
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConfiguration- and concentration-dependent hybrid white light generation using red, green, and blue quantum dots embedded in DNA thin films
Velu
Arasu
a,
Deoksu
Jo
ab,
Heeyeop
Chae
 *ab,
Ho Kyoon
Chung
*a and
Sung Ha
Park
*ac
*ab,
Ho Kyoon
Chung
*a and
Sung Ha
Park
*ac
aSungkyunkwan Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University, Suwon 16419, Korea. E-mail: hchae@skku.edu; hokchung@skku.edu; sunghapark@skku.edu
bSchool of Chemical Engineering, Sungkyunkwan University, Suwon 16419, Korea
cDepartment of Physics, Sungkyunkwan University, Suwon 16419, Korea
First published on 9th October 2018
Abstract
Artificial white-light production from primary red, green, and blue (RGB) colours is in high demand in the lighting, information technology, and wearable display industries, and it requires a simple device structure and efficient templates with stable luminophores. To realize efficient and stable optoelectronic devices, relevant materials and device structures need to be identified. Therefore, we report the construction of a simple hybrid white-light optoelectronic device with a single excitation source with efficient RGB colours on a stable optical platform. Emission wavelength-tunable R, G, and B quantum dots (QDs) with specific ligands and cetyltrimethylammonium chloride (CTMA)-modified DNA (CDNA) are prepared for the fabrication of QDs embedded in CDNA thin films with mixed and orthogonally stacked configurations. Fourier transform infrared, photoluminescence quantum yield (PLQY), ultraviolet (UV)-vis absorbance, photoluminescence, and electroluminescence (excited by blue and UV LEDs) spectra of the QDs embedded in CDNA thin films are analyzed to investigate their ligand attachment, luminescence efficiency, optical excitation, spectral emission, and hybrid white-light properties. In addition, the dispersion and photostability of QDs in the CDNA matrix were analyzed. The colour rendering index (CRI) values and colour gamut of the QDs embedded in CDNA thin films are studied for evaluating the light quality. The results show that the ligands on the QDs enhance PLQY up to 95 and 25% in liquid and solid phases, respectively. The optical properties of the QDs in the CDNA thin films are not significantly affected by phase changes, which implies the effective hosting of QDs within CDNA. The CRI values of the mixed and stacked configuration-dependent QDs embedded in CDNA thin films are 21 and 80%, respectively, which suggest the relatively stronger self-absorption of R QDs in the mixed configuration than in the stacked configuration. In addition, CRI values and colour gamut are affected by different R, G, and B QD concentrations in CDNA. These findings are important for solid-state lighting, information display systems, flexible displays, and wearable displays.
1. Introduction
Information display systems are in high demand in the field of information technology, with their applications varying from flight information display screens to ticket vending machines. The information perceived by light emitters is displayed by optoelectronic devices such as light-emitting diodes (LEDs) and organic LEDs. Bright luminaires with continuous and uninterrupted emissions require energy efficient and pure-colour emission capability. Among the various light-emitting materials, nanocrystal quantum dots (QDs) are well known because of their high energy efficiency and wide colour gamut caused by their high photoluminescence quantum yield (PLQY) and narrow emission profile.1,2 The detailed photophysical properties of QDs have been reported as well.3 Colloidal QDs have applications in artificial lighting, QD LEDs, fluorescence-based microscopy, QD spectrometers, flexible lighting and displays, food science and biology, and other optoelectronic fields.4–9 At present, white-light generation using LEDs and phosphors has been widely reported, and colour quality is achieved to some extent.10–12 However, achieving high colour purity and wide gamut practically is challenging.Conventionally, two-dimensional synthetic polymer-based thin films with organic red, green, and blue (RGB) dyes are used for white-light generation. However, organic emitters cannot preserve their optical properties in synthetic polymers due to low stability and agglomeration.13 Furthermore, an organic emitter requires multiple photo-excitation sources for light emission due to its narrow excitation bandwidth or Stokes shift. Consequently, the use of multiple photo-excitation sources and the range of different organic emitters for different colours complicate the processing and stability of white-light-emitting devices over time.14,15
The major goal of this study is to construct a simple hybrid white-light-emitting optoelectronic device structure containing a single excitation source with efficient RGB emission embedded on a stable optical platform. The combination of RG with a blue LED source as well as the combination of RGB emitters with an ultraviolet (UV) LED source can significantly simplify the device system. Different combinations and arrangements of the R, G, and B emitters in the thin films also significantly affect the white-light spectral quality.16
QDs have the potential to replace phosphors and can provide vivid colours and sharp contrast for future information display technology.17–19 A previous computational study has reported that QD-based lighting is more efficient than phosphor-based lighting.20 Moreover, because of the large Stokes shift range of QDs, it is possible to replace multiple excitation sources with a single excitation source in QD-based devices, which leads to simple and efficient hybrid (combination of electric and photonic) white-light emission.21
The luminescence properties of QDs are good in the liquid phase due to their colloidal stability. However, a suitable embedding matrix or platform is desirable to preserve the optical properties of QDs in the solid phase. Deoxyribonucleic acid (DNA) molecules, which have intrinsic optical characteristics (e.g., wide optical bandgap) and molecular recognition, provide a rigid platform for hosting various functional nanomaterials.22–25 Owing to the high aspect ratio and helical structure of DNA, it can provide more room for embedding luminophores with less agglomeration.26 In addition, DNA is advantageous for bio-optoelectronic applications with QD luminophores because of its biocompatible nature.27
Chemical composition-tunable gradient core/alloy/shell structured R, G, and B QDs with ligands (trioctylphosphine (TOP), tributylphosphine (TBP), and 1-dodecanethiol (DDT)) are prepared by a hot-injection single-pot synthesis method. In addition, DNA with cetyltrimethylammonium chloride (CTMA), represented as CDNA, is used for the fabrication of QDs embedded in CDNA thin films with two distinct QD configurations: mixed and orthogonally stacked QDs in CDNA. In addition, we measure the Fourier transform infrared (FTIR), PLQY, UV-vis absorbance, and photoluminescence (PL) spectra of the QDs embedded in CDNA thin films to study the ligand attachment, efficiency, excitation, and emission of the QDs, respectively. The dispersion and photostability of QDs in the CDNA matrix were analyzed.
Also, electroluminescence (EL) with different concentrations and configurations of QDs in CDNA are investigated to understand the characteristics of the white light generated by blue LEDs with RG QD films and UV LEDs with RGB QD films. Finally, the colour rendering index (CRI) and International Commission on Illumination (CIE) coordinates of the QDs embedded in CDNA thin films are discussed.
2. Experimental
2.1 Preparation of TOP, TBP, and DDT ligands attached to QDs
Luminescent R, G, and B QDs were prepared by a one-pot hot-injection method (Fig. 1(a) and Table 1).26 Cadmium oxide (CdO), zinc acetate (Zn(Ac)2), selenium (Se), sulfur (S), TOP, TBP, DDT, OA, and ODE (Sigma-Aldrich, St. Louis, USA) were used for QD synthesis. The entire process was conducted under nitrogen gas to avoid any oxidation. CdO and Zn(Ac)2 (molar quantities in Table 1) were loaded into a three-necked round bottom flask with 5 mL of OA and heated to 150 °C while stirring. We prepared two different stock solutions: Se and S dissolved in 3 mL TOP for R and G QDs (for relatively larger and medium-sized cores) and S dissolved in 5 mL ODE for B QDs (for smaller cores). At 150 °C, 15 mL ODE was added and heated further to 300 °C, and the stock solution was injected rapidly. After 8 min of stock solution injection, 3.6 mM S dissolved in 2 mL TBP was slowly injected into the solution for the B QDs. Consequently, TOP (for R and G) and TBP (for B) ligands were attached on the surface of the QDs (named as QDT). In addition, 0.5 mL DDT ligand was slowly injected at 290 °C after 15 min of stock solution injection, forming TOP + DDT or TBP + DDT ligands on the QD surface (QDTD). Finally, chemical composition-tunable R, G, and B QDs with distinct ligands were obtained, QDT(R), QDTD(R), QDT(G), QDTD(G), QDT(B), and QDTD(B), which were purified and dispersed in 50 mg mL−1 benzene. | ||
| Fig. 1 Schematics of the photophysical process of chemical composition-tunable QDs, comparison of Stokes shifts of QDs and organic dyes, pristine DNA and CTMA-modified DNA (CDNA) structures, fabrication of QDs embedded in CDNA thin films and excitation by blue and UV LEDs. (a) Graphical representation of composition-tunable photonic emissions emerging from QDs of different core sizes: the larger the core size, the longer the wavelength. (b) Comparison of Stokes shifts of QDs (QDBs and QDRs) and organic dyes (CMR and R6G). The QDBs and QDRs have two PL bands from the 1st (weak) and nth (strong) excitation bands and wider Stokes shifts. These bands are not applicable for dyes. (c) Structures of DNA and CDNA. (d) Schematics of QDs embedded in CDNA thin films fabricated from mixed RGB QDs (QD(RGB)-CDNA) and stacked RGB QDs (QD(R/G/B)-CDNA) formed by drop casting. (e) Schematic of hybrid white-light generation using QDs embedded in CDNA thin films excited by electrically driven blue and UV LEDs. | ||
| QD RGB colours | CdO (mM) | Zn(Ac)2 (mM) | Se (S) (mM) | DDT (mL) | Abs. λmax (nm) | PL λmax (nm) | FWHM (nm) |
|---|---|---|---|---|---|---|---|
| QDT(R) | 1.6 | 4 | 0.4 (1) | 0 | 590 | 607 | 35 |
| QDTD(R) | 1.6 | 4 | 0.4 (1) | 0.5 | 590 | 608 | 35 |
| QDT(G) | 0.3 | 4 | 0.2 (3.4) | 0 | 510 | 531 | 45 |
| QDTD(G) | 0.3 | 4 | 0.2 (3.4) | 0.5 | 510 | 529 | 45 |
| QDT(B) | 0.5 | 5 | 0 (0.9) | 0 | 440 | 454 | 25 |
| QDTD(B) | 0.5 | 5 | 0 (0.9) | 0.5 | 440 | 455 | 25 |
2.2 Preparation of DNA and CDNA
In 10 mL deionized (DI) water, 0.05 g DNA obtained from salmon (molecular weight of ∼8000 kDa, GEM Corporation, Shiga, Japan) was added. A homogeneous mixture of 0.5 wt% DNA solution was achieved by stirring this solution overnight at 1000 rpm for 6 h.2.3 Preparation of CDNA
The CDNA complex was prepared according to a previously reported process with slight modifications.28 Equal concentration (0.4 wt%) solutions of CTMA surfactant and DNA were prepared by dissolving them in DI water separately. Then CTMA was added dropwise into the DNA solution while stirring. This process allowed the attachment of the CTMA surfactant to the surface of the DNA duplexes via ionic interaction, forming a white precipitate. The precipitate was stirred for 3 h, and then filtered and washed with excess DI water to remove unbound CTMA. Finally, the CDNA complex was obtained by washing the precipitates and drying them overnight. The CDNA solution was obtained by adding 0.3 g of the CDNA complex in a 10 mL benzene and butanol (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40) mixture to disperse the QDs and CDNA. The final concentration of CDNA dissolved in the organic solvent was 3.0 wt% (Fig. 1(c)).
:40) mixture to disperse the QDs and CDNA. The final concentration of CDNA dissolved in the organic solvent was 3.0 wt% (Fig. 1(c)).
2.4 Fabrication of R, G, and B QDs embedded in CDNA thin films
R, G, and B QD solutions (50 mg mL−1) were dropped into a CDNA solution (3.0 wt%) in ratios of 1:20 (for R1, G1 and B1, reference concentration) and 2:20 (for R2, G2 and B2, double concentration). For homogeneous mixing, the solution was vortexed and left for 1 h. The QDs embedded in CDNA solutions were drop-cast (with 15 μL drop volume) on a given substrate (glass for PL, fused silica for FTIR, PLQY, and UV-vis, and blue and UV LEDs for EL). QDs embedded in CDNA thin films fabricated from mixed RGB QDs (QD(RGB)-CDNA) and stacked RGB QDs (QD(R/G/B)-CDNA) were constructed by drop casting. In addition, QDs embedded in CDNA thin films were fabricated by varying the concentration ratio of R, G, and B QDs. Thin layers of 0.5 wt% DNA (marked as /) were placed in QD(R/G/B)-CDNA formed by spin coating (7000 rpm) to prevent the dissolution of the bottom layers (Fig. 1(d) and Table 2).
| Blue LEDs (λEL max 460 nm) + R and G QD-CDNA | UV LEDs (λEL max 365 nm) + R, G, and B QD-CDNA | ||||
|---|---|---|---|---|---|
| R to G ratio | CIE (x, y) | CRI % | R to G to B ratio | CIE (x, y) | CRI % |
| Mixed QDs embedded in single layered CDNA configuration | |||||
| QDTD(R1G1)-CDNA | (0.46, 0.24) | 4 | QDTD(R1G1B1)-CDNA | (0.63, 0.34) | 71 |
| QDTD(R2G1B1)-CDNA | (0.66, 0.33) | 61 | |||
| QDTD(R2G1)-CDNA | (0.59, 0.30) | 30 | QDTD(R1G2B1)-CDNA | (0.62, 0.36) | 79 |
| QDTD(R1G1B2)-CDNA | (0.62, 0.34) | 70 | |||
| QDTD(R1G2)-CDNA | (0.49, 0.26) | 21 | QDTD(R2G2B1)-CDNA | (0.66, 0.34) | 64 |
| QDTD(R1G2B2)-CDNA | (0.59, 0.37) | 79 | |||
| QDTD(R2G1B2)-CDNA | (0.66, 0.33) | 58 | |||
|
|||||
| Orthogonally stacked QDs embedded in CDNA configuration | |||||
| QDTD(R1/G1)-CDNA | (0.56, 0.34) | 64 | QDTD(R1/G1/B1)-CDNA | (0.54, 0.40) | 81 |
| QDTD(R2/G1/B1)-CDNA | (0.62, 0.37) | 78 | |||
| QDTD(R2/G1)-CDNA | (0.65, 0.34) | 64 | QDTD(R1/G2/B1)-CDNA | (0.51, 0.46) | 74 |
| QDTD(R1/G1/B2)-CDNA | (0.55, 0.37) | 78 | |||
| QDTD(R1/G2)-CDNA | (0.53, 0.39) | 80 | QDTD(R2/G2/B1)-CDNA | (0.58, 0.41) | 80 |
| QDTD(R1/G2/B2)-CDNA | (0.49, 0.42) | 87 | |||
| QDTD(R2/G1/B2)-CDNA | (0.62, 0.38) | 82 | |||
2.5 Measurement of FTIR and PLQY spectra
The light–matter interaction was measured by FTIR spectroscopy with an MIR_ATR detector (ZnSe) (TENSOR 27, Bruker Inc., MA, USA) in the range 3600–600 cm−1 for the QDs in the liquid phase and CDNA thin films. The QD-CDNA thin films were fabricated on a fused silica substrate to minimize the absorbance by the substrate at a relatively higher energy. The scans were averaged (32 scans) with a resolution of 4 cm−1 (Fig. 2(c)). The PLQY of the QDs in the liquid phase and CDNA thin films was measured using an integrating sphere (C9920-20, Hamamatsu Photonics, Shizuoka, Japan) via the absolute method. The PL spectra were recorded in the wavelength range 200–950 nm at a fixed photoexcitation wavelength of 365 nm (Fig. 2(d)).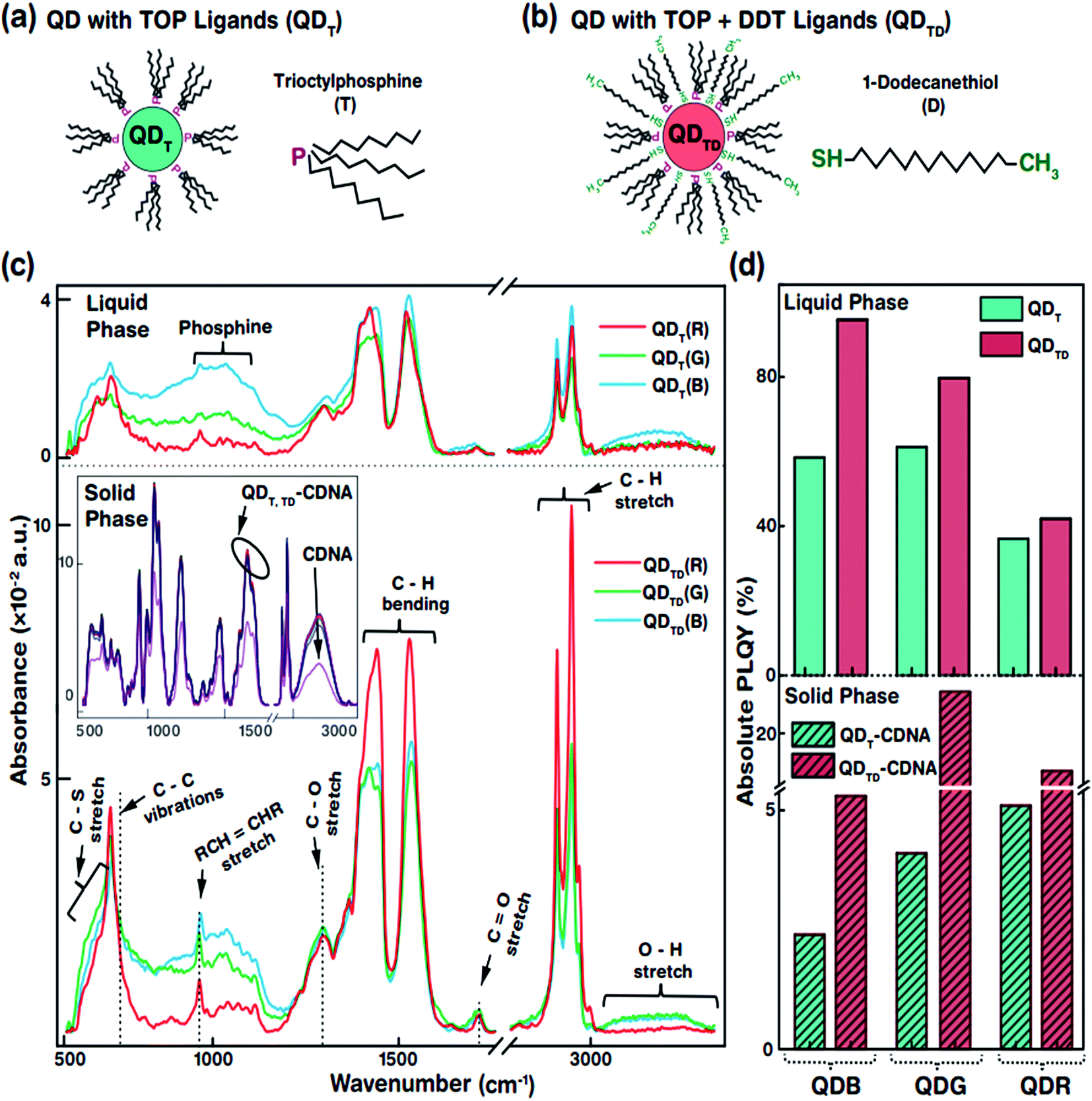 | ||
| Fig. 2 Representations of T and TD ligands on QD surfaces and FTIR and PLQY bar graphs of QDs (R, G, and B) in the liquid phase and in CDNA thin films. (a and b) Schematic views of T and TD ligands attached to QDs (represented as QDT and QDTD, respectively). (c) FTIR absorbance spectra of QDT (top) and QDTD (bottom) in the liquid phase and of QDT and QDTD embedded in CDNA thin films (QDT-CDNA and QDTD-CDNA, respectively) in the solid phase (inset). Functional groups, fingerprints of the different ligands, and their corresponding absorbance peaks are shown. The FTIR absorbance of CDNA is not noticeably altered by the QDs (inset). (d) PLQY of QDT and QDTD in the liquid phase and of QDT-CDNA and QDTD-CDNA in the solid phase. The PLQY values of QDTD are relatively higher than those of QDT in both phases. | ||
2.6 Measurement of UV-vis absorbance and PL
The absorbance of QDs in the liquid phase and CDNA thin films fabricated on fused silica was measured using a UV-vis spectrophotometer (Cary 5G, Varian, CA, USA) by passing frequency-dependent light through the QDs (Fig. 3(a)). Also, the PL spectra of the QDs in the liquid phase and CDNA thin films fabricated on glass substrates were obtained at a fixed photoexcitation wavelength of 365 nm. The PL excitation and emission spectra were measured at room temperature using a 25 W Xe arc lamp equipped with a fluorometer (FS-2, Scinco, Seoul, Korea) (Fig. 3(b)).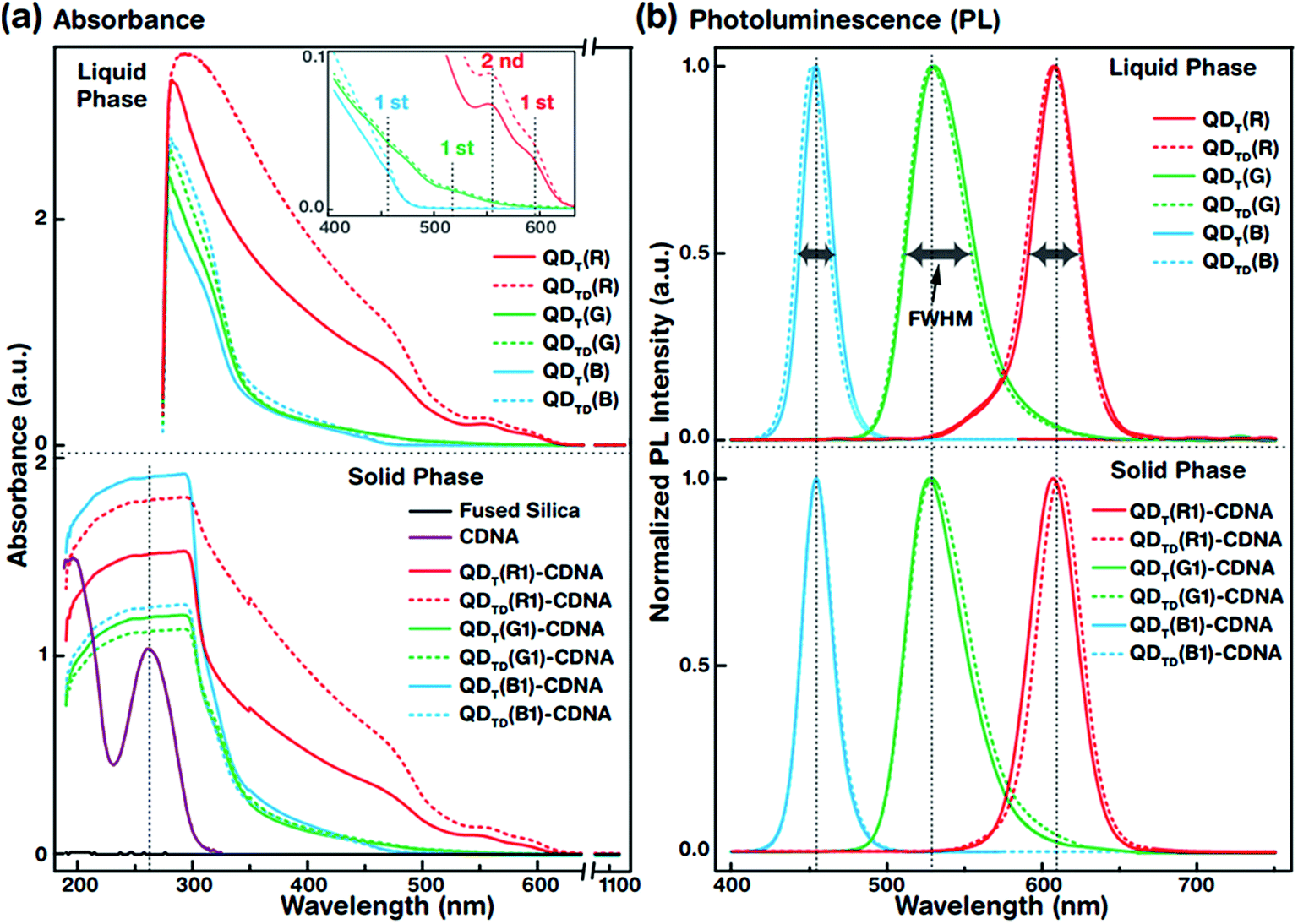 | ||
| Fig. 3 Optical spectra of QDs in the liquid phase and of QDs embedded in DNA thin films. UV-vis absorbance and PL spectra of R, G, and B QDs with either T or TD ligands (QDT or QDTD, respectively) and QDs embedded in DNA thin films with either T or TD ligands (QDT-CDNA or QDTD-CDNA, respectively) are shown by solid and dotted lines, respectively (details in the Experimental section). (a) UV-vis absorbance spectra of QDT and QDTD and QDT-CDNA and QDTD-CDNA. The characteristic DNA absorbance peak at around 260 nm is shown by a vertical dotted line (bottom) (inset). The first absorbance maximum peaks of QDT,TD(R), QDT,TD(G), and QDT,TD(B) are observed at wavelengths of 590, 510, and 440 nm, respectively. Interestingly, the second absorbance peak of QDT,TD(R) is clearly visible at 560 nm. The QD absorbance peak positions in QDT-CDNA and QDTD-CDNA are similar to those in QDT and QDTD in the liquid phase. (b) Normalized PL spectra of QDT and QDTD and QDT-CDNA and QDTD-CDNA. QDT,TD(G) has a relatively wider FWHM than the other samples. Reference concentrations of QDs for R, G, and B in CDNA are used and marked as R1, G1, and B1, respectively. | ||
2.7 HRTEM, FESEM and photostability measurements
QDRs dispersed in hexane (5 mg mL−1) and QDRs in CDNA (3.0 and 6.0 wt%) solution were drop-cast on a copper mesh grid of a high-resolution transmission electron microscope (HRTEM), followed by drying for 1 h at 60 °C. The grids with QDRs and QDRs embedded in CDNA were placed inside the HRTEM (Cs-corrected/EDS/EELS, JEM ARM 200F, JEOL Corp., Tokyo, Japan) under an ultra-vacuum for imaging. The surface degradation of QDRs embedded in CDNA thin films was measured using a field emission scanning electron microscope (FE-SEM) (JSM 7401F, JEOL Corp., Tokyo, Japan). Before the measurement, the QDR-CDNA thin film surface was coated with a platinum conductive layer (∼10 nm) by sputtering to reduce thermal damage and improve the secondary electron signals.The photostability of QDRs embedded in CDNA was measured by exposing them to UV light (VL-6.LC, Vilber Lourmat, Suebia, Germany) having a wavelength of 254 nm (6 W, 610 μW cm−2) at room temperature. The sample was placed 5 cm away from the surface of the lamp to avoid direct thermal degradation. The setup was isolated from ambient light by placing it in a black enclosure (Fig. 4).
 | ||
| Fig. 4 Dispersion and UV light degradation analysis of QDRs embedded in CDNA thin films. (a) HRTEM images of QDRs without and with the CDNA matrix: (left) QDRs without CDNA show aggregation and (right) QDRs in CDNA show moderate dispersion. The inset in the left image indicates the particle size of QDRs (∼6.7 nm). (b) Surface degradation analysis of (top) CDNA and (bottom) QDRs embedded in a CDNA thin film by FESEM. The FESEM images show minute surface degradation after 20 h of UV light exposure. | ||
2.8 Measurement and analysis of EL and white-light spectra
EL characteristics were measured in the wavelength range 360–700 nm by illuminating blue or UV LEDs without and with the QD-CDNA thin films. The blue or UV LEDs were driven by a current source of 5 mA, which was controlled by a sourcemeter (Keithley Instruments Inc., OH, USA). The LEDs with the QD-CDNA thin films were mounted on an array spectrometer (MCPD-9800, Otsuka Electronics, Japan). EL data (radiation intensity, W sr−1) were collected through a half-integrating sphere. Finally, CIE colour coordinates and CRI values were calculated from the EL spectral data using the ColorCalculator simulation software (Osram Sylvania, MA, USA) (Fig. 5–7 and Table 2).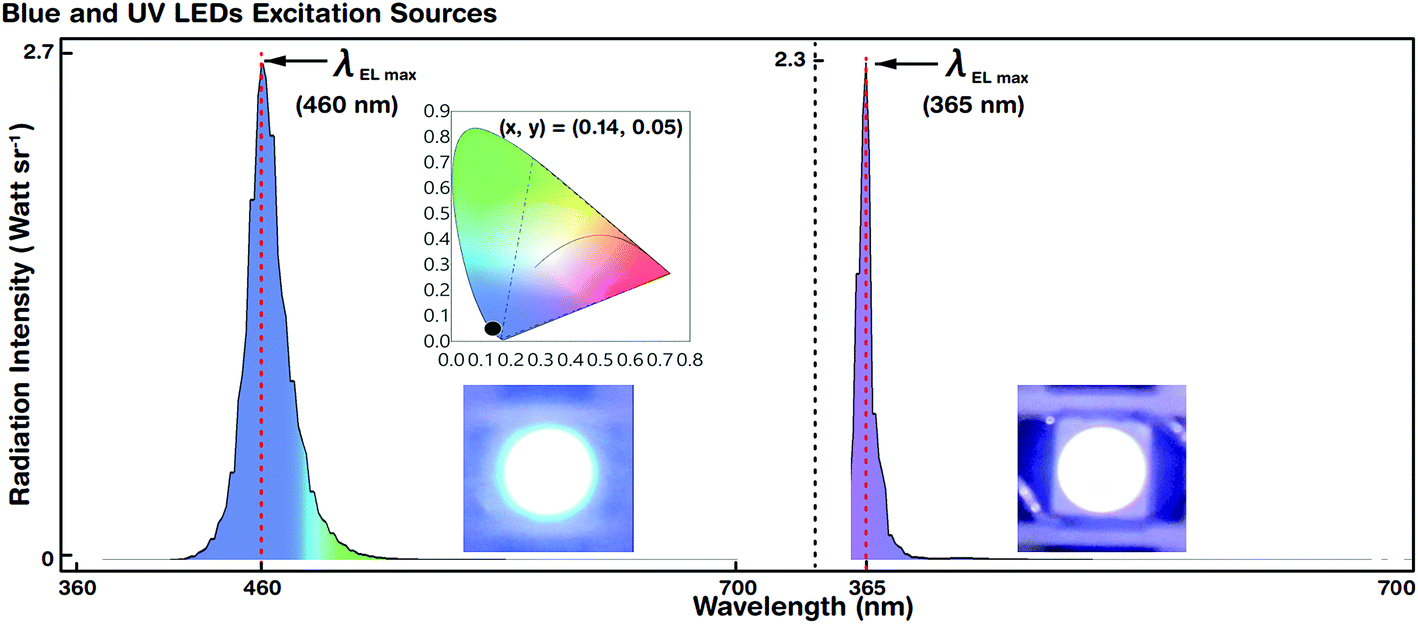 | ||
| Fig. 5 EL spectra and colour coordinates of reference blue and UV LEDs. The reference blue (left) and UV (right) excitation sources and CIE colour coordinates are shown in the insets. The maximum EL peak wavelengths (λEL max) of the blue and UV LEDs are observed at 460 and 365 nm, respectively. FWHM values of blue and UV LEDs are 27 nm and 15 nm, respectively. CIE colour coordinates of blue LEDs are (0.14, 0.05). | ||
 | ||
| Fig. 6 EL spectra of blue LEDs with QDs embedded in CDNA thin films with mixed and orthogonally stacked configurations. The white-light luminescence spectra and CIE colour coordinates are shown in the insets. White-light radiation intensities emitted by (left) a mixture of reference concentration of R (R1) and double concentration of G (G2) QDs embedded in CDNA (QDTD(R1G2)-CDNA) and (right) stacked R1 and G2 QDs embedded in CDNA thin films (QDTD(R1/G2)-CDNA) integrated with a blue LED. Although the R to G QD concentration ratio (1:2) in QDTD(R1G2)-CDNA and QDTD(R1/G2)-CDNA is the same, their CRI values are considerably different. This indicates that the self-absorption of QDs in the QDTD(R1G2)-CDNA configuration is greater than that in the QDTD(R1/G2)-CDNA configuration. | ||
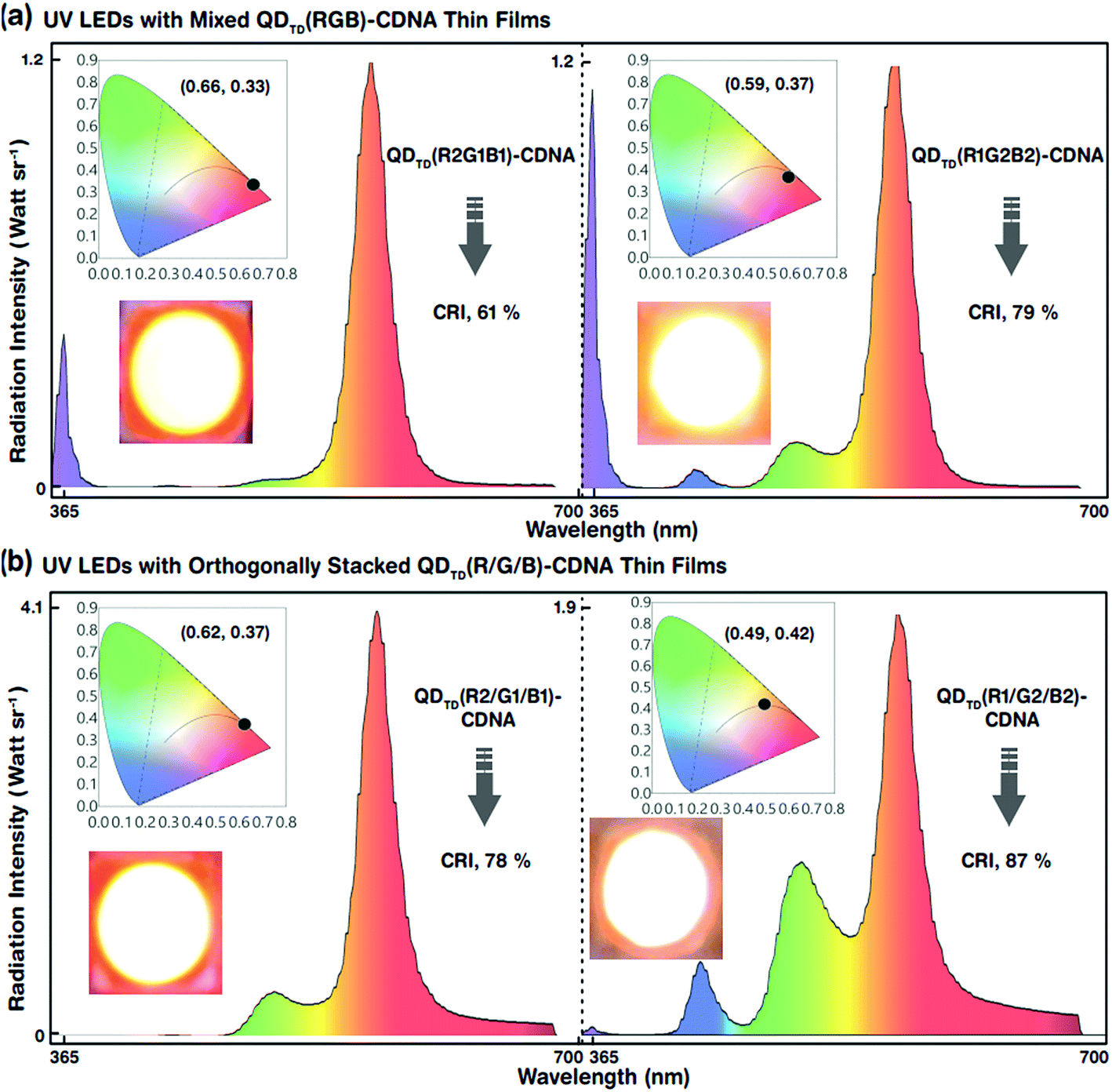 | ||
| Fig. 7 White-light radiation intensities emitted by mixed R, G, and B QDs embedded in CDNA and stacked R, G, and B QDs embedded in CDNA thin films integrated with a UV LED. Two different sets of QD concentrations (R2, G1, B1 and R1, G2, B2) are used in the mixed and stacked configurations. In addition, the device white-light luminescence photographs and CIE colour coordinates are shown in the insets. (a) White-light spectra of QDTD(R2G1B1)-CDNA and QDTD(R1G2B2)-CDNA excited by the UV LED. Even though the configuration is the same, different R, G, and B QD concentrations in QDTD(R2G1B1)-CDNA and QDTD(R1G2B2)-CDNA lead to an ∼18% CRI increase because of the significant change in self-absorption of R QDs. (b) White-light spectra of QDTD(R2/G1/B1)-CDNA and QDTD(R1/G2/B2)-CDNA excited by the UV LED. The CRI values of QDTD(R2/G1/B1)-CDNA and QDTD(R1/G2/B2)-CDNA are 78% and 87%, respectively. The CRI increases because of the change in R, G, and B QD concentrations. Qualitatively, the device luminescence photographs agree well with the CIE colour coordinates. | ||
3. Results and discussion
3.1 Emission wavelength-tunable QDs and Stokes shift
The optical properties of QDs (which are treated as artificial atoms) can be easily tuned by controlling their structural parameters such as core size, chemical composition, shell thickness, and surface ligands. The crystalline interface between the core and the shell is important to reduce the lattice mismatch and enhance the stability of the QD system. Although a well-defined core–shell QD structure constructed by a multi-pot synthesis process has been widely studied, this method produces a relatively large lattice mismatch and is time consuming, leading to an unstable QD system. Hence, we adopted a single-pot, hot-injection method, which allows tunable chemical composition, gradient core–shell structures, less lattice mismatch, and controllable core size. This provided a wide emission wavelength from blue to red (due to the chemical composition and core size) and a relatively stable QD system (due to the gradient core–shell interface). A schematic representation of the tunable emission properties derived from the different core sizes (related to the conduction band (CB) and valence band (VB)) and chemical compositions (e.g., CdZn) of QDs is shown in Fig. 1(a). A larger core led to a longer emission wavelength, i.e., red, green and blue photon emissions. The core size was controlled by tuning the ratio of the cadmium and selenium precursors (see the Experimental section and Table 1); increased amounts of cadmium in the presence of selenium yielded a relatively larger QD core rapidly.Fig. 1(b) shows the absorbance spectra of QDs (red and blue QDs, expressed as QDRs and QDBs) and organic dyes (coumarin (CMR) and rhodamine 6G (R6G)), which can be used to determine the difference in Stokes shifts of the QDs and dyes. The magnitude of the Stokes shift (difference between the emission (solid line) and absorption (dotted line) maximum peak wavelengths) was relatively larger for the QDs than for the organic dyes due to dissimilar absorbance characteristics. The QDBs and QDRs have two PL bands from the 1st (weak) and nth (strong) excitation bands and wider Stokes shifts. The continuous absorbance characteristics of the QDs allowed luminescence excitation using a UV or green-light excitation source,29 which indicated that the absorbance spectra of QDRs ranged from 300 to 590 nm. Consequently, QDRs and QDGs could be excited by a single blue LED and all QDs (QDRs, QDGs, and QDBs) could be excited by a UV LED. In contrast, the organic dyes rarely showed continuous absorbance properties in the high-energy region and hence had limited Stokes shifts.
QDs embedded in CDNA thin films fabricated from mixed RGB QDs (named as QD(RGB)-CDNA) and stacked RGB QDs (QD(R/G/B)-CDNA) are shown in Fig. 1(d). Thin layers of DNA (marked as /) formed by spin coating were sandwiched between the QD(R/G/B)-CDNA layers to avoid the dissolution of the bottom layers (a detailed discussion can be found in our previous report).30Fig. 1(e) shows a schematic of the hybrid white light generated by the QDs embedded in CDNA thin films.
3.2 Ligand-dependent PLQY characteristics of individual R, G, and B QDs in the liquid phase and CDNA thin films
The specific morphological and photophysical properties of colloidal QDs strongly depend on the ligands attached to them. Ligands act as stabilizing agents (to prevent agglomeration and regulate the diffusion kinetics during synthesis) and dispersing agents, i.e., they facilitate dispersion in liquid and solid media to prevent the self-aggregation of QDs. The ligands attached to the QD surface interact with the dispersion medium or template (CDNA thin films) via electrostatic and van der Waals forces. In particular, the dispersion of QDs with thiol ligands in a solid medium has been reported to lead to improved stability and PLQY due to the optimal dispersion of QDs.31,32 Consequently, appropriate ligands on the QD surface can significantly enhance the photophysical characteristics of QDs in liquid and solid phases. In this study, we used oleic acid (OA) and 1-octadecene (ODE) ligands and TOP for R and G or TBP for B (both TOP and TBP are represented as T) and 1-dodecanethiol (DDT is represented as D) ligands for the synthesis and dispersion of QDs in both chloroform solution and CDNA thin films (Fig. 2(a and b)). D ligands, composed of a thiol group at the end of the chain, served as additional passivation ligands after T stabilized the QDs in the liquid and solid phases.33 CDNA could be used as an efficient template for QDs in solid films due to its unique polymeric nature (rigidity, transparency, and biocompatibility).
Fig. 2(c) shows the Fourier transform infrared (FTIR) absorbance spectra of QDT and QDTD in the liquid phase and of QDT and QDTD embedded in CDNA thin films (represented as QDT-CDNA and QDTD-CDNA) in the solid phase. As expected, the FTIR absorbance peak intensities varied with functional groups and alkane chains, confirming the presence of ligands on the QDs. The functional groups originating from OA, ODE, T, and D ligands were identified as follows. The medium absorbance peaks at 630–660 cm−1 corresponded to the C–S stretching of thiol, presumably originating from D. Broad peaks corresponding to inorganic elemental phosphine were observed at 1000–1100 cm−1. The broad peaks at 700–1300 cm−1 corresponded to the C–C skeletal vibrations of methyne. The narrow short peaks at 965–975 cm−1 were attributed to the RCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR (R refers to methyl) vibrations of OA. Ligands possessing C–H chains showed relatively strong absorbance peaks. The strong peaks at 1445–1485 cm−1 and 2848 and 2921 cm−1 corresponded to the C–H bending of methylene and the C–H asymmetric stretching of methyl and methylene, respectively. In contrast, the minor peaks at 1310 and 1720 cm−1 corresponded to the C–O and CO stretching of OA. Finally, the OH stretching of OA was observed as a broad peak at 3200–3570 cm−1.34 The FTIR absorbance spectra of the QDs embedded in CDNA thin films in the solid phase are shown in the inset of Fig. 2(c). FTIR absorbance of the QD-CDNA thin films exhibited the characteristic behaviour of pristine CDNA. The FTIR absorbance of CDNA clearly showed characteristic peaks for sugar, phosphate groups (1250–600 cm−1), and nucleotide bases (1800–1300 cm−1); detailed mode assignments are shown in our previous report.24 As expected, D ligands formed a stronger bond on the QD surface because they had more free electrons (about 2 times) than T. The optical properties of QDs with different ligands were found to be different due to the degree of surface passivation and dispersion in liquid and solid media.
CHR (R refers to methyl) vibrations of OA. Ligands possessing C–H chains showed relatively strong absorbance peaks. The strong peaks at 1445–1485 cm−1 and 2848 and 2921 cm−1 corresponded to the C–H bending of methylene and the C–H asymmetric stretching of methyl and methylene, respectively. In contrast, the minor peaks at 1310 and 1720 cm−1 corresponded to the C–O and CO stretching of OA. Finally, the OH stretching of OA was observed as a broad peak at 3200–3570 cm−1.34 The FTIR absorbance spectra of the QDs embedded in CDNA thin films in the solid phase are shown in the inset of Fig. 2(c). FTIR absorbance of the QD-CDNA thin films exhibited the characteristic behaviour of pristine CDNA. The FTIR absorbance of CDNA clearly showed characteristic peaks for sugar, phosphate groups (1250–600 cm−1), and nucleotide bases (1800–1300 cm−1); detailed mode assignments are shown in our previous report.24 As expected, D ligands formed a stronger bond on the QD surface because they had more free electrons (about 2 times) than T. The optical properties of QDs with different ligands were found to be different due to the degree of surface passivation and dispersion in liquid and solid media.
The PLQY of QDT and QDTD in the liquid phase and of QDT-CDNA and QDTD-CDNA in the solid phase is shown in Fig. 2(d). The PLQY values of QDRs, QDGs, and QDBs with T (TD) ligands in the liquid phase were about 37, 61, and 58% (42, 80, and 95%), respectively. Similarly, the PLQY values of QDRs, QDGs, and QDBs with T (TD) ligands in the CDNA thin films were about 5, 4, and 3% (17, 25, and 6%), respectively. Although QDBs showed a higher PLQY than QDRs in the liquid phase, QDGs showed a higher PLQY than QDBs in the CDNA thin films. This might be due to the variation in the QD shell thickness. Interestingly, QDs with both T and D ligands (QDTD) showed an enhanced PLQY in both phases compared to QDs with only T ligands (QDT) because of the efficient passivation and removal of dangling bonds.35 CDNA duplexes (with a high aspect ratio) served as good passivation materials as they provided stable surroundings for QDs because of their flexible duplex networks. Consequently, the CDNA templates prevented aggregation of QDs by acting as effective spacers and increased the loading efficiency of QDs.12,36
3.3 Optical properties of individual R, G, and B QDs in the liquid phase and CDNA thin films
The UV-vis absorbance spectra of QDT (solid lines) and QDTD (dotted lines) in the liquid phase (top) and the CDNA thin films (bottom) are shown in Fig. 3(a). The absorbance spectra were recorded in the wavelength range 190–1100 nm. The absorbance peaks of the QDs (inset) and CDNA (around 260 nm) thin films are shown by vertical dotted lines. Although the characteristic peak positions did not change, QDT and QDTD showed different absorbance intensities in the liquid phase and in CDNA thin films. As shown in the inset, the spectra of QDT,TD(R), QDT,TD(G), and QDT,TD(B) showed the first absorbance maximum at wavelengths of around 590, 510, and 440 nm (second absorbance maximum of QDT,TD(R) at 560 nm), respectively. The absorbances of QDs in the liquid phase gradually increased toward shorter wavelengths up to 280 nm. The absorbance of QD-CDNA thin films exhibited the characteristic behavior of QDs and pristine CDNA. The QD absorbance characteristic peak positions in QDT-CDNA and QDTD-CDNA were similar to those in QDT and QDTD in the liquid phase. Here, the reference concentrations of R, G, and B QDs in the CDNA thin films were used and marked as R1, G1, and B1, respectively (detailed in the Experimental section).Fig. 3(b) displays the normalized PL spectra of QDT and QDTD in the liquid phase (top) and QDT-CDNA and QDTD-CDNA thin films (bottom) measured in the range 400–750 nm. The PL characteristic emission peaks and full width at half maximum (FWHM) values are shown by vertical dotted lines and horizontal arrows, respectively. QDT,TD(G) in the liquid phase and in CDNA showed a relatively wider FWHM than the other samples (details in Table 1). The PL peak positions of QDT and QDTD in the liquid phase and QDT-CDNA and QDTD-CDNA thin films did not change significantly (less than 2 nm). The PL emission area (proportional to PLQY) of QDT, QDTD, QDT-CDNA, and QDTD-CDNA varied with individual QDs and was strongly dependent on the ligands.
3.4 Dispersion and photostability analysis of QDRs embedded in CDNA thin films
Fig. 4 shows the dispersion and UV light degradation analysis of QDRs embedded in CDNA thin films. The HRTEM image of QDRs without CDNA clearly showed the aggregation of QDRs. On the other hand, QDRs with the CDNA matrix revealed a moderate and uniform distribution of QDRs in the CDNA thin film. Here, CDNA (which provided surface passivation of QDs) could effectively reduce the surface energy and kinetic motion of QDs. Consequently, CDNA prevents their aggregation. The better dispersion of QDs in CDNA obviously leads to higher PLQY, which is in good agreement with the discussion of PLQY in Section 3.2.FESEM analysis is carried out to understand the potential surface damage and degradation of CDNA and QDs embedded in CDNA thin film surfaces under UV light exposure (i.e., harsh environment). The FESEM images indicated the stability of QDs embedded in CDNA thin films after 20 h of exposure to UV light (Fig. 4b). There is no visible damage (e.g., cracking of the surface) in the FE-SEM images for both CDNA and QDs embedded in CDNA thin films. Hence, CDNA acts as an efficient matrix (i.e., host) for QDs even under the accelerated photodegradation conditions.
3.5 EL spectra and colour coordinates of reference blue and UV LEDs
Blue and UV LED photoexcitation sources for down-conversion of white light were used for EL spectral characterization. Fig. 5 shows the luminescence spectra and colour coordinates of the reference blue and UV LEDs. The LEDs emitted at a maximum EL peak wavelength (λEL max) of 460 and 365 nm with a driving voltage (current) of 2.6 V (@5 mA) and 3.2 V (@5 mA), respectively. The FWHM of blue and UV LEDs was 27 and 15 nm, respectively.For blue LEDs, the CIE colour coordinates were (0.14, 0.05). We designed the experiment such that white light was generated by R, G, and B primary colours. The blue LED as the B emitting source also served as a photoexcitation source for R and G QDs. However, the UV LED merely acted as a photoexcitation source for R, G, and B QDs to produce hybrid (combination of EL and PL) white light by the down-conversion phenomenon (via Stokes shift). The blue and UV LEDs were stable and showed Gaussian-like spectral emission distributions.
3.6 EL spectra of QDs embedded in CDNA thin films with mixed and orthogonally stacked configurations integrated on blue LEDs
Two configurations, mixed and orthogonally arranged R and G QD structures, were obtained with different concentrations of R and G QDs. R and G QDTD embedded in CDNA thin films at three different concentration ratios (1:1, 2:1, and 1:2) of QDs were obtained either by mixing, denoted as QDTD(R1G1)-, QDTD(R2G1)-, and QDTD(R1G2)-CDNA, respectively, or by orthogonally stacking, denoted as QDTD(R1/G1)-, QDTD(R2/G1)-, and QDTD(R1/G2)-CDNA, respectively, and were integrated on blue LEDs for EL measurement in the wavelength range 360–700 nm. Although a CDNA thin film with a mixture of R and G QDs could form a single layer, layers formed with the orthogonal stacking configuration for R and G QDs could form double layers too. However, a DNA layer without CTMA was introduced between the CDNA layers to avoid the dissolution of the bottom layers by the 1-butanol solvent in CDNA during stacking. Here, the hydrophilic DNA layer effectively separated the R and G QDTD embedded in CDNA layers.
The white light spectra and CIE colour coordinates of the mixed QDTD(R1G2)-CDNA and orthogonally stacked QDTD(R1/G2)-CDNA thin films integrated on blue LEDs are shown in Fig. 6. Although the concentration ratio (1:2) of R to G was the same, their CRI (CIE) values were significantly different, 21% (0.49, 0.26) and 80% (0.53, 0.39), respectively. This difference may originate because of suppression of the G QD intensity. Although the G QD concentration was twice that of the R QDs, the spectral wavelengths of the latter were relatively dominant in QDTD(R1G2)-CDNA. This indicated the self-absorption nature of the R QDs. Consequently, the R QDs absorbed G light more in the mixed R and G QDs than in the stacked structures. The R QDs in the mixed structures showed high-energy continuous absorbance involving G spectral energy emission and inter-particle resonance absorption.37,38 In contrast, the self-absorption of R QDs in the stacked configuration was relatively smaller than that in the mixed configuration. Similar self-absorption trends were observed at the other R and G QD concentration ratios of 1:1 and 2:1 with mixed and stacked QDTD-CDNA thin films (detailed CRI values and CIE colour coordinates are shown in Table 2). We concluded that the configuration and concentration ratio of the QDs, which could be easily controlled, also significantly affected the spectral distribution.
3.7 EL spectra of CDNA thin films with different R, G, and B QD concentrations integrated on UV LEDs
White-light spectra of the QDs embedded in CDNA thin films with seven different R, G, and B QD concentration ratios (1:1:1, 2:1:1, 1:2:1, 1:1:2, 2:2:1, 1:2:2, and 2:1:2) with mixed, QDTD(R1G1B1)-, QDTD(R2G1B1)-, QDTD(R1G2B1)-, QDTD(R1G1B2)-, QDTD(R2G2B1)-, QDTD(R1G2B2)-, and QDTD(R2G1B2)-CDNA, and orthogonally stacked, QDTD(R1/G1/B1)-, QDTD(R2/G1/B1)-, QDTD(R1/G2/B1)-, QDTD(R1/G1/B2)-, QDTD(R2/G2/B1)-, QDTD(R1/G2/B2)-, and QDTD(R2/G1/B2)-CDNA, configurations integrated on UV LEDs were measured in the wavelength range 360–700 nm (Fig. 7). The white light was generated by the combination of R, G, and B primary colours via excitation of the R, G, and B QDs using UV LEDs. The UV LEDs functioned as excitation sources of the R, G, and B QDs and produced hybrid white light by the down-conversion process.
The white-light spectra, CRI, and CIE colour coordinates of the mixed-layered QDTD(R2G1B1)- and QDTD(R1G2B2)-CDNA thin films excited by the UV LED are shown in Fig. 7(a). Their CRI values were 61 and 79%, respectively. Although the configuration was the same, the different R, G, and B QD concentration ratios of 2:1:1 and 1:2:2 in CDNA led to an approximately 18% CRI increase because of the significant change in the self-absorption of R QDs. This implied that the self-absorption of the R QDs could be minimized using excess G and B QDs. Consequently, the CRI efficiency could be improved easily by controlling the R, G, and B QD concentrations. The EL, CRI, and CIE colour coordinates of the triple-layered QDTD(R2/G1/B1)- and QDTD(R1/G2/B2)-CDNA thin films excited by the UV LED are shown in Fig. 7(b). We observed that the CRI of the orthogonally stacked thin films was enhanced compared to that of the mixed thin films, which indicated that the R QD intensity could be controlled by curtailing the self-absorption. The CRI values for the QDTD(R2/G1/B1)- and QDTD(R1/G2/B2)-CDNA thin films were 78 and 87%, which corresponded to roughly 17 and 8% increments compared to the CRI of QDTD(R2G1B1)- and QDTD(R1G2B2)-CDNA thin films, respectively.
Similar trends of EL for both mixed and stacked configurations were observed for the other R, G, and B QD concentration ratios of 1:1:1, 2:2:1, 1:1:2, 1:2:1, and 2:1:2 (CRI values and CIE colour coordinates are shown in Table 2). This demonstrated that the reduction in R concentration significantly decreased the self-absorption and hence increased the CRI.
4. Conclusions
We prepared ligands (TOP/TBP and DDT) attached to R, G, and B QDs and DNA with CTMA (CDNA) for fabrication of QDs embedded in CDNA thin films with two distinct configurations (mixed and stacked), and measured their FTIR, PLQY, UV-vis absorbance, PL, and EL characteristics. Continuous absorption characteristics of QDs with TOP/TBP and DDT ligands (QDTD) provided large Stokes shifts and showed the benefits of single excitation via the down-conversion process. The PLQY values of QDTD in the liquid phase and in CDNA thin films were enhanced up to 95% and 25%, respectively. Although the UV-vis absorbance and PL intensity of QDT- and QDTD-CDNA thin films varied, their characteristic peak positions were not significantly altered. The dispersion of QDs in the CDNA matrix effectively stabilized the QDs, consequently leading to high PLQY. In addition, the accelerated photostability measurement revealed the stability of QD-CDNA thin films. EL spectra of the QDTD-CDNA thin films with two different configurations (mixed, QDTD(R1G2)-CDNA, and stacked, QDTD(R1/G2)-CDNA) integrated on blue LEDs were obtained. Although the concentration ratio of R to G was the same, their CRI values were significantly different, 21 and 80%, respectively, due to suppression of the G QD intensity. This indicated the self-absorption characteristics of R QDs. Consequently, the R QDs absorbed G light more in the mixed structures than in the stacked structures. Similarly, we studied white-light luminescence with a UV LED source for QDTD(RGB)-CDNA and QDTD(R/G/B)-CDNA thin films having different R, G, and B QD concentrations; these concentrations noticeably affected CRI efficiency and CIE colour coordinates. The proposed approach showed the benefits of a single excitation source due to the continuous absorbance of QDs toward higher energy and simplified RGB spectral generation of white light.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea (2012M3A6A7054855 and 2018R1A2B6008094).References
- T. Erdem and H. V. Demir, Nanophotonics, 2013, 2, 57–81 CAS.
- B. Xie, R. Hu and X. Luo, J. Electron. Packag., 2016, 138, 20803–20813 CrossRef.
- B. C. Fitzmorris, Y.-C. Pu, J. K. Cooper, Y.-F. Lin, Y.-J. Hsu, Y. Li and J. Z. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 2893–2900 CrossRef CAS PubMed.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photonics, 2013, 7, 13–23 CrossRef CAS.
- E. Petryayeva, W. R. Algar and I. L. Medintz, Appl. Spectrosc., 2013, 67, 215–252 CrossRef CAS PubMed.
- J. Bao and M. G. Bawendi, Nature, 2015, 523, 67–70 CrossRef CAS PubMed.
- Y. Altıntas, S. Genc and M. Y. Talpur, Nanotechnology, 2016, 27, 295604 CrossRef PubMed.
- J. C. Bonilla, F. Bozkurt, S. Ansari, N. Sozer and J. L. Kokini, Trends Food Sci. Technol., 2016, 53, 75–89 CrossRef CAS.
- S. V. Kershaw, L. Jing, X. Huang, M. Gao and A. L. Rogach, Mater. Horiz., 2017, 4, 155–205 RSC.
- S. Ye, F. Xiao, Y. X. Pan, Y. Y. Ma and Q. Y. Zhang, Mater. Sci. Eng., R, 2010, 71, 1–34 CrossRef.
- C. Xu and K. M. Poduska, J. Mater. Sci.: Mater. Electron., 2015, 26, 4565–4570 CrossRef CAS.
- Y.-C. Pu and Y.-J. Hsu, Nanoscale, 2014, 6, 3881 RSC.
- J. E. Lee, E. D. Do, U. R. Lee, M. J. Cho, K. H. Kim, J.-I. Jin, D. H. Shin, S.-H. Choi and D. H. Choi, Polymer, 2008, 49, 5417–5423 CrossRef CAS.
- E. F. Schubert and J. K. Kim, Science, 2005, 308, 1274–1278 CrossRef CAS PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- D. Cai, X. Yuan, D. Zhu, H. Zhou, H. Li and J. Zhao, Mater. Res. Bull., 2017, 85, 222–227 CrossRef CAS.
- V. Wood and V. Bulović, Nano Rev., 2010, 1, 5202 CrossRef PubMed.
- J. H. Oh, S. J. Yang and Y. R. Do, Light: Sci. Appl., 2014, 3, e141 CrossRef CAS.
- L. Zan, D. Lin, P. Zhong and G. He, Opt. Express, 2016, 24, 7643 CrossRef PubMed.
- T. Erdem, S. Nizamoglu and H. V. Demir, Opt. Express, 2012, 20, 3275 CrossRef CAS PubMed.
- S. Abe, J. J. Joos, L. IDJ Martin, Z. Hens and P. F. Smet, Light: Sci. Appl., 2017, 6, e16271 CrossRef CAS PubMed.
- A. J. Steckl, H. Spaeth, H. You, E. Gomez and J. Grote, Opt. Photonics News, 2011, 22, 34 CrossRef CAS.
- E. F. Gomez, V. Venkatraman, J. G. Grote and A. J. Steckl, Adv. Mater., 2015, 27, 7552–7562 CrossRef CAS PubMed.
- V. Arasu, S. R. Dugasani, J. Son, B. Gnapareddy, S. Jeon, J.-H. Jeong and S. H. Park, J. Phys. D: Appl. Phys., 2017, 50, 415602 CrossRef.
- M. R. Kesama, S. R. Dugasani, S. Yoo, P. Chopade, B. Gnapareddy and S. H. Park, ACS Appl. Mater. Interfaces, 2016, 8, 14109–14117 CrossRef CAS PubMed.
- V. Arasu, S. R. Dugasani, M. R. Kesama, H. K. Chung and S. H. Park, Sci. Rep., 2017, 7, 11567 CrossRef PubMed.
- Y. Park, S. Jeong and S. Kim, J. Photochem. Photobiol., C, 2017, 30, 51–70 CrossRef CAS.
- L. Wang, J. Yoshida, S. Sasaki, T. Kajiyama and N. Ogata, Chem. Mater., 2001, 13, 1273–1281 CrossRef CAS.
- J. Kundu, Y. Ghosh, A. M. Dennis, H. Htoon and J. A. Hollingsworth, Nano Lett., 2012, 12, 3031–3037 CrossRef CAS PubMed.
- V. Arasu, S. Jeon, J.-H. Jeong and S. H. Park, J. Mater. Chem. C, 2018, 6, 5199–5209 RSC.
- H. Woo, J. Lim, Y. Lee, J. Sung, H. Shin, J. M. Oh, M. Choi, H. Yoon, W. K. Bae and K. Char, J. Mater. Chem. C, 2013, 1, 1983 RSC.
- C. Yoon, T. Kim, M.-H. Shin, Y.-G. Song, K. Shin, Y.-J. Kim and K. Lee, J. Mater. Chem. C, 2015, 3, 6908 RSC.
- S. Ananthakumar, J. Ram Kumar and S. Moorthy Babu, Mater. Sci. Semicond. Process., 2017, 67, 152–174 CrossRef CAS.
- B. D. Mistry, A Handbook of Spectroscopic Data Chemistry, Oxford Book Company, Jaipur, 2009 Search PubMed.
- L. S. Sohn, S. Unithrattil and W. B. Im, ACS Appl. Mater. Interfaces, 2014, 6, 5744–5748 CrossRef PubMed.
- M. Zhu, X. Peng, Z. Wang, Z. Bai, B. Chen, Y. Wang, H. Hao, Z. Shao and H. Zhong, J. Mater. Chem. C, 2014, 2, 10031 RSC.
- C.-J. Chen, C.-C. Lin, J.-Y. Lien, S.-L. Wang and R.-K. Chiang, J. Mater. Chem. C, 2015, 3, 196 RSC.
- H. Chen, Z. Luo, R. Zhu, Q. Hong and S.-T. Wu, Opt. Express, 2015, 23, 13060 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |