
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailoring the stability, photocatalysis and photoluminescence properties of Au11 nanoclusters via doping engineering†
Zhaoxian
Qin‡
ab,
Dan
Zhao‡
ce,
Li
Zhao
a,
Qian
Xiao
a,
Tingting
Wu
bc,
Jiangwei
Zhang
 *b,
Chongqing
Wan
*ad and
Gao
Li
*b
*b,
Chongqing
Wan
*ad and
Gao
Li
*b
aBeijing Key Laboratory for Optical Materials and Photonic Devices, Department of Chemistry, Capital Normal University, Beijing 100048, P. R. China. E-mail: wancq@cnu.edu.cn
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: jwzhang@dicp.ac.cn; Web: https://publons.com/a/1297379gaoli@dicp.ac.cn
cSchool of Marine Technology and Environment, Dalian Ocean University, Dalian 116023, P. R. China
dKey Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China
eState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China
First published on 8th May 2019
Abstract
Dopants in gold nanoclusters have been proved to mediate the intrinsic electronic properties of homo-clusters. In this work, we report the precise synthesis of atomically precise Au8Ag3(PPh3)7Cl3 alloy nanoclusters with multiple Ag dopants for the first time. Their structure was resolved by single-crystal X-ray crystallography. Au8Ag3(PPh3)7Cl3 nanoclusters possessed a similar structure topology to the well-known Au11(PPh3)7Cl3 nanoclusters. It is observed that the three Ag atoms were fixed at the cluster surface and bound selectively with the chlorine ligands in a C3-axis manner. The alloy nanoclusters exhibited a closed-shell electronic structure (i.e., 8(Au 6s1) + 3(Ag 5s1) − 3(Cl) = 8e), as evidenced by electrospray ionization-mass spectrometry (ESI-MS). The photothermodynamic stability of alloy clusters was remarkably improved (e.g., full decomposition after 7 days under sunlight irradiation vs. 3 days for Au11(PPh3)7Cl3 clusters). DFT calculations indicated that the Ag dopants in a C3-axis manner could obviously delocalize the electrons of Au to the orbitals of P atoms and then mediate the electronic property of the clusters. Shrinkage of the HOMO–LUMO gap to 1.67 eV of Au8Ag3(PPh3)7Cl3 was observed as compared with that of homo-nanoclusters of Au11(PPh3)7Cl3 (2.06 eV). The electrochemical gap of Au8Ag3(PPh3)7Cl3 alloy nanoclusters was 1.272 V, which was higher than that of Au11(PPh3)7Cl3 nanoclusters, which indicated higher electrochemical stability, as evidenced by the differential pulse voltammetry (DPV) method. Au8Ag3(PPh3)7Cl3 clusters exhibited three specific photoluminescence peaks at 405, 434 and 454 nm. AuAg alloy clusters exhibited twofold greater activity than homo gold clusters in the photooxidation of benzylamine, which was mainly due to the unique electronic properties of the alloy clusters. Controllable heteroatom doping engineering is a powerful method to tune the electronic properties of clusters, and then improve their photothermodynamic and electrochemical stability simultaneously for potential photocatalytic applications.
Introduction
Gold nanoclusters with atomic precision have emerged as a novel, robust class of nanomaterials over the last decade.1–3 These gold nanoclusters are usually protected by thiolate, phosphine and alkyne ligands, which are exploited widely in the areas of photosensitizers, electronics, biomedicine, and catalysis.4–10 These nanoclusters exhibit a nonmetallic nature due to electron energy quantization arising from a quantum size effect, which exerts a strong influence on their physical and chemical properties. With regard to phosphine-stabilized gold nanoclusters, Au11(PPh3)7/8X3/2 (X = SCN, Cl, or Br) nanoclusters are the most well-known and investigated nanoclusters, which were discovered in the 1980s. They are composed of an 11-atom incomplete icosahedral kernel, which is derived from the icosahedral Au13 synthon through etching and removal of two gold atoms.11,12 The major drawback of these phosphine-stabilized Au nanoclusters is very poor stability because the electron-rich phosphine groups oxidized readily and detached from the surface of gold nanoclusters. Subsequently, the phosphine-capped Au nanoclusters will decompose rapidly to Au(0) species (e.g., gold mirrors) in a few days, especially under light irradiation in air, which limits their practical applications significantly. An effective strategy is urgently required to enhance the stability of these nanoclusters.Recently, several scholars have discovered that doping of heteroatom metal atoms in Au nanoclusters can mediate their electronic properties (e.g., optical property, stability) even though these nanoclusters with heteroatom metal dopants often exhibit similar topological frameworks.13–16 It is well known that Pd and Pt heteroatoms in the gold nanoclusters can improve the stability of nanoclusters considerably.17,18 For example, Negishi and colleagues synthesized mono-Pd-doped Pd1Au10(PPh3)8Cl2 nanoclusters, and showed much better stability against the homo-nanocluster Au11(PPh3)8Cl2.17 Also, the photoluminescence (PL) property of such alloy nanoclusters was modified. Similarly, the Au24Pt1(SR)18 nanoclusters improved the thermal and antioxidation stabilities considerably compared with homo Au25(SR)18.18 In such Pd and Pt doping modes, heteroatoms are located in the center of the metal core mode with only mono-atom.
An Ag doping strategy in Au25 nanoclusters has also been investigated intensively.3 However, in all cases, gold nanoclusters with Ag dopants have been shown to decrease their stability.19–21 In our previous study, the AgxAu25−x(SR)18 (where, x: 1 to 13, and SR represents thiolate) showed very poor stability upon exposure to sunlight. The alloy clusters quickly decomposed to Au(I):SR and Ag(I):SR complexes in a CH2Cl2 solution in a few hours.20 Similar phenomena have been observed in AgxAu25−x(PPh3)10(SR)5Cl2, AgxAu38−x(SR)24 and AgxAu144−x(SR)60 systems. Thus, multiple doping of Ag atoms into Au nanoclusters in a precise manner and maintaining/improving their stability (e.g., under sunlight irradiation) are big challenges. Doping multiple Ag dopants into Au11 nanoclusters in a precise manner has not been achieved yet.
Herein, we developed a novel synthetic protocol to afford Au8Ag3(PPh3)7Cl3 (abbreviated as Au8Ag3) alloy nanoclusters with multiple Ag dopants. Its precise crystal frameworks were determined fully by single crystal X-ray crystallography. We found the surface gold (7) and silver (3) atoms to be bound selectively with the seven phosphine and three chlorine ligands, respectively. The three AgCl motifs located in the C3 axis in the Au8Ag3 alloy nanoclusters. Experimental evaluation showed that Au8Ag3 alloy nanoclusters were more stable than the corresponding homo-Au11(PPh3)7Cl3 nanoclusters under sunlight irradiation. Density functional theory (DFT) calculations indicated that the AgCl motifs in the C3 axis could obviously delocalize the electrons of Au to the orbitals of P atoms in PPh3 ligands and mediate the electronic property of Au8Ag3. A decrease in the highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) gap would ensure easier excitation of the electrons of Au8Ag3(PMe3)7Cl3 at a lower energy, resulting in PL enhancement. Au8Ag3 clusters exhibited specific three PL peaks at 405, 434 and 454 nm. The electrochemical gap of the Au8Ag3(PPh3)7Cl3 alloy nanoclusters was 1.272 V, which was higher than that of Au11(PPh3)7Cl3, which suggested higher electrochemical stability as evidenced by differential pulse voltammetry (DPV). Besides, these Au8Ag3(PPh3)7Cl3 alloy nanoclusters exhibited twofold greater activity of the HOMO gold clusters in the photooxidation of benzylamine, which was due mainly to the unique electronic properties of the alloy nanoclusters (Scheme 1).
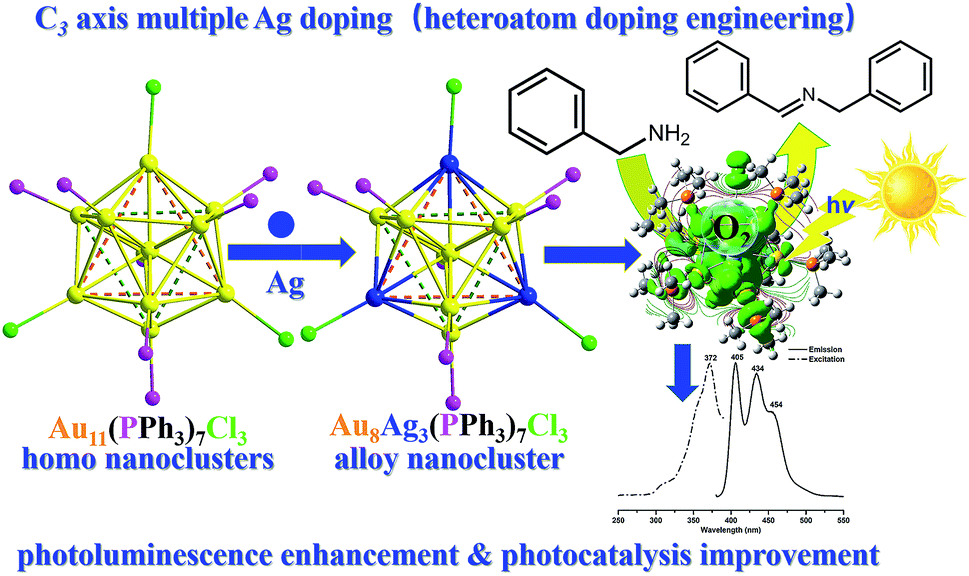 | ||
| Scheme 1 Au8Ag3(PPh3)7Cl3 alloy nanoclusters synthesis through controllable multiple Ag doping of C3 axes with photoluminescence enhancement and photocatalysis improvement. | ||
Experimental
Chemicals
All chemicals were commercially available in reagent grade and used as received without further purification. Hydrogen tetrachloroaurate(III) tetrahydrate (HAuCl4·4H2O, 99.95%) was purchased from HWG. AgSbF6 (99.95%), dimethyl sulfide (Me2S, 99%), sodium borohydride (NaBH4, 98%), triphenylphosphine (98%), benzylamine (98%), P25, ethyl ether (99.5%), ethanol (99.5%), methanol (99.5%), acetonitrile (99%), and dichloromethane (99.5%) were purchased from Adamas-beta. Ultrapure water (resistance, 18.2 MΩ cm) was purified with a Barnstead Nanopure Di-water™ system. All glassware was cleaned thoroughly with aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of hydrochloric acid and nitric acid), rinsed with copious amounts of ultrapure water, and then stored in an oven at 100 °C before use.
:1 mix of hydrochloric acid and nitric acid), rinsed with copious amounts of ultrapure water, and then stored in an oven at 100 °C before use.
Synthesis of Au8Ag3 nanoclusters
All procedures were carried out in the dark under an air atmosphere. Ph3PAuCl was synthesized via the reaction of HAuCl4 and two equivalent PPh3 in an ethanolic solution. Then, 25 mg of Ph3PAuCl (0.05 mmol) was dissolved in 1 mL of CH2Cl2 solution, followed by addition of AgSbF6 (17.2 mg in 1 mL of CH2Cl2). The mixture was stirred for 10 min at room temperature. Then, the white AgCl solid obtained was filtrated and removed. The filtrate was reduced by NaBH4 (0.5 mg in 1 mL of methanol) in an ice bath. After 36 h of stirring, the red-brown solution was filtered and concentrated to 1 mL. Diethyl ether was added and the solution volatilized for 1 week. The Au8Ag3 clusters (∼5 mg) were collected as a red plate crystal. The cluster yield was ∼24% based on Ph3PAuCl consumption.X-ray crystallography
Suitable single crystals were selected and data from synchrotron radiation X-ray diffraction were collected at the BL17B beamline of the National Facility for Protein Science and Shanghai Synchrotron Radiation Facility, China, using a constant wavelength of λ = 0.65248 Å and a large Debye–Scherrer camera. Data reduction, cell refinement and experimental correction of absorption were undertaken with software packages (HKL-3000 and Bruker APEX3). The cluster structure was solved by intrinsic phasing and refined against F2 by the full-matrix least-squares method. Non-hydrogen atoms were refined anisotropically. All calculations were carried out by SHELX22 and Olex2 v1.2.10.23Characterization
The UV-vis spectra of Au8Ag3 clusters (dissolved in CH2Cl2) were analyzed on a diode array spectrophotometer (8453) from Agilent Technologies. Electrospray ionization (ESI) mass spectra were obtained using a Waters quadrupole time-of-flight (Q-TOF) mass spectrometer equipped with a Z-spray source. The alloy clusters were dissolved in ethanol with CsOAc adducts. PL spectra were recorded on a spectrofluorometer (Fluorolog 3; Horiba) between 800 nm and 1500 nm using a liquid N2-cooled InGaAs detector, an excitation wavelength of 350 nm, slits of 14.7 nm, and a long-pass optical filter at 550 nm between the sample and detector to block overtones from the excitation source. DPV measurements were made on a CHI 760E electrochemical station at room temperature under a N2 atmosphere with TBABF4 as the electrolyte and methanol as the solvent. A Pt wire (counter electrode), Pt working electrode, and Ag/AgCl quasi-reference electrode were used for DPV. Thermal gravimetric analysis (TGA) was undertaken on a Mettler Toledo TGA/SDTA851 in air flow of 50.0 mL min−1 with heating rate of 20 °C min−1.Computational details
All DFT calculations were carried out with a Gaussian 09 program. Au8Ag3(PPh3)7Cl3 and Au11(PPh3)7Cl3 clusters were simplified using Au8Ag3(PMe3)7Cl3 and Au11(PMe3)7Cl3 molecular models, respectively. The geometries of Au8Ag3(PMe3)7Cl3 and Au11(PMe3)7Cl3 were optimized fully via the DFT method with the Lee–Yang–Parr gradient-corrected correction function (B3LYP)24–26 in combination with two basis sets: the SDD basis set for Au and Ag atoms and the 6-31G(d) basis set for other elements. Calculated absorption spectra were obtained using the time-dependent density functional theory (TDDFT) method with the same functional and basis sets as mentioned above. To be consistent in experiments, the solvent (dichloromethane) was represented based on the polarizable continuum model using the integral equation formalism variant (IEF-PCM).27–30Photocatalysis of benzylamine
A 5 mg cluster was dissolved in 12 mL of dichloromethane, and then 1 g of P25 was added. After stirring for 1 h, the obtained solids were collected by centrifugation and washed thrice by dichloromethane. The solids were dried in a vacuum at 60 °C overnight to prepare the 0.5 wt% Au8Ag3/P25 and Au11/P25 photocatalysts. Typically, 5 mg of photocatalysts, 20 mg of benzylamine, 0.1 mmol of p-xylene and 1 mL of CH3CN was added in a 5 mL quartz tube with a magnetic stirrer. The system was fully furnished with O2. The tube was sealed and irradiated with a LED light (455 nm; Ceaulight) for 1 h. After the reaction, the catalysts were separated by a membrane filter. The product solution was quantified by gas chromatography-mass spectrometry (GC-MS; 7890B GC system with 5977A MSD; Agilent Technologies). The conversion of benzylamine and selectivity for amine products were determined using internal standards. The photocatalysts were recovered by filtration and reused in the recyclability test with fresh solvent and reactants under identical conditions.Results and discussion
Synthesis and structure determination of Au8Ag3 alloy nanoclusters
The Au8Ag3 alloy clusters were obtained by simultaneous NaBH4 reduction of the mixture of Ph3PAuCl and AgSbF6 in a CH2Cl2 solution at 0 °C. A block-like crimson crystal was obtained via evaporation and resolved by X-ray crystallography.31 The Au8Ag3 clusters crystallized in the monoclinic group of P21/n, and the full framework is shown in Fig. 1a. The cluster exhibited a similar structure of the reported Au11(PPh3)7Cl3 (hereafter referred to as Au11) clusters12 and a C3 axis along the Ag3 plane (Fig. 1b). The three chlorine and seven phosphine ligands were coordinated selectively with the three Ag and seven Au atoms, leaving only one central Au atom in the M11 kernel (Fig. 1). Such coordination selectivity arose because gold is substantially more electronegative than silver. The more electron-donating phosphine ligands prefer the more electronegative Au atoms, whereas the more electron-withdrawing halide ligands prefer the more electropositive Ag atoms.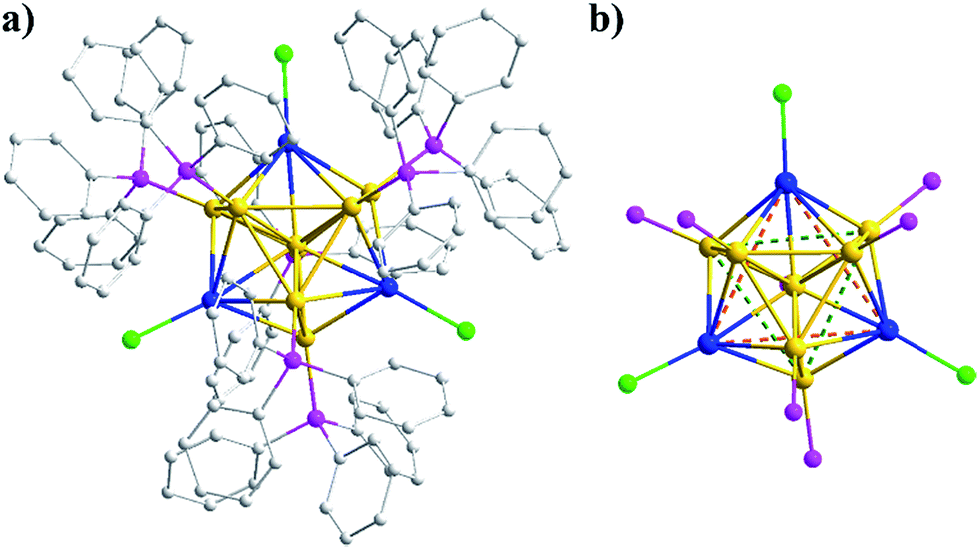 | ||
| Fig. 1 (a) Complete structure of Au8Ag3(PPh3)7Cl3 clusters. (b) Atomic arrangement of the Au8Ag3 kernel seen along the C3 axis. Au: orange; Ag: blue; P: purple; Cl: green; C: gray. All the H atoms have been omitted for clarity. The chlorine and phosphine ligands bond the silver and gold atoms selectively, respectively. | ||
A fixed number of three Ag atoms were doped controllably and precisely into the Au11 framework in a C3-axis manner in specific positions instead of a statistical distribution resembling AgxAu25−x nanoclusters reported previously.14a This phenomenon could be attributed to specific coordination selectivity in which the three “origin” Au atoms coordinated with three Cl atoms in the Au11(PPh3)7Cl3 framework are more prone to substitution by three Ag atoms to form Au8Ag3 alloy nanoclusters. Besides doping along the C3 axis, Ag could also maximally delocalize the electrons of Au in the Au8Ag3 framework to enhance its stability, as supported by related DFT calculations (see below).
The selected bond lengths of Au8Ag3 are compiled in Table 1. Compared with Au11, the average bond length of Au(staple)–Au(central) in Au8Ag3 clusters was shorter than that in Au11 clusters (2.647 vs. 2.672 Å, Table 1, entry 1). However, the average of Ag(Cl)–Au(central) bonds in the Au8Ag3 cluster was much longer than the Au(Cl)–Au(central) of the Au11 (2.754 vs. 2.701 Å, Table 1, entry 2). These data suggested that the Au sphere in Au8Ag3 was reduced slightly (−0.49%) compared with Au11 clusters. However, the Ag segment was stretched considerably (+1.96%). The kernel of M11 was mainly responsible for the HOMO–LUMO electronic transition of the clusters.32 The average Ag–Cl and Au–P bonds in Au8Ag3(PPh3)7Cl3 were also much longer than these bonds in Au11(PPh3)7Cl3 (2.418 vs. 2.374 Å and 2.290 vs. 2.275 Å, Table 1, entries 3 and 4). Overall, the Ag dopants modified the topological framework of Au11 clusters.
| Entry | Lengths (Å) | Au11 | Au8Ag3 | Ref. (%) |
|---|---|---|---|---|
| 1 | Au(PPh3)–Au(center) | 2.660 (2.608–2.678) | 2.647 (2.605–2.669) | −0.49 |
| 2 | M(Cl)–Au(center) | 2.701 (2.700–2.704) | 2.754 (2.749–2.757) | 1.96 |
| 3 | M(Au/Ag)–Cl | 2.374 (2.372–2.377) | 2.418 (2.412–2.422) | 1.85 |
| 4 | Au–P | 2.275 (2.269–2.285) | 2.290 (2.273–3.295) | 0.66 |
Characterization of Au8Ag3 nanoclusters
The final product of Au8Ag3 at a large scale (e.g., 10 mg) was analyzed further by UV-vis spectroscopy, and its purity was determined by ESI-MS. As seen in the Fig. 2a, the Au8Ag3 clusters exhibited three main UV-vis peaks at 417, 500 and 656 nm in the range 300–800 nm, which was remarkably different to the optical property of Au11 clusters (black profile vs. red profile). The optical energy gap (derived from UV-vis spectroscopy) was 1.69 and 2.05 eV for Au8Ag3 and Au11, respectively (Fig. S1†).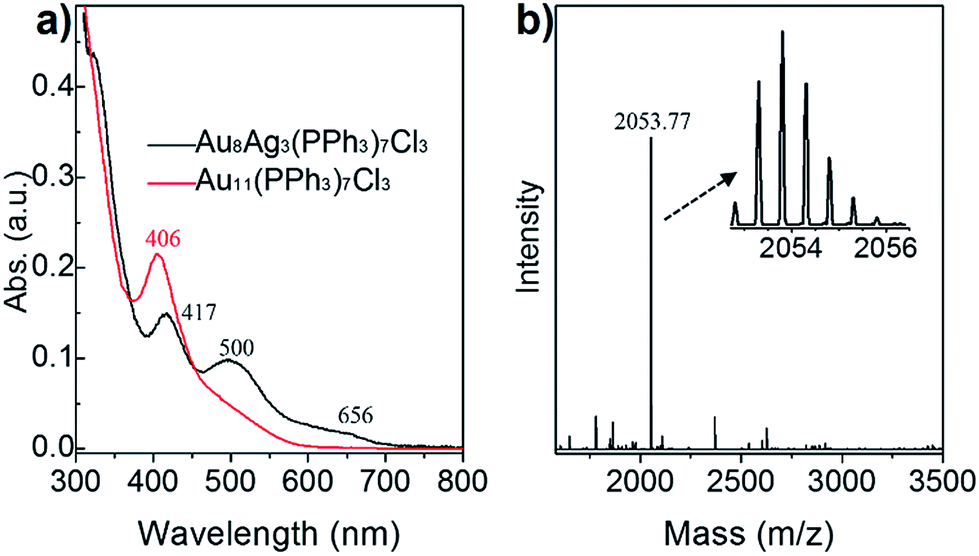 | ||
| Fig. 2 (a) Comparison of the optical spectra of Au11 and Au8Ag3 clusters. (b) Positive-mode ESI-MS of Au8Ag3; inset shows a close-up of the intense mass peak. | ||
Next, the as-obtained nanoclusters were characterized by ESI-MS (Fig. 2b). No counter ion (e.g., Na+ and Cl−) was found in the single-crystal analysis, thereby implying the electrical neutrality of the nanocluster (vide supra). Therefore, cesium acetate was added to impart charges to the metal clusters to form [(cluster)Csz]z+ (z = 1, 2, 3, etc.) adducts before ESI-MS. Only one expected intense peak at m/z = 2053.77 Da was found in the positive-mode spectrum in the range of 1500 Da to 3500 Da (Fig. 2b). The spacing of the isotope patterns was 0.5 Da (Fig. 2b, inset), confirming that the adducts were double charged (i.e., z = 2). After detailed calculations, the molecular mass of the adducts was determined to be 3841.72 Da (i.e., [2053.77 × 2] − [132.91 × 2] = 3841.72 Da) and, accordingly its chemical formula was determined to be Au8Ag3(PPh3)7Cl3 (Au8Ag3P7C126H105Cl3, theoretical m/z: 3841.69 Da, deviation: +0.03 Da). Of note, the singly charged adduct (i.e., [Au8Ag3(PPh3)7Cl3]Cs+, m/z: 3974.64 Da) was beyond the limit of detection. The experimental isotope pattern matched well with the simulated one. Thus, the Au8Ag3 nanocluster had a closed-shell electronic structure (i.e., 8(Au 6s1) + 3(Ag 5s1) − 3(Cl) = 8e), implying that the alloy nanoclusters should exhibit good stability.
Thermal and photothermodynamic stability of the Au8Ag3(PPh3)7Cl3 nanoclusters
To compare the thermal stability between Au8Ag3(PPh3)7Cl3 alloy nanoclusters and Au11(PPh3)7Cl3 nanoclusters, TGA was conducted from 30 °C to 600 °C, respectively. Au11(PPh3)7Cl3 nanoclusters remained intact up to 250 °C. Complete desorption of ligands from nanocluster surfaces began from 250 °C to 600 °C with ∼50% weight loss, which was highly consistent with the theoretical value (51.5 wt%). We found that Au8Ag3(PPh3)7Cl3 remained intact up to 200 °C, and complete desorption of ligands from nanocluster surfaces began from 200 °C to 600 °C with ∼55% weight loss, which was highly consistent with the theoretical value (53.7 wt%) (Fig. S3†). The thermal stability of Au11(PPh3)7Cl3 was slightly better than that of Au8Ag3(PPh3)7Cl3. Thus, they should be thermally stable at room temperature with sunlight exposure. Under sunlight irradiation, their photothermodynamic stability is important.Hutchison et al. reported that Au11 clusters were not stable in CH2Cl2 solution in the presence of air; the clusters decomposed in a few hours.33 Recently, we found that the stability of Au11 nanoclusters was improved when using diphenyl-2-pyridylphosphine (PPh2Py) as capping ligands.34 Here, we evaluated and compared the photothermodynamic stability of Au8Ag3 and Au11 clusters under atmospheric condition with sunlight irradiation at room temperature for 1 week, which was monitored by UV-vis spectroscopy. Interestingly, an obvious gold mirror was seen on the wall of the glass beaker. Also, the CH2Cl2 solution became colorless, which was also evidenced by UV-vis spectroscopy (the spectrum became straight, Fig. 3a, red line). These results indicated clearly that the Au11 clusters were not stable and decomposed to Au(0) species under sunlight irradiation, data that are consistent with the literature.33 Au8Ag3 alloy nanoclusters showed much greater stability under identical conditions. The intensity of the optical peaks decreased after 3 days (∼66% clusters survived), and disappeared after 7 days under sunlight irradiation (Fig. 3b). These data strongly demonstrated that the Ag dopants in the Au11 clusters improved the photothermodynamic stability considerably.
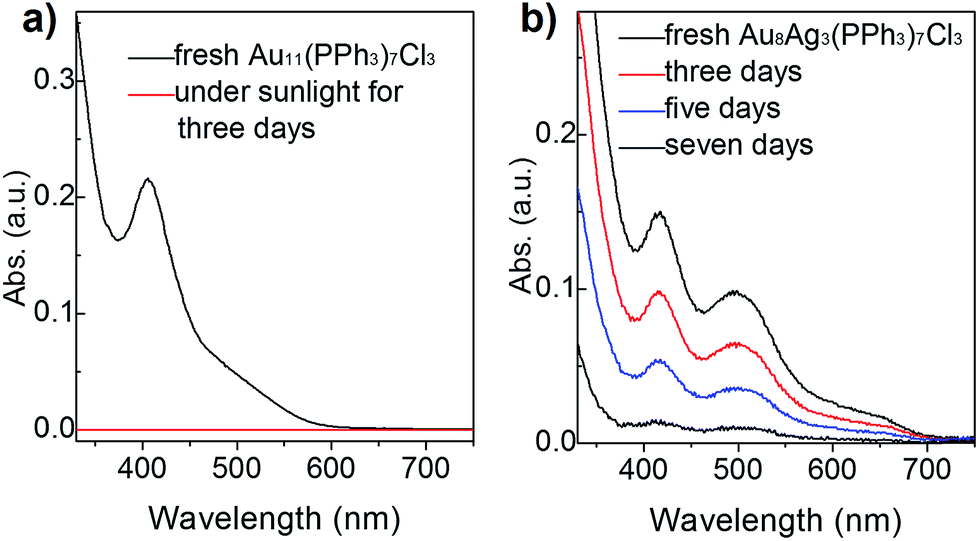 | ||
| Fig. 3 Stability of (a) Au11 and (b) Au8Ag3 clusters (dissolved in CH2Cl2 solution) under sunlight at room temperature for 1 week according to UV-vis spectroscopy. | ||
Computational simulation
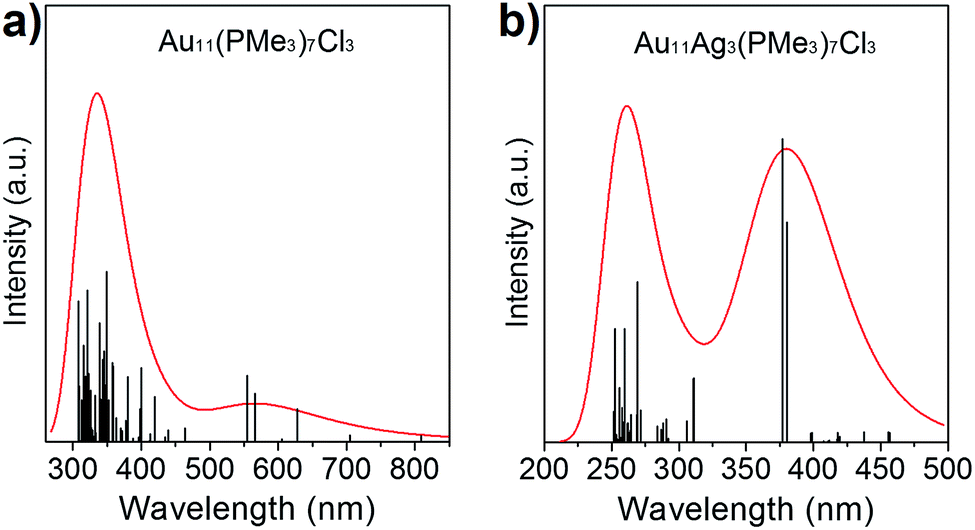
The absorption spectra of Au11(PMe3)7Cl3 and Au8Ag3(PMe3)7Cl3 have been calculated based on ground-state geometries using the TDDFT method. The calculated results are compiled in Fig. 5. Au11(PMe3)7Cl3 gave maximum absorption at 343.9 nm, and Au8Ag3(PMe3)7Cl3 showed two absorption bands at 261.4 and 382.5 nm, implying that more shells of electrons of Au8Ag3(PMe3)7Cl3 were excited in a shorter wavelength range. The delocalization effect of clusters induced decreasing energy. In addition, it would be easier for π electrons to be excited. For further analyses of the stability of Au11(PMe3)7Cl3 and Au8Ag3(PMe3)7Cl3, the energies and frequencies of Au11(PMe3)7Cl3, Au8Ag3(PMe3)7Cl3 and β-Au8Ag3(PMe3)7Cl3 were calculated. Three PMe3 ligands were grafted on Ag atoms whereas three Cl ligands was grafted on Au atoms in β-Au8Ag3(PMe3)7Cl3 (Fig. S2†).As shown in Table 2, the energies of Au8Ag3(PMe3)7Cl3 and β-Au8Ag3(PMe3)7Cl3 were lower than that of Au11(PMe3)7Cl3, implying that the former were more stable. The frequencies of β-Au8Ag3(PMe3)7Cl3 were negative, indicating that β-Au8Ag3(PMe3)7Cl3 was not stable. As stated above, the electronegative nature of metals contributes to such coordination selectivity.
| Clusters | G (a.u.) | HF (a.u.) | Frequency (cm−1) |
|---|---|---|---|
| Au8Ag3(PMe3)7Cl3 | −6135.69 | −6136.35 | 8.21 |
| Au11(PMe3)7Cl3 | −6101.81 | −6102.47 | 3.46 |
| β-Au8Ag3(PMe3)7Cl3 | −6135.65 | −6136.31 | −9.93 |
We wished to further illustrate the mediation of electronic property and HOMO–LUMO gap with regard to Ag dopants in a C3-axis manner in Au8Ag3(PMe3)7Cl3 alloy nanoclusters. Hence, the electron density map isocontours of fully optimized Au8Ag3(PMe3)7Cl3 and Au11(PMe3)7Cl3 were calculated (Fig. 6). With Ag dopants, the electrons of Au were obviously delocalized to the orbitals of P atoms in PPh3 ligands, whereas the electrons of Au assembled in the kernels of Au11(PMe3)7Cl3. Therefore, Au8Ag3(PMe3)7Cl3 possessed higher stability. The corresponding HOMO–LUMO gap of Au8Ag3(PMe3)7Cl3 alloy nanoclusters was reduced to 1.67 eV, compared with that of Au11(PMe3)7Cl3 of 2.06 eV (Fig. 6), data that were consistent with the optical gaps (Fig. S1†). The shrinkage of the HOMO–LUMO gap would make excitation of the electrons in Au8Ag3(PMe3)7Cl3 alloy nanoclusters easier at a lower energy, which may result in PL enhancement.8
We wished to further investigate mediation of electrochemical redox properties and electrochemical gap (EG) with regard to Ag dopants in a C3-axis manner in Au8Ag3(PPh3)7Cl3 alloy nanoclusters. Hence, we compared the DPV data of Au11 and Au8Ag3 nanoclusters (Fig. 4).
 | ||
| Fig. 4 DPV behaviour of (a) Au11 and (b) Au8Ag3 nanoclusters (dissolved in methanol/0.1 M TBABF4) on a Pt electrode at room temperature (measurement conditions: pulse cycle, 0.2 s; scan rate in either direction, 0.05 V s−1). | ||
 | ||
| Fig. 5 Calculated UV-vis spectra of (a) Au8Ag3(PMe3)7Cl3 and (b) Au11(PMe3)7Cl3 nanoclusters in CH2Cl2 solution. | ||
The EG is defined as the gap between the first oxidation potential and the first reduction potential, which correlates to some fundamental physicochemical properties of materials, including electrochemical stability.35 The first oxidation wave (O1) and first reduction wave (R1) of Au11 nanoclusters were at +0.708 and −0.372 V (versus the quasi-reference Ag electrode), respectively (Fig. 4a). Hence, the EG of Au11 was 1.180 V. The O1, R1 and R2 of Au8Ag3 nanoclusters were at +0.664, −0.508 and −0.978 V, respectively (Fig. 4b). The EG of Au8Ag3 was 1.272 V (i.e., (+0.664 V) − (−0.508 V)), which was larger than that for Au11. These data indicated that Ag dopants in a C3-axis manner in Au8Ag3(PPh3)7Cl3 alloy nanoclusters dramatically mediated its corresponding electrochemical redox property compared with homo-nanocluster Au11(PPh3)7Cl3 with a similar structure. The lower EG of Au11(PPh3)7Cl3 nanoclusters indicated that Au11(PPh3)7Cl3 nanoclusters were more prone to undergo redox processes, whereas the higher EG of Au8Ag3(PPh3)7Cl3 alloy nanoclusters suggested that they had higher electrochemical stability. Heteroatom dopants could obviously mediate the corresponding electrochemical redox property.
PL properties of Au8Ag3(PPh3)7Cl3
In recent years, the luminescence properties of gold clusters have motivated researchers due to their potential utility as new types of quantum dots.36–38 It has been reported that the PL of gold nanoclusters can be mediated by protecting ligands and other heteroatom metal dopants.17,39 For example, Negishi et al. demonstrated that PdAu10(PPh3)8Cl2 clusters exhibit strong PL at 950 nm and that Au11(PPh3)8Cl2 clusters do not show luminescence.17 The unique PL properties of Au8Ag3 clusters are presented in Fig. 7. The Au11 clusters gave near-infrared PL, centered at ∼1074 nm (1.16 eV), consistent with the reported result.8 The PL spectrum of Au8Ag3 clusters contained three main emission peaks centered at ca. 405 (∼3.06 eV), 434 (2.86 eV), and 454 nm (2.73 eV), respectively, which were distinctly different compared with Au11. Overall, these results suggested that Ag dopants definitely mediated the electronic structure of Au8Ag3 alloy clusters, thereby leading to PL enhancement.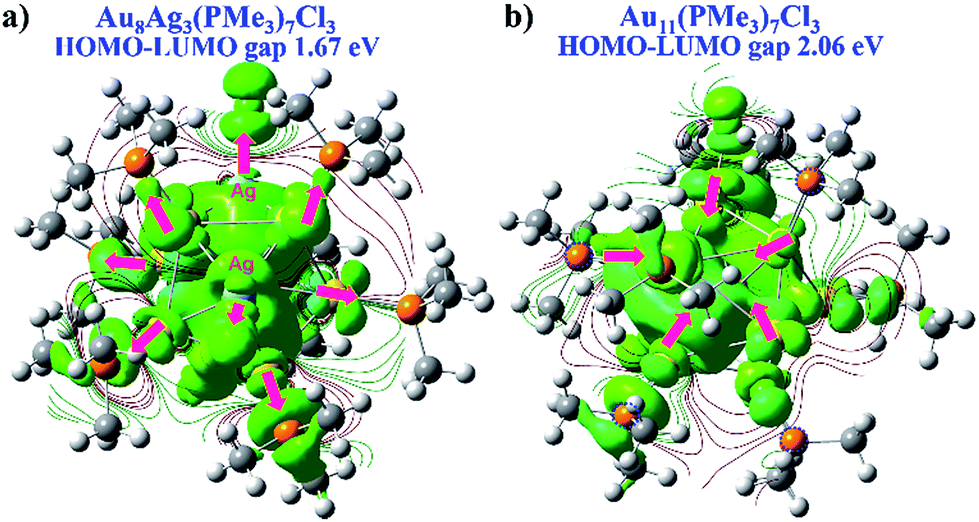 | ||
| Fig. 6 DFT electron density map isocontours and HOMO–LUMO gap of (a) Au8Ag3(PMe3)7Cl3 and (b) Au11(PMe3)7Cl3 from the same perspective. | ||
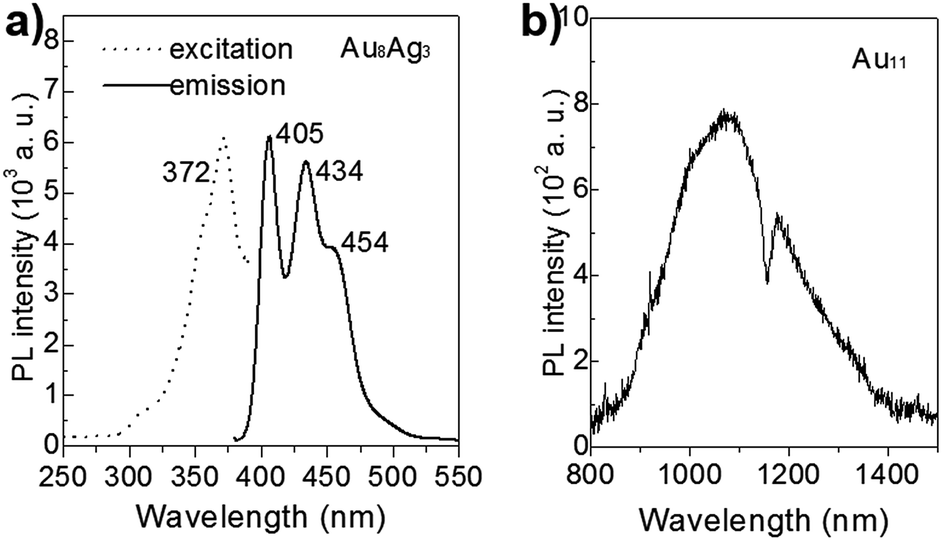 | ||
| Fig. 7 PL spectra of (a) Au8Ag3 (excited at 372 nm) and (b) Au11 clusters (excited at 350 nm), dissolved in CH2Cl2 solution. | ||
Catalytic activity in the photocatalytic oxidation of amines
Very recently, free and TiO2-supported Au nanocluster catalysts have been applied in the photocatalytic oxidation of amines to imines, which are applied widely in dye industries.40–43 Such photocatalytic oxidation involved activation of oxygen molecules.8,40 Imines are important versatile intermediates for fine chemicals and pharmaceuticals. The nature of the photothermodynamic stability (especially under light irradiation) and electrochemical stability of Au8Ag3(PPh3)7Cl3 nanoclusters with a suitable lower HOMO–LUMO gap (1.67 eV) located in the sunlight range (1.55–2.06 eV) makes them promising material for amine photocatalysis. Thus, we explored and compared the photocatalytic properties of Au8Ag3 and Au11 nanoclusters (supported on P25) via a one-step chemical transformation of benzylamine to N-(phenylmethylene)benzenemethanamine under LED light (λ ∼ 455 nm) in the presence of molecular oxygen at room temperature.The results of the photocatalysis are compiled in Table 3. The Au8Ag3/P25 catalysts gave 72.5% benzylamine conversion under irradiation for 1 h (Table 3, entry 2). Low conversion (37.8%) was found when using Au11/P25 as catalysts under identical reaction conditions (Table 3, entry 3). The selectivity towards the N-(phenylmethylene)benzenemethanamine product was >99%. In a control experiment, the P25 support gave only 13.8% conversion with >99% selectivity (Table 3, entry 6). The turnover frequency of the Au8Ag3/P25 photocatalyst (turnover frequency = (reacted mol of amine)/[(mol of cluster) × (reaction time in second)]) was calculated to be 2.92 s−1. This value was ∼2 times that for Au11/P25 (1.51 s−1), which was caused mainly by the unique electronic property of Au8Ag3 alloy clusters. Furthermore, Au8Ag3/P25 and Au11/P25 photocatalysts showed no activity in the absence of light irradiation, and gave <1% conversion under a N2 atmosphere (Table 3, entries 4–7). Meanwhile, conversion was not observed in the blank experiment without TiO2 and gold clusters (Table 3, entry 8). Next, we investigated the reusability of Au8Ag3/P25 photocatalysts under identical reaction conditions. During the second and third cycles, appreciable loss of catalytic activity or product selectivity were not observed (Table 1, entries 9 and 10). Overall, these catalysis results clearly demonstrated that photocatalytic oxidation was promoted by oxygen and was associated with the specific electronic property through heteroatom Ag dopants in gold nanoclusters as Au8Ag3(PPh3)7Cl3 alloy nanoclusters. Actually, shrinkage of the HOMO–LUMO gap to 1.67 eV of Au8Ag3(PPh3)7Cl3, as compared with 2.06 eV for homo-nanoclusters of Au11(PPh3)7Cl3, through Ag doping, was the reason for the higher photocatalytic activity of Au8Ag3. Metal nanoclusters with a suitably low HOMO–LUMO gap located at the sunlight range (1.55–2.06 eV) are more prone to generating 1O2 molecules via photoexcitation of metal nanoclusters,8,40 which involves Dexter-type electron-exchange coupling between the metal nanocluster (serving as an excited photosensitizer) and ground-state 3O2 molecules. This would greatly enhance their corresponding photocatalytic-oxidation activity. For such metal nanoclusters with a HOMO–LUMO gap >2.06 eV that are out of the sunlight range, generation of 1O2 molecules is not possible, so they would not show photocatalytic oxidation.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conversionb (%) | Selectivityb (%) |
| a Reaction conditions: 20 mg of benzylamine, 0.1 mmol of p-xylene, 1 mL of acetonitrile, 10 mg of Au8Ag3/P25 or Au11/P25 or P25 in the presence of O2 at 30 °C under LED light centered at λ ∼ 455 nm for 1 h. b The conversion of benzylamine and selectivity for imine were determined by GC-MS. c In absence of light. d Under a N2 atmosphere. e Second reuse of the Au8Ag3/P25 photocatalysts recovered from entry 1. f Third reuse of the Au8Ag3/P25 photocatalysts recovered from entry 1. | |||
| 1 | Au8Ag3/P25 | 72.5 | >99 |
| 2 | Au11/P25 | 37.8 | >99 |
| 3 | P25 | 13.8 | >99 |
| 4c | Au8Ag3/P25 | — | — |
| 5c | Au11/P25 | — | — |
| 6d | Au8Ag3/P25 | <1 | — |
| 7d | Au11/P25 | <1 | — |
| 8 | — | — | — |
| 9e | Au8Ag3/P25 | 70.8 | >99 |
| 10f | Au8Ag3/P25 | 71.7 | >99 |
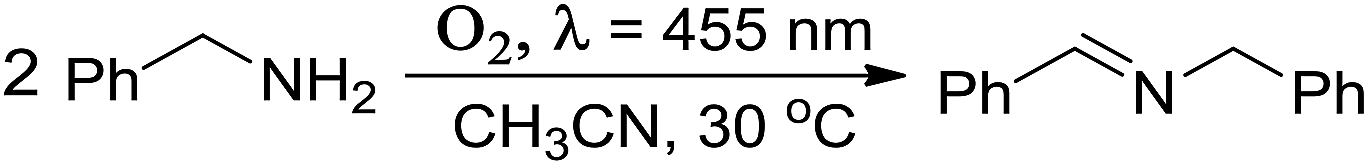
Conclusions
In summary, a novel synthetic protocol to afford Au8Ag3(PPh3)7Cl3 alloy nanoclusters with precise Ag dopants in a controllable manner was developed. The precise atomic framework was determined by single-crystal X-ray diffraction. Au8Ag3(PPh3)7Cl3 nanoclusters possessed a closed-shell electronic structure and a similar framework to Au11(PPh3)7Cl3 nanoclusters. The alloy clusters exhibited significant photothermodynamic stability (e.g., under sunlight irradiation) and electrochemical stability as evidenced by UV-vis spectroscopy and DPV, respectively, by comparison with homo-nanocluster Au11(PPh3)7Cl3 with a similar structure. DFT calculations indicated that the Ag dopants in a C3-axis manner obviously delocalized the electrons of Au to the orbitals of P atoms, and induced shrinkage of the HOMO–LUMO gap to 1.67 eV. These actions made excitation of the electrons of alloy clusters easier at a lower energy, resulting in PL enhancement. The EG of Au8Ag3(PPh3)7Cl3 alloy nanoclusters was 1.272 V, which is higher than that of Au11(PPh3)7Cl3 nanoclusters, which indicated higher electrochemical stability. Finally, the Au8Ag3(PPh3)7Cl3 clusters exhibited twofold greater values for turnover frequency for Au11(PPh3)7Cl3 nanoclusters in the photocatalysis of benzylamine. Heteroatom doping could obviously mediate the intrinsic electronic properties and frontier molecular orbitals of alloy nanoclusters, leading to shrinkage of the HOMO–LUMO gap, EG modification and the corresponding PL enhancement and photocatalytic activity improvement. This study provides a new strategy to synthesize alloy clusters with both high photothermodynamic and electrochemical stability simultaneously for potential photocatalysis applications through heteroatom doping engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Grant no. 21701168), Liaoning Natural Science Foundation (No. 20170540897, 20180510050). Natural Science Foundation of Beijing Municipality (Grant no. 2172014). Key Laboratory of Life Organic Phosphorus Chemistry and Chemical Biology, Ministry of Education, Tsinghua University and open project Foundation of State Key Laboratory of Physical Chemistry of Solid Surfaces, Xiamen University (No. 201709). We gratefully acknowledge the National Facility for Protein Science and Shanghai Synchrotron Radiation Facility, Shanghai, China for providing the BL17B beamline beam time.Notes and references
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346 CrossRef CAS PubMed.
- J. Fang, B. Zhang, Q. Yao, Y. Yang, J. Xie and N. Yan, Coord. Chem. Rev., 2016, 322, 1 CrossRef CAS.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749 CrossRef CAS PubMed.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816 CrossRef CAS PubMed.
- K. Zheng, J. Zhang, D. Zhao, Y. Yang, Z. Li and G. Li, Nano Res., 2019, 12, 501 CrossRef CAS.
- Q. Li, Y. Pan, T. Chen, Y. Du, H. Ge, B. Zhang, J. Xie, H. Yu and M. Zhu, Nanoscale, 2018, 10, 10166–10172 RSC.
- J. Zhang, Y. Zhou, K. Zheng, H. Abroshan, D. R. Kauffman, J. Sun and G. Li, Nano Res., 2018, 11, 5787 CrossRef CAS.
- W. Zhou, Y. Cao, D. Sui, W. Guan, C. Lu and J. Xie, Nanoscale, 2016, 8, 9614–9620 RSC.
- Z. Li, W. Li, H. Abroshan, Q. Ge, Y. Zhou, C. Zhang, G. Li and R. Jin, Nanoscale, 2018, 10, 6558–6565 RSC.
- M. McPartlin, R. Mason and L. Malatesta, Chem. Commun., 1969, 334 RSC.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13 Search PubMed.
- (a) R. Jin, S. Zhao, C. Liu, M. Zhou, G. Panapitiya, Y. Xing, N. L. Rosi, J. P. Lewis and R. Jin, Nanoscale, 2017, 9, 19183 RSC; (b) S. Wang, H. Abroshan, C. Liu, T.-Y. Luo, M. Zhu, H. J. Kim, N. L. Rosi and R. Jin, Nat. Commun., 2017, 8, 848 CrossRef PubMed.
- (a) S. Wang, Q. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2018, 51, 2784 CrossRef CAS PubMed; (b) Z. Li, X. Yang, C. Liu, J. Wang and G. Li, Prog. Nat. Sci.: Mater. Int., 2016, 26, 477 CrossRef CAS.
- (a) S. Takano, S. Hasegawa, M. Suyama and T. Tsukuda, Acc. Chem. Res., 2018, 51, 3074 CrossRef CAS PubMed; (b) M. Kim, Q. Tang, A. V. N. Kumar, K. Kwak, W. Choi, D. Jiang and D. Lee, J. Phys. Chem. Lett., 2018, 9, 982 CrossRef CAS PubMed.
- J. Koivisto, S. Malola, C. Kumara, A. Dass, H. Hakkinen and M. Pettersson, J. Phys. Chem. Lett., 2018, 3, 3076 CrossRef PubMed.
- W. Kurashige and Y. Negishi, J. Cluster Sci., 2012, 23, 365 CrossRef CAS.
- H. Qian, D. Jiang, G. Li, C. Gayathri, A. Das, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2012, 134, 16159 CrossRef CAS PubMed.
- C. Kumara, C. M. Aikens and A. Dass, J. Phys. Chem. Lett., 2014, 5, 461 CrossRef CAS PubMed.
- W. Li, C. Liu, H. Abroshan, Q. Ge, X. Yang, H. Xu and G. Li, J. Phys. Chem. C, 2016, 120, 10261 CrossRef CAS.
- S. Yamazoe, W. Kurashige, K. Nobusada, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2014, 118, 25284 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200 CrossRef CAS.
- B. Mennucci, E. Cances and J. Tomasi, J. Phys. Chem. B, 1997, 101, 10506 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS.
- R. Cammi and J. Tomasi, J. Comput. Chem., 1995, 16, 1449 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, J. Chem. Phys., 1981, 55, 117 CAS.
- Crystal data: monoclinic, P21/n, a = 17.93 Å, b = 26.24 Å, c = 227.00 Å, β = 91.6646°, V = 12701 Å3, Z = 4, T = 173 K, 21589 reflections measured, R1 = 0.0454, wR2 = 0.1232. CCDC-1873307 contains the supplementary crystallographic data.†.
- K. Spivey, J. I. Williams and L. Wang, Chem. Phys. Lett., 2006, 432, 163 CrossRef CAS.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426 CrossRef CAS PubMed.
- C. Liu, H. Abroshan, C. Y. Yan, G. Li and M. Haruta, ACS Catal., 2016, 6, 92 CrossRef CAS.
- L. Liao, S. Zhuang, P. Wang, Y. Xu, N. Yan, H. Dong, C. Wang, Y. Zhao, N. Xia, J. Li, H. Deng, Y. Pei, S.-K. Tian and Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 12644 CrossRef CAS PubMed.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- S. Wang, X. Zhu, T. Cao and M. Zhu, Nanoscale, 2014, 6, 5777–5781 RSC.
- Z. Wang, L. Wu, W. Cai and Z. Jiang, J. Mater. Chem., 2012, 22, 3632–3636 RSC.
- G. Zhang, R. Wang and G. Li, Chin. Chem. Lett., 2018, 29, 687–693 CrossRef CAS.
- S. Naya, K. Kimura and H. Tada, ACS Catal., 2013, 3, 10–13 CrossRef CAS.
- H. Chen, C. Liu, M. Wang, C. Zhang, N. Luo, Y. Wang, H. Abroshan, G. Li and F. Wang, ACS Catal., 2017, 7, 3632–3638 CrossRef CAS.
- Z. Li, C. Liu, H. Abroshan, D. R. Kauffman and G. Li, ACS Catal., 2017, 7, 3368–3374 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00234k |
| ‡ These authors contributed equally to this publication. |
| This journal is © The Royal Society of Chemistry 2019 |