
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the good and poor cell targeting activity of gold nanostructures functionalized with molecular units for the epidermal growth factor receptor†
Claudia
Mazzuca‡
 a,
Benedetta
Di Napoli‡
a,
Francesca
Biscaglia‡
b,
Giorgio
Ripani
a,
Senthilkumar
Rajendran
c,
Andrea
Braga
b,
Clara
Benna
c,
Simone
Mocellin
cd,
Marina
Gobbo
b,
Moreno
Meneghetti
*b and
Antonio
Palleschi
*a
a,
Benedetta
Di Napoli‡
a,
Francesca
Biscaglia‡
b,
Giorgio
Ripani
a,
Senthilkumar
Rajendran
c,
Andrea
Braga
b,
Clara
Benna
c,
Simone
Mocellin
cd,
Marina
Gobbo
b,
Moreno
Meneghetti
*b and
Antonio
Palleschi
*a
aDepartment of Chemical Science and Technology, University of Rome “Tor Vergata” and CSGI Unit, Via della Ricerca Scientifica, 00133 Rome, Italy. E-mail: antonio.palleschi@uniroma2.it
bDepartment of Chemical Sciences, University of Padova, Via Marzolo 1, 35131, Padova, Italy. E-mail: moreno.meneghetti@unipd.it
cDepartment of Surgery, Oncology and Gastroenterology, University of Padova, Via Giustiniani, 2, 35124 Padova, Italy
dVeneto Institute of Oncology IOV-IRCCS, Via Gattamelata, 64, 35128 Padova, Italy
First published on 29th March 2019
Abstract
Nanostructures can strongly interact with cells or other biological structures; furthermore when they are functionalized with targeting units, they are of great interest for a variety of applications in the biotechnology field like those for efficient imaging, diagnosis and therapy and in particular for cancer theranostics. Obtaining targeting with good specificity and sensitivity is a key necessity, which, however, is affected by the complexity of the interactions between the nanostructures and the biological components. In this work we report the study of specificity and sensitivity of gold nanoparticles functionalized with the peptide GE11 for the targeting of the epidermal growth factor receptor, expressed on many cells and, in particular, on many types of cancer cells. We show how a combination of spectroscopic measurements and molecular dynamics simulations allows the comprehension of the targeting activity of peptides linked to the surface of gold nanostructures and how the targeting is tuned by the presence of polyethylene glycol chains.
Introduction
Nanoparticles are promising tools in cancer therapy as they can act as contrast agents in optical imaging, sensitizers for Raman-based diagnostic probes, vectors for drugs, and tools for photothermal therapy.1–9 However, gold nanoparticles (AuNPs) must be functionalized with ad hoc coating to achieve efficient cell targeting.10–12 More precisely, in order to recognize cancer cells, the ligand on nanoparticles must be designed with high specificity for receptors that are overexpressed on malignant cells, but minimally expressed on healthy cells. These targeting ligands allow nanoparticles to remain within the tumor and enable their transport across the cell membranes. It should be noted that, despite the importance of having sufficiently active loaded nanoparticles, the rationalization and a better understanding of the organization of the cell targeting units on the surface of the nanoparticles are still lacking. In this context, the study regarding the dodecapeptide YHWYGYTPQNVI (GE11), a ligand of the epidermal growth factor receptor (EGFR), revealed features not easily predictable on gold nanostructures.13 It was also shown that AuNPs loaded with GE11 show more efficient targeting against EGFR if GE11 is covalently linked to a polyethylene glycol (PEG) chain through a peptide spacer (KKKGG) than if it is directly linked to PEG.13 GE11 is an interesting targeting ligand because it binds without activating EGFR mediated signaling, like the natural EGF ligand does, avoiding, therefore, contributing to tumor growth.14 Due to this property GE11 has been used for targeted gene delivery,14,15 as a drug carrier to deliver organic nanoparticles and for imaging.16–20 Moreover, this peptide has been linked to liposomes, micelles, polymers as well as drugs.14,18,19,21,22In this work the GE11 peptide has been covalently linked to Au nanostructures through a short 8-amino-3,6-dioxaoctanoic acid ending with a cysteine to exploit the sulfur–gold bond (GE11-C, Scheme 1). The targeting activity of this nanostructure (AuNP@GE11-C) has been evaluated, using surface enhanced resonance Raman scattering (SERRS), against two different colorectal tumor cells lines, expressing (Caco-2, EGFR+) or not expressing (SW620, EGFR−) EGFR. The functionalized AuNPs show high sensitivity (percentage of positive cells which are observed as positive), but very low specificity (percentage of negative cells which are observed as negative). To overcome this drawback, we have prepared Au nanostructures covered by a mixture of GE11 and a thiolated polyethylene glycol methyl ether (PEG-SH; MW = 800 Da). The presence of PEG-SH chains in addition to the peptide reduced non-specific interactions, leading to an enhancement of specificity and sensitivity of the nanostructures. However, the amount of PEG-SH strongly influenced the targeting capability of these nanostructures. The best results (sensitivity = 93%; specificity = 86%) were obtained with GE11-C/PEG-SH = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. These results are rationalized in the present work by studying the interaction between PEG-SH and GE11-C, the aggregation property of GE11-C and the organization of both GE11-C and PEG-SH molecules on the surface of the AuNP through a combination of spectroscopic measurements and molecular dynamics (MD) simulations.
:1. These results are rationalized in the present work by studying the interaction between PEG-SH and GE11-C, the aggregation property of GE11-C and the organization of both GE11-C and PEG-SH molecules on the surface of the AuNP through a combination of spectroscopic measurements and molecular dynamics (MD) simulations.
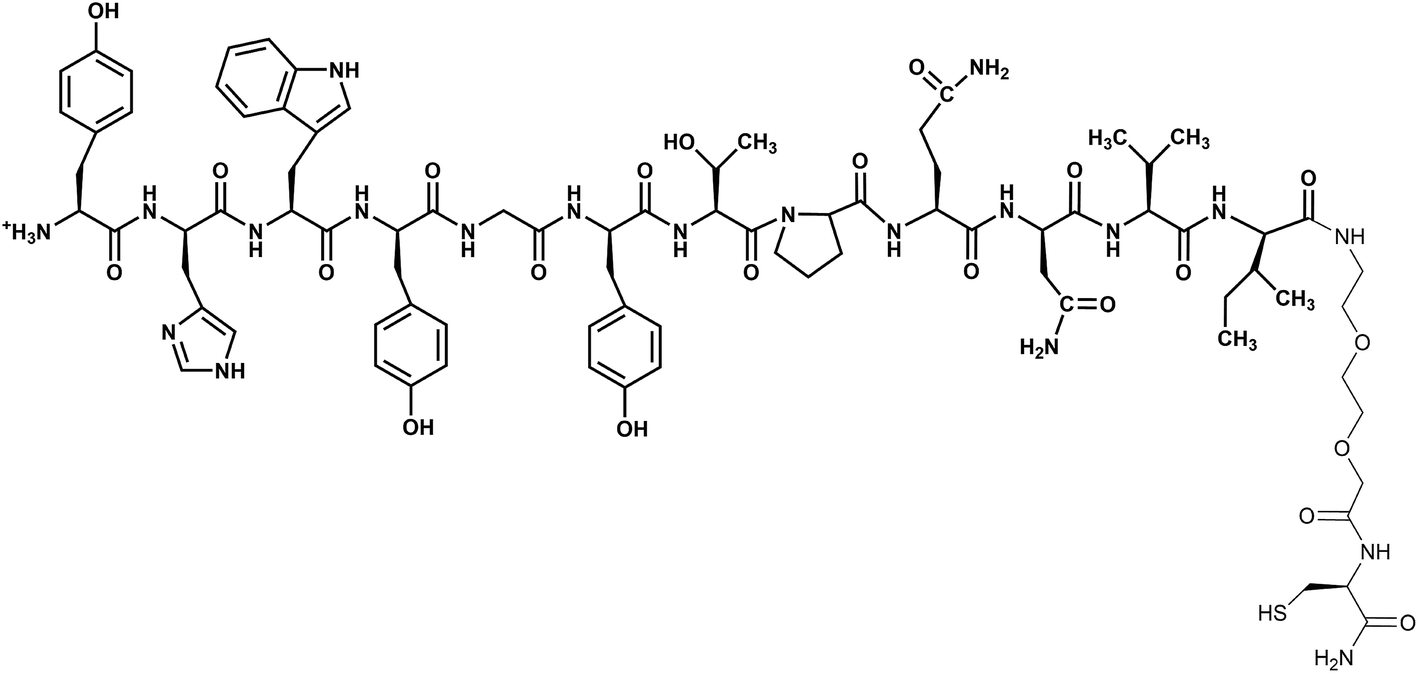 | ||
| Scheme 1 Structure of GE11-C. The peptide GE11 is in bold. | ||
Experimental section
Materials
Unless otherwise specified, all chemicals were commercial products and were used without further purification. Bovine Serum Albumin (BSA), polyethylene glycol methyl ether thiol (PEG-SH; MW = 800 Da) and 3-carboxy-4-nitrophenyl disulfide were provided by Sigma-Aldrich. The GE11-C peptide was synthesized as previously described13 and characterized as reported in the ESI (Table S1 and Fig. S1†). Sulforhodamine 101-bis-cysteamide (TR-SH) was prepared according to a previously published procedure.11Preparation of SERRS nanostructures
000g for 10 min and the supernatant containing the SERRS reporter in excess was discarded. The functionalized nanoaggregates were redispersed in PBS buffer at nanomolar concentration. The extent of aggregation and the presence of the characteristic SERRS signals were controlled with the UV-Vis-NIR and Raman spectra of the resulting colloidal solution.11 PEG-SH coated nanosystems were prepared according to a previously published procedure.13 Briefly, 1 mL of the colloidal solution of nanoaggregates (4 nM) was mixed with 0.1 mL of a 0.75 mM solution of PEG-SH in water. The mixture was shaken for 3 h at RT, the unbound ligands were removed by centrifugation (25000g, 10 min) and the resulting AuNP@PEG-SH was redispersed in PBS (1 mL). The number of ligands covering, on average, each nanoparticle was estimated by performing the Ellman test for thiols before and after removal of nanoparticles by centrifugation (see Table 1). At each step, UV-Vis-NIR and Raman spectra were recorded (Fig. S2†). DLS and ζ-potential results are reported in Table 1. BSA (5 mg) was added to the solution just before incubation with cells.
| Nanostructure | GE11-C per AuNp | PEG-SH per AuNp | GE11-C/PEG-SH molar ratio | Hydrodynamic diameter of AuNPs (nm) | ζ-Potential (mV) |
|---|---|---|---|---|---|
| AuNP@GE11-C | 5500 | 0 | 1:0 |
86.6 ± 3.8 | −24.2 ± 2.7 |
| AuNP@GE11-C/PEG-SH_1 | 9180 | 492 | 19:1 |
122.8 ± 4.3 | −36.2 ± 0.3 |
| AuNP@GE11-C/PEG-SH_2 | 1850 | 1016 | 1.7:1 |
98.9 ± 16.8 | −31.9 ± 8.0 |
| AuNP@GE11-C/PEG-SH_3 | 810 | 2860 | 0.3:1 |
51.3 ± 1.1 | −27.4 ± 7.1 |
| AuNP@PEG-SH | 0 | 5700 | 0:1 |
63.7 ± 1.1 | −14.6 ± 1.5 |
Conjugation with ligands
The colloidal solution of gold nanoaggregates (1 mL, 4 nM) was centrifuged at 25000g for 10 min and redispersed in 1 mL of PBS containing the peptide GE11-C and PEG-SH. By changing the concentration of the ligands in the micromolar range, nanostructures functionalized with different molar ratios of the peptide GE11-C and PEG-SH were obtained (see Table 1). The mixture was shaken for 3 h at room temperature and stored at 4 °C overnight. The excess ligand was removed by centrifugation and the nanoaggregates were re-dispersed in 1 mL of PBS. The number of ligands covering, on average, each nanoparticle was estimated by measuring the absorbance at 280 nm of the ligand solution (ε(GE11-C) 5350 M−1 cm−1; ε(PEG-SH) 86 M−1 cm−1) and by performing the Ellman test for thiols before and after removal of nanoparticles by centrifugation (see Table 1) using reference samples for the spectroscopic measurements. At each step, UV-Vis-NIR and Raman spectra were recorded (see Fig. S2†) and the fully functionalized nanostructures were also characterized by DLS and ζ-potential measurements (see Table 1). BSA (5 mg) was added to the solution just before incubation with cells.
Cell lines and receptor expression assay
Caco-2 (EGFR+) and SW620 (EGFR−) colorectal cancer cell lines were purchased from CLS Cell Lines Service GmbH (Eppelheim, Germany) and were cultured in specific culture media suggested by the manufacturer and following standard aseptic procedures. In brief, Caco-2 cells were cultured in Eagle's minimum essential medium (EMEM) (Sigma-Aldrich, Irvine, UK) supplemented with L-glutamine, 1% nonessential amino acids, 1 mM sodium pyruvate, and 10% fetal bovine serum. SW620 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma-Aldrich, Irvine, UK) supplemented with 4.5 g L−1 glucose, 2 mM L-glutamine, and 10% fetal bovine serum. All the above-mentioned media were supplemented with 100 U mL−1 penicillin and 100 μg mL−1 streptomycin (Gibco). All the cell cultures were maintained at 37 °C with 5% CO2. As described in a previously published procedure13 flow cytometry was used to verify the EGFR expression.Incubation with nanoparticles and SERRS measurements
Caco-2 and SW620 cells were seeded at a density of 50000 per well in 8 well chamber slides (Sigma-Aldrich, Irvine, UK) and allowed to grow overnight. On day 2, the normal growth medium was aspirated and the medium containing different concentrations of nanoparticles (160, 80 and 40 pM) was added and incubated for 2 h at 37 °C. After incubation, the medium was removed and the cells were washed with PBS and fixed with 2% paraformaldehyde for 15 min at 4 °C. The fixed cells were then washed with sterile water three times and subjected to SERRS analysis.13 SERRS spectra were collected for each single cell (20× objective) using an automated homemade procedure. Two replicates were performed for each experiment choosing 100 cells randomly among about 50000 cells present in a well. SERRS spectra were collected from four different positions on each cell. The presence of the characteristic Raman signals was evaluated with a Pearson correlation coefficient of 0.6. The error bars in Fig. 1 indicate 95% confidence intervals.
 | ||
| Fig. 1 Targeting activity of the nanosystems (a) AuNP@GE11-C, (b) AuNP@GE11-C/PEG-SH_1, (c) AuNP@GE11-C/PEG-SH_2, (d) AuNP@GE11-C/PEG-SH_3 and (e) AuNP@PEG-SH toward SW620 (black line) and Caco-2 (red line) cells. | ||
Statistical analysis
The dataset, consisting of SERRS spectra recorded for each point, was evaluated using the Pearson correlation coefficient and the reference SERRS spectra of the Au nanostructures. Each spectrum was preliminarily corrected by subtracting the baseline with the WiRE 4 built-in routines of the micro-Raman instrument. The Pearson correlation coefficients were calculated using the built-in function corrcoef of Matlab software. Only the spectra with a correlation coefficient above 0.6 were considered to have a positive sign of the presence of the nanostructures, and thus positive targeting activity. Data are reported in Fig. 1 as percentages of positive cells among the 100 cells considered for each experiment and the error bars are 95% confidence intervals. The homogeneity of the results of the two replicates was checked using a two-tailed test. A p-value < 0.01 was considered to be statistically significant. All statistical analyses were performed using R software (R Development Core Team, version 3.4; R Foundation for Statistical Computing, Vienna, Austria).Fluorescence experiments and fluorescence anisotropy experiments
Emission spectra and anisotropy values were obtained by steady-state fluorescence experiments on a thermostatted Spex Fluorolog III spectrofluorimeter (Horiba, Japan) operated in single photon counting mode. The excitation wavelength (λexc) was 295 nm while the emission range was 300–450 nm. Anisotropy experiments (λexc = 295 nm; λem = 370 nm) were performed using Glan–Thompson polarizer prisms.Time-resolved experiments were performed on a Lifespec-ps fluorescence lifetime spectrometer (Edinburgh Instruments, U.K.) operating in single photon counting mode. Nanosecond pulse excitation was obtained with a NanoLED light source (298 nm, pulse excitation width: 1.0 ns, 0.9 MHz repetition rate). Fluorescence intensity decays were acquired until a peak value of 104 counts was reached and analyzed with the software provided by Edinburgh Instruments.25,26
From anisotropy values (r) it is possible to estimate the hydrodynamic radius (rH) of the samples using the Perrin equation27 (eqn (1)):
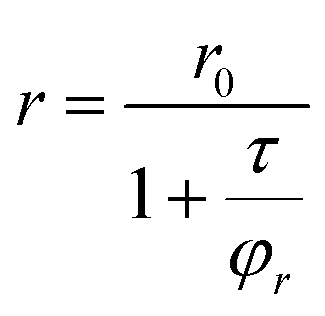 | (1) |
 | (2) |
In our case, r0 is close to the value of 0.15–0.20,28τ is 1.7 ns (from time resolved fluorescence lifetime experiments), and η is the viscosity of water at 25 °C, that is, 0.89 cp.29
FTIR-ATR experiments
Fourier-transform infrared (FTIR) absorption spectra were measured on a Thermo Fisher FTIR iS50 spectrometer (Thermo Fisher Scientific Co., Madison, WI, USA) in the attenuated total reflection (ATR) mode using a ZnSe cell on peptide films obtained by casting a peptide solution on an Al plate. Each spectrum was obtained averaging over 128 scans with a resolution of 2 cm−1. Peptide secondary structures were identified by performing a deconvolution of bands in the amide I and amide II (1800–1500 cm−1) regions of the FTIR-ATR spectra, using OMNIC software. The analysis was performed considering the regions of the spectra as a sum of Gaussian functions. Optimization of band position, FWHH and amplitude was performed using the Fletcher–Powell–McCormick algorithm.30Molecular dynamics calculations
MD simulations were carried out using the GROMACS v5.0.7 software package31 and the GROMOS 53A6 force field parameters were used for the peptide32 while parameters needed for PEG-SH were taken from Fuchs et al.33 A constant pressure and temperature ensemble (NPT) was used for all simulations performed, with periodic boundary conditions. Following our protocol, for each system there was a two-step energy minimization: first, only the solvent was energy minimized and then the solute (GE11-C and PEG-SH) was energy minimized. Subsequently, the solvent was equilibrated for 150 ps at 50 K (time steps of 0.5 fs). All the systems were gradually heated from 50 to 300 K in a 1.1 ns MD, and then production runs were performed, using a time step of 2 fs. The particle mesh Ewald (PME) algorithm34,35 was used for the electrostatic interactions (cut-off = 1.4 nm). Explicit simple point-charge (SPC) water molecules36 were used to solvate the simulation box and chloride ions were added to ensure the electroneutrality. A cut-off was used for the van der Waals interactions (1.4 nm). In all the simulations, the velocity rescale scheme37 was used for keeping constant the temperature (coupling constant τT = 0.6 ps) and the Berendsen algorithm38 under isotropic conditions for pressure coupling (τP = 1 ps), except if specified.All the results reported were obtained when the systems were equilibrated. Secondary structure analyses were performed using the DSSP (define secondary structures of proteins) GROMACS tool.39 The visual molecular dynamics (VMD) program40 was used for the structure visualization.
MD calculations in solution
The simulated systems were obtained with different numbers of peptides (n = 1, 2, 5, 8, 9 and 20) and in the absence or presence of one molecule of PEG-SH. A cubic box of 8 nm length was filled with water molecules. Both peptides and PEG-SH were placed randomly in the box, in an extended conformation as the starting configuration. For each system three replicates of 250 ns long were performed. The features of the aggregates were analyzed using the gyrate (for radius of gyration and moments of inertia calculations), rms (for root mean square deviations) and mindist tools of GROMACS. To characterize the shape of peptide aggregates, symmetry factor (Is) was used. It is defined as: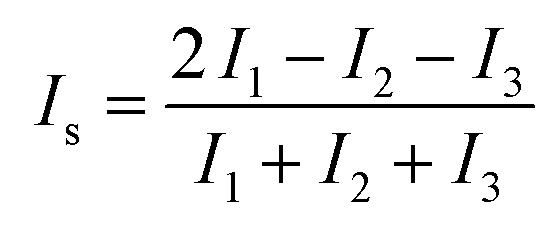 | (3) |
MD calculations on the Au surface
Simulations were performed by placing all the molecules in extended conformation, perpendicular to the Au surface. The positions of S atoms were restrained,41 only for the z-coordinate, at 2.95 Å from the gold surface to mimic the Au–S covalent bonds during simulations. Aligned molecules were placed on the simulation box of 5 × 5 × 12 nm3, filled with water molecules. For all the simulations three replicates of at least 200 ns long were performed. The Lennard–Jones parameters for the Au atom were taken from Pu et al.42 In order to study the interaction between bulk peptides and the monolayer, additional simulations were performed placing 4 peptides randomly in the same simulation box, at a distance greater than 2 nm from the monolayer already formed, without any restraint (as previously reported). All the features were analyzed using the density, rms and mindist tools of GROMACS.MD calculations with wall potential
To study the diffusion process of the molecules from the solution to the surface, a wall potential along the z-axis was used. The atom type of the wall (z = 0) was chosen according to Wright et al.43 (no explicit Au atoms are needed using this approach). A Lennard–Jones potential was added for S atoms only (C6 = 68.5929 10−3 kJ mol−1 nm,6 C12 = 36.7580 10−6 kJ mol−1 nm (ref. 12)), to mimic the chemical bond between Au and S atoms. The periodic boundary conditions were applied only to the x- and y-coordinates, due to the defined wall along the z-axis. A semi-isotropic Berendsen algorithm38 was chosen for pressure coupling. Two replicates of at least 100 ns long were performed for each simulated system, characterized by different numbers of peptides (n = 8, 9) with and without one molecule of PEG-SH. The box of 6 × 6 × 12 nm3 was filled with water molecules. Both peptides and PEG-SH molecules were placed randomly in the box, in an extended conformation as the starting configuration. The system features were analyzed using the sasa (for solvent accessible surface analysis) and mindist tools of GROMACS.Results
Gold nanostructures reported in Table 1 were prepared according to established protocols13 also reported in the Experimental section. Nanostructures were obtained by aggregation of naked nanoparticles (AuNPs) synthesized by laser ablation of bulk gold under water. By mixing the Au colloidal solution with a tiny amount of a SERRS reporter, a thiolated Texas Red molecule, the aggregation of nanostructures was obtained and controlled by UV-Vis-NIR spectra, after removal of the excess of Texas Red by centrifugation. The aggregated AuNP are SERRS active because of the presence of hot spots where strong enhancements of the Raman signal of Texas Red can be predicted and observed (see Fig. S2†). The functionalization of the nanostructures with different cell targeting units is obtained by mixing the colloidal solution of the aggregated AuNPs with a solution of the targeting molecules (see Experimental section). Five different functionalized nanostructures were obtained using only GE11-C (AuNP@GE11-C), only PEG-SH (AuNP@PEG-SH) or variable contents of the two ligands, GE11-C and PEG-SH (AuNP@GE11-C/PEG-SH_1, 2 and 3) (see Table 1).In Fig. 1 the targeting activity of the five different nanostructures against two different lines of colorectal tumor cells (SW620 (EGFR−) and Caco-2 (EGFR+)) is reported. As expected, nanostructures with PEG-SH only (AuNP@PEG-SH) exhibit a sensitivity (the percentage of positive cells, Caco-2, which are observed as positive) below 10%, at the highest nanostructure concentration. Nanostructures functionalized with GE11-C alone (AuNP@GE11-C) show high sensitivity (above 90%), but a specificity (percentage of negative cells, SW620, which are observed as negative) below 60%. The best results are obtained when GE11-C is in strong excess with respect to PEG-SH (AuNP@GE11-C/PEG-SH_1, GE11-C/PEG-SH = 19:1 molar ratio), in terms of both specificity (above 90%) and sensitivity (above 85%) at the highest nanostructure concentration. For lower GE11-C/PEG-SH ratios, both sensitivity and specificity values approach those obtained with the nanostructures functionalized with PEG-SH only (AuNP@PEG-SH).
Interestingly, it should be noted that on varying the ratio between the two ligands the total number of molecules per AuNP will change (see Table 1). In particular, in the presence of a single ligand, about 5000 molecules per nanoparticle can be bound on the nanostructures, regardless of the type of ligand (GE11-C or PEG-SH). When the two ligands are mixed, the number of total molecules on AuNPs changes significantly, indicating that the co-presence of both ligands plays a pivotal role in the AuNP coating. It should be pointed out that in AuNP@GE11-C/PEG-SH_1, characterized by a 19:1 GE11-C/PEG-SH ratio, the number of GE11-C molecules linked on AuNPs is almost double with respect to GE11-C alone (AuNP@GE11-C).
The value of 5000 molecules per nanoparticle agrees with that obtained by means of models based on the steric hindrance of molecules on the surface of a nanoparticle of 20 nm (the average diameter of the nanoparticles obtained by LASiS). Therefore, the number (almost double) of total attached molecules for AuNP@GE11-C/PEG-SH_1 (GE11-C/PEG-SH = 19:1 ratio) exceeds the number of molecules that can be covalently linked to the surface.
In order to investigate the molecular organization that makes it possible to allocate such a high number of molecules on AuNPs, we hypothesized that peptides in solution (bulk peptides) can interact with an already formed monolayer on the Au surface.
This hypothesis was studied by performing MD simulations, monitoring the density profile along the z-axis of peptides in solution approaching the AuNP monolayer constituted by GE11-C only or by GE11-C/PEG-SH at a 1:1 molar ratio. The features of the already formed monolayers of GE11-C only or PEG-SH only, obtained by MD simulations, are reported in Fig. S3.†
The density profile along the z-axis, when GE11-C molecules only are linked on the Au surface (Fig. 2A), shows that peptides approaching from the solution (bulk peptides) avoid the monolayer on the surface. A better inspection, reported in Fig. 3, reveals that peptides can approach the peptide coated AuNP surface, but this interaction is not stable.
 | ||
| Fig. 2 Partial density values (along the z-axis) of GE11-C in solution (black), of the monolayer (red) and of PEG-SH (blue), when only GE11-C (A) and both GE11-C and PEG-SH (molar ratio 1:1, (B)) are linked to the Au surface. | ||
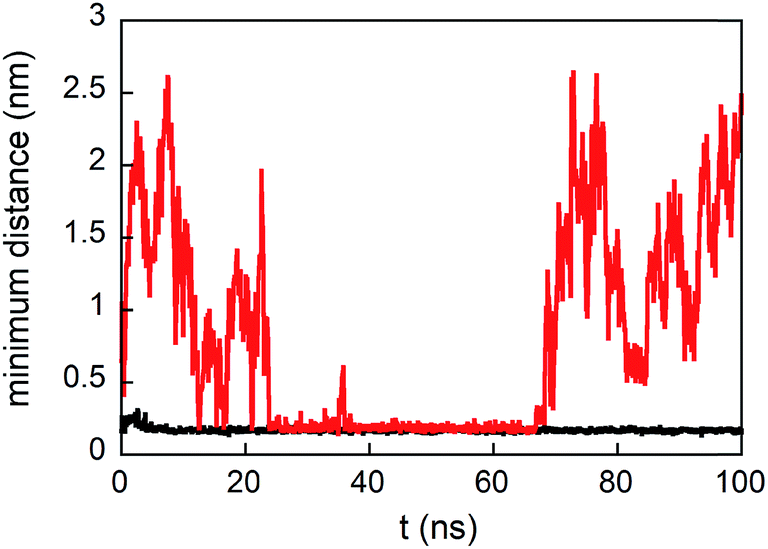 | ||
| Fig. 3 Minimum distance from any atoms of peptides in solution and any atoms of the monolayer formed by only GE11-C (red line) and both GE11-C and PEG-SH (molar ratio 1:1, black line) during simulations. | ||
However, when a monolayer constituted of GE11-C and PEG-SH (molar ratio 1:1) is present on the Au surface, peptides from the solution not only approach the monolayer surface but can also interact with both peptides and polymers. This is evident from the low values of distance between bulk peptides and the monolayer reported in Fig. 3. In detail, the interaction of bulk peptides with PEG-SH on the gold surface is suggested by the overlapping of density profiles along the z-axis (Fig. 2B) of peptides (black line) and PEG-SH (blue line) in the 2.5–5 nm range.
In Fig. 4, a picture of the process is shown; it shows that bulk peptides present in solution (red) are anchored by one of the PEG-SH chains (orange) in the monolayer formed by PEG-SH and GE11-C molecules.
 | ||
| Fig. 4 Side-view snapshot of a GE11-C aggregate (red) lying on the monolayer constituted by PEG-SH (light green) and GE11-C (blue) in a molar ratio of 1:1. One PEG-SH (orange) interacts with the aggregate. | ||
This result suggests, therefore, that AuNPs can coordinate with a higher number of GE11-C molecules than those present in a monolayer, as shown by experimental data (Table 1).
The high density of surface molecules that characterizes AuNP@GE11-C/PEG-SH_1, which is the nanostructure with the best targeting activity, was further investigated by characterizing in detail the interaction between GE11-C and PEG-SH by using different spectroscopic techniques and MD simulations.
First of all, we analyzed the behavior of the peptide and PEG-SH in solution with the aim of understanding whether the AuNP coating derives from an initial aggregative process already present in solution or not.
We experimentally determined the aggregation of GE11-C at different concentrations with and without the presence of PEG-SH by means of fluorescence anisotropy. As shown in Fig. 5A, the anisotropy value is nearly constant in the 0–50 μM range and increases slightly at a relatively high concentration (308 μM).
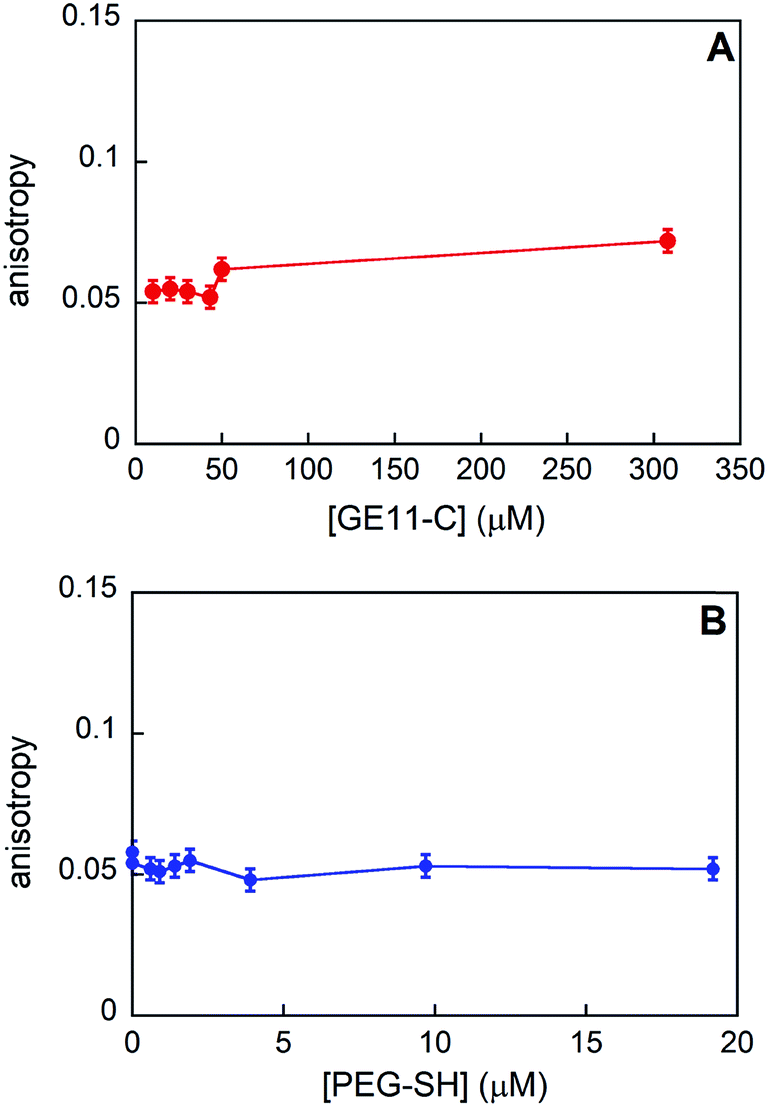 | ||
| Fig. 5 Anisotropy values obtained for GE11-C when increasing the peptide concentration (A) and at a fixed peptide concentration (20 μM) when increasing the PEG-SH one (B). | ||
From the experimental anisotropy data, the hydrodynamic radius (rH) of the samples has been estimated (as shown in the Experimental section) and it is found to be in the range of 0.9–1.1 nm. Data obtained from MD simulations showed that these rH values are compatible with the presence of aggregates composed by 5–8 peptides (generally, less than 10) in all the concentration ranges investigated.
Furthermore, this finding suggests that the dimensions of the aggregates in solution do not increase while increasing the concentration of peptide in solution.
When PEG-SH is added to the peptide solution, anisotropy values (see Fig. 5B) do not change, indicating that the peptide aggregates are approximately of the same dimensions, even at relatively high values of peptide/polymer molar ratio (1:1).
From steady state fluorescence experiments (see Fig. 6) one can see that the emission intensity of the GE11-C, due to the presence of a tryptophan in the peptide sequence, is almost the same regardless of the presence or absence of PEG-SH. This suggests that the environment surrounding the tryptophan in the peptides is very similar and thus not influenced by the presence of the polymer.
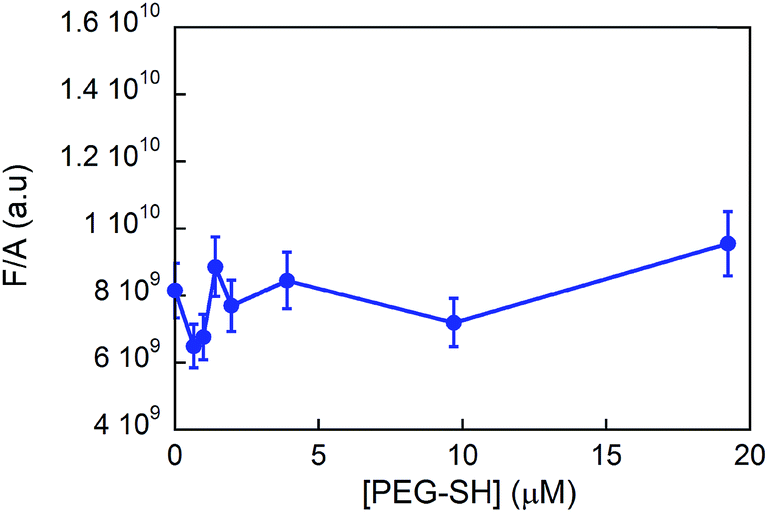 | ||
| Fig. 6 Fluorescence intensity values (normalized to the absorbance ones) of the tryptophan residue in GE11-C (λexc = 295 nm; λem = 300–450 nm) in the presence of PEG-SH ([GE11-C] = 20 μM). | ||
Conformational studies using circular dichroism (CD) spectroscopy in solution and FTIR-ATR experiments on SAM were also performed.
CD spectra reported in Fig. S4† indicate that the peptide conformation does not change substantially on addition of PEG-SH and that GE11-C does not adopt a unique conformation, spanning different structures. FTIR analysis performed in the amide I region of the spectra confirmed these results. Indeed, as shown in Table 2, the peptide can adopt several possible structures, and the related populations do not vary significantly on adding PEG-SH. These experiments show that small clusters of peptides are present in solution, and that this aggregation is not influenced by the presence of PEG-SH, even at relevant concentrations. Details concerning the interaction between GE11-C and PEG-SH in solution and on the AuNP surface were obtained by MD simulations.
| Band maximum (cm−1) | Secondary structure | GE11-C relative intensity (%) | GE11-C/PEG-SH relative intensity (%) |
|---|---|---|---|
| 1620 | β-Aggregates | 9 | 10 |
| 1639/1678 | Antiparallel β-sheets | 21 | 22 |
| 1660–1680 | Coil/bend/bridge/helix/turn structures | 53 | 50 |
| 1695 | Coils | 17 | 18 |
Simulations were performed placing a number of peptides (n) in the simulation box over the Au surface equal to n = 2, 5, 8, 9, and 20, with and without the presence of PEG-SH molecules. The results indicate that a unique aggregate is formed in the cases of n up to 9; however, when n = 20 two or more separate aggregates are formed. Moreover, simulations concerning the presence in the simulation box of two aggregates each constituted by 9 GE11-C and 1 PEG-SH show that they remain separated during the simulation time of 100 ns (see Fig. 7), supporting the idea that large aggregates are not favored. These results agree with the above reported experimental results.
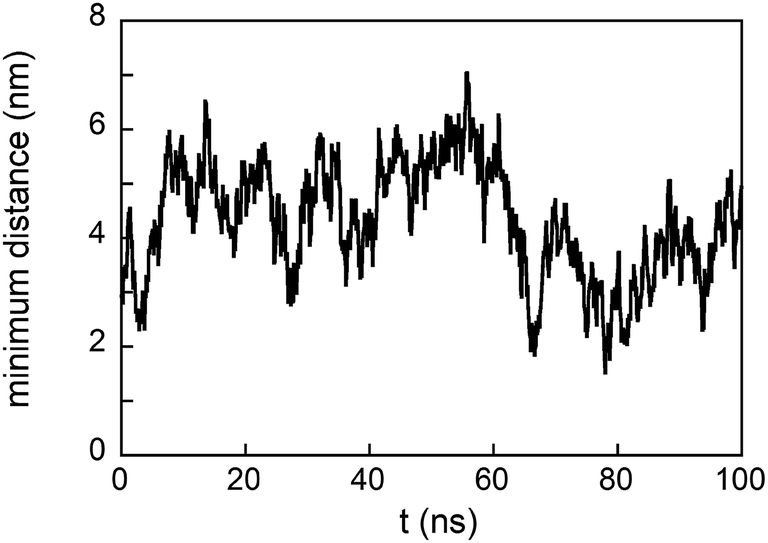 | ||
| Fig. 7 Minimum distance between two aggregates each constituted by 9 GE11-C and 1 PEG-SH. | ||
Calculated gyration radii (rg) and symmetry factors (Is) of aggregates with n = 1, 2, 5, 8, and 9 are reported in Table 3. These values agree with those obtained from fluorescence anisotropy values, thus confirming that in solution aggregates are formed, on average, by less than 10 peptides.
| n° peptides | Without PEG-SH | With PEG-SH | ||||||
|---|---|---|---|---|---|---|---|---|
| 〈rg〉 (nm) | σ | 〈Is〉 | σ | 〈rg〉 (nm) | σ | 〈Is〉 | σ | |
| 1 | 0.88 | 0.17 | 0.35 | 0.05 | 0.94 | 0.12 | 0.34 | 0.04 |
| 2 | 1.01 | 0.10 | 0.32 | 0.05 | 1.10 | 0.11 | 0.30 | 0.11 |
| 5 | 1.26 | 0.07 | 0.22 | 0.08 | 1.26 | 0.02 | 0.25 | 0.04 |
| 8 | 1.45 | 0.02 | 0.20 | 0.01 | 1.48 | 0.04 | 0.19 | 0.02 |
| 9 | 1.77 | 0.04 | 0.31 | 0.01 | 1.58 | 0.03 | 0.19 | 0.01 |
The presence of PEG-SH does not substantially modify the features of these aggregates. However, the distribution of their gyration radius values in the presence of PEG-SH is found to be less broad than in its absence, whereas Is does not change; the greater difference in rg obtained in the case of n = 9 is due to a casual change in the form of the aggregate, as demonstrated by the change in Is.
The reason for the formation of small aggregates only can be clarified by an inspection of their structural features. Indeed, as shown in the snapshot of the final frame of simulation for n = 5 (see Fig. 8A), the aggregate surface is positively charged due to presence of charged NH3+ terminal groups exposed to water. The high surface charge density thus inhibits the growth of the already formed small aggregates because of electrostatic interactions. As shown in Fig. 8B, the presence of PEG-SH does not substantially change this feature. Interestingly, it is important to note a peculiar structural characteristic of the peptide cluster with a PEG-SH chain; in all the simulations, indeed, the PEG-SH molecule is located on the surface of the cluster.
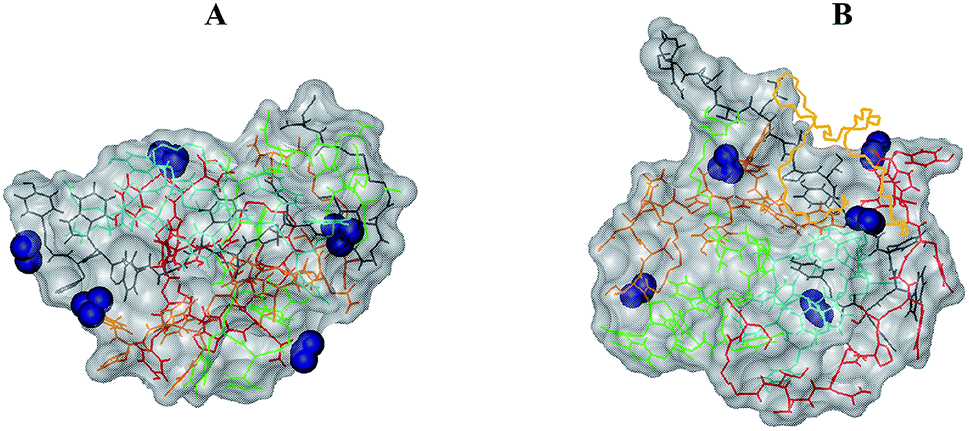 | ||
| Fig. 8 GE11-C aggregates (n = 5) in the absence (A) and in the presence (B) of 1 PEG-SH. In blue are represented the positively (NH3+) charged residues. PEG-SH is in yellow, while peptides are in green, gray, red, light blue and orange, using bond representation in the VMD program. | ||
From the analysis of the conformational features of the peptides in the last 50 ns of MD simulations, it should be noted that the whole peptide can assume several different conformations and that there is no unique dominant conformation. This finding is in agreement with the above reported spectroscopic experiments.
In all the cases, the conformation of the first part of the sequence (residues 1–4, in which there are 4 of the 5 aromatic amino acids of GE11-C) is preserved (RMSD values less than 0.7 Å). As shown in Fig. 9, both in monomers and in aggregates, the central segment of the peptide (residues 4–9) adopts mainly two conformations (total population: 70%), despite the presence of the Gly amino acid responsible for great conformational flexibility. When GE11-C peptides are covalently linked to the AuNP, they assume a different conformation (see Fig. S3†) that may explain the low selectivity of AuNP@GE11-C with respect to AuNP@GE11-C/PEG-SH_1.
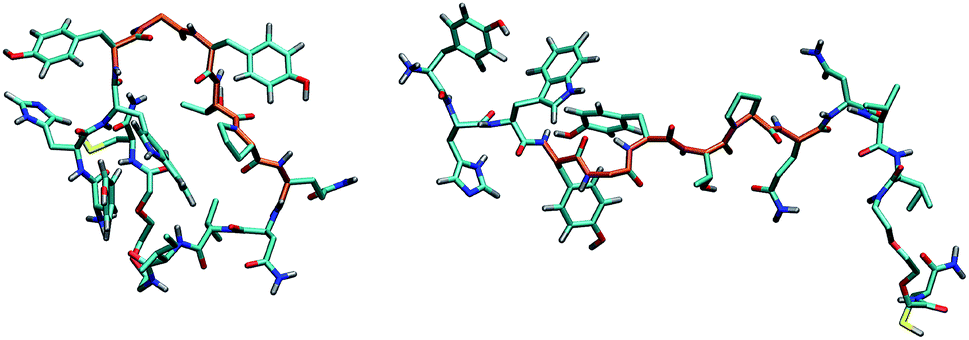 | ||
| Fig. 9 Principal structures adopted by GE11-C. The central tract of the peptide is shown in orange. C, N, O, H and S atoms are colored in cyan, blue, red, gray and yellow, respectively, using licorice representation in VMD. | ||
In agreement with spectroscopic experiments, MD calculations show that, in solution, small aggregates exist, whose conformational characteristics are due to the coexistence in defined peptide segments of several well preserved conformations. Furthermore, these characteristics do not change substantially with the presence of PEG-SH.
Additional MD simulations were performed to evaluate the linking process of GE11-C and/or PEG-SH on the AuNP surface. Simulations of a cluster of 9 GE11-C and 1 PEG-SH show that the aggregate (already constituted in solution) is able to reach the AuNP surface and to form, during the simulation time, several Au–S bonds (see Fig. 10). As a consequence, a change of the morphology and of the shape of the aggregate itself occurs; importantly, the bonding of the aggregate to the Au surface is not influenced by the presence of PEG-SH (data not shown).
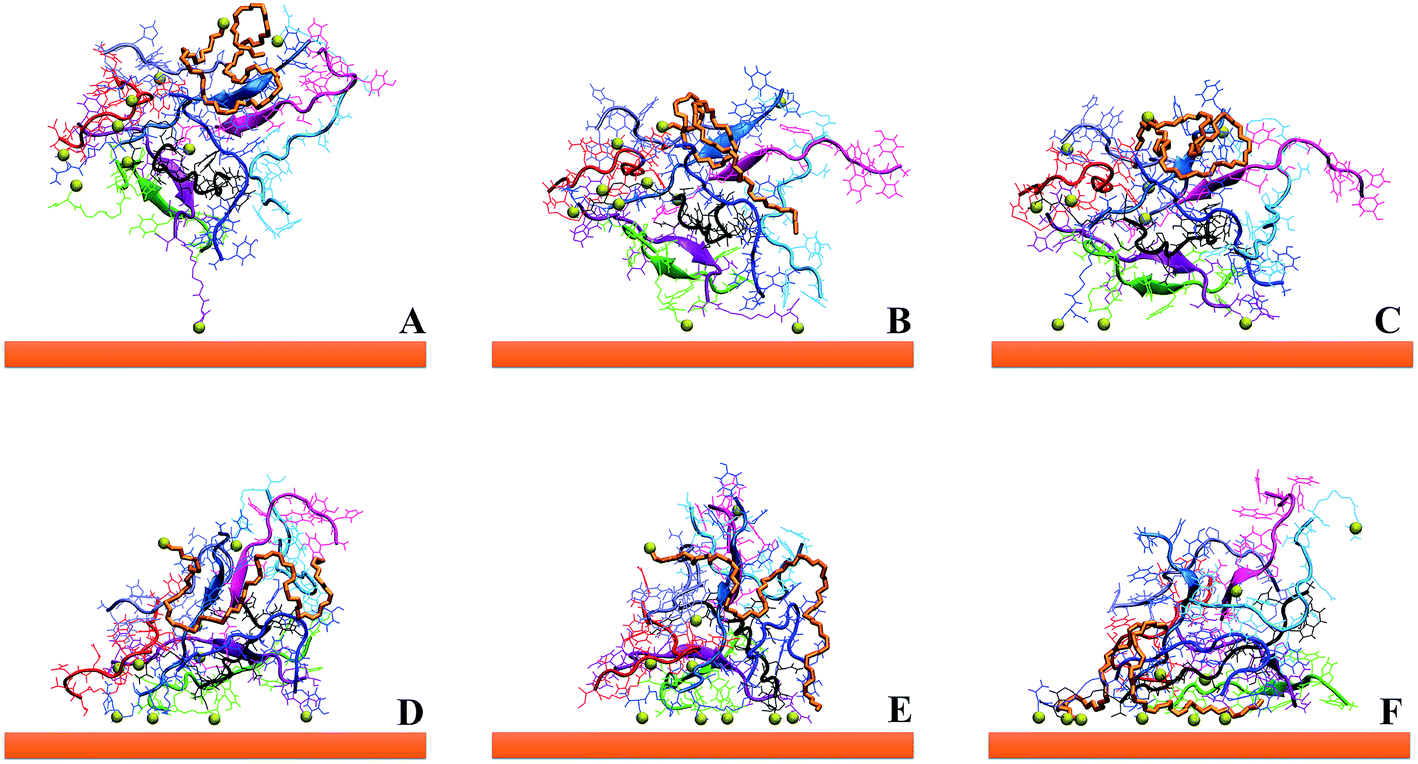 | ||
| Fig. 10 Subsequent steps (from A to F) concerning the formation of several Au–S bonds on the Au surface of an aggregate of 9 GE11-C and 1 PEG-SH during the MD simulation time. S atoms are in yellow using vdW representation in VMD. | ||
The formation of a peptide monolayer on the gold surface has been monitored following the interactions and the coalescence between two aggregates of 9 GE11-C. Interestingly, when only one aggregate is linked to the Au surface, the second one, from the solution, is not able to merge with it. For this reason, we considered two aggregates initially placed relatively far (d > 1.4 nm, that is the cut-off distance of dispersion forces) linked on a AuNP with a single Au–S bond (see Fig. 11, left). As shown in Fig. 11 (right), the two aggregates interact during the simulation, forming a noticeable interconnection only in the presence of PEG-SH. In this assembly, one PEG-SH is found located between the two peptide aggregates allowing their coalescence and thus, the formation of a dense monolayer. These results also agree with the above reported simulations (see Fig. 2 and 3) in which peptides in solution were approaching a functionalized surface. This feature is confirmed by the solvent accessible surface values obtained in the presence (151 ± 5 nm2) and in the absence (170 ± 4 nm2) of PEG-SH.
 | ||
| Fig. 11 Features of 2 aggregates (colored in blue and purple) on the Au surface, constituted by 9 GE11-C, without (top) and with 1 molecule of PEG-SH per aggregate (bottom), in the starting (left) and final (right) frames of simulations. Lines, NewCartoon and Quicksurface representations have been used for peptides (VMD); PEG-SH is in orange, with bonds style in VMD representation. For the sake of clarity, water molecules are not represented. | ||
This result strongly suggests that the presence of PEG-SH molecules in the solution and on the external surface of the aggregates plays a pivotal role in the monolayer formation.
The features of the aggregates reported in Fig. 10 and 11 also show that not all the thiolate groups of the peptides are covalently linked to the Au surface. This is an important characteristic of the organization of the peptides on the AuNPs because it means that many peptides are partially exposed, forming an external layer. It is possible to speculate that the more exposed peptides can be involved in the interactions with receptors on cells because they are free to adapt themselves to the receptors. This feature can justify the good targeting activity of AuNP@GE11-C/PEG-SH_1 where a very large number of peptides, above those directly linked to the AuNP, is present, as evidenced above (see Table 1). Clearly, a higher concentration of PEG-SH, as present on the other nanostructures (AuNP@GE11-C/PEG-SH_2 and AuNP@GE11-C/PEG-SH_3) limits the number of GE11-C on the surface of the nanostructures and, therefore, decreases the targeting activity.
The presence of a limited number of PEG-SH is very important also because they are found mostly on the surface of the GE11-C aggregates. This is another important characteristic of the organization of the molecules on the AuNP surface, since it can be responsible for the decrease of the non-specific interactions with cells without the receptors. This result can explain the higher cell selectivity of AuNP@GE11-C/PEG-SH_1 compared to AuNP@GE11-C nanostructures (see Fig. 1).
Conclusions
AuNP nanostructures functionalized with different amounts of GE11-C and of PEG-SH show different specificity and sensitivity when they target Caco-2 (EGFR+) and SW620 (EGFR−) colorectal cancer cells. Using spectroscopic techniques and MD simulations, a picture of the organization of the targeting units on the surface of the nanostructures emerges. Small peptide aggregates were found to be present in solution, stabilized by external positive charges that also inhibit their growth. Results indicate that peptide assembly in solution is maintained when they interact with the Au surface and that the presence of PEG-SH chains has a small influence on their features, both in solution and on the Au surface. The role of PEG-SH chains, however, is very important for the organization of small aggregates of GE11-C on the Au surface, because they allow anchoring of molecules from the bulk, forming an external assembly. These more exposed peptides are responsible for the specific interactions with receptors on cells because they are able to adapt themselves for good targeting.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AP acknowledges Prof. G. Bocchinfuso for helpful discussion and Dr M. Mansueti for technical support. MM acknowledges the financial contributions of the Strategic Program of the University of Padova NAMECA, the Project P-DiSC #04BIRD2016-UNIPD, and the project P-DiSC #08SID2017-UNIPD.References
- J. Giner-Casares, M. Henriksen-Lacey, M. Coronado-Puchau and L. M. Liz-Marzan, Mater. Today: Proc., 2016, 19, 19 CrossRef CAS.
- M. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307 CrossRef CAS PubMed.
- Y. Cheng, Q. Dai, R. A. Morshed, X. Fan, M. L. Wegscheid, D. A. Wainwright, Y. Han, L. Zhang, B. Auffnger, A. L. Tobias, E. Rincon, B. Thaci, A. U. Ahmed, P. C. Warnke, C. He and M. S. Lesniak, Small, 2014, 10, 5137 CAS.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Laser Med. Sci., 2008, 23, 217 CrossRef PubMed.
- K. Sokolov, M. Follen, J. Aaron, I. Pavlova, A. Malpica, R. Lotan and R. Richards-Kortum, Cancer Res., 2003, 63, 1999 CAS.
- L. C. Kennedy, L. R. Bickford, N. A. Lewinski, A. J. Coughlin, Y. Hu, E. S. Day, J. L. West and R. A. Drezek, Small, 2011, 7, 169 CrossRef CAS PubMed.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188 RSC.
- C. Fasolato, F. Domenici, S. Sennato, F. Mura, L. De Angelis, F. Luongo, F. Costantini, F. Bordi and P. Postorino, Appl. Phys. Lett., 2014, 105, 073105 CrossRef.
- C. W. Moon, J. Park, S.-P. Hong, W. Sohn, D. M. Andoshe, M. Shokouhimehr and H. W. Jang, Chem. Soc. Rev., 2018, 8, 18442 CAS.
- M. Meneghetti, A. Scarsi, L. Litti, G. Marcolongo, V. Amendola, M. Gobbo, M. Di Chio, A. Boscaini, G. Fracasso and M. Colombatti, Small, 2012, 24, 3733 CrossRef PubMed.
- C. Fasolato, S. Giantulli, I. Silvestri, F. Mazzarda, Y. Toumia, F. Ripanti, F. Mura, F. Luongo, F. Costantini, F. Bordi, P. Postorino and F. Domenici, Nanoscale, 2016, 8, 17304 RSC.
- F. Biscaglia, S. Rahendran, P. Conflitti, C. Benna, R. Sommaggio, L. Litti, S. Mocellin, G. Bocchinfuso, A. Rosato, A. Palleschi, D. Nitti, M. Gobbo and M. Meneghetti, Adv. Healthcare Mater., 2017, 6, 1700596 CrossRef PubMed.
- Z. Li, R. Zhao, X. Wu, Y. Sun, M. Yao, J. Li, Y. Xu and J. Gu, FASEB J., 2005, 19, 1978 CrossRef CAS PubMed.
- A. Schaefer, A. Pahnke, D. Schaffert, W. M. van Weerden, C. M. A. de Ridder, W. Roedl, A. Vetter, C. Spitzweg, R. Kraaij, E. Wagner and M. Ogris, Hum. Gene Ther., 2011, 22, 1463 CrossRef CAS PubMed.
- M. Fan, D. Yang, X. Liang, J. Ao, Z. Li, H. Wang and B. Shi, Biomed. Pharmacother., 2015, 70, 268 CrossRef CAS PubMed.
- S. Song, D. Liu, J. Peng, Y. Sun, Z. Li, J. Gu and Y. Xu, Int. J. Pharm., 2008, 363, 155 CrossRef CAS PubMed.
- A. M. Master, Y. Qi, N. L. Oleinick and A. Sen Gupta, Nanomed. Nanotechnol. Biol. Med., 2012, 8, 655 CrossRef CAS PubMed.
- H. Tang, X. Chen, M. Rui, W. Sun, J. Chen, J. Peng and Y. Xu, Mol. Pharmaceutics, 2014, 11, 3242 CrossRef CAS PubMed.
- P. L. Chariou, K. L. Lee, A. M. Wen, N. M. Gulati, P. L. Stewart and N. F. Steinmetz, Bioconjugate Chem., 2015, 26, 262 CrossRef CAS PubMed.
- K. Klutz, M. J. Willhauck, C. Dohmen, N. Wunderlich, K. Knoop, C. Zach, R. Senekowitsch-Schmidtke, F. Gildehaus, S. Ziegler, S. Fuerst, B. Goeke, E. Wagner, M. Ogris and C. Spitzweg, Hum. Gene Ther., 2011, 22, 1563 CrossRef CAS PubMed.
- S. Dissoki, A. Hagooly, S. Elmachily and E. Mishani, J. Labelled Compd. Radiopharm., 2011, 54, 693 CrossRef CAS.
- L. Litti, A. Ramundo, F. Biscaglia, G. Toffoli, M. Gobbo and M. Meneghetti, J. Colloid Interface Sci., 2019, 533, 621 CrossRef CAS PubMed.
- J. M. Romo-Herrera, R. A. Alavrez-Puebla and L. M. Liz-Marzan, Nanoscale, 2011, 4, 1304 RSC.
- M. Caruso, E. Placidi, E. Gatto, C. Mazzuca, L. Stella, G. Bocchinfuso, A. Palleschi, F. Formaggio, C. Toniolo and M. Venanzi, J. Phys. Chem. B, 2013, 117, 5448 CrossRef CAS PubMed.
- G. Bocchinfuso, P. Conflitti, S. Raniolo, M. Caruso, C. Mazzuca, E. Gatto, E. Placidi, F. Formaggio, C. Toniolo, M. Venanzi and A. Palleschi, J. Pept. Sci., 2014, 20, 494 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, NY, 2006 Search PubMed.
- B. Valeur, Molecular fluorescence, Wiley-VCH, Verlag, GmbH, 2002 Search PubMed.
- D. R. Lide, Handbook of Chemistry and Physics, CRC press, Boca Raton, FL, USA, 2008 Search PubMed.
- K. Eeva-Liisa and B. Andreas, J. Phys. Chem. B, 2012, 116, 4448 CrossRef PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19 CrossRef.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656 CrossRef CAS PubMed.
- P. F. J. Fuchs, H. S. Hansen, P. H. Huenenberger and B. A. C. Horta, J. Chem. Theory Comput., 2012, 8, 3943 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, and J. Hermans, Intermolecular Forces, ed. B. Pullman, Kluwer/Reidel, Dordrecht, Netherlands, 1981, pp. 331–342 Search PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 14101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. Di Nola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- M. G. L. Messina, B. Di Napoli, M. De Zotti, C. Mazzuca, F. Formaggio, A. Palleschi and G. Marletta, Langmuir, 2019 DOI:10.1021/acs.langmuir.8b03895.
- Q. Pu, Y. Leng, X. Zhao and P. T. Cummings, Nanotechnology, 2007, 18, 4007 CrossRef PubMed.
- L. B. Wright, P. M. Rodger, S. Corni and T. R. Walsh, J. Chem. Theory Comput., 2013, 9, 1616 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00096h |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |