
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCore-in-cage structure regulated properties of ultra-small gold nanoparticles†
Nirmal
Goswami
 *a,
Richard
Bright
ac,
Rahul Madathiparambil
Visalakshan
ab,
Bhabananda
Biswas
b,
Peter
Zilm
c and
Krasimir
Vasilev
*ab
*a,
Richard
Bright
ac,
Rahul Madathiparambil
Visalakshan
ab,
Bhabananda
Biswas
b,
Peter
Zilm
c and
Krasimir
Vasilev
*ab
aSchool of Engineering, University of South Australia, Mawson Lakes Campus, SA 5095, Australia. E-mail: Nirmal.Goswami@unisa.edu.au
bFuture Industries Institute, University of South Australia, Mawson Lakes Campus, SA 5095, Australia. E-mail: Krasimir.Vasilev@unisa.edu.au
cAdelaide Dental School, Faculty of Health and Medical Sciences, University of Adelaide, SA, Australia
First published on 2nd May 2019
Abstract
Understanding the structure–property relationships of novel materials is pivotal for the advances in science and technology. Thiolate ligand protected ultra-small gold nanoparticles (AuNPs; diameter below 3 nm) constitute an emerging class of nanomaterials with molecule-like properties that make them distinct from their larger counterparts. Here we provide new insights into the structure–property relationships of these nanomaterials by developing a series of ultra-small AuNPs, having comparable size and surface functionalities, but with different core-in-cage structures. We identified the density of metallic core and cage containing Au(I)–thiolate motifs, as well as cage rigidity as crucial factors that can significantly modulate the optical and biological properties of these AuNPs. In particular, AuNPs having a longer motif with a more rigid cage structure exhibited stronger luminescence while those containing a high percentage of loosely bound oligomeric Au(I)–thiolate motifs in the cage (semi-rigid structure) had better antibacterial activity. We also studied for the first time the inflammatory response to these NPs and revealed the importance of cage structure. We envisage that the finding reported in this paper can be applied not only to ultra-small AuNPs but also to other nanomaterials to develop new pathways to exciting future applications in electronics, sensing, imaging and medicine.
Introduction
The transition from “periodic table of elements” to “nanomaterials” through the guiding principle of nanochemistry has led to the discovery of several unique physical, chemical and biological properties which are distinctly different from those displayed by bulk materials.1–3 These nanoscale properties are the cornerstones of many revolutionary and existing applications in areas such as optoelectronics,4 catalysis5 and biomedicine.6,7 The intrinsic properties of a nanomaterial are governed by its size, shape, composition and structure. In the last two decades, considerable progress has been made in regard to controlling the properties of nanomaterials by defining the size,8 shape9 and composition.10 In contrast, nanomaterials with tailored structures as an important means of defining functions have rarely been considered. This is mainly due to the difficulty in manipulating the structure without compromising size, composition and surface chemistry. Thus, uncovering the structure regulated properties of nanomaterials is of major significance. To achieve this goal, novel materials and approaches that allow for more precise structural control are required.In this context, thiolate ligand protected, ultra-small (diameter below 3 nm) gold nanoparticles (AuNPs) could be an ideal platform to uncover new structure regulated properties because they are well studied.11–14 Typically, they have core-in-cage like architecture where the core consists of metallic gold and the cage/shell comprises oligomeric units (e.g. RS–[Au(I)–SR]x where –SR is the thiolate ligand and ‘x’ is the number of units). Together with their distinctive architecture, these materials exhibit many potential advantages over larger size NPs (>3 nm), as already demonstrated in the observation of molecule-like properties such as HOMO–LUMO transitions,15,16 chirality,17–19 magnetism20 and photoluminescence (PL).21–25 These unique properties are governed by size diminution and have led to dramatic improvements of synthesis methods and to novel applications in various fields including catalysis,26,27 sensing,28,29 imaging30 and medical therapies.31–33 Recently, great improvement has been made in understanding the properties of these ultra-small AuNPs. This was achieved either by tailoring their core composition or by changing the nature of surface ligands. For example, the PL properties of well-defined, AuNPs have been improved significantly by doping silver atoms in the core.34,35 The PL can also be enhanced by rigidifying the ligand shell as well as connecting the ligand shell with a polymer.36–40 The stability as well as antibacterial properties of AuNPs have also been shown to be affected by the surface ligands.41–43 These studies have resulted in several AuNPs with impressive properties with great potential for a broad range of applications. However, the role of nanomaterial structure in determining the properties of these ultra-small AuNPs remains largely unexplored. We hypothesize that molecular control over the material structure can be the sole determining factor for the optical properties of these NPs, their PL and the correlation with their biological properties. To address these gaps, the sole impact of the core and cage structure needs to be elucidated.
One strategy to tackle these scientific challenges is to develop a series of AuNPs with comparable size and surface chemistry, but with different core-in-cage structures. The successful synthesis of such AuNPs would improve our understanding on unresolved issues related to their properties. For example, AuNPs with variable cage contents could specifically reveal the contribution of Au(I)–thiolate motifs as well as the ligand density to their PL properties. The correlation between cage structures (rigidity) and the PL of NPs could also be recognized. In addition, these tailored core-in-cage AuNPs can also serve as a great model system for exploring the relationships between the material structure and its biological performance, which could provide innovative pathways for devising new biomedical applications.
The aim of this work is to shed light on how structural manipulation of a nanomaterial in a confined dimension can be used to regulate its properties. Here, we first report a strategy for designing ultra-small AuNPs with tailored and tunable core-in-cage structures. The synthesis of these AuNPs was realized by controlling the size and structure of the in situ generated Au(I)–thiolate complexes and manipulating the reduction power of the thiolate ligands. These NPs with distinct core-in-cage structures were used as a model system to investigate the effect of nanomaterial structure on both its optical and biological properties. Our study identified that a longer motif with a more rigid cage structure is responsible for the strong PL of NPs while the antibacterial activity of these NPs was governed by the density of Au(I)–thiolate motifs. We further provide the first experimental evidence of the capacity of ultra-small AuNPs to reduce inflammation.
Results and discussion
Synthesis of AuNPs with tunable core-in-cage structures
In this study, we selected to work with thiol ligand passivated ultra-small gold nanoparticles (AuNPs), as they are known to be highly stable in solution and have unique core-in-cage like structures (Fig. 1a). Thiol ligand passivated AuNPs having comparable size and surface properties, but with different core-in-cage structures, could be developed by tailoring their synthesis routes. The general route for the synthesis of these ultra-small AuNPs involves a two-step process, whereby Au(I)–SR complexes (also known as precursors) are initially formed followed by the reduction of these precursors in a subsequent step i.e., a reduction assisted growth process.44 By controlling reaction parameters such as pH, temperature and thiol-to-gold ratio, the size and composition of the AuNPs can be easily and precisely controlled. In this study, we hypothesized that delicate control of the selection of precursors together with right balance in the reduction process could enable the synthesis of AuNPs with different core-in-cage structures. To achieve that, the selection of the appropriate thiol ligand with desirable properties is crucial. Glutathione (GSH), a naturally occurring tri-peptide, could be an ideal choice for this study as it is known to have rich chemistry (i.e., consists of multiple functional groups such as thiol, amine and carboxylate) (Fig. 1b), and can be used as both reducing and capping agent with the opportunity to tune the molecule reduction potential by adjusting the reaction parameters. It is highly biocompatible and an important ligand for biological applications which indeed makes it a perfect choice for exploring the biological properties of ultra-small AuNPs.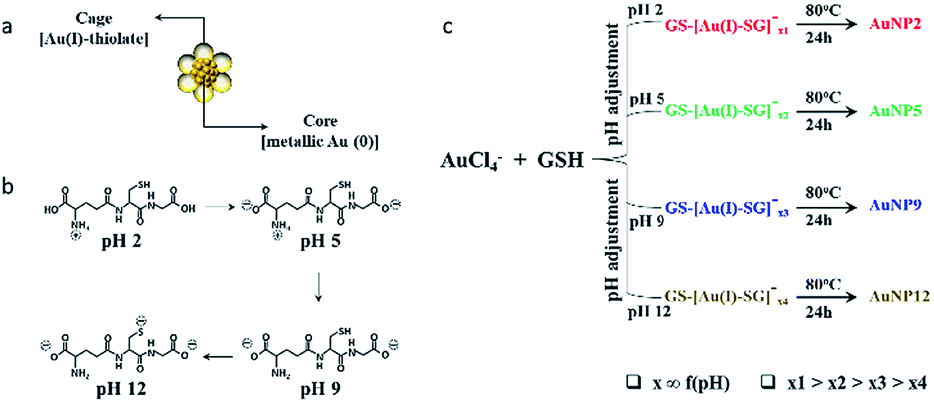 | ||
| Fig. 1 (a) Schematic illustration of the core-in-cage structure of AuNPs. (b) pH dependent ionization of GSH. (c) Schematic showing the synthesis of AuNP2, AuNP5, AuNP9, and AuNP12 by adjusting the pH of the reaction solution, followed by reduction at elevated temperature. | ||
Benefiting from the intriguing features of the GSH ligand, we first describe our synthesis route for achieving a series of AuNPs with similar size but different core-in-cage structures. The formation of Au(I)–SG complexes (also referred to as GS–[Au(I)–SG]x complexes) occurred by the reaction between the aqueous solution of GSH and HAuCl4 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio) at room temperature. The size and structure of the precursors were controlled by adjusting the pH of the solution immediately (∼1 min) after the formation of Au(I)–SG complexes. The solution temperature was then elevated to 80 °C to enhance the rate of decomposition of Au(I)–SG complexes by the GSH ligands as well as the subsequent formation of ultra-small AuNPs (Fig. 1c).
:2 molar ratio) at room temperature. The size and structure of the precursors were controlled by adjusting the pH of the solution immediately (∼1 min) after the formation of Au(I)–SG complexes. The solution temperature was then elevated to 80 °C to enhance the rate of decomposition of Au(I)–SG complexes by the GSH ligands as well as the subsequent formation of ultra-small AuNPs (Fig. 1c).
Four types of AuNPs were synthesized and studied here after purification by dialysis: AuNP2, AuNP5, AuNP9 and AuNP12, respectively (NPs were named based on the solution pH during synthesis). The surface functionalities of all these NPs remained the same as we used the same ligand (GSH) for their synthesis. Since the size is an important aspect to test our hypothesis, we extensively characterized the particle size by transmission electron microscopy (TEM) and dynamic light scattering (DLS). TEM studies showed the presence of monodisperse AuNPs of comparable interior core size (Fig. 2a–d). All the AuNPs exhibited similar hydrodynamic diameters of ∼2 nm (Fig. 2e). The size obtained from DLS was slightly larger than that obtained from TEM for all NPs due to the additional consideration of the ligand shell and hydration layer in the DLS measurement. Nevertheless, both studies confirmed that all four particles had nearly identical size.
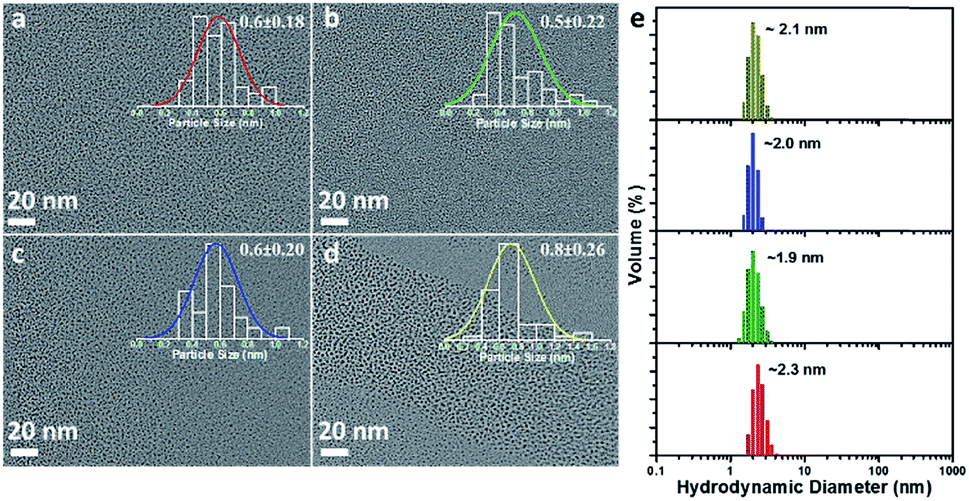 | ||
| Fig. 2 TEM micrograph of (a) AuNP2, (b) AuNP5, (c) AuNP9 and (d) AuNP12. The overlaid inset of (a–d) shows the corresponding particle size histogram with Gaussian fit. (e) DLS data showing the comparable hydrodynamic diameter of AuNP2 (red), AuNP5 (green), AuNP9 (blue) and AuNP12 (dark yellow). | ||
To gain a structural perspective of these AuNPs, we first carried out X-ray photoelectron spectroscopy (XPS) studies (Fig. 3a). The normalized Au 4f spectra of all four AuNPs were compared with that of Au(I)–SG complexes (black, dashed line). The purified Au(I)–SG complex that only consisted of Au(I) had an Au 4f7/2 binding energy (BE) of ∼84.6 eV. The XPS spectra of all four AuNPs remained under the spectrum of Au(I)–SG complexes which suggested the presence of a high percentage of Au(I)–SG complexes on the surface of all these NPs. However, the variation of Au 4f BE peak position among these AuNPs is clearly evident (Fig. 3a). The Au 4f BEs vary in the following order: AuNP5 (green) > AuNP2 (red) ≥ AuNP9 (blue) > AuNP12 (dark yellow). The Au 4f7/2 peak position of AuNP5 was ∼0.4 eV higher than that of AuNP12, which is significant especially for these ultra-small AuNPs. The shift in BE can be attributed to the presence of different percentages of Au(I) and Au(0) components in each type of NP (Table S1†). Since the Au(I)-component is likely to be present as GS–[Au(I)–SG]x motifs in the cage structures of these AuNPs, we also anticipated significant changes in their ligand density. Indeed, this was evident from our thermogravimetric analysis (TGA) (Fig. 3b). According to TGA, the thiol ligand content was highest for AuNP5 (∼64%) and lowest for AuNP12 (∼52%). These results were fully consistent with our XPS studies, which reflects the fact that AuNPs with highly dominant Au(I)-content in their cage had greater ligand density than other AuNPs.
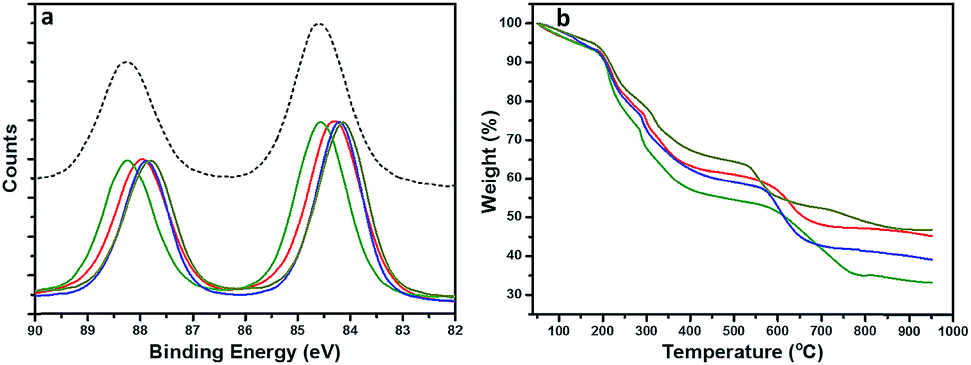 | ||
| Fig. 3 (a) High resolution XPS spectra showing the shift in the Au 4f spectral region. (b) TGA spectra show the difference in the presence of various thiolate ligands. Sample codes are as follows: AuNP2 (red), AuNP5 (green), AuNP9 (blue) and AuNP12 (dark yellow). | ||
Optical properties
Since all these AuNPs had comparable size and surface functionalities, yet different core and cage contents, we decided to compare their optical properties. The optical absorption spectra of AuNP2, AuNP5, AuNP9 and AuNP12 are shown in Fig. 4a. The absence of the plasmon resonance peak in the UV-vis spectra ruled out the possibility of the presence of any plasmonic AuNPs (∼3–100 nm AuNPs). All NPs showed a high energy optical band in the ∼400–450 nm region of the UV-vis absorption spectrum, which was similar to that of Au(I)–SG complexes (Fig. 4b). However, a slight variation in the onset of UV-vis spectra of these NPs was indeed observed, which could be due to the difference in their core densities. In particular, NPs with the highest motif contents i.e. AuNP5 showed the maximum spectral similarity to Au(I)–SG complexes (doublet UV-vis peak with the onset at ∼400 nm; Fig. S1†), while more metallic NPs (evidenced by the presence of the highest percentage of Au(0)) i.e., AuNP12 had a significantly different UV-vis spectrum (onset at ∼550 nm with a shoulder peak at ∼450 nm) than that of Au(I)–SG complexes. The aqueous solution of these NPs also displayed a clear colour appearance under visible light (Fig. 4a, inset), the difference being easily seen by the naked eye.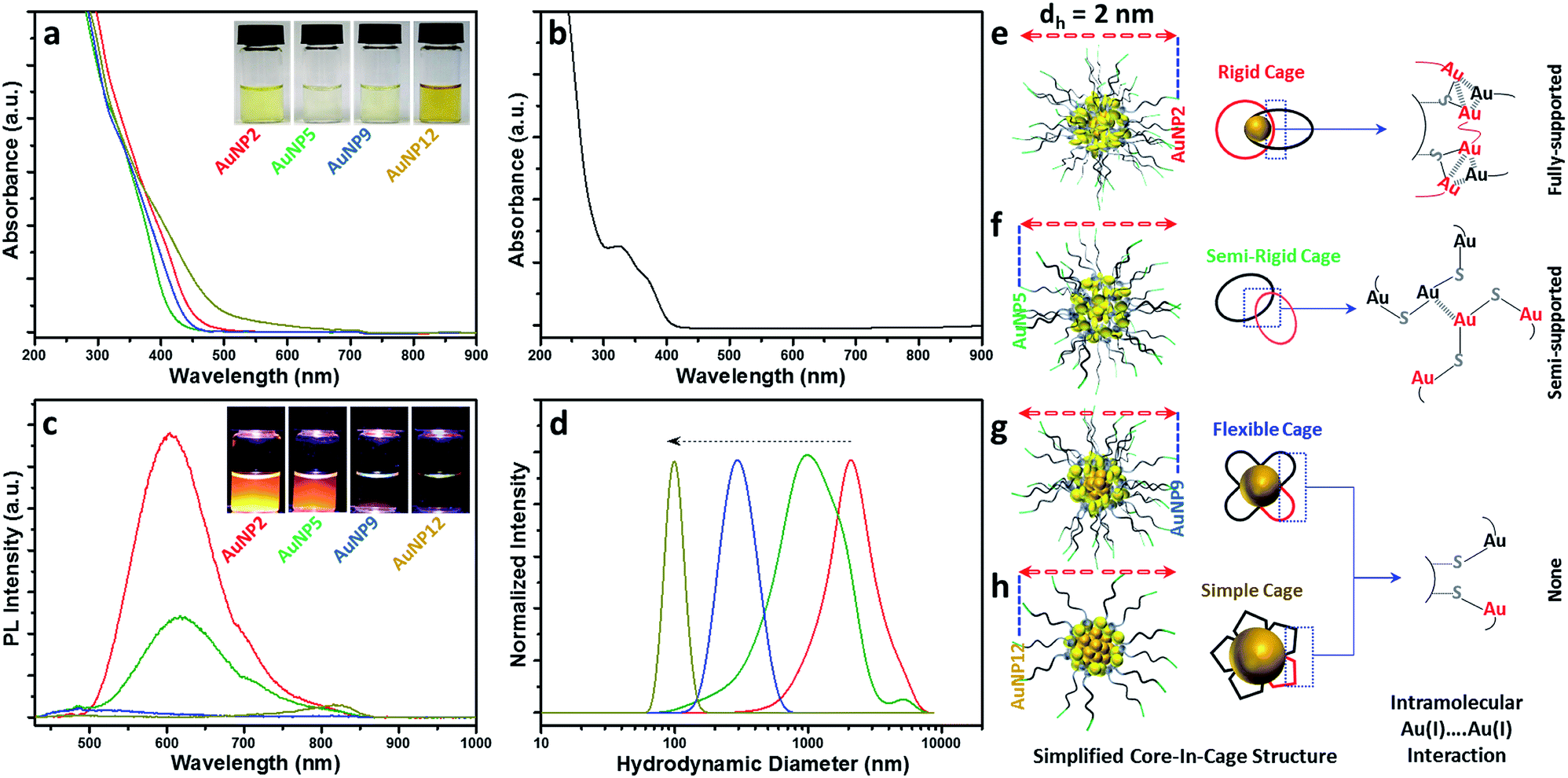 | ||
| Fig. 4 (a) UV-vis spectra of AuNP2 (red), AuNP5 (green), AuNP9 (blue) and AuNP12 (dark yellow). Inset shows the corresponding photograph of the NPs under visible light. (b) UV-vis spectra of the Au(I)–SG complex. (c) Photoluminescence spectra of AuNP2 (red), AuNP5 (green), AuNP9 (blue) and AuNP12 (dark yellow). Excitation wavelength (λex) was at 409 nm. Inset shows the photographs of the NPs under UV light. (d) DLS spectra of Au(I)–SG complexes at pH 2 (red), 5 (green), 9 (blue) and 12 (dark yellow). The dotted arrow indicates the reduction of complex size with different pH. (e–h) Simplified core-in-cage structure model shows the presence and absence of intramolecular aurophilic interaction in different AuNPs. | ||
One of the most distinctive features of ultra-small AuNPs that sets them apart from their plasmonic counterpart is their PL properties. For this reason, the PL properties of the AuNPs prepared in this work were thoroughly investigated. The PL spectra of the AuNPs are shown in Fig. 4c. Interestingly, both AuNP2 and AuNP5 displayed strong emission maxima at 610 nm, while AuNP9 was completely non-luminescent. AuNP12, however, showed poor PL at the emission maximum at 810 nm (Fig. S2†). The excitation spectrum of each of these AuNPs was closely alike to their corresponding UV-vis spectrum (Fig. S3†). A Large Stokes shift (>200 nm) was observed for all three luminescent NPs (AuNP2, AuNP5 and AuNP12). Bright orange PL colours from AuNP2 and AuNP5 were observed under UV light with the naked eye (excitation at 365 nm, Fig. 4c, inset).
This remarkable difference in the optical properties clearly demonstrates that structural manipulation is indeed an important consideration to regulate the properties of these ultra-small AuNPs. Note that the characteristics of our AuNPs i.e. high energy optical bands, high Au(I)–thiolate content, and large Stokes shift in the PL spectrum closely resembled those of previously reported Au nanoclusters (NCs), where the mechanism of aggregation-induced emission (AIE) was proposed.22,45 From that viewpoint, we expected that the origin of PL for our AuNPs could be attributed to AIE. However, amongst all four types of NPs studied here, only AuNP2 displayed strong PL like other reported AIE-type NCs.45 This was quite surprising since few other AuNPs, especially AuNP9, which had almost similar characteristics to AuNP2, did not exhibit any PL. Such a discrepancy in the PL properties of various AuNPs has raised an important question: is the presence of a high percentage of Au(I)–thiolate complexes in the cage adequate to justify the strong PL of AIE-type AuNCs? We anticipated that the size and structure of the Au(I)–SG complexes in the cage might be critical for the generation of strong PL. To address this, we measured the size of the Au(I)–SG complexes (motifs) that were formed during the synthesis of the AuNPs (Fig. 4d). The hydrodynamic diameter of the complexes became shorter with increasing the pH of the solution. Based on these results, it can be concluded that the cage structure of each type of AuNP consisted of different motif sizes.
Previous studies as well as our control PL experiments clearly defined the correlation between the Au(I)⋯Au(I) interactions and the extent of aggregation of Au(I)–SG complexes (Fig. S4a†).45 Based on the available information, we then proposed a simplified structural model for all these AuNPs which suggested a core-in-cage like structure with substantial difference in their cage assembly (Fig. 4e–h). In the case of AuNP2, longer motifs with a relatively smaller core favoured the formation of more interlocked cage assemblies, producing an increased probability of fully supported intra-molecular Au(I)⋯Au(I) interactions (Fig. 4e) which in turn generates strong PL. On the other hand, both the extent of aggregation of the motifs in the cage structure and the metallic core content of AuNP5 was lower than that of AuNP2. This may favour the formation of a semi-rigid cage structure with a reduced number of intra-molecular Au(I)⋯Au(I) interactions (Fig. 4f). The presence of different extents of Au(I)⋯Au(I) interactions in the cage structures also clarified the basis of stronger PL for AuNP2 than that of AuNP5. The presence of relatively shorter motifs together with the lack of PL for AuNP9 supported the presence of non-interactive motifs in the cage structure of these NPs (Fig. 4g). A similar cage structure was also demonstrated for AuNP12 (Fig. 4h). In both cases, the intra-molecular Au(I)⋯Au(I) interactions in their respective cage structures were absent.
Additional experimental support for the proposed structural model is given in Fig. S4.† We assumed that addition of ethanol could induce inter particle aggregation and there will be even higher PL than the corresponding well dispersed NPs. A schematic of such an experimental design was highlighted using AuNP9 as a model system (Fig. S5†). The motifs within the individual NP cage might not interact well through intra-molecular Au(I)⋯Au(I) interactions but in the aggregated state, motifs from one particle will interact with those of other particles and result in the generation of inter-molecular Au(I)⋯Au(I) interactions. The presence of such aurophilic interactions could make the aggregated AuNP9 luminescent. Indeed, PL was detected when AuNP9 was incubated in 90% (v/v) ethanol and excited at 400 nm. However, the PL maximum was ∼21 nm blue shifted (λmax = 589 nm) when compared to the PL maximum of AuNP2 (λmax = 610 nm) (Fig. S4†). This could be due to the difference in the type of Au(I)⋯Au(I) interactions present in their respective cage structures/aggregated state. For the aggregated Au(I)–SG complexes, where inter-molecular Au(I)⋯Au(I) interactions are likely to be more dominant than intra-molecular Au(I)⋯Au(I) interactions, the PL maximum was found to be ∼578 nm, which was ∼32 nm blue shifted compared to the PL maxima of AuNP2 and AuNP5. The large blue shift as well as the close proximity of PL maxima between the aggregated complex and AuNP9 suggested the existence of inter-molecular Au(I)⋯Au(I) interactions in the aggregated state of AuNP9. The small blue shift and high PL enhancement for aggregated AuNP2 and AuNP5 offered another piece of evidence that the existence of Au(I)⋯Au(I) interactions determines the PL properties of the AIE type of Au NCs.
Antibacterial properties
Next, we explored the effect of core-in-cage structures on the biological properties of all four AuNPs. It was reported recently that AuNPs in the confined dimensions (∼1 nm) can be used as an antimicrobial agent against both Gram-positive and Gram-negative bacteria.46,47 This antimicrobial activity was a result of the internalization of Au NCs and the subsequent generation of intracellular reactive oxygen species (ROS). These results were indeed fascinating, especially from the nanomaterial safety perspective, as gold is expected to have much better biocompatibility than other commonly used metals e.g., Ag, Cu, etc. In a later study, the same group demonstrated that the antimicrobial activity of these well-defined AuNPs could be enhanced further by tuning their surface properties.41,48 Alternatively, ultra-small AuNPs functionalized by rationally selected ligands have also been proven to be potent nano-antibiotics against multi-drug resistant bacteria in vivo.49In this study, we aimed to resolve an outstanding question about the exclusive contribution of the core-in-cage structures of the ultra-small AuNPs to their antibacterial effects. To address this challenge, we first evaluated the antibacterial properties of all four NPs using Escherichia coli as a model bacterium (Fig. 5a). A disk diffusion method was used to compare the antibacterial potency of the AuNPs against E. coli. The concentration of each type of NP was kept fixed (300 μg mL−1; on the basis of Au-atoms) and kanamycin was included as a positive control. Surprisingly, only AuNP2 and AuNP5 showed antibacterial activity while both AuNP9 and AuNP12 remained inactive against E. coli. However, the zone of inhibition for AuNP5 was considerably larger than that of AuNP2 which suggested that AuNP5 had a much stronger antibacterial activity compared with AuNP2 (Fig. 5a). Furthermore, the effect of concentration on the antibacterial activity on all AuNPs was also studied. The size of the inhibition zone became larger with the increase in concentration of both AuNP2 and AuNP5. We found that the minimum concentration of NPs required to obtain a clear zone of inhibition was less than 150 μg mL−1 (Fig. S6†). In contrast, no inhibition zone was observed for AuNP9 and AuNP12, even when the same bacteria were treated with a concentrated NP solution (up to 1500 μg mL−1) (Fig. S7†). These results clearly demonstrated that the antibacterial activity of these ultra-small AuNPs was highly related to their core-in-cage structures.
 | ||
| Fig. 5 (a) Antibacterial efficacy of AuNP2, AuNP5, AuNP9, AuNP12 and kanamycin against E. coli. Live-dead assay: representative fluorescence images of the E. coli from the growth zone (b–d) and from the zone of inhibition (e–g). Morphological changes of E. coli before (h and i) and after the treatment with AuNP5 (j and k). (l) Antibacterial effect of AuNP2, AuNP5, AuNP9, AuNP12 and kanamycin against P. aeruginosa. Live-dead assay: representative fluorescence images of the P. aeruginosa from the growth zone (m–o) and from the zone of inhibition (p–r). Morphological changes of P. aeruginosa before (s and t) and after (u and v) the treatment with AuNP5. | ||
We further examined whether these AuNPs with varied core-in-cage structures could exhibit a similar trend in antibacterial activity against other bacterial species. When we tested their efficacy against Pseudomonas aeruginosa with various concentrations of NPs, we observed a similar trend, however, with a smaller inhibition zone compared to that in E. coli (Fig. S8†). Furthermore, we also noted that a higher dose of AuNPs was required to inhibit the growth of P. aeruginosa (750 μg mL−1 for AuNP2 and 300 μg mL−1 for AuNP5) (Fig. S9†). An inhibition assay experiment with the same concentration of each type of AuNP (600 μg mL−1) revealed that only AuNP5 showed a clear inhibition zone while all other NPs remained inactive against P. aeruginosa (Fig. 5l).
To get a better understanding of the bacterial viability after treatment with our AuNPs, we utilized two different staining agents that can differentiate between viable and nonviable bacteria. Green fluorescent SYTO9 green nucleic acid stain, which enters live and dead bacterial cells, was used for identifying the total bacterial population. Red fluorescent propidium iodide (PI), which can penetrate only bacterial cells with disrupted membranes, was utilized for the identification of dead bacteria. By using a sterile loop, we removed bacteria from two different zones surrounding the disk treated with AuNP5 and assessed their viability with the above mentioned live/dead staining agents. As an example, AuNP5 was chosen since it showed the highest antibacterial activity among all four AuNPs. Confocal laser scanning microscopy (CLSM) imaging indicated that E. coli collected from the growth zone showed strong green fluorescence intensity, which was retained after counterstaining with PI (Fig. 5b–d). On the contrary, almost all bacteria collected from the zone of inhibition exhibited strong red fluorescence after PI counterstaining (Fig. 5e–g) indicating a strong bactericidal effect of the NPs. To verify the loss of viability, bacteria collected from the growth and inhibition zone were imaged by scanning electron microscopy (SEM). While clear rod-shaped bacteria with distinct cellular integrity were evident in untreated bacteria, most of the NP-treated bacteria displayed disrupted cell walls (Fig. 5h–k). In the case of P. aeruginosa, both the live/dead staining experiments and SEM images also support the effect of AuNP5 on the bacterial integrity (Fig. 5m–v). The fact that among all four AuNPs, AuNP5 with the highest percentage of loosely bound oligomeric motifs showed the best antibacterial efficacy implies that the antibacterial properties of thiol ligand passivated ultra-small AuNP are cage structure dependent.
Biocompatibility
In view of the promising antibacterial properties of the ultra-small AuNPs synthesized in this work, it is important to evaluate their biocompatibility. Thus, in a next step, we tested for any potential cytotoxicity of these nanomaterials to Primary Human Dermal Fibroblast cells (HDFs). The results of the resazurin assay are shown in Fig. 6a. Treatment with 150 μg mL−1 of AuNPs did not cause any cytotoxicity as the viability of the cells was the same or even greater compared to the control. Further analysis was conducted by fluorescence microscopy imaging of cells grown on a tissue culture plate (TCP) without and with added AuNPs. The images are shown in Fig. 6b and demonstrate that all cells were well confluent and had normal fibroblast morphology. Combined with the antibacterial properties of AuNP5, the excellent biocompatibility of the ultra-small AuNPs reported here presents potential for development of new nano-antibiotics that could be useful for treating infections.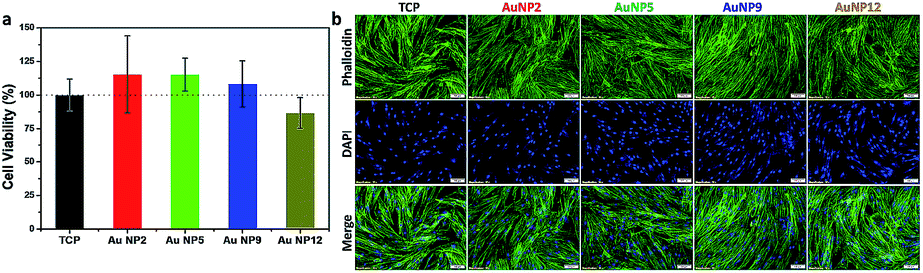 | ||
| Fig. 6 (a) Viability of HDFs and (b) fluorescence microscopy images of HDFs with and without the treatment of 150 μg mL−1 AuNP2, AuNP5, AuNP9 and AuNP12. | ||
Inflammatory properties
Inflammation is important in any medical application. We therefore assessed the changes in macrophage response to all four AuNPs in the presence of lipopolysaccharide (LPS), which is known to trigger strong inflammatory responses (Fig. 7a). We measured the secretion of three key pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α), interleukin-8 (IL-8) and interleukin-1β (IL-1β), following addition of an equal amount of all four AuNPs to LPS-stimulated macrophages. An enzyme-linked immunosorbent assay (ELISA) was performed to quantify the inflammatory cytokines from the culture supernatants. Our results reveal that all four NPs reduced the secretion of TNF-α. No significant variation was observed amongst different NPs (Fig. 7b). The secretion of IL-8 was also significantly reduced for all NPs relative to the control (Fig. 7c). The highest reduction of IL-8 secretion was observed for AuNP5 (>16 fold). In contrast, AuNP12 only reduced IL-8 secretion by ∼8 fold under similar experimental conditions. AuNP5 and AuNP2 also suppressed the expression of IL-1β (Fig. 7d). However, AuNP9 did not affect the expression of this cytokine while AuNP12 appears to lead to its slight overexpression (Fig. 7d). While the mechanisms of this dependency require further investigation, the data show a structure dependent inflammatory response to nanomaterials. With the rapid intake of nanomaterials in different aspects of medicine, our results demonstrate the need for closer evaluation of the effect of nanomaterial structure on inflammatory consequences. On one hand, such evaluation is required from the safety point of view. On the other hand, these structure dependent properties may lead to new therapeutic applications. For example, AuNP5 may be useful for treating inflammatory conditions.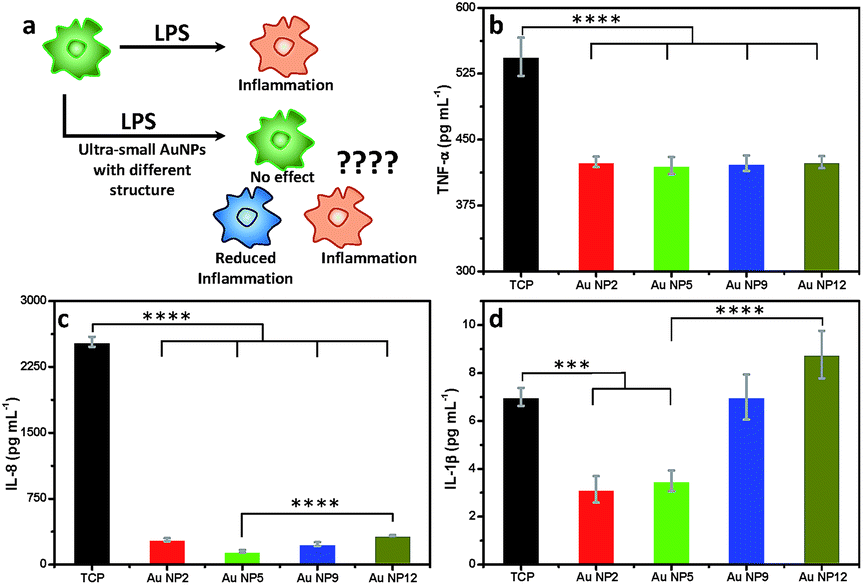 | ||
| Fig. 7 (a) Schematic representation of the inflammatory challenge by LPS and impact of AuNPs; generation of (b) TNF-α, (c) IL-8 and (d) IL-1β. Cells were treated with 150 μg mL−1 AuNPs. Statistical significance is assessed by one-way ANOVA software; ****p < 0.0001, ***p < 0.001. | ||
Conclusions
In summary, we synthesized a series of AuNPs which had four distinct core-in-cage structures but identical particle size and surface chemistry. These unique core-in-cage structures offer new possibilities to control the properties of these AuNPs by variation of the core and cage contents instead of changing the size, composition and surface properties. Our results suggest that when GSH is used as the ligand, lower pH favours the formation of longer Au(I)–thiolate complexes, which in turn favour the formation of highly luminescent AuNPs. By comparing the cage structure of all four NPs, we identified that the extent of intra-molecular Au(I)⋯Au(I) interactions in the cage dictates their PL properties. In fact, some of the recent examples of highly luminescent AuNPs reported by other groups such as AIE type Au(0)@Au(I)–thiolate core–shell NCs and Au22(SG)18 also rely on low pH where a relatively large size of the corresponding Au(I)–thiolate complexes was expected.45,50 These results together with our experimental observations imply that such long size motifs could trigger the formation of a more rigid cage structure, where the possibility of intra-molecular aurophilic interactions is high. This in turn generates high PL.We also revealed the importance of NP core-in-cage structures to their antibacterial properties. AuNPs with a high percentage of loosely bound Au(I)–thiolate complexes in their cage showed stronger antibacterial activity than NPs with a shorter motif and larger core. We should acknowledge that the MIC of the AuNPs used as a model in this study to correlate with the core-in-cage structure is relatively high. One probable reason for this could be the presence of GSH ligands, which are known to be an antioxidant. GSH ligand protected AuNPs are also proven to scavenge ROS,51 which are considered as a primary factor for high antibacterial activity of other agents. We expect that the direct structural correlation which we have identified here can be translated to other thiolated ligands for obtaining highly potent gold-based antibacterial agents.
In this work, we have also examined for the first time the inflammatory response to ultra-small AuNPs. We have found unique structure dependent inflammatory consequences. Strikingly, AuNP5, which consisted of loosely bound Au(I)–thiolate complexes, had the greatest potential to reduce the expression of the three pro-inflammatory cytokines examined in this study. The observation of such anti-inflammatory properties caused by ultra-small AuNPs offers new opportunities for potential applications of these exciting nanomaterials in controlling and suppressing inflammatory conditions.
Taken together, the structure–property relationship insights offered in this work provide opportunities for future directions of research where the target controls and tailors the nanomaterial structure and leads to the discovery of exciting new properties and applications. For example, in the case of ultra-small AuNPs the discovery of anti-inflammatory and antibacterial properties may stimulate various areas of nanomedicine and lead to development of new nano-antibiotics, diagnostic and imaging tools and treatments for inflammatory diseases. However, understanding the structure regulated properties of nanomaterials can also have a significant impact on other fields such as catalysis and electronics.
Experimental
Materials
Hydrogen tetrachloroaurate (HAuCl4·xH2O) and glutathione (GSH) from Sigma Aldrich, and sodium hydroxide (NaOH) and ethanol (EtOH) from Merck were used as received without further purification. All aqueous solutions were prepared with ultrapure Millipore water.Materials characterization
Solution pH was monitored using a pH meter from SmartCHEM, TPS Australia. UV-vis absorbance spectra were recorded on a Shimadzu spectrophotometer. TEM images were obtained using a JEOL-2100F microscope operating at an accelerating voltage of 200 kV. All hydrodynamic diameters were recorded on a Malvern-Zetasizer Nano ZS. Photoluminescence (PL) spectra were measured on a Varian Cary Eclipse Fluorescence Spectrometer. XPS measurements were performed on a Kratos AxisUltra XPS. CasaXPS software was used for data analysis. The C 1s carbon peak at 285 eV was kept as a reference for the calibration of all Au 4f binding energies. Thermogravimetric analysis (TGA) was conducted on TA Discovery Instruments under a N2 atmosphere (flow rate of 50 mL min−1). Live dead imaging for the antibacterial experiments was performed using an Olympus FV3000 confocal laser scanning microscope. SEM for antibacterial studies was done using a Philips XL30 FEGSEM.Synthesis of AuNPs
In a typical synthesis, a freshly prepared aqueous solution of HAuCl4 (20 mM, 1.0 mL) and GSH (100 mM, 0.3 mL) was mixed in water (8.70 mL) under stirring (500 rpm), leading to the formation of Au(I)–SG complexes. After 1 min of stirring, the solution pH was adjusted to 2, 5, 9 and 12 by adding an appropriate amount of sodium hydroxide or hydrochloric acid. Each of these solutions was then kept on a hotplate for 24 h at 80 °C. The solution colour changed from colourless to yellow or dark yellow. After synthesis, all four NPs were purified by dialysis (Pur-A-Lyzer™ Maxi Dialysis Kit; MWCO 3.5 kDa) and used for characterization.Antibacterial study
000 magnification and a voltage of 10.0 kV.
Cell culture
Human monocyte cell line THP-1 and primary human dermal fibroblast (HDF) were used in this study. Primary derived HDFs were gifted by Dr Louise Smith, University of South Australia. HDFs were harvested and grown, according to the published protocol.52 RPMI 1640 (Sigma Aldrich) was used as growth medium for THP-1 cells and Dulbecco's Modified Eagle Medium DMEM (Life Technologies) was used for HDF along with 10% heat-inactivated fetal bovine serum (FBS, Thermo Scientific) and 1% (v/v) penicillin/streptomycin (Life Technologies). The cells were maintained in a humidified atmosphere containing 5% CO2 at 37 °C.Cell viability
The cell viability was determined using the resazurin assay (Sigma Aldrich). HF cells were trypsinized and seeded on a 96 well plate at a density of 5 × 104 cells per well. After overnight growth, the medium was removed and the cells were washed with PBS. Fresh medium was then added to the wells along with various AuNPs. The cells were allowed to grow for 24 hours, after which the culture medium was replaced with medium containing 10% resazurin and incubated for an hour. 100 μL of the medium was pipetted into a 96 well plate and the fluorescence intensity was measured using a microplate spectrophotometer (λex = 544 nm and λem = 590 nm). The percentage cell viability was calculated as:| Cell viability % = ((fluorescence intensity of treated)/(fluorescence intensity of control)) × 100 |
Inflammation studies
THP-1 cells were differentiated into macrophages dTHP-1 using PMA (phorbol-12-myristate 13-acetate) according to a previously reported protocol,53 and seeded into a 96 well plate at a density of 5 × 104 cells per well. After overnight growth, the medium was removed, and the cells were washed with PBS. Fresh medium was then added to the wells along with various AuNPs. Experiments were conducted with LPS (1 μg mL−1) to activate inflammatory macrophages and to give an infectious or inflammatory environment. After 6 hours incubation conditioned medium was collected and centrifuged to remove the cell debris and AuNPs. Supernatants were collected and analysed for pro-inflammatory cytokines of TNFα, IL-1β, and IL-8 using ELISA kits (BioLegend, San Diego, CA, USA) following the manufacturer's instructions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. V. thanks ARC for DP15104212, NHMRC for the Fellowship APP1122825 and the Project grant APP1032738, and the Alexander von Humboldt Foundation for the Fellowship for Experienced Researchers.Notes and references
- M. Auffan, J. Rose, J.-Y. Bottero, G. V. Lowry, J.-P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634 CrossRef CAS PubMed.
- G. A. Ozin and L. Cademartiri, Small, 2009, 5, 1240–1244 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- D. V. Talapin, J.-S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- B. Pelaz, C. Alexiou, R. A. Alvarez-Puebla, F. Alves, A. M. Andrews, S. Ashraf, L. P. Balogh, L. Ballerini, A. Bestetti, C. Brendel, S. Bosi, M. Carril, W. C. W. Chan, C. Chen, X. Chen, X. Chen, Z. Cheng, D. Cui, J. Du, C. Dullin, A. Escudero, N. Feliu, M. Gao, M. George, Y. Gogotsi, A. Grünweller, Z. Gu, N. J. Halas, N. Hampp, R. K. Hartmann, M. C. Hersam, P. Hunziker, J. Jian, X. Jiang, P. Jungebluth, P. Kadhiresan, K. Kataoka, A. Khademhosseini, J. Kopeček, N. A. Kotov, H. F. Krug, D. S. Lee, C.-M. Lehr, K. W. Leong, X.-J. Liang, M. Ling Lim, L. M. Liz-Marzán, X. Ma, P. Macchiarini, H. Meng, H. Möhwald, P. Mulvaney, A. E. Nel, S. Nie, P. Nordlander, T. Okano, J. Oliveira, T. H. Park, R. M. Penner, M. Prato, V. Puntes, V. M. Rotello, A. Samarakoon, R. E. Schaak, Y. Shen, S. Sjöqvist, A. G. Skirtach, M. G. Soliman, M. M. Stevens, H.-W. Sung, B. Z. Tang, R. Tietze, B. N. Udugama, J. S. VanEpps, T. Weil, P. S. Weiss, I. Willner, Y. Wu, L. Yang, Z. Yue, Q. Zhang, Q. Zhang, X.-E. Zhang, Y. Zhao, X. Zhou and W. J. Parak, ACS Nano, 2017, 11, 2313–2381 CrossRef CAS PubMed.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225–4241 CrossRef CAS.
- J. Piella, N. G. Bastús and V. Puntes, Chem. Mater., 2016, 28, 1066–1075 CrossRef CAS.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- M. P. Mallin and C. J. Murphy, Nano Lett., 2002, 2, 1235–1237 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- R. Jin, Nanoscale, 2015, 7, 1549–1565 RSC.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- S. Knoppe and T. Bürgi, Acc. Chem. Res., 2014, 47, 1318–1326 CrossRef CAS PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Häkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. Qian, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2009, 131, 2490–2492 CrossRef CAS PubMed.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- J. Liu, P. N. Duchesne, M. Yu, X. Jiang, X. Ning, R. D. Vinluan, P. Zhang and J. Zheng, Angew. Chem., Int. Ed., 2016, 55, 8894–8898 CrossRef CAS PubMed.
- A. Yahia-Ammar, D. Sierra, F. Mérola, N. Hildebrandt and X. Le Guével, ACS Nano, 2016, 10, 2591–2599 CrossRef CAS PubMed.
- Z. Gan, Y. Lin, L. Luo, G. Han, W. Liu, Z. Liu, C. Yao, L. Weng, L. Liao, J. Chen, X. Liu, Y. Luo, C. Wang, S. Wei and Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 11567–11571 CrossRef CAS PubMed.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816–824 CrossRef CAS PubMed.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed.
- L.-Y. Chen, C.-W. Wang, Z. Yuan and H.-T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS PubMed.
- X. Yuan, Z. Luo, Y. Yu, Q. Yao and J. Xie, Chem.–Asian J., 2013, 8, 858–871 CrossRef CAS PubMed.
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS.
- X.-D. Zhang, Z. Luo, J. Chen, X. Shen, S. Song, Y. Sun, S. Fan, F. Fan, D. T. Leong and J. Xie, Adv. Mater., 2014, 26, 4565–4568 CrossRef CAS PubMed.
- X.-D. Zhang, J. Chen, Z. Luo, D. Wu, X. Shen, S.-S. Song, Y.-M. Sun, P.-X. Liu, J. Zhao, S. Huo, S. Fan, F. Fan, X.-J. Liang and J. Xie, Adv. Healthcare Mater., 2014, 3, 133–141 CrossRef CAS PubMed.
- N. Goswami, Z. Luo, X. Yuan, D. T. Leong and J. Xie, Mater. Horiz., 2017, 4, 817–831 RSC.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS PubMed.
- G. Soldan, M. A. Aljuhani, M. S. Bootharaju, L. G. AbdulHalim, M. R. Parida, A.-H. Emwas, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 5749–5753 CrossRef CAS PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- Y.-T. Tseng, Z. Yuan, Y.-Y. Yang, C.-C. Huang and H.-T. Chang, RSC Adv., 2014, 4, 33629–33635 RSC.
- S. E. Crawford, C. M. Andolina, A. M. Smith, L. E. Marbella, K. A. Johnston, P. J. Straney, M. J. Hartmann and J. E. Millstone, J. Am. Chem. Soc., 2015, 137, 14423–14429 CrossRef CAS PubMed.
- N. Goswami, F. Lin, Y. Liu, D. T. Leong and J. Xie, Chem. Mater., 2016, 28, 4009–4016 CrossRef CAS.
- K. Zheng, M. I. Setyawati, D. T. Leong and J. Xie, Chem. Mater., 2018, 30, 2800–2808 CrossRef CAS.
- X. Yuan, N. Goswami, I. Mathews, Y. Yu and J. Xie, Nano Res., 2015, 8, 3488–3495 CrossRef CAS.
- W. Kurashige, M. Yamaguchi, K. Nobusada and Y. Negishi, J. Phys. Chem. Lett., 2012, 3, 2649–2652 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, T. Chen and J. Xie, Coord. Chem. Rev., 2016, 329, 1–15 CrossRef CAS.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- K. Zheng, M. I. Setyawati, D. T. Leong and J. Xie, ACS Nano, 2017, 11, 6904–6910 CrossRef CAS PubMed.
- W.-Y. Chen, J.-Y. Lin, W.-J. Chen, L. Luo, E. W.-G. Diau and Y.-C. Chen, Nanomedicine, 2010, 5, 755–764 CrossRef CAS PubMed.
- W.-Y. Chen, H.-Y. Chang, J.-K. Lu, Y.-C. Huang, S. G. Harroun, Y.-T. Tseng, Y.-J. Li, C.-C. Huang and H.-T. Chang, Adv. Funct. Mater., 2015, 25, 7189–7199 CrossRef CAS.
- Y. Xie, Y. Liu, J. Yang, Y. Liu, F. Hu, K. Zhu and X. Jiang, Angew. Chem., Int. Ed., 2018, 57, 3958–3962 CrossRef CAS PubMed.
- Y. Yu, Z. Luo, D. M. Chevrier, D. T. Leong, P. Zhang, D.-e. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 1246–1249 CrossRef CAS PubMed.
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed.
- S. MacNeil, J. Shepherd and L. Smith, in 3D Cell Culture. Methods in Molecular Biology (Methods and Protocols), ed. J. Haycock, Humana Press, 2011, vol. 695, pp. 129–153 Search PubMed.
- W. Chanput, J. J. Mes, H. F. J. Savelkoul and H. J. Wichers, Food Funct., 2013, 4, 266–276 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00211a |
| This journal is © The Royal Society of Chemistry 2019 |