioned transformations in aqueous media.50,51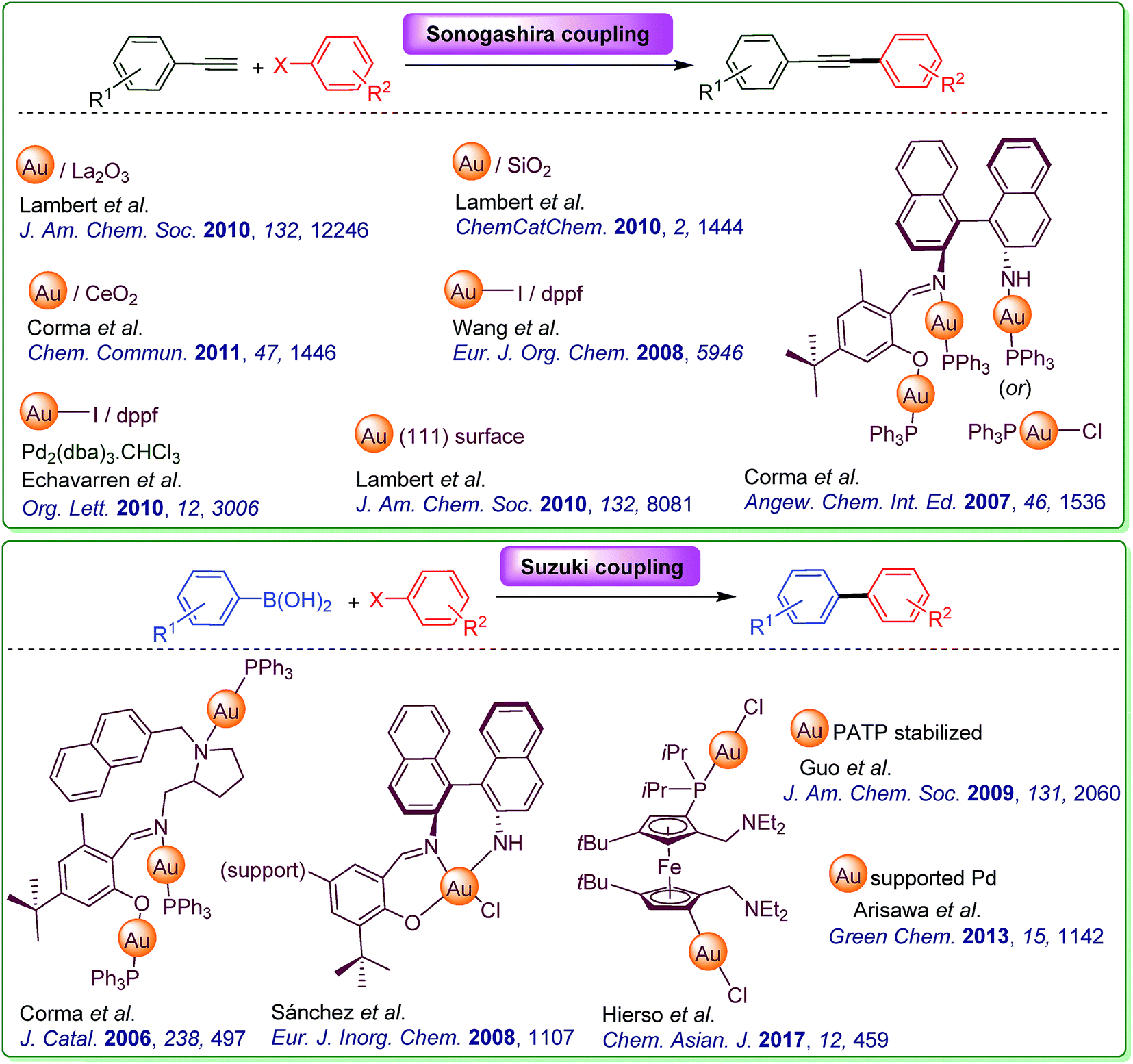 | ||
| Fig. 1 Literature precedents of gold catalyzed Sonogashira and Suzuki coupling. | ||
As far as the preparative method is concerned, huge consideration has been paid in recent years for the synthesis of nanomaterials with precisely defined morphology and dimensions using supercritical fluid (SCF) processing. In this approach, the temperature and pressure of fluids/solvents are increased to their critical points to attain a homogeneous SCF medium, wherein the SCF phase has both gas and liquid-like properties. This condition allows the rapid and facile preparation of nanomaterials in high yields in a shorter reaction time.52–54 Moreover, SCF exhibits interesting physicochemical properties such as high diffusion coefficient, excellent wetting of surfaces, low interfacial tension and low viscosity. Indeed, these are some of the critical parameters for the control of nucleation and growth of nanomaterials.55 Hence, nanomaterials with the desired size and shape without using any surfactants can be accomplished by varying the fluid/temperature/pressure. Recently, AuNPs have been shown to deposit on the surface of graphene oxide nanosheets,56 on supported silica57 and inside the pores of MCM-48 using supercritical carbon dioxide (scCO2)58 for various important applications. In pursuance of our continuing efforts in SCF promoted nanoparticle/sheet synthesis,59–64 we envisaged the supercritical fluid assisted direct conversion of tetrachloroauric acid trihydrate (HAuCl4·3H2O) to AuNPs using water as the green solvent. Our idea was to employ water both as a fluid as well as a base catalyst for the preparation of AuNPs by sequential reduction and decomposition of aqueous HAuCl4·3H2O by SCF processing. This is because of the fact that supercritical water can form a homogeneous phase with inorganic/organic substances and virtually serves as a base catalyst under supercritical conditions.65 Under these conditions the critical temperature, pressure and density of water molecule are 374 °C, 22.06 MPa and 0.32 g mL−1 respectively. Herewith, we report the sustainable preparation of AuNPs from HAuCl4·3H2O using water as a green solvent without using any reducing agents. The as-prepared AuNPs were thoroughly characterized by PXRD, XPS, UV-vis, FE-SEM and HR-TEM analysis followed by the demonstration of their catalytic potential towards Sonogashira and Suzuki cross-coupling reactions under aqueous conditions. To highlight the synthetic utility of the developed methodology, we also established the one-pot synthesis of π-semiconductors along with optoelectronics investigation and all results are compiled in this paper.
2. Experimental section
2.1 Chemicals, methods and instruments
HAuCl4·3H2O (99.995% trace metal basis), halobenzenes, arylboronic acids, and terminal acetylenes were purchased from Sigma-Aldrich. All chemicals and solvents were used as received without further purification. All reactions were carried out in ultra-clean glass vials and stir bars. Analytical TLC was performed on precoated plastic sheets of silica gel G/UV-254 of 0.2 mm thickness (Macherey-Nagel, Germany) using analytical grade solvents and visualized with iodine spray (10% w/w I2 in silica gel), UV light (λ = 254 and 365 nm) and alkaline KMnO4 solution. Column chromatography was performed using thick-walled glass columns along with a mixture of petroleum ether and ethyl acetate on silica gel (100–200 mesh, SRL, India). The relative proportions of solvents in chromatography solvent mixtures refer to the volume to volume ratio (v/v). FT-IR spectra were recorded on a Bruker TENSOR 27 spectrometer in the frequency range from 400 to 4000 cm−1 using the KBr pellet technique. 1H and 13C NMR spectra were obtained in CDCl3 on a BRUKER spectrometer at 400 and 100 MHz respectively. Proton chemical shifts (δ) are relative to tetramethylsilane (TMS, δ = 0.00) as internal standard and expressed in parts per million (ppm). The number of protons (n) for a given resonance is indicated as nH. Spin multiplicities are given as s (singlet), d (doublet), t (triplet) and m (multiplet). Coupling constants (J) are given in hertz (Hz). High resolution mass spectra (HRMS) were obtained using a Q-TOF and a JEOL-GCMate-II mass spectrometer. GC-MS spectra were obtained using a PerkinElmer instrument. Inductively coupled plasma (ICP) measurements were done using Agilent Technologies 5110-ICP-OES equipped with an auto sampler capacity of 240 tubes. For ICP-OES analysis, 1.77 g of Et3PAuCl (based on the calculation of molecular weight of Et3PAuCl/atomic weight of gold) was made up to 1 L using 2% HNO3 solution. This corresponds to 1000 ppm of gold standard and was used as the stock solution. 1 mL of filtrate obtained after removing the solid AuNP catalyst was made up to 10 mL using a standard gold solution in a Teflon vessel and subjected to microwave digestion. This was again made up to 50 mL and used for ICP-OES analysis. UV-visible spectra for all solutions and thin film phases were recorded as chloroform solutions and a thin film respectively on a SHIMADZU UV-3600 plus UV-VIS-NIR spectrometer. Photoluminescence spectra were recorded on a Jasco spectrofluorometer FP-8500. Cyclic voltammetry (CV) experiments were performed with an Autolab potentiostat galvanostat (Model PGSTAT-30). All CV measurements were carried out at room temperature with a conventional three-electrode configuration employing glassy carbon as the working electrode, silver wire as the pseudo-reference electrode and platinum wire as the counter electrode. Tetrabutylammonium perchlorate (0.1 M) as the supporting electrolyte was used in argon-purged acetonitrile at a scan rate of 50 mV s−1. A thermal evaporator with a sputtering unit (model no. VRSU04D) and thickness monitor (model no. CTM-200) was used for chemical vapour deposition. The OPV device performance was measured by recording the current–voltage characteristics using a potentiostat (Keithley 2400) under illumination from a Solar Simulator (Science-Tech) with an AM1.5G filter set and an illumination intensity of 100 mW cm−2.2.2 Catalyst characterization
The formation of AuNPs was confirmed using powder X-ray diffraction (PXRD) analysis, which was performed on a BRUKER D8 ADVANCE X-ray diffractometer with Cu Kα radiation (α = 1.5418 Å) from 2θ = 10 to 80° in steps of 0.02° and a count time of 0.2 s. UV-visible absorption spectra were measured using a VARIAN (Cary 500 Scan) UV-vis-NIR spectrophotometer system. The surface morphology of AuNPs was characterized by field emission scanning electron microscopy (FE-SEM) and high-resolution transmission electron microscopy (HR-TEM) analysis. SEM and HR-TEM images were captured at different locations of the AuNP coated substrate using a Carl Zeiss AG, Supra 55VP with an accelerating voltage of 0.1–30 kV and Tecnai G2 F20S-Twin working at an accelerating voltage of 200 kV, respectively. The formation of AuNPs was further confirmed by X-ray photoelectron spectroscopy (XPS) analysis using a Thermo Scientific MULTILAB 2000 base system with X-ray, Auger and ISS attachments using Mg Kα (1253.6 eV) as the X-ray source.3. Results and discussion
3.1 Catalyst preparation and characterization
At the outset, we intended to prepare AuNPs by reducing highly pure HAuCl4·3H2O (99.995% trace metal basis).66–68 In a typical synthesis, 250 mg of HAuCl4·3H2O was dissolved in 25 mL of distilled water and sonicated for 5 min at room temperature. This homogeneous solution was filled in a stainless-steel supercritical reactor (35 mL volume capacity) and both ends of the reactor were tightly closed (Fig. 2). Then, the sealed supercritical reactor was placed in a preheated tubular furnace at 400 °C for 60 min and subsequently the hot reactor was cooled in an ice-cold water bath. The resulting AuNPs formed were collected by washing the precipitate with distilled water and ethanol using repeated centrifugation. The powder sample obtained was dried in a hot air oven at 80 °C for 12 h. It is worth mentioning that there was no product formation when the reaction time was less than 60 min. If the reaction time was allowed for >60 min at high temperature (>400 °C), doping of chromium and iron (leached out from the supercritical reactor) elements on the surface of AuNPs increases. Therefore, we preferred a reaction condition of 60 min at 400 °C for the synthesis of AuNPs.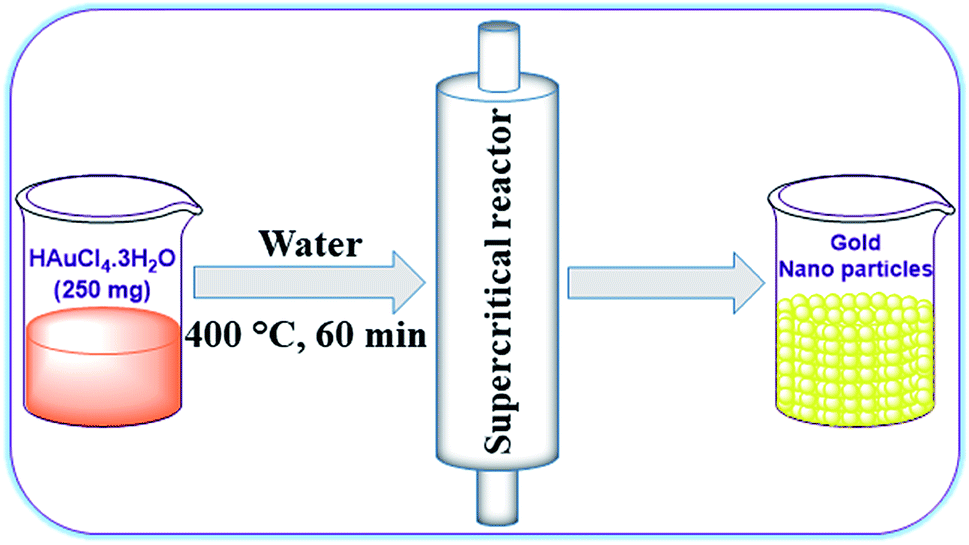 | ||
| Fig. 2 Preparation of AuNPs in supercritical water. | ||
Initially, powder X-ray diffraction (PXRD) analysis was used to confirm the formation of AuNPs. Fig. 3a shows the XRD pattern of as-synthesized AuNPs (bottom panel) and the standard pattern of Au (top panel). The obtained well-defined diffraction lines of the as-synthesized material are in good agreement with the reference pattern of the face centred cubic (fcc) structure of Au (JCPDS card no. 04-0784), which revealed the formation of AuNPs. The diffraction lines also demonstrate that the obtained AuNPs are highly crystalline in nature even without applying any external thermal treatments to improve crystallinity. The observed diffraction lines at 2θ = 38.19°, 44.39°, 64.58° and 77.55° correspond to the (111), (200), (220) and (311) lattice planes of the cubic phase of AuNPs respectively. It is pertinent to note that no other reflection lines can be found along with the AuNP diffraction peaks, which confirms the phase purity of the prepared AuNPs. Thus, these findings emphasize the advantage of the SCF approach in producing phase pure nanomaterials with high crystallinity in a short reaction time. The well-known Scherrer formula (D = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) has been used to calculate the crystallite size of AuNPs. Here, D is the crystallite size of the synthesized sample, K is the dimensionless shape factor, λ is the wavelength of the X-ray beam, β is the full width half maximum (FWHM) and θ is the diffraction angle. The calculated crystallite size at 2θ = 38.19°, 44.39°, 64.58° and 77.55° was found to be 29.15, 23.08, 39.04 and 35.27 nm, respectively. Thus, the average crystallite size of the synthesized AuNPs is 31.6 nm.
cosθ) has been used to calculate the crystallite size of AuNPs. Here, D is the crystallite size of the synthesized sample, K is the dimensionless shape factor, λ is the wavelength of the X-ray beam, β is the full width half maximum (FWHM) and θ is the diffraction angle. The calculated crystallite size at 2θ = 38.19°, 44.39°, 64.58° and 77.55° was found to be 29.15, 23.08, 39.04 and 35.27 nm, respectively. Thus, the average crystallite size of the synthesized AuNPs is 31.6 nm.
 | ||
| Fig. 3 (a) PXRD pattern of synthesized AuNPs (bottom) and its reference pattern (top); (b) Au 4f high resolution XPS of AuNPs; (c) UV-vis absorption spectrum of AuNPs dispersed in aqueous medium. | ||
The chemical state of the synthesized AuNPs was explicitly confirmed by X-ray photoelectron spectroscopic (XPS) analysis. Fig. 3b represents the deconvoluted Au 4f spectra of AuNPs which show their characteristic doublets at 83.7 and 87.4 eV which are in good agreement with the binding energies of the Au 4f7/2 and Au 4f5/2 spin orbit pair respectively.69–73 These values clearly correspond to the metallic Au(0) state. It also ascertains that supercritical water serves the purpose of both fluid as well as base to effectively reduce the Au(III) ions to Au(0) metal. XPS results unambiguously confirmed that under SCF conditions, the formation of AuNPs can be accomplished without using any reducing agents. Hence, the SCF approach can be considered as an alternative energy-efficient and greener method to reduce other noble metal ions for sustainable energy and environmental related applications.
In general, the absorption peak position for the surface plasmon resonance (SPR) band depends on the size and shape of the AuNPs, i.e. the band can shift either bathochromically or hypsochromically.74 Therefore, electronic absorption spectroscopy was used to further elucidate the formation of AuNPs. The as-prepared AuNP powder was dispersed in water by ultrasonication for 15 min prior to recording UV-vis spectra (Fig. 3c). Interestingly, AuNPs exhibited a broad SPR band around 578 nm, which matches well with the literature reports on AuNPs with particle size of 3–100 nm.75,76 In addition, the broadness of the SPR band suggests the large particle size distribution, which was later confirmed by SEM and HR-TEM analysis.
The surface morphology of AuNPs was characterized using electron microscopic analysis. Fig. 4 shows the FE-SEM images of AuNPs coated over a silica wafer substrate at different magnifications. It clearly indicates that AuNPs are relentlessly stacked together, in which agglomeration of ultra-small individual AuNPs can be clearly seen in high magnification. It also suggests that the assembling of AuNPs may not be controlled by SCF processing, since no reducing agents or surfactants have been employed during the synthetic stage. It is worth mentioning that the surface hydroxyl groups are mainly accountable for the aggregation of any nanoparticles, wherein the rate of aggregation depends on the nature of the fluid medium. In other words, the aggregation will be more in the aqueous compared to the non-aqueous medium.77 However, the nucleation and growth of AuNPs can be significantly hampered by the rapid reaction time and cooling of the supercritical reactor in an ice-cold water bath after SCF processing. Hence, ultra-small AuNPs with controlled size can be achieved which may be suitable as a heterogeneous catalyst for organic reactions.
 | ||
| Fig. 4 (a–d) FE-SEM images of AuNPs at different magnifications. | ||
To identify the presence of elements in the AuNPs, SEM-EDX analysis was performed (Fig. 5). A high intense elemental peak corresponding to gold atoms can be seen along with the absence of elemental chloride peak, which confirms the complete reduction of HAuCl4·3H2O to AuNPs. However, elemental peaks corresponding to chromium and iron were observed along with the Au peak although in very less quantity. This could be attributed to the leaching out of alloys of the supercritical reactor by the highly acidic HAuCl4·3H2O and also by in situ generated HCl. As a consequence, the leached-out Cr and Fe possibly deposit on the surface of AuNPs under low pH at high temperature/pressure SCF conditions. It is appropriate to mention that this unwanted reaction can be excluded by choosing suitable Au precursors. Nevertheless, these elemental impurities were not detected in the XRD analysis due to their much lower concentration and also because they are spread over AuNPs only as a very thin layer. Yet, these elements are not expected to contribute much in the subsequent reactions because of their catalytic inertness in cross-coupling chemistry.
 | ||
| Fig. 5 SEM-EDX analysis of the synthesized AuNPs. | ||
On the basis of the above results, the formation of AuNPs under SCF conditions can be explained as follows: AuNPs are formed by high temperature/pressure assisted decomposition of HAuCl4·3H2O, in which supercritical water (scH2O) acts both as a fluid as well as a base catalyst. It is pertinent to mention that, Montero et al. recently disclosed the direct thermal decomposition of HAuCl4·nH2O under atmospheric conditions to prepare AuNPs along with the formation of H2O, HCl and Cl2 as by-products.78 Based on this report, we propose a reasonable chemical reaction for the synthesis of AuNPs under SCF conditions (Scheme 1).
 | ||
| Scheme 1 Reduction of HAuCl4·3H2O under supercritical conditions. | ||
The formation of size-reduced AuNPs with insignificant agglomeration without using any capping agents has been our expectation based on the unique physico-chemical properties of SCFs. These properties include diffusion coefficient, low viscosity, thermal conductivity, molecular density, excellent wetting of surfaces, low surface tension, high degree of super saturation and higher nucleation rate. Indeed, these are some of the critical parameters to control the size and morphology of the metal nanoparticles. However, we obtained AuNPs only with large particle-size distribution which may be because of the low density of the supercritical water (0.32 g mL−1). Under this condition, the formed nanoparticles settle down easily and might have longer residence time which facilitates the formation of large particles by crystal growth. To further confirm the surface morphology and size of AuNPs, HR-TEM analysis at different magnifications was conducted (Fig. 6). It can be clearly seen that AuNPs are accumulated at different regions, in which the lateral size of individual AuNPs ranges from 10 to 30 nm and are found to be in parallel agreement with FE-SEM analysis. It is worth mentioning that the well distributed AuNPs with 10–30 nm in size are observed throughout the HR-TEM analysis. The lattice fringe value for the AuNPs was calculated to be 0.24 nm which corresponds to the (111) crystal plane of the cubic phase of AuNPs.79 The electron diffraction pattern of a selected area reveals the crystalline nature of AuNPs which were in agreement with the PXRD results.
 | ||
| Fig. 6 (a–c) HR-TEM images and (d) SAED pattern of AuNPs. | ||
3.2 Catalytic tests towards cross-coupling reactions
Having prepared and characterized the requisite AuNPs, we next set out to investigate their scope towards Suzuki and Sonogashira coupling reactions. At the outset, it is appropriate to mention that all reactions were performed in ultraclean glass vials and stir bars not previously used for Pd-catalyzed reactions. The purpose of this precautionary measure is to exclude the possibility of participation of Pd species, which otherwise would catalyze the reaction even in ppm levels.40,41 It is important to note that there are several gold catalyzed methodologies disclosed in the literature for cross-coupling reactions and are either limited to Sonogashira38–45 or Suzuki.46–49 Our approach differs from these protocols with an objective of establishing a generalized protocol that can accommodate an array of substrates suitable for both Suzuki as well as Sonogashira coupling. Towards this end, we initiated our studies by refluxing a mixture of iodobenzene (0.01 mol), phenylboronic acid (0.01 mol), NaOH (0.02 mol) and AuNPs (0.5 mol%) in water for 1 h (Table 1). To our delight, we obtained the expected biphenyl 3a in 61% yield (entry 1). Switching over to K3PO4 as the base in place of NaOH, the yield improvised to 83% (entry 2). Instead of K3PO4, a mild base like K2CO3 afforded the cross-coupled product in an excellent 98% yield (entry 3). This is quite contradictory to some of the related reports, wherein K3PO4 was reported to be an effective base compared to K2CO3.80,81 Nevertheless, it is to be remembered that these two reports deal with heterogenized Au-NHC complexes and not bare gold nanoparticles. Reducing the catalytic amount of AuNPs from 0.5 to 0.1 mol% still affords more or less similar yield (entry 4). Performing a blank reaction devoid of AuNPs resulted in no product formation at all (entry 5), which evidently indicates the catalytic role of AuNPs in promoting this transformation. As a final screen, a room temperature reaction yielded only a trace amount of product suggesting the need for high temperature (entry 6).
As per entry 4, the best conditions for the prototype reaction are as follows: heating a reaction mixture comprising 0.01 mol each of iodobenzene and phenylboronic acid, K2CO3 (0.02 mol), AuNPs (0.1 mol%) in demineralized water for 1 h. To this end, we utilized the same reaction conditions for other structurally and electronically different substrates (Table 2). At first, the influence of electronically different substituents such as 4-Me and 4-Ac was assessed, which revealed that our protocol can accommodate both electron releasing as well as electron withdrawing substituents equally (entries 1 and 2). Modifying the steric environment from the C4 to the C2-position also led to identical reactivity with a yield of 90% (entry 3). At this point of our investigation, we also conducted the reaction by varying the substituents at the boronic acid reacting partner. Hence, testing with 4-MeO and 4-F phenylboronic acid afforded the coupled products with only a slight diminution in yield (entries 4 and 5). As exemplified in entries 6 and 7, the catalytic activity of AuNPs is not only limited to the iodo precursor, but also can be extended to less reactive bromides and chlorides with the same efficacy (entries 6 and 7). The catalytic behaviour towards these halo derivatives can be attributed to the precisely controlled size of AuNPs in conjunction with the absence of any stabilizer, which otherwise could act as a catalyst inhibitor for cross-coupling reactions.82 It is to be remembered that all the tested reactions resulted only in cross-coupling with no formation of undesired homocoupling products, which highlights the excellent site-selectivity of the present methodology. This is indeed advantageous from a synthetic chemist perspective, since nanostructured gold was also reported to catalyze the self-coupling of aryl iodides (Ullman coupling)83,84 and aryl boronic acids (homocoupling)49,71,84–88 to form different products. Thus, the observed hetero-selectivity is possibly due to the absence of the immobilizer in AuNPs which leads to sterically unhindered facets that allow easy access of both reactants towards the catalyst surface.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Product | Isolated yielda (g) | Yieldb (%) |
| a 0.01 mol each of aryl boronic acid and haloarene were used for the reaction. b Isolated yield after flash chromatography. | ||||||
| 1 | H | 4-Me | I | 3b | 1.61 | 96 |
| 2 | H | 4-Ac | I | 3c | 1.72 | 88 |
| 3 | H | 2-MeO | I | 3d | 1.65 | 90 |
| 4 | 4-MeO | H | I | 3e | 1.49 | 81 |
| 5 | 4-F | H | I | 3f | 1.53 | 89 |
| 6 | H | H | Br | 3a | 1.41 | 92 |
| 7 | H | H | Cl | 3a | 1.29 | 84 |
It is to be remembered that trace amounts of Cr and Fe contents were present in the AuNPs as indicated by the SEM-EDX analysis. Therefore, to exclude the possibility of Cr or Fe in catalyzing the cross-coupling reaction, we conducted two control experiments for the reaction between 1a and 2a. In these experiments, we used commercially available Cr (∼70 nm) and Fe (∼25 nm) nanopowder instead of AuNPs, which resulted in no reaction at all with quantitative recovery of 2a (Scheme 2). In order to exclude any potential effect of Cr or Fe, a control reaction with AuNPs completely devoid of these metal impurities would be a confirmatory indication. Therefore, we prepared AuNPs (∼40 nm) with no metal contamination by the sonolysis of an aqueous solution of HAuCl4.17 AuNPs prepared in this way afforded 3a in almost quantitative yields similar to our catalytic system. These experimental findings clearly revealed the catalytic inertness of Cr and Fe towards cross-coupling and confirm AuNPs as the actual catalytic species.
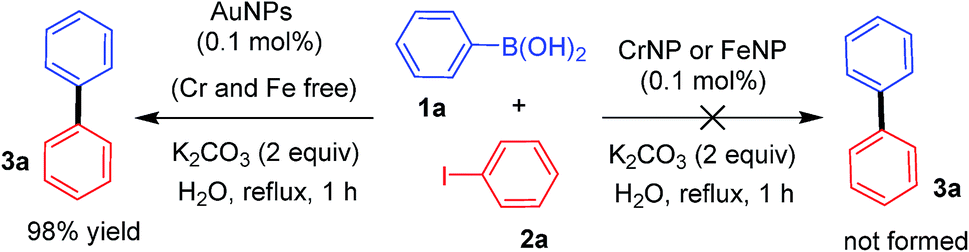 | ||
| Scheme 2 Control reactions using CrNPs and FeNPs. | ||
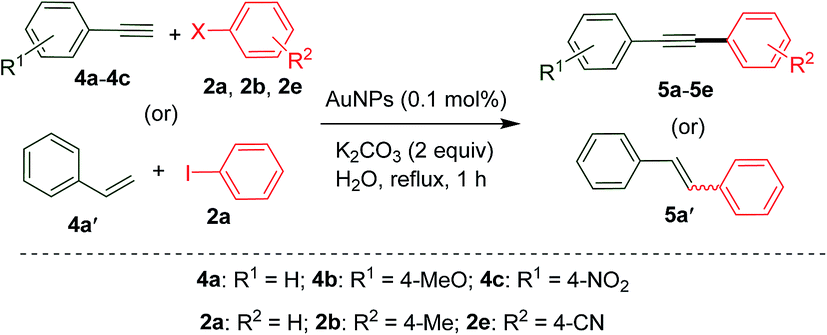
As per our objective, we also look into the possibility of exploring our AuNP catalysis for Sonogashira coupling by including terminal alkynes as coupling partners instead of boronic acids. Gratifyingly, the desired cross-coupling proceeds well for the tested acetylenes (4a–4c) and haloarenes (2a, 2b and 2e) of different electronic natures (Table 3). However, a slight diminution in yield was observed for these couplings compared to Suzuki. Nevertheless, no formation of homo-coupling side products is suggestive of the good site-selectivity of the AuNP catalysis. This could be attributed to the influence of the large particle size of AuNPs (∼10–30 nm) in controlling the selectivity of the heterogeneous cross-coupling chemistry and is in sharp agreement with the findings of Lambert et al.42 As evidenced by crude NMR analysis, some of the iodoarene and alkyne remains unreacted even after 1 h of the reaction, which accounts for the lack of complete conversion. Interestingly, replacing the alkyne with an olefin such as styrene 4a′ to effect Mizoroki–Heck coupling with iodobenzene 2a under our established conditions resulted only in 22% yield of stilbene 5a′. We attribute this poor reactivity of alkenes to the low acidity compared to alkynes. As far as the selectivity is concerned, both Suzuki as well as Sonogashira resulted exclusively in cross-coupling and no homocoupling was observed.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Product | Isolated yielda (g) | Yielda,b (%) |
| a Isolated yield after flash chromatography. b Trace amount of the homo-coupled product was detected in all cases by crude NMR analysis. | ||||||
| 1 | H | H | I | 5a | 1.41 | 79 |
| 2 | H | H | Br | 5a | 1.21 | 68 |
| 3 | 4-MeO | H | I | 5b | 1.68 | 81 |
| 4 | 4-NO2 | H | I | 5c | 1.36 | 61 |
| 5 | H | 4-Me | I | 5d | 1.44 | 75 |
| 6 | H | 4-CN | I | 5e | 1.40 | 69 |
| 7 | H | H | I | 5a′ | 0.39 | 22 |
In order to evaluate the conversion of different substrates under the optimized conditions, individual reaction solutions were subjected to HPLC analysis in a time interval of every 15 minutes (Fig. 7a and b). As depicted in Fig. 7a, time-dependent behaviour in the conversion for Suzuki coupling was observed, where complete or nearly complete conversion was possible only after allowing the reaction for 1 h. Similar conversion with respect to time was observed in the case of Sonogashira coupling, but not all substrates went onto complete conversion (Fig. 7b). In these reactions, complete conversion was hampered by the formation of trace quantities of homo-coupled by-products through a competition of hetero-coupling vs. homo-coupling. Consequently, the Sonogashira reaction of all substrates led to cross-coupled products with slightly lower turnover numbers (TONs = ∼3 to 4) than that of Suzuki coupling (TONs = ∼4 to 5). Nevertheless, the findings of high conversion efficiency are consistent with a heterogeneous catalysis pathway by AuNPs.42,45
 | ||
| Fig. 7 Conversion of substrates in (a) Suzuki coupling and (b) Sonogashira coupling. | ||
3.3 Catalyst recycling
Another significant advantage of our methodology is the ease with which the catalyst can be recovered and reused after the coupling reaction. The recyclability test of the catalyst was proved through a gram-scale synthesis of 3a in a reaction between 1a (0.1 mol, 12.2 g), 2a (0.1 mol, 20.4 g) and AuNPs (0.1 mmol, 19.7 mg) (Fig. 8a). After completion of the reaction, the catalyst was recovered by simple centrifugation followed by filtration from an ethanol suspension. This was followed by sequential washing with ethanol, water, acetone and finally dried at 40 °C for 1 h. Thus, the recovered gold after each reaction cycle was found to be 18.71 mg (95%), 18.51 mg (94%), 18.12 mg (92%), 17.73 mg (90%), 17.33 mg (88%) and 16.55 mg (84%). It was found that up to six cycles, the recovered AuNPs can be reused, after which the catalytic activity reduced gradually owing to the lower mass recovery in each step. This means, the amount of recycled catalyst available for subsequent reactions is lower compared to the previous cycle. This gradual loss of catalyst after each cycle could account for the slight decrease in the yield after each cycle. To quantify the amount of any leached-out gold in the filtrate, ICP-OES analysis was performed. The process involves the introduction of product solution into the plasma using a nebulizer and a spray chamber. To this end, freshly prepared standard gold solutions at concentrations ranging between 0.05 and 10 ppm were initially analyzed, in which gold was detected at an emission wavelength of 242.794 nm (Fig. 8b). The standard linear curve exhibited a positive correlation between the concentration and intensity with a regression coefficient of 0.9995 (Fig. 8c). The gold catalyst recovered after four catalytic cycles was used for the trace element analysis, the result of which indicates no significant peak at 242.794 nm, clearly revealing the absence of gold content (Fig. 8d). As depicted in Fig. 8e, spiking (standard addition method) was carried out to confirm the above results. Two levels of concentrations such as 1.02 ppm and 2.01 ppm were spiked in the first recycled sample and the content of gold was found to be 1.02 μg mL−1 and 2.06 μgmL−1 respectively with 100.0% and 101.9% recoveries, which confirmed that the leaching of gold is negligibly small. Nevertheless, a filtrate test was performed by adding 1a (0.01 mmol), 2a (0.01 mmol) and K2CO3 (0.02 mmol) to the obtained filtrate and refluxed. After several intervals, it was found that no conversion to product 3a was detected which confirms that the reaction does not proceed in the absence of the solid AuNP catalyst (Fig. 8f). This could possibly be because of the absence of AuNPs in the filtrate and thus validates the heterogeneity of the catalytic process. Additionally, a hot filtration test was carried out to prove the heterogeneity of AuNPs in a reaction between 1a and 2a under standard conditions.89 When the reaction was stopped at 35 min, the isolated yield of 3a was found to be 55%. Then, the catalyst was removed by centrifugation and filtered-off from the solution and the reaction was allowed to continue for another 1 h. However, no increase in product yield was observed which testifies to the catalyst's heterogeneous nature. | ||
| Fig. 8 (a) Recyclability of AuNPs tested in the synthesis of 3a; (b) ICP-OES curve for the gold standard at different concentrations; (c) linearity curve for the gold standard; (d) ICP-OES curve of first four recycled samples; (e) spiked curve; (f) filtrate test of Suzuki coupling. | ||
In another series of experiments, the spent catalyst obtained after six catalytic cycles was subjected to PXRD, XPS, SEM and FE-SEM analysis (Fig. 9). As depicted in Fig. 9a, the XRD peak positions of the recovered catalyst at 2θ values of 38.21°, 44.41°, 64.62° and 77.60° [JCPDS card no. 01-089-3697] correspond to the (111), (200), (220) and (311) lattice planes respectively. Since the 2θ values match well with the fcc phase of the as-prepared sample, it clearly indicates no change in phase purity after recycling. The calculated crystallite sizes corresponding to the different 2θ values were found to be 29.40, 30.11, 40.22 and 43.94 nm respectively. Thus, the average crystallite size of the recycled catalyst is calculated to be 35.9 nm, which is 4.3 nm greater than that of the as-prepared sample (31.6 nm). This increase in crystallite size after recycling is attributed to the agglomeration during catalytic tests which is in parallel agreement with the gradual decrease in catalytic activity after every recycling. It is to be remembered at this juncture that during recycling studies, a gradual loss in catalyst amount was observed after each catalytic run, which was considered to be the reason for the slight decrease in yield after each cycle. We believe that probably both the increase in crystallite size as well as loss in catalyst amount after each cycle could contribute for the gradual loss of yield. To exclude the possibility of oxidation of Au(0) to Au(I) and/or Au(III) during the course of catalytic tests or recycling, XPS analysis was carried out on the recovered catalyst (Fig. 9b). The spectra featured a large 4f peak as a doublet with 4f7/2 and 4f5/2 spin orbit split peaks centred at 84.1 and 87.7 eV respectively. These binding energy values are in good agreement with gold in its zerovalent state, thus revealing no change in the oxidation state during catalysis. This in turn confirms that Au(0) is indeed the catalytic species. Moreover, the surface morphology of the spent catalyst was assessed by SEM and FE-SEM analysis (Fig. 9c). Notably, the recovered AuNP catalyst maintains its unique morphology even after six catalytic cycles, which is a clear indication of the stability of the catalyst.
 | ||
| Fig. 9 Recovered AuNPs after six catalytic cycles: (a) PXRD pattern, (b) Au 4f high resolution XPS, (c) SEM image, (d) FE-SEM image. | ||
Based on the above results, a mechanistic basis representing the probable sequence of events is provided in Fig. 10. For Suzuki coupling, the reaction is initiated by the interaction of aryl boronic acids 1 and haloarenes 2 on the catalyst surface to form transient species I, which further reorganize to form organometallic intermediates II. The latter in the presence of K2CO3 and water deliver the biaryl products 3 along with KHCO3, KX and B(OH)3 as byproducts.90 In the case of Sonogashira coupling, the reaction is likely to proceed through the chemisorption of terminal alkynes 4 and haloarenes 2 on the catalyst surface to form adsorbed species III. The adsorbed molecules then undergo reorganization on the surface to form intermediate IV. The latter intermediate under the action of K2CO3 affords the desired coupling products 5 accompanied by KX and KHCO3 with the regeneration of the catalyst.
 | ||
| Fig. 10 Plausible mechanism for the AuNP catalyzed Suzuki and Sonogashira coupling. | ||
3.4 Applications in the one-pot synthesis of optoelectronic chromophores
As an application of AuNP catalysis coupled with our pursuance on photoactive functional chromophores,28–30 we envisaged the synthesis of optoelectronic π-semiconductors by taking advantage of our sustainable conditions (Scheme 3). In this connection, 3g,91,92 an important thiophene oligomer intermediate frequently encountered in dye-sensitized solar cells (DSSCs) has been prepared by the Suzuki reaction between 1d and 2f, albeit in low yield (28%). Sonogashira coupling of 4d with 2g and 2h respectively affords the photoactive alkynylated adducts 5f and 5g in moderate yields (41 and 52%). At this point of our investigation, the feasibility of preparing D–π–A (push–pull type) chromophores under one-flask conditions by applying our standard conditions was also investigated (Scheme 3, middle panel). For this study, we chose 4-aminoarylboronic acid (1e–1f), iodo-arylcarboxaldehyde (2i and 2j) and malononitrile as the donor (D), π-spacer (π) and acceptor (A) respectively. All the screened reactions were carried out initially by coupling the boronic acid with iodo precursors for 1 h. Without isolating the coupled products (3h–3k), malononitrile was subsequently added to the same flask (in situ) and the reaction was continued for the next 30 min under reflux, which allowed the one-flask preparation of the targeted D–π–A chromophores 6a–6d in good to excellent yields (77–91%). Moreover, separate experiments were performed to check the conversion of the precursors (1e–1g, 2i and 2j) to the intermediates 3h–3k by discontinuing the reaction at the coupling stage, which afforded an isolated yield of 84 to 97%. These intermediates were characterized by 1H and 13C NMR, which clearly revealed that the cross-coupling step proceeds well before the ultimate condensation step. It is to be remembered that all these reactions were carried out in EtOH/H2O medium (1:1 v/v) to enhance the solubility of the reactants and products. Finally, to investigate the catalytic activity of AuNPs in the preparation of bithiophene (3k), 2-halothiophenes (2k–2m) were subjected to Suzuki coupling with thiophene 2-boronic acid (1f). Under identical conditions, the yields gradually reduced from iodo (78%) to bromo (59%) to chloro (22%) precursors, which is in accordance with the electronic difference of the halogens (Scheme 3, bottom panel).
 | ||
| Scheme 3 AuNP catalyzed synthesis of functional chromophores. | ||
3.5 Application of synthesized chromophores
Having prepared the desired chromophores 6a–6d under one-pot conditions, we next set out to investigate their photophysical, electrochemical and photovoltaic properties in view of exploring their possible application in organic photovoltaics (Table 4). UV-visible electronic spectra recorded for these chromophores in solution and solid state are depicted in Fig. 11a and S1† respectively. All chromophores in solution phase exhibited an intense π–π* absorption band in the blue region of the visible spectrum with λmax ranging from 462 to 506 nm. The molar absorptivity (ε) of all chromophores was found to be in between 2600 and 27400 M−1 cm−1 which is indicative of their good light harvesting properties. Thin-film spectra recorded on a quartz plate revealed an extended absorption edge (λonset = 595 to 611 nm) compared to the solution state spectra (λonset = 532 to 571 nm). This bathochromic shift can be attributed to the solid-state J-aggregation of the chromophores, thus leading to a decrease in the optical band gap (Eoptg = 2.08 to 2.02 eV), which in turn is essential to enhance the short circuit current (Jsc) in organic solar cells.93 Prompted by the change in colour with respect to solvent polarity, we also evaluated the absorption behavior of all chromophores in different solvents (viz. hexane, THF and MeOH), which led to the finding of a hypsochromic shift in their respective absorption spectra compared to CHCl3 (Fig. S2 and S3†). Intrigued by the intense colour of all chromophores under UV-vis illumination, we also recorded their emission fluorescence (Fig. S4†). All chromophores displayed an orange to red emission maxima (λem = 616 to 667 nm) with Stokes shift ranging between 60200 and 89300 cm−1. This is an indication of the high molecular flexibility of these π-semiconductors which allows the excited molecules to rapidly relax to the lower frequency vibrational modes of the first excited state (S1) by dissipating excess energy. The cyclic voltammetry of all chromophores was carried out to evaluate the formal redox potential and HOMO–LUMO energy levels of 6a–6d (Fig. 11b). With reference to the Fc+/Fc couple, all chromophores showed one oxidation wave with the onset anodic potential (Eox) between 0.59 and 1.0 V and one reduction potential with the onset cathodic potential (Ered) between −0.91 and −1.07 V. The anodic potential values are characteristic of electron rich aryl/alkyl amine groups, while the cathodic potential corresponds to the nitrile reduction.94,95 By applying the Pommerehne empirical relationship,85 the Frontier molecular orbital energies (EHOMO and ELUMO) were calculated from the respective Eox and Ered values. The results revealed that the HOMOs (ca. −4.94 to −5.35 eV) and LUMOs (ca. −3.28 to −3.44 eV) are appropriately located to enable the chromophores to act as hole-transporting materials (p-type) against a standard n-type material like PCBM (Fig S5†).96–98 Therefore, a preliminary bulk heterojunction (BHJ) solar cell was fabricated using these chromophores in conjunction with a standard n-type material (PCBM) in a cell configuration of ITO/ZnO/BHJ/V2O5/Ag (Fig. 11c). The photovoltaic performance revealed that all chromophores displayed similar open circuit voltage (Voc = 0.47 to 0.53 V) and fill-factor (FF = 0.40 to 0.60). However, there is a notable difference in the short circuit-current density (Jsc = 4.83 to 9.20 mAcm−2), which in turn is influential in enhancing the power conversion efficiency (η) of the overall device. Among the tested chromophores, 6b emerged as the best in terms of its high η (2.19%). The efficiencies reported are merely from unoptimized device structures and can be improved further by varying the cell configuration, optimizing the surface morphology and understanding the charge-carrier mobility which falls outside the realm of the present work.99,100 Nevertheless, these initial results highlight the catalytic potential of AuNPs in preparing small molecule organic solar cells (SMOSCs) essentially under one-pot conditions.
| Abs. (solution) λmax/λonseta (nm) | ε (M−1 cm−1) | Abs. (film) λmax/λonsetb (nm) | E optg (eV) | E ox (V) | E red (V) | E HOMO (eV) | E LUMO (eV) | E cvg (eV) | V oc (V) | J sc (mA cm−2) | FF | η (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a Absorption in chloroform solution (2 × 10−5 M).
b Absorption in thin film.
c Optical band gap was derived from λonset of thin film absorption using the formula, Eoptg (eV) = 1240/λonset.
d Anodic potential onset.
e Cathodic potential onset.
f
E
HOMO was calculated using the relationship, EHOMO = −[(Eox − E1/2(ferrocene)) + 4.8] eV.
g
E
LUMO was calculated using the relationship, ELUMO = −[(Ered − E1/2(ferrocene)) + 4.8] eV, given that E1/2(ferrocene) = 0.45 V.
h
E
cvg (eV) calculated from ELUMO − EHOMO.
i Current characteristics of photovoltaic devices fabricated from a solution of 6a–6d:PCBM (1:1 blend) in o-dichlorobenzene and thermally annealed at 70 °C for 5 min.
j Efficiencies of inverted OPVs under AM1.5G illumination (100 mW cm−2).
|
|||||||||||||
| 6a | 462/532 | 21300 |
506/595 | 2.08 | 1.00 | −1.03 | −5.35 | −3.32 | 2.03 | 0.52 | 6.84 | 0.48 | 1.70 |
| 6b | 506/571 | 27400 |
507/605 | 2.04 | 0.90 | −1.07 | −5.25 | −3.28 | 1.97 | 0.53 | 9.20 | 0.45 | 2.19 |
| 6c | 472/563 | 2600 | 468/607 | 2.04 | 0.84 | −0.91 | −5.19 | −3.44 | 1.75 | 0.47 | 5.85 | 0.4 | 1.09 |
| 6d | 479/546 | 12000 |
497/611 | 2.02 | 0.59 | −1.02 | −4.94 | −3.33 | 1.61 | 0.48 | 4.83 | 0.6 | 1.39 |
 | ||
| Fig. 11 (a) Normalized UV-vis absorption spectra of 6a–6d in CHCl3 (5 × 10−5 M); (b) CV of 6a–6d in dichloroethane (2 × 10−3 M) using n-Bu4ClO4 (0.1 M) as the supporting electrolyte at a scan rate of 100 mV s−1; (c) current–voltage (J–V) characteristics of fabricated devices of 6a–6d. | ||
4. Conclusions
To summarize, we have developed a supercritical processing method for the preparation of AuNPs using environmentally benign water as the solvent. The formation of phase pure and crystalline AuNPs in a facet of 10–30 nm was characterized by PXRD, XPS, UV-VIS, FE-SEM and HR-TEM analysis. Unlike chemical methods, the current protocol neither requires any reducing agents for AuNP preparation nor a surfactant or rare earth metal oxide as a stabilizing agent. Application of the as-prepared AuNPs as a heterogeneous recyclable catalyst was exemplified in Suzuki as well as Sonogashira cross-coupling reactions under sustainable conditions. To delineate the synthetic scope, one-pot preparation of π-conjugated small molecular chromophores was also highlighted. Following their optoelectronic assessment by UV-vis and CV, a preliminary organic photovoltaic device was constructed which resulted in efficiencies ranging between 1.09 and 2.19%. We are currently engaged in unravelling the mechanistic insights of the AuNP catalyzed coupling processes, which will be disclosed shortly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
One of the authors P. T. thanks UGC-CSIR, India for the Senior Research Fellowship (SRF). C. P. acknowledges the Science and Engineering Research Board (SERB) and Department of Science and Technology (DST), India for providing the fast-track grant (SB/FT/CS-001/2015) and INSPIRE faculty award (IFA14-MS27) respectively.References
- Y. Lua and W. Chen, Chem. Soc. Rev., 2012, 41, 3594–3623 RSC.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS.
- T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636–2654 RSC.
- X. Liu, L. He, Y.-M. Liu and Y. Cao, Acc. Chem. Res., 2014, 47, 793–804 CrossRef CAS.
- R. Ye, A. V. Zhukhovitskiy, C. V. Deraedt, F. D. Toste and G. A. Somorjai, Acc. Chem. Res., 2017, 50, 1894–1901 CrossRef CAS.
- G. Li and R. Jin, Nanotechnol. Rev., 2013, 2, 529–545 CAS.
- B. S. Takale, M. Bao and Y. Yamamoto, Org. Biomol. Chem., 2014, 12, 2005–2027 RSC.
- Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467–2505 CrossRef CAS.
- M. Padilla, F. Peccati, J. L. Bourdelande, X. S. Monfort, G. Guirado, M. Sodupe and J. Hernando, Chem. Commun., 2017, 53, 2126–2129 RSC.
- G. Bharath, A. Naldoni, K. H. Ramsait, A. A. Wahab, R. Madhu, E. Alsharaeh and N. Ponpandian, J. Mater. Chem. A, 2016, 4, 6385–6394 RSC.
- Y. C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- I. O. Jiménez, F. M. Romero, N. G. Bastús and V. Puntes, J. Phys. Chem. C, 2010, 114, 1800–1804 CrossRef.
- C. Deraedt, L. Salmon, S. Gatard, R. Ciganda, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 14194–14196 RSC.
- P. Pati, S. McGinnis and P. J. Vikesland, Environ. Eng. Sci., 2014, 31, 1–11 CrossRef.
- T. Sakai, S. Miwa, T. Okada and S. Mishima, AIP Conf. Proc., 2012, 1474, 167–170 CrossRef CAS.
- R. S. Varma, Curr. Opin. Chem. Eng., 2012, 1, 123–128 CrossRef CAS.
- T. Baran, Catal. Lett., 2019, 149, 1496–1503 CrossRef CAS.
- T. Baran, Carbohydr. Polym., 2018, 195, 45–52 CrossRef CAS.
- T. Baran, Ultrason. Sonochem., 2018, 45, 231–237 CrossRef CAS.
- T. Baran, Catal. Lett., 2019, 149, 1721–1729 CrossRef CAS.
- N. Y. Baran, T. Baran and A. Menteş, Carbohydr. Polym., 2018, 181, 596–604 CrossRef.
- T. Baran, N. Y. Baran and A. Menteş, Appl. Organomet. Chem., 2018, 32, e4075 CrossRef.
- M. B. Gawande, A. K. Rathi, J. Tucek, K. Safarova, N. Bundaleski, O. M. N. D. Teodoro, L. Kvitek, R. S. Varma and R. Zboril, Green Chem., 2014, 16, 4137–4143 RSC.
- M. Vadivelu, S. Sugirdha, P. Dheenkumar, Y. Arun, K. Karthikeyan and C. Praveen, Green Chem., 2017, 19, 3601–3610 RSC.
- K. Parthasarathy, C. Praveen, K. Saranraj, C. Balachandran and P. S. Kumar, Med. Chem. Res., 2016, 25, 2155–2170 CrossRef CAS.
- A. A. Raheem, S. Kamaraj, V. Sannasi and C. Praveen, Org. Chem. Front., 2018, 5, 777–787 RSC.
- A. A. Raheem, S. Gopi, M. Kathiresan and C. Praveen, RSC Adv., 2019, 9, 1895–1902 RSC.
- C. Kumar, A. A. Raheem, K. Pandian, V. Nandakumar, R. Shanmugam and C. Praveen, New J. Chem., 2019, 43, 7015–7027 RSC.
- C. Praveen and D. B. Babu, Bioorg. Med. Chem. Lett., 2016, 26, 2507–2512 CrossRef CAS.
- C. Praveen and P. T. Perumal, Synthesis, 2016, 48, 855–864 CrossRef CAS.
- K. Parthasarathy, C. Praveen, J. C. Jeyaveeran and A. A. M. Prince, Bioorg. Med. Chem. Lett., 2016, 26, 4310–4317 CrossRef CAS.
- J. C. Jeyaveeran, C. Praveen, Y. Arun, A. A. M. Prince and P. T. Perumal, J. Chem. Sci., 2016, 128, 787–802 CrossRef CAS.
- J. C. Jeyaveeran, C. Praveen, Y. Arun, A. A. M. Prince and P. T. Perumal, J. Chem. Sci., 2016, 128, 73–83 CrossRef CAS.
- C. Praveen, Coord. Chem. Rev., 2019, 392, 1–34 CrossRef CAS.
- C. Praveen, Catal. Rev.: Sci. Eng., 2019, 61, 1–41 CrossRef.
- C. González-Arellano, A. Abad, A. Corma, H. García, M. Iglesias and F. Sánchez, Angew. Chem., Int. Ed., 2007, 46, 1536–1538 CrossRef.
- S. K. Beaumont, G. Kyriakou and R. M. Lambert, J. Am. Chem. Soc., 2010, 132, 12246–12248 CrossRef CAS.
- T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS.
- A. Corma, R. Juárez, M. Boronat, F. Sánchez, M. Iglesias and H. García, Chem. Commun., 2011, 47, 1446–1448 RSC.
- G. Kyriakou, S. K. Beaumont, S. M. Humphrey, C. Antonetti and R. M. Lambert, ChemCatChem, 2010, 2, 1444–1449 CrossRef CAS.
- V. K. Kanuru, G. Kyriakou, S. K. Beaumont, A. C. Papageorgiou, D. J. Watson and R. M. Lambert, J. Am. Chem. Soc., 2010, 132, 8081–8086 CrossRef CAS.
- P. Li, L. Wang, M. Wang and F. You, Eur. J. Org. Chem., 2008, 5946–5951 CrossRef CAS.
- C. González-Arellano, A. Corma, H. García, M. Iglesias and F. Sánchez, Eur. J. Inorg. Chem., 2008, 1107–1115 CrossRef.
- J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060–2061 CrossRef CAS.
- M. Al-Amin, M. Akimoto, T. Tameno, Y. Ohki, N. Takahashi, N. Hoshiya, S. Shuto and M. Arisawa, Green Chem., 2013, 15, 1142–1145 RSC.
- N. Dwadina, J. Roger, N. Pirio, H. Cattey, R. B. Salem and J.-C. Hierso, Chem.–Asian J., 2017, 12, 459–464 CrossRef.
- C. González-Arellano, A. Corma, M. Iglesias and F. Sanchez, J. Catal., 2006, 238, 497–501 CrossRef.
- M.-O. Simon and C.-J. Li, Chem. Soc. Rev., 2012, 41, 1415–1427 RSC.
- C.-J. Li, Chem. Rev., 2005, 105, 3095–3166 CrossRef CAS PubMed.
- T. Adschiri, Y. Hakuta and K. Arai, Ind. Eng. Chem. Res., 2000, 39, 4901–4907 CrossRef CAS.
- R. S. Oakes, A. A. Clifford and C. M. Rayner, J. Chem. Soc., Perkin Trans. 1, 2001, 0, 917–941 RSC.
- K. P. Johnston and P. S. Shah, Science, 2004, 303, 482–483 CrossRef CAS.
- T. Adschiri, Y.-W. Lee, M. Goto and S. Takami, Green Chem., 2011, 13, 1380–1390 RSC.
- Y. Meng, F. Su and Y. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 39549–39559 CrossRef CAS.
- B. Wong, S. Yoda and S. M. Howdle, J. Supercrit. Fluids, 2007, 42, 282–287 CrossRef CAS.
- M. Chatterjee, Y. Ikushima, Y. Hakuta and H. Kawanami, Adv. Synth. Catal., 2006, 348, 1580–1590 CrossRef CAS.
- P. Thangasamy and M. Sathish, J. Mater. Chem. C, 2016, 4, 1165–1169 RSC.
- M. Sathish, S. Mitani, T. Tomai and I. Honma, J. Mater. Chem. A, 2014, 2, 4731–4738 RSC.
- P. Thangasamy, K. Selvakumar, M. Sathish, S. M. Senthil Kumar and P. Thangamuthu, Catal. Sci. Technol., 2017, 7, 1227–1234 RSC.
- P. Thangasamy, T. Partheeban, S. Sudanthiramoorthy and M. Sathish, Langmuir, 2017, 33, 6159–6166 CrossRef CAS PubMed.
- P. Thangasamy, V. Maruthapandian, V. Saraswathy and M. Sathish, Catal. Sci. Technol., 2017, 7, 3591–3597 RSC.
- P. Thangasamy, M. Santhanam and M. Sathish, ACS Appl. Mater. Interfaces, 2016, 8, 18647–18651 CrossRef CAS PubMed.
- D. Rangappa, K. Sone, M. Wang, U. K. Gautam, D. Golberg, H. Itoh, M. Ichihara and I. Honma, Chem.–Eur. J., 2010, 16, 6488–6494 CrossRef CAS PubMed.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS.
- S. S. Zalesskiy, A. E. Sedykh, A. S. Kashin and V. P. Ananikov, J. Am. Chem. Soc., 2013, 135, 3550–3559 CrossRef CAS.
- A. Moragues, F. Neaţu, V. I. Pârvulescu, M. D. Marcos, P. Amoros and V. Michelet, ACS Catal., 2015, 5, 5060–5067 CrossRef CAS.
- K. Nobusawa, N. Okamoto, K. S. L. Chong, X. Lin, K. Iwahori and I. Yamashita, ACS Omega, 2017, 2, 1416–1424 CrossRef.
- X. Zhang, H. Shi and B.-Q. Xu, Angew. Chem., Int. Ed., 2005, 44, 7132–7135 CrossRef CAS PubMed.
- H. Tsunoyama, H. Sakurai, N. Ichikuni, Y. Negishi and T. Tsukuda, Langmuir, 2004, 20, 11293–11296 CrossRef CAS.
- Z. Zhong, J. Lin, S.-P. Teh, J. Teo and F. M. Dautzenberg, Adv. Funct. Mater., 2007, 17, 1402–1408 CrossRef CAS.
- B. Vilhanová, J. Václavík, L. Artiglia, M. Ronocchiari, A. Togni and J. A. van Bokhoven, ACS Catal., 2017, 7, 3414–3418 CrossRef.
- V. Amendola, R. Pilot, M. Frasconi, O. M. Maragò and M. A. Iatì, J. Phys.: Condens. Matter, 2017, 29, 203002 CrossRef PubMed.
- D. Liu, C. Li, F. Zhou, T. Zhang, H. Zhang, X. Li, G. Duan, W. Cai and Y. Li, Sci. Rep., 2015, 5, 7686 CrossRef CAS.
- C. Li, K. L. Shuford, M. Chen, E. J. Lee and S. O. Cho, ACS Nano, 2008, 2, 1760–1769 CrossRef CAS.
- T. He, D. Chen and X. Jiao, Chem. Mater., 2004, 16, 737–743 CrossRef CAS.
- M. A. Montero, M. R. Gennero de Chialvo and A. C. Chialvo, J. Mater. Chem., 2009, 19, 3276–3280 RSC.
- Y. Tan, Y. Liu, L. Kong, L. Kang and F. Ran, J. Power Sources, 2017, 363, 1–8 CrossRef CAS.
- A. Corma, C. González-Arellano, M. Iglesias, S. Pérez-Ferreras and F. Sánchez, Synlett, 2007, 1771–1774 CrossRef CAS.
- A. Corma, E. Gutiérrez-Puebla, M. Iglesias, A. Monge, S. Pérez-Ferreras and F. Sánchez, Adv. Synth. Catal., 2006, 348, 1899–1907 CrossRef CAS.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 173, 249–274 CrossRef CAS.
- K. Layek, H. Maheswaran and M. L. Kantam, Catal. Sci. Technol., 2013, 3, 1147–1150 RSC.
- A. Monopoli, P. Cotugno, G. Palazzo, N. Ditaranto, B. Mariano, N. Cioffi, F. Ciminale and A. Nacci, Adv. Synth. Catal., 2012, 354, 2777–2788 CrossRef CAS.
- L. Wang, H. Wang, W. Zhang, J. Zhang, J. P. Lewis, X. Meng and F.-S. Xiao, J. Catal., 2013, 298, 186–197 CrossRef CAS.
- A. Primo and F. Quignard, Chem. Commun., 2010, 46, 5593–5595 RSC.
- C. González-Arellano, A. Corma, M. Iglesias and F. Sánchez, Chem. Commun., 2005, 1990–1992 RSC.
- S. Carrettin, J. Guzman and A. Corma, Angew. Chem., Int. Ed., 2005, 44, 2242–2245 CrossRef CAS.
- S. Hemmati, L. Mehrazin, M. Pirhayati and H. Veisi, Polyhedron, 2019, 158, 414–422 CrossRef CAS.
- J. Sun, Y. Fu, G. He, X. Sun and X. Wang, Appl. Catal., B, 2015, 165, 661–667 CrossRef CAS.
- T.-H. Kwon, V. Armel, A. Nattestad, D. R. MacFarlane, U. Bach, S. J. Lind, K. C. Gordon, W. Tang, D. J. Jones and A. B. Holmes, J. Org. Chem., 2011, 76, 4088–4093 CrossRef CAS.
- J. Frey, A. D. Bond and A. B. Holmes, Chem. Commun., 2002, 2424–2425 RSC.
- H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS.
- O. R. Luca, J. L. Gustafson, S. M. Maddox, A. Q. Fenwick and D. C. Smith, Org. Chem. Front., 2015, 2, 823–848 RSC.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- L.-L. Chua, J. Zaumseil, J.-F. Chang, P. E. C.-W. Ou, K.-H. Ho, H. Sirringhaus and R. H. Friend, Nature, 2005, 434, 194–199 CrossRef CAS.
- B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS.
- H. Spanggaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2004, 83, 125 CrossRef CAS.
- W. J. Potscavage, A. SharmaJr and B. Kippelen, Acc. Chem. Res., 2009, 42, 1758–1767 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00240e |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |