
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe impact of processing on the cytotoxicity of graphene oxide†
Valerie
Gies
a,
Gregory
Lopinski
a,
Jerry
Augustine‡
ab,
Timothy
Cheung‡
ab,
Oltion
Kodra
c and
Shan
Zou
 *ad
*ad
aMetrology Research Centre, National Research Council Canada, 100 Sussex Drive, Ottawa, Ontario K1A 0R6, Canada. E-mail: shan.zou@nrc-cnrc.gc.ca
bDepartment of Chemistry and Chemical Biology, McMaster University, 1280 Main St. W., Hamilton, Ontario L8S 4L8, Canada
cEnergy, Mining and Environment Research Centre, National Research Council Canada, M-12, Ottawa, Ontario K1A 0R6, Canada
dDepartment of Chemistry, Carleton University, 1125 Colonel By Drive, Ottawa, Ontario K1S 5B6, Canada
First published on 23rd November 2018
Abstract
In-house prepared graphene oxide (GO) was processed via base washing, sonication, cleaning and combinations of these processing techniques to evaluate the impact on the flake morphology, composition and cytotoxicity of the material. The flakes of unprocessed GO were relatively planar, but upon base washing, the flakes became textured exhibiting many folds and creases observed by AFM. In addition to the pronounced effect on the topography, base washing increased the C/O ratio and increased the cytotoxicity of GO on all four cell lines studied determined via the WST-8 assay. Sonicating the unprocessed and base washed samples resulted in smaller flakes with a similar topography; the base washed flakes lost the texture previously observed upon sonication. The sonicated samples were more toxic than the unprocessed sample, attributed to the smaller flake size, but were interestingly less toxic than the base washed, unsonicated sample despite the base washed unsonicated sample having a larger flake size. This unexpected finding was confirmed by a second analyst using the same, and a different source of GO and resulted in the conclusion that the morphology of GO greatly impacts the cytotoxicity. Cleaning the GO reduced the amount of nitrogen and sulfur impurities in the sample but had no significant impact on the cytotoxicity of the material. It was observed that nutrient depletion via nanomaterial adsorption was not the route of cytotoxicity for the GO samples studied.
Introduction
Graphene oxide (GO) has been recognized for its solubility in aqueous media, ease of functionalization and production in large quantities,1 electroactivity,2 and fluorescence.3 In order to meet the requirements of many applications, physical and chemical processing is often necessary. Processing GO is often used to enhance characteristics of the material and possibly achieve improved capabilities for applications.4–7 Processing GO may induce changes in flake size, morphology and chemical composition modification.The structure of GO remains elusive though it is generally accepted that GO sheets are composed of sp2 hybridized carbon atom sheets with oxygen containing functional groups.1 The Lerf–Klinowski model describes GO to be composed of regions of aromatic, unoxidized benzene rings and aliphatic six membered rings, the ratio of which depends on the extent of oxidation.8–10 The highly oxidized regions are described by the presence of alcohols, epoxides and double bonds. Carboxylic acids are also postulated to exist on the periphery of the flakes. One explanation for the ill-defined structure of GO may lie in the variation in the materials itself. The structure and composition of GO has been shown to vary depending on the starting material and the oxidation conditions.11,12 Additionally, it has been suggested that GO sheets are indeed a composite material comprising of smaller, highly oxidized disks, commonly referred to as oxidative debris (OxD), complexed to large, lightly oxidized flakes.13
Graphene oxide is commonly processed to yield materials with different and/or augmented properties. Introduced as a green synthetic route to graphene preparation, base washing of GO is now a common processing technique.14 Base washing of GO with NaOH is known to increase the carbon to oxygen ratio, possibly by removing the OxD.13,14 It has been demonstrated that unprocessed and base washed GO have different properties. Base washed flakes were demonstrated to be 75% more effective to adsorb organic pollutants15 and demonstrated enhanced catalytic activity for the oxidative coupling of amines to imines.16 The fluorescence of bulk GO has been largely attributed to the OxD,3 as has its inherent electrical properties.2 One downside to base washing is that GO loses much of its solubility once the OxD is removed13 which in part may be a manifestation of OxD carrying 10–25% of the total surface charge despite being a fraction of the total mass.17 It should be noted that this two component model has been widely contested.18,19
Sonication is another processing technique used to manipulate the flake size of GO.20 Varying flake sizes are needed to meet different applications. For example, when producing electronic devices with reduced GO, it is ideal to have large flakes to reduce the number of junctions formed by overlapping flakes, as this may reduce the overall electron mobilities across a device.21 When GO is applied in bio-imaging, it is important to have control over the flake size as the photoluminescence and absorbance is impacted by the flake size.22
As processed GO is used in a variety of applications, it is important to evaluate how the toxicity of the processed GO may vary from parent GO for safe handling and application development. The cytotoxicity of GO has been evaluated by many groups and the results vary, likely as a result of the variation in the material itself, the cell lines studied and the method used to evaluate toxicity. Some groups have also been interested in the cytotoxicity of processed GO. Chang et al. observed a cytotoxicity dependence on the lateral dimension of the flakes of in-house produced GO.23 This finding was shared by Das et al. and previously by our group using commercially available sources of graphene oxide.24,25
In regards to the impact of oxygen content on the cytotoxicity of GO, Das et al. also found reducing graphene oxide with hydrazine mitigated the cytotoxic effects.24 In contrast, Contreras-Torres et al. reported that GO reduced under mild conditions had a higher toxicity than unprocessed GO.26 By evaluating the cytotoxicity of GO produced via different oxidation methods, Chng and Pumera found that increased oxygen content decreased the toxicity of GO determined via the MTT assay, but found the opposite when the WST-8 assay was used.27 Pelin et al. evaluated the cytotoxicity of commercial and research grade GO and observed that the GO which had the greatest cytotoxic effect also had the highest oxygen content.28 These reports provide valuable insight into how oxygen content may impact the cytotoxicity of GO; however, there is little reported knowledge on the cytotoxicity of base washed GO. Toxic effects on zebra fish embryos imparted from base washed GO has been evaluated by the group of Clemente et al.29 They observed that base washing GO mitigated adverse effects on the health of the embryos. Finally, Faria et al. investigated what role the presence of oxidative debris on GO had on the material's antimicrobial properties.30 They found that upon base washing, the toxicity of GO to Escherichia coli cells significantly decreased.
In the present study, it was set out to characterize and quantify the changes in the properties of in-house made graphene oxide (IHGO) pre and post processing for the purpose of correlating changes in cytotoxicity to changes in the materials' properties as well as determine which processing technique results in a material that is the most cytotoxic. The processing approaches performed on IHGO were base treatment, sonication, and cleaning – which was the process of precipitating and resuspending the GO in water in an attempt to remove soluble impurities. Materials produced from a combination of these three processing techniques were also evaluated. Fig. 1 outlines the processes which were performed on IHGO. The processed and unprocessed materials were then analyzed via AFM, DLS (when possible) and XPS. Assay and cell line selections are key to obtaining reliable cytotoxicity data. In a previous work, we compared the effectiveness of MTT, MTS, trypan blue and WST-8.25 It was found that WST-8 was the assay best suited to evaluate the cytotoxicity of GO. WST-8 was the ideal assay due to its sensitivity, simplicity and reproducibility over trypan blue and MTS as well as due to the complications observed with the MTT assay. Fluorescence assays were not considered as it has been previously demonstrated that GO quenches fluorescence and may lead to erroneous results.31,32 The chick chorioallantoic membrane assay in fertilized eggs (CAM assay)33 was not considered due to its major drawback that a large number eggs are required to obtain consistent results. Also CAM is a non-mammalian system which would be complicated when interpreting results, using mammalian cell lines. Previously, fibroblasts (NIH 3T3 cell line) and phagocytic cells (Raw 264.7 cell line) were used to measure the cytotoxicity (for GOs with concentrations of 5, 10, 20, 30, 50, 75, 100 and 200 μg mL−1) in addition to the presently used A549, U-87 MG, HepG2 and HL-60 cell lines. These cell lines represent different target areas within the body. A549 cells are human epithelial lung cells, U-87 MG cells are human epithelial brain cells, HepG2 are human epithelial liver cells and HL-60 lung cells are human promyeloblast peripheral blood cells. It is believed that the benefits of using immortal cancerous cells outweighs the possibility of the cells behaving differently from their non-cancerous counterparts. Using these commonly used cell lines provides the additional benefit of ease of comparison between other works. Using more than one cell line is important as different cell lines have different responses to GO.25,34 The WST-8 assay was used to measure the cytotoxicity of GO. This assay provides a method for the quantification of viable cells.25,35 In short, a tetrazolium salt is reduced by metabolically active cells to a water soluble formazan, the optical density of the sample at relevant wavelengths may then be used as a surrogate for cell viability.
 | ||
| Fig. 1 Summary of the processes performed on IHGO. GO was first synthesized via a modified Hummer's method in which natural flake graphite is oxidized and exfoliated to produce GO. The IHGO underwent three additional processes: it was ‘cleaned’ in which the suspending solution was replaced with fresh Milli-Q water via centrifugation. The IHGO underwent sonication to produce s-IHGO, a sample of the s-IHGO was then cleaned to produce s-c-IHGO. Finally, the IHGO was washed with 1 M NaOH and dialyzed to produce bw-IHGO. The bw-IHGO was further processed via sonication to produce bw-s-IHGO. | ||
Methods and materials
Synthesis of GO
All chemicals and solvents (HPLC grade) were purchased from Sigma-Aldrich and used as received, unless otherwise noted. IHGO was synthesized via the Hummers method.36,37 1.0 g of flake graphite (7782-42-5, Graphene Supermarket, USA) and 1.0 g KNO3 (P6083-500G, Milipore Sigma, USA) were added to 35 mL H2SO4 (58780, ACP, Canada) over ice. 4.5 g of KMnO4 was added to the solution over 75 minutes with stirring. The solution was stirred for 24 h, and was then allowed to sit for another 4 days. The solution was added to 110 mL of 5 wt% H2SO4 in water over 75 minutes and then allowed to sit for 3 h. 9 mL of 30 wt% H2O2 in water was added over 5 min to react Mn into MnO2. The solution was stirred for 2 h and then was diluted with 100 mL of 3 wt% H2SO4, 0.5 wt% H2O2 in H2O. The solution was divided into six 50 mL centrifuge tubes and centrifuged for 20 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, the supernatant was decanted and replaced with 3 wt% H2SO4, 0.5 wt% H2O2 in H2O. The precipitate was washed 3 more times with 3 wt% H2SO4, 0.5 wt% H2O2 in H2O for a total of four washes. Such acidic washes were performed to remove MnO2 which is soluble in acidic solutions. Following the acidic washes, the precipitate was washed an additional 8 times with water to remove H2SO4 and H2O2. The centrifuge time was increased to 25, 30 and 40 minutes for the last three respective washes. New centrifuge tubes were used for every two washes; when transferring, black solid particulate collected at the bottom of the tubes (unreacted graphite) was not transferred. The resulting approximately 100 mL of translucent brown ‘gel’ was measured to have a concentration of 8 mg mL−1 – labelled as IHGO. See the ESI for details on observations and images of the reaction stages in Fig. S1.†
000 rpm, the supernatant was decanted and replaced with 3 wt% H2SO4, 0.5 wt% H2O2 in H2O. The precipitate was washed 3 more times with 3 wt% H2SO4, 0.5 wt% H2O2 in H2O for a total of four washes. Such acidic washes were performed to remove MnO2 which is soluble in acidic solutions. Following the acidic washes, the precipitate was washed an additional 8 times with water to remove H2SO4 and H2O2. The centrifuge time was increased to 25, 30 and 40 minutes for the last three respective washes. New centrifuge tubes were used for every two washes; when transferring, black solid particulate collected at the bottom of the tubes (unreacted graphite) was not transferred. The resulting approximately 100 mL of translucent brown ‘gel’ was measured to have a concentration of 8 mg mL−1 – labelled as IHGO. See the ESI for details on observations and images of the reaction stages in Fig. S1.†
Processing of GO
000 rpm for 1 h which completely pelleted the GO. The supernatant was removed and replaced with Milli-Q H2O. The cleaned IHGO was labelled as c-IHGO and the cleaned s-IHGO was labelled as s-c-IHGO.
DLS measurements
DLS measurements of GO were performed on the Zetasizer Nano ZS particle size analyzer (Malvern Instruments, UK) in a semi-micro polystyrene disposable cuvette (VWR, USA). Samples for DLS were diluted to 4 μg mL−1 in Milli-Q water. Measurements were performed at 25 °C with 180 s of equilibration time. The measurement angle was set to 173° with 0.4 to 10000 nm as the detectable particle range. The instrument output Zavg was taken to represent the intensity-weighted arithmetic-average equivalent sphere hydrodynamic diameter, which was used as the descriptor for the flake size of the GO. Each measurement was performed in triplicate with an instrument optimized number of runs (typically 12 runs). The reported error represents the standard deviation.
AFM measurements
Samples for AFM were made by spin-coating diluted GO dispersions (40 μL, 0.05 mg mL−1) onto freshly cleaved 1 cm by 1 cm squares of mica substrate (Ted Pella Inc.) at 500 rpm for 30 s followed by 2000 rpm for one minute. AFM height/topography images were recorded using MultiMode NanoScope V with PeakForce QNM mode (Bruker Nano Surfaces Division, USA). ScanAsyst-Air probes with a typical spring constant of 0.4 N m−1 and a resonance frequency of 50–90 kHz were used. The peak force was always kept at the lowest stable imaging level of 200–500 pN. The scan ranges of the images were typically 2 μm by 2 μm, 30 μm by 30 μm or 120 μm by 120 μm. Images were processed (flattened with a third-order polynomial fit) using Gwyddion 2.38 (Czech Metrology Institute, Czech Republic).XPS
Gold substrates (Arrandee metal, Germany) were cleaned with acetone, chloroform, isopropyl alcohol and water before being submerged into a piranha bath for 2 minutes. After thorough rinsing, 100 μL of the GO samples were drop casted onto the surface at a concentration of 500 μg mL−1. The samples were dried under nitrogen. A gold substrate without material was taken as a blank. Spectra were obtained using a Kratos Analytical Axis Ultra DLD spectrometer with monochromatized Al Kα X-rays. Data was acquired at three points on two replicates of each sample. At each point survey scans were obtained in order to determine the relative atomic composition of the sample and detect any impurities that may be present. In addition, high resolution spectra in regions corresponding to the major elements present on these samples (C 1s, O 1s, N 1s and S 2p) were also measured, with the pass energy of 20 eV. Data analysis was carried out with the CasaXPS analysis software using relative sensitivity factors for the Kratos instrument. The Shirley method was used for background subtraction.38 For all the samples prepared by the method above, no Au signal was observed, indicating that the GO films were sufficiently thick as to completely attenuate the substrate signal. This eliminated the need to correct for possible oxygen and carbon signals arising from the substrate.Cell culture specifics
U-87 MG and HepG2 cells were cultured in Gibco™ DMEM supplemented with 10% Gibco™ heat-inactivated fetal bovine serum and Gibco™ 1× penicillin–streptomycin in an incubator at 37 °C with 5% CO2 and a 95% relative humidity. A549 cells were cultured under the same conditions in Gibco™ FK12 culture medium and HL-60 in Gibco™ IMDM with 20% FBS. Cells were passaged once to twice a week depending on confluence. Adherent cells were trypsinized to suspend the cells for passaging. Cell media, fetal bovine serum and penicillin–streptomycin were all purchased from Life Technologies (USA) and cells were purchased from Cedarlane Laboratories (Canada).WST-8 assay
The WST-8 measurements were performed in a 96 well, flat bottom plate and the cells were seeded to a density of 0.7–2.5 × 104 cells per well, depending on the cell line. The cells were treated with 0, 10, 50, 100 and 200 μg mL−1 of GO. A cell free control was prepared for every concentration of GO. To minimize the impact of the different rates of vaporization of the interior vs. exterior wells on the plate, the outer wells were not used to collect data points. The cells were incubated for 24 h after which the WST-8 assay reagent (VistaLab Technologies, Brewster, USA) was added. After 4 h of incubation with the assay reagent, the optical densities of the WST-8 was measured at 450 nm. Optical densities were recorded on the FLUOstar Omega microplate reader (BMG Labtech, Germany).Results
Material properties
IHGO and c-IHGO dispersions had a similar appearance, they were a translucent brown solution with visible brown flakes. s-IHGO and s-c-IHGO were also similar in appearance except flakes were no longer visible. The visible flakes present in IHGO and c-IHGO settled over a week when no obvious precipitates were observed for s-IHGO. s-c-IHGO was observed to be stable for months. Base washing the IHGO had a pronounced effect on the colour and the stability of the material. The solution changed from a comparably stable, translucent, light brown solution to an unstable, dark brown opaque solution upon treatment. The unsonicated sample, bw-IHGO was the least stable and had visible black flakes. bw-s-IHGO did not have visible black flakes, but was still observed to settle over time. See Fig. S2† for images of the samples. In order to quantify the chemical and physical characteristics of the materials, the samples were measured by XPS and AFM as well as DLS when possible. | ||
| Fig. 2 AFM images of (A) IHGO, (B) c-IHGO, (C) bw-IHGO, (D) s-IHGO, (E) s-c-IHGO and (F) bw-s-IHGO. Images (A) and (B) have a z-scale of 7 nm; (C) 10 nm; (D–F) 5 nm. | ||
Upon sonication, the IHGO flakes were greatly reduced in size as s-IHGO is composed of vastly smaller flakes than IHGO. AFM images show few differences between s-IHGO and s-c-IHGO, the two materials have planar flakes of similar size. Interestingly, the bw-s-IHGO flakes were not crumpled as were the bw-IHGO flakes. The bw-s-IHGO flakes were similar in size and morphology to the s-IHGO and s-c-IHGO. The sonicated materials are less polydisperse compared to the unsonicated materials.
DLS measurements which yield the equivalent hydrodynamic diameter of the flakes were recorded for s-IHGO and s-c-IHGO and were measured to be 154 ± 4 nm and 150 ± 4 nm, respectively with a polydispersity index (PDI) less than 0.4. The remaining solutions were not suitable for DLS measurements; IHGO and c-IHGO were too polydisperse with a PDI greater than 0.8, bw-IHGO and bw-s-IHGO were insufficiently stable.
| Sample | C 1s, % | O 1s, % | N 1s, % | S 2p, % | C/O |
|---|---|---|---|---|---|
| IHGO | 66.1 ± 1.6 | 30.1 ± 0.6 | 1.6 ± 0.6 | 2.2 ± 0.9 | 2.39 ± 0.10 |
| c-IHGO | 68.4 ± 0.5 | 29.6 ± 0.5 | 0.9 ± 0.2 | 1.0 ± 0.1 | 2.36 ± 0.07 |
| s-IHGO | 65.1 ± 4.7 | 29.9 ± 2.2 | 2.0 ± 0.8 | 2.9 ± 2.0 | 2.41 ± 0.16 |
| s-c-IHGO | 67.8 ± 1.1 | 29.7 ± 0.6 | 1.1 ± 0.2 | 1.3 ± 0.5 | 2.36 ± 0.08 |
| bw-IHGO | 73.6 ± 0.3 | 25.2 ± 0.1 | 0.8 ± 0.1 | 0.2 ± 0.1 | 2.94 ± 0.02 |
| bw-s-IHGO | 74.1 ± 0.6 | 24.7 ± 0.5 | 0.8 ± 0.1 | 0.2 ± 0.1 | 3.07 ± 0.11 |
| GGO | 68.3 ± 1.2 | 30.6 ± 0.6 | 0.3 ± 0.1 | 0.8 ± 0.6 | 2.27 ± 0.09 |
| bw-GGO | 75.1 ± 0.2 | 24.2 ± 0.3 | 0.6 ± 0.1 | 0.1 ± 0.1 | 3.15 ± 0.11 |
Comparing the C/O ratios across all the samples it is clear that the base washed IHGO samples have higher C/O ratios than IHGO or other processed forms of IHGO. These higher C/O ratios are indicative of the presence of less oxygen containing functional groups. A similar increase in the C/O ratio upon base washing was also observed for the commercial sample: the C/O ratio of GGO increased from 2.27 to 3.15.
Significant impurities present in the sample include nitrogen and sulfur which were likely introduced into the sample from the reactants in the oxidation process. As seen in Table 1, the levels of nitrogen and sulfur are significantly reduced in c-IHGO and s-c-IHGO samples, clearly indicating that the washing steps remove some of these impurities. The cleaning process did not significantly impact the C/O ratios. Similar reduction in impurities is observed for the base washed samples, likely as a result of the dialysis step. It may also be noted that the commercial sample exhibited a lower level of impurities compared to the in-house prepared counterparts.
Cytotoxicity
The impact of base washing on the cytotoxicity of IHGO was thus evaluated. Base-washed IHGO was observed to consistently increase the cytotoxicity of the material on all cell lines studied. As demonstrated in Fig. 3(i)(a–d) it can be seen that fewer viable cells remained post exposure to bw-IHGO compared to IHGO for all four cell lines studied. In the cases of the HepG2 and HL-60 cells, there was little difference in cytotoxicity at the lowest concentration studied, 10 μg mL−1, but the difference in toxicity was obvious at the three higher concentrations studied. The increase in toxicity of the bw-IHGO compared to the IHGO was reasonably constant across all concentrations studied for A549 and U-87 MG cells.
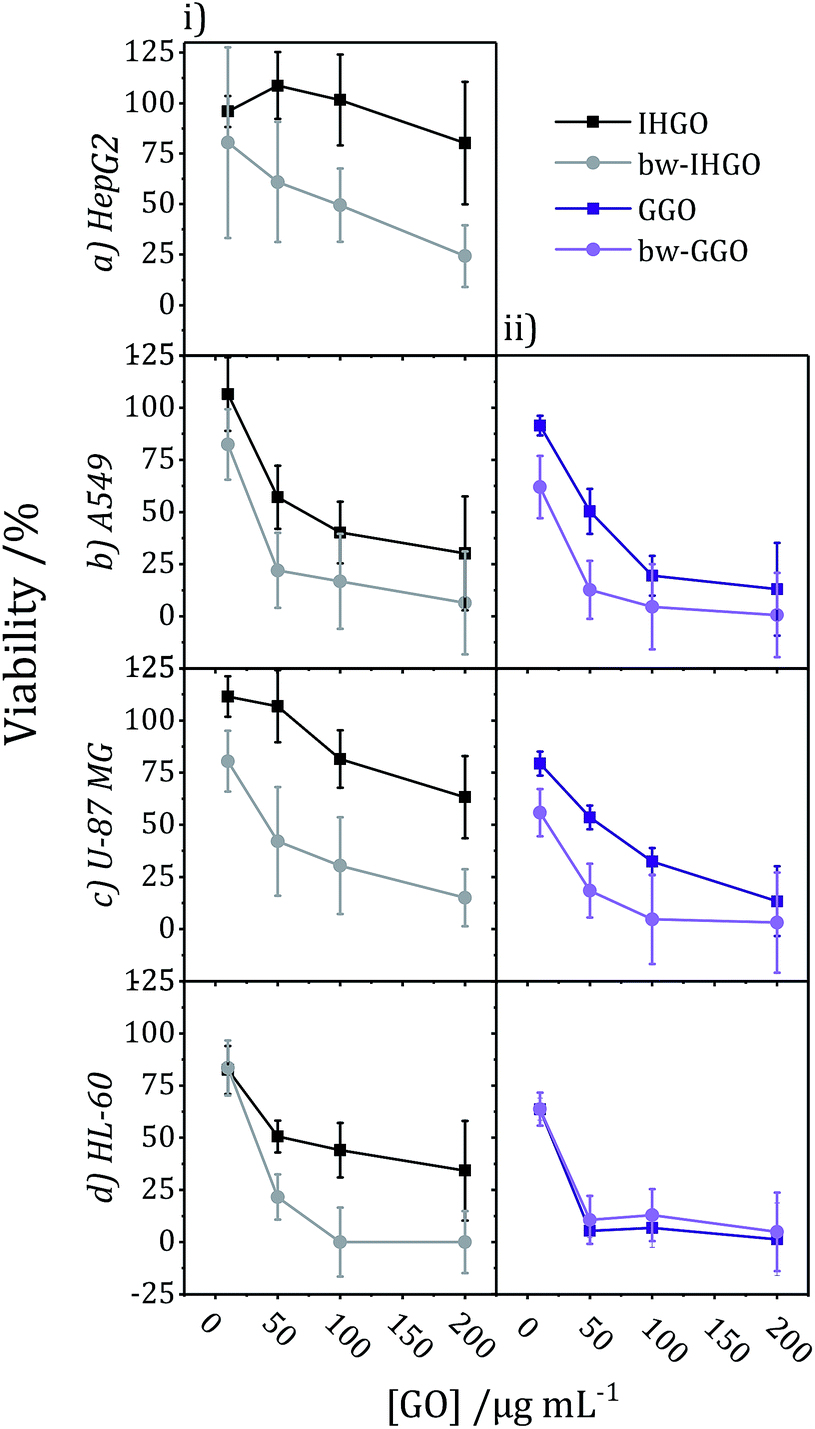 | ||
| Fig. 3 Comparison of cytotoxicity tested on (a) HepG2, (b) A549, (c) U-87 MG, and (d) HL-60 cell lines between (i) IHGO to bw-IHGO and (ii) GGO to bw-GGO after 24 + 4 h exposure as determined by the WST-8 assay (GGO data is previously published25). | ||
In order to verify the observation that base washed GO is indeed more cytotoxic than GO, GGO, a commercial source of GO, was base washed and identical experimental conditions were used to evaluate the cytotoxicity of the resulting material. The cytotoxicity of unprocessed GGO was previously reported by our group25 and the results are displayed in Fig. 3(ii)(b–d). Just as observed with IHGO, the results demonstrate that post base washing, the material has increased cytotoxicity. Interestingly, the starting material GGO was more toxic than IHGO, and the bw-GGO was similarly more toxic than bw-IHGO for A549 and U-87 MG cells at the lower concentrations. HL-60 showed little to no difference in viability post treatment to GGO and bw-GGO. HL-60 is the only suspension cell line studied, and we have previously shown that suspension cell lines show a higher susceptibility to toxicity induced by GO.25 At the lowest concentration studied, no difference was also observed between the IHGO and bw-IHGO samples. At higher concentrations few (<10%) cells remained viable post treatment to GGO, it was therefore not possible for bw-GGO to induce a much greater cytotoxicity. AFM images of the GGO and bw-GGO samples may be found in the ESI, Fig. S4.†
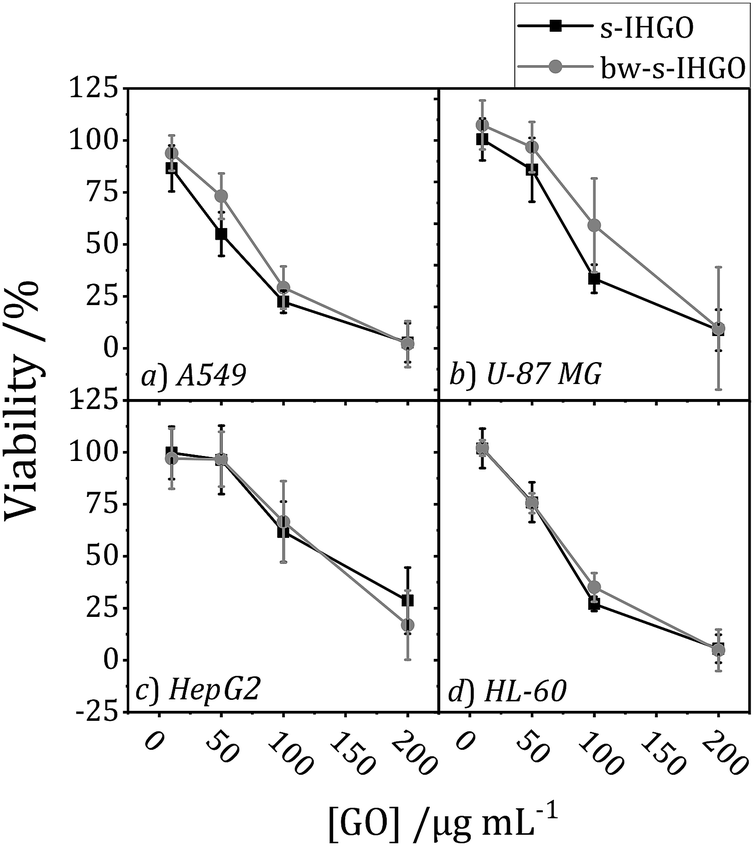 | ||
| Fig. 4 Cytotoxicity of s-IHGO and bw-s-IHGO tested on (a) A549, (b) U-87 MG, (c) HepG2, and (d) HL-60 cell lines after 24 + 4 h exposure as determined by the WST-8 assay. | ||
Despite the differences in nitrogen and sulfur content, the overall cytotoxicity of the un-cleaned and cleaned samples follow the same trend; cleaning was not observed to impact the cytotoxicity of IHGO and s-IHGO. The results of the cytotoxicity experiments may be found in the ESI Fig. S5.† Since no statistically meaningful difference was observed in the cytotoxicity of the cleaned and un-cleaned samples, this suggests that the sulfur and nitrogen impurities which were removed during washing, at the level to which they were removed, do not significantly impact the overall cytotoxicity of IHGO and s-IHGO.
Nanomaterial adsorption
One potential route of cytotoxicity is nutrient depletion induced by nanomaterial adsorption. This indirect route of cytotoxicity was observed for carbon nanotubes on A549 cells.40 In order to determine if this is a contributing factor to the route of toxicity for processed and unprocessed GO, F12-K (media for A549 cells), IMDM (media for HL-60 cells) and DMEM (media for U-87 MG and HepG2 cells) were treated with IHGO, s-IHGO and bw-IHGO at a concentration of 200 μg mL−1. The media were incubated with the GO samples for 24 h at 37 °C. The solutions were then centrifuged (600 × g for 10 min) and the supernatant media were collected. A549, HepG2, U-87 MG and HL-60 cells were treated with their respective treated media and incubated for another 24 h. The WST-8 reagent was then used to measure the viability of the cells and the results are displayed in Fig. 5. It is clear that nutrient depletion via nanomaterial adsorption is not the route of toxicity as the differences between the control (media treated with water) and the treated media led to the same number of viable cells.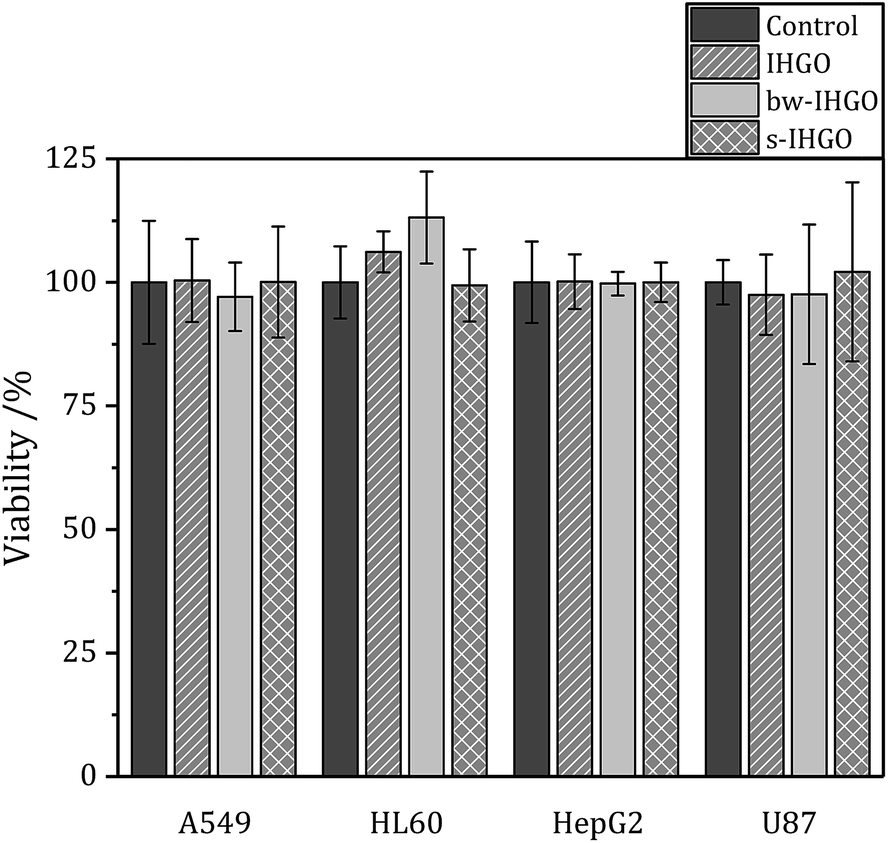 | ||
| Fig. 5 Viability of A549, HL-60, HepG2 and U87 cells after 24 + 4 h culture with media treated with IHGO, bw-IHGO and s-IHGO determined by the WST-8 assay. | ||
Discussion
As base washing, sonication and a combination of the two techniques resulted in materials unique to each other, a comparison of the cytotoxicity of the materials may provide information on which material property has the greatest impact on the cytotoxicity. In order to facilitate comparison between the cytotoxicity of the materials, a box plot comparing the processed materials to the unprocessed materials was prepared and is displayed in Fig. 6. The box plot displays the percentage point difference between the average cell viability post treatment with IHGO and the average cell viability post treatment with the processed materials across all concentrations studied. To obtain the values used for comparison, an average viability was taken across all concentrations studied for IHGO and for the processed GO materials, and simple subtraction of the average viability of the processed GO material treated cells from the average viability of IHGO treated cells was used. The dotted line across the graph indicates zero difference in cytotoxicity compared to IHGO, values greater than zero indicate increased cytotoxicity and values less than zero indicate decreased cytotoxicity. By compressing the data in such a way, trends become more obvious. From Fig. 6, it is clear that bw-IHGO resulted in the greatest increase of cytotoxicity across all cell lines studied. The median of the average cell viability post exposure to bw-IHGO was reduced by the greatest amount for all cell lines studied. s-IHGO and bw-s-IHGO also reduced the average cell viability relative to IHGO on the A549, U-87 MG and HepG2 cell lines, but to a lesser extent than bw-IHGO. As already eluded to in Fig. 4, it can again be seen that the toxicity of s-IHGO and bw-s-IHGO are similar.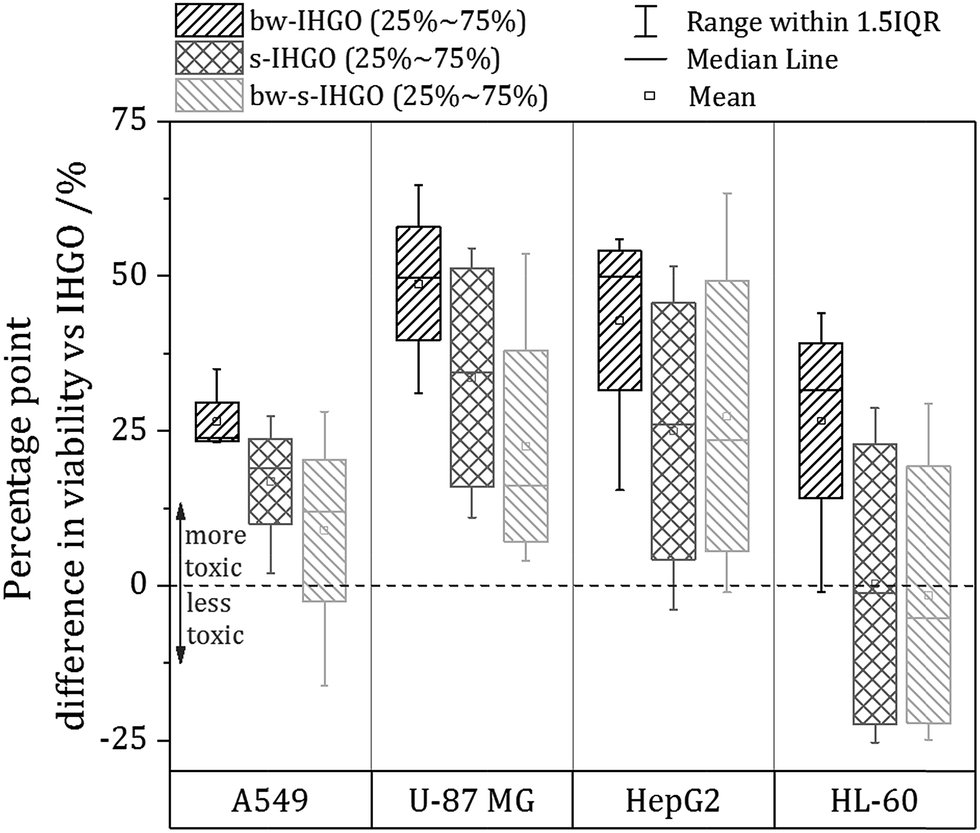 | ||
| Fig. 6 Average percentage point difference in cytotoxicity between bw-IHGO, s-IHGO or bw-s-IHGO and IHGO across the four concentrations studied on A549, U-87 MG, HepG2 and HL-60 cells as determined by the WST-8 assay for 24 h treatment. | ||
The high toxicity of the base washed material may be attributed to the properties which define bw-IHGO from IHGO: the base washed material had a reduced C/O ratio as well as a substantially different morphology. The IHGO flakes were planar when the bw-IHGO flakes were wrinkled. Other groups have evaluated the impact of the C/O ratio and conflicting results are reported. Das et al. evaluated the cytotoxicity of GO and reduced GO and found that reduced GO was less toxic than GO of the same size.24 They attributed this observation to the fewer oxygen containing functional groups on the reduced material and thus less oxidative stress induced toxicity. Faria et al. had a complementary finding observing base washed GO to be less toxic to Escherichia coli cells30 as did the group of Clemente et al. who demonstrated base washing GO mitigated the materials cytotoxicity on zebra fish embryos.29 Chng and Pumera evaluated the cytotoxicity of GO produced by different oxidation methods with different oxygen contents and observed that a lower C/O ratio resulted in a higher toxicity via the MTT assay but the opposite via the WST-8 assay.41 Contreras-Torres et al. observed GO reduced under mild conditions was five times more toxic than the un-reduced material.26 Though these works process the materials differently and may use different cell lines/organisms and assays, it is clear that there is not a strong consensus regarding the impact of the C/O on the cytotoxicity.
As for the impact of size on the cytotoxicity, it is generally accepted that a reduced flake size results in an increased cytotoxicity. Chang et al. and Das et al. are examples of other teams who also observed an increased activity of the toxicity of GO in response to decreased flake size.23,24 This was indeed the trend observed when comparing IHGO and s-IHGO on A549, U-87 MG and HepG2 cells; IHGO was more toxic than s-IHGO and IHGO had a larger flake size than s-IHGO. The HL-60 cell line, the only suspension cell line, did not show much difference as a result of the decreased flake size across all concentrations studied, only at the higher concentrations were the smaller flake sized materials more toxic. This result was not surprising as it is the only suspension cell line and our group has demonstrated that different cell lines are impacted differently by GO.25 Despite the accepted relationship between flake size and cytotoxicity, an increase in toxicity as a result of decreased flake size was not observed when comparing bw-IHGO to bw-s-IHGO; bw-IHGO has much larger flakes than bw-s-IHGO but was observed to be more toxic across the cell lines studied. As this was a surprise, other properties of the materials must be taken into account to look for an explanation for this unexpected result.
By comparing the materials' unique properties, interesting conclusions regarding the impact of the materials properties on the cytotoxicity may be made. bw-IHGO differs from IHGO by two observed material properties: the C/O ratio and the flake morphology. Since bw-IHGO was much more cytotoxic than IHGO, it is likely that one or a combination of these two defining properties results in the higher toxicity. By comparing s-IHGO and bw-s-IHGO which had different C/O ratios, similar morphologies and were observed to have a similar toxicity, it may be concluded that the C/O ratio does not significantly independently impact the cytotoxicity. From these observations, the impact of flake morphology was observed to have the greatest impact on the cytotoxicity of the material. The impact of size was also observed to impact the cytotoxicity as the smaller materials were more toxic than the unprocessed materials when they had the same morphology. The C/O ratio was not observed to have a measurable, independent impact on the cytotoxicity of IHGO.
So far, all measurements were performed by one analyst using the same source of IHGO (prepared in duplicate). In order to provide more evidence to support the findings of this series of experiments, two sets of experiments were repeated by a second analyst on a select cell line, A549. In the first set of experiments, the same source of IHGO was separately processed to prepare s-IHGO, bw-IHGO and bw-s-IHGO and the toxicity of the resulting materials were evaluated. In a second set of experiments, the same processing and toxicity measurements were performed on a separate, independently prepared source of IHGO. The results of these experiments are presented in the ESI, Fig. S6.† The same trends in cytotoxicity were observed, bw-IHGO was measured to be the most cytotoxic, IHGO was the most cytocompatible and bw-s-IHGO and s-IHGO imparted a toxicity greater than IHGO, but less than bw-IHGO.
Additional evidence to support the relationship between toxicity and flake morphology was observed; in an attempt to prepare a sample of bw-IHGO, a material was obtained that was observed by AFM to be an intermediate material between IHGO and bw-IHGO in which a portion of the flakes were beginning to become wrinkled. This material was thus observed to have a toxicity intermediate to IHGO and bw-IHGO. The cytotoxicity measurements and an AFM image of the intermediate base washed material are presented in Fig. S7.†
In this work, a relationship between the morphology of GO flakes and the cytotoxicity of the material was observed. It is recognized that the different morphology of the GO flakes may be a product of another material characteristic which was not measured in the present study, such as surface charges. Future work should further evaluate the route of the increased cytotoxicity as a result of the folded morphology.
Conclusions
The morphology, size, C/O ratio and presence of impurities of IHGO processed via sonication, base washing, a combination of sonication and base washing, and cleaning were evaluated via AFM and XPS. The materials' properties were then related to the materials' cytotoxicity to compare the impact of the properties to the cytotoxicity of the material. Sonication, base washing and a combination of the two processes resulted in materials that were distinctly different from the parent IHGO, in both properties and cytotoxicity, while the impact of cleaning was comparatively minor.Base washing IHGO changed both the morphology of the flake from planar to wrinkled as revealed by AFM, as well as increased the C/O ratio as revealed by XPS. Sonication of IHGO and bw-IHGO greatly reduced the flake size of both materials. Sonication of bw-IHGO also resulted in the loss of the wrinkled texture. Cleaning IHGO had a comparatively small impact on the sample and reduced the amounts of sulfur and nitrogen from ca. 2% to 1%. Similarly, the materials resulted from sonication, base washing and a combination of the two processes had different cytotoxicities than IHGO but the ‘cleaned’ material, did not.
The cytotoxicity of all samples was evaluated via the WST-8 assay on four different cell lines, A549, U-87 MG, HepG2 and HL-60 for a 24 h exposure period. bw-IHGO was observed to consistently be the most cytotoxic across all cell lines studied. The two smaller flake materials studied, s-IHGO and bw-s-IHGO, had similar cytotoxicities and a size dependent toxicity was observed as IHGO was generally less toxic than s-IHGO and bw-s-IHGO. Interestingly, bw-IHGO was more toxic than s-IHGO and bw-s-IHGO despite having a larger flake size. The high cytotoxicity of bw-IHGO was attributed to the morphology of the flakes which was unique to bw-IHGO. As s-IHGO and bw-s-IHGO were so similar in cytotoxicity but different in C/O ratio, the C/O ratio was not observed to independently impact the cytotoxicity. Cleaning reduced the amounts of nitrogen and sulfur impurities but was not observed to impact the cytotoxicity of IHGO and s-IHGO on the level to which they were removed. These results indicate that flake morphology, followed by flake size has the greatest impact on the cytotoxicity of the material.
Toxicity due to depletion of nutrients from nanomaterial adsorption was evaluated as a possible route of toxicity for IHGO, bw-IHGO and s-IHGO. Cells grown in media treated with the GO samples showed little or no difference versus untreated media so it was determined that nanomaterial adsorption is not the route of toxicity. Future work should focus on evaluating other routes of cytotoxicity and determine which property of the wrinkled GO results in the high observed cytotoxicity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge and thank Dr Maohui Chen for his support in AFM image analysis. J. A. and T. C. were supported by a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada (NSERC) to S. Z.References
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- A. Bonanni, A. Ambrosi, C. K. Chua and M. Pumera, ACS Nano, 2014, 8, 4197–4204 CrossRef CAS PubMed.
- H. R. Thomas, C. Vallés, R. J. Young, I. A. Kinloch, N. R. Wilson and J. P. Rourke, J. Mater. Chem. C, 2013, 1, 338–342 RSC.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Nat. Chem., 2010, 2, 1015–1024 CrossRef CAS PubMed.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- H. He, T. Riedl, A. Lerf and J. Klinowski, J. Phys. Chem., 1996, 100, 19954–19958 CrossRef CAS.
- C. K. Chua, Z. Sofer and M. Pumera, Chem.–Eur. J., 2012, 18, 13453–13459 CrossRef CAS PubMed.
- Z.-L. Chen, F.-Y. Kam, R. G. Goh, J. Song, G.-K. Lim and L.-L. Chua, Chem. Mater., 2013, 25, 2944–2949 CrossRef CAS.
- J. P. Rourke, P. A. Pandey, J. J. Moore, M. Bates, I. A. Kinloch, R. J. Young and N. R. Wilson, Angew. Chem., Int. Ed., 2011, 50, 3173–3177 CrossRef CAS PubMed.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490–4493 CrossRef CAS.
- V. R. Coluci, D. S. f. T. Martinez, J. G. Honório, A. i. F. de Faria, D. A. Morales, M. S. Skaf, O. L. Alves and G. A. Umbuzeiro, J. Phys. Chem. C, 2014, 118, 2187–2193 CrossRef CAS.
- C. Su, M. Acik, K. Takai, J. Lu, S.-j. Hao, Y. Zheng, P. Wu, Q. Bao, T. Enoki and Y. J. Chabal, Nat. Commun., 2012, 3, 1298 CrossRef PubMed.
- Z. Guo, S. Wang, G. Wang, Z. Niu, J. Yang and W. Wu, Carbon, 2014, 76, 203–211 CrossRef CAS.
- A. M. Dimiev and T. A. Polson, Carbon, 2015, 93, 544–554 CrossRef CAS.
- A. Naumov, F. Grote, M. Overgaard, A. Roth, C. E. Halbig, K. Nørgaard, D. M. Guldi and S. Eigler, J. Am. Chem. Soc., 2016, 138, 11445–11448 CrossRef CAS PubMed.
- B. R. Coleman, T. Knight, V. Gies, Z. J. Jakubek and S. Zou, ACS Appl. Mater. Interfaces, 2017, 9, 28911–28921 CrossRef CAS PubMed.
- G. Eda, G. Fanchini and M. Chhowalla, Nat. Nanotechnol., 2008, 3, 270–274 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- Y. Chang, S.-T. Yang, J.-H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- S. Das, S. Singh, V. Singh, D. Joung, J. M. Dowding, D. Reid, J. Anderson, L. Zhai, S. I. Khondaker and W. T. Self, Part. Part. Syst. Charact., 2013, 30, 148–157 CrossRef CAS.
- V. Gies and S. Zou, Toxicol. Res., 2018, 7, 93–101 RSC.
- F. F. Contreras-Torres, A. Rodríguez-Galván, C. E. Guerrero-Beltrán, E. Martínez-Lorán, E. Vázquez-Garza, N. Ornelas-Soto and G. García-Rivas, Mater. Sci. Eng., C, 2017, 73, 633–642 CrossRef CAS PubMed.
- E. L. K. Chng, C. K. Chua and M. Pumera, Nanoscale, 2014, 6, 10792–10797 RSC.
- M. Pelin, L. Fusco, V. León, C. Martín, A. Criado, S. Sosa, E. Vázquez, A. Tubaro and M. Prato, Sci. Rep., 2017, 7, 40572 CrossRef CAS PubMed.
- Z. Clemente, V. L. S. Castro, L. S. Franqui, C. A. Silva and D. S. T. Martinez, Environ. Pollut., 2017, 225, 118–128 CrossRef CAS PubMed.
- A. F. Faria, F. Perreault and M. Elimelech, ACS Appl. Nano Mater., 2018, 1, 1164–1174 CrossRef CAS.
- M. Wu, R. Kempaiah, P. J. Huang, V. Maheshwari and J. Liu, Langmuir, 2011, 27, 2731–2738 CrossRef CAS PubMed.
- J. Farkas and A. M. Booth, Nanotoxicology, 2017, 11, 569–577 CrossRef CAS PubMed.
- H. Yue, W. Wei, Z. Yue, B. Wang, N. Luo, Y. Gao, D. Ma, G. Ma and Z. Su, Biomaterials, 2012, 33, 4013–4021 CrossRef CAS PubMed.
- Y. Wang, S. Wu, X. Zhao, Z. Su, L. Du and A. Sui, Bio-Med. Mater. Eng., 2014, 24, 2007–2013 CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- H. R. Thomas, S. P. Day, W. E. Woodruff, C. Vallés, R. J. Young, I. A. Kinloch, G. W. Morley, J. V. Hanna, N. R. Wilson and J. P. Rourke, Chem. Mater., 2013, 25, 3580–3588 CrossRef CAS.
- J. Végh, J. Electron Spectrosc. Relat. Phenom., 2006, 151, 159–164 CrossRef.
- X. Qi, T. Zhou, S. Deng, G. Zong, X. Yao and Q. Fu, J. Mater. Sci., 2014, 49, 1785–1793 CrossRef CAS.
- A. Casey, E. Herzog, F. Lyng, H. Byrne, G. Chambers and M. Davoren, Toxicol. Lett., 2008, 179, 78–84 CrossRef CAS PubMed.
- E. L. K. Chng and M. Pumera, Chem.–Eur. J., 2013, 19, 8227–8235 CrossRef CAS PubMed.
- S. Mukherjee, P. Sriram, A. K. Barui, S. K. Nethi, V. Veeriah and S. Chatterjee, Adv. Healthcare Mater., 2015, 4, 1722–1732 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00178b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |