
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing an RNA aptamer probe for super-resolution imaging of native EGFR†
Qiuyan
Yan
ac,
Mingjun
Cai
 a,
Lulu
Zhou
ac,
Haijiao
Xu
ac,
Yan
Shi
a,
Jiayin
Sun
a,
Junguang
Jiang
a,
Jing
Gao
*a and
Hongda
Wang
*ab
a,
Lulu
Zhou
ac,
Haijiao
Xu
ac,
Yan
Shi
a,
Jiayin
Sun
a,
Junguang
Jiang
a,
Jing
Gao
*a and
Hongda
Wang
*ab
aState Key Laboratory of Electroanalytical Chemistry, Research Center of Biomembranomics, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, P. R. China. E-mail: hdwang@ciac.ac.cn; gaojing@ciac.ac.cn
bLaboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Wenhai Road, Aoshanwei, Jimo, Qingdao, Shandong 266237, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 28th August 2018
Abstract
Aptamers, referred to as “chemical antibodies”, are short single-stranded oligonucleotides that bind to targets with high affinity and specificity. Compared with antibodies, aptamers can be designed, developed and modified easily. Since their discovery, aptamers have been widely used in in vitro diagnostics and molecular imaging. However, they are relatively less studied and applied in advanced microscopy. Here we used an RNA aptamer in dSTORM imaging and obtained a high-quality image of EGFR nanoscale clusters on live cell membranes. The results showed that the cluster number and size with aptamer labeling were almost the same as those with labeling with the natural ligand EGF, but the morphology of the clusters was smaller and more regular than that with cetuximab labeling. Meanwhile, dual-color imaging demonstrated sufficient fluorophore labeling, highly specific recognition and greatly accurate clustering information provided by aptamers. Furthermore, the aptamer labeling method indicated that active EGFR formed larger clusters containing more molecules than resting EGFR, which was hidden under the antibody labeling. Our work suggested that aptamers can be used as versatile probes in super-resolution imaging with small steric hindrance, opening a new avenue for detailed and precise morphological analysis of membrane proteins.
Introduction
The epidermal growth factor receptor (EGFR), as a first identified receptor of the ErbB family, plays an important role in regulating cell proliferation, differentiation, and migration.1,2 It has an ectodomain composed of four sub-domains, two of which are ligand binding domains and two of which are cysteine-rich domains.3,4 When ligands, such as epidermal growth factor (EGF) or transforming growth factor-α (TGF-α),5 bind to the EGFR ligand binding domains, the EGFR conformation changes3 and forms homo- and hetero-dimers with other ErbB family members.6It has been widely recognized that exploring the spatial organization of EGFR on cell surfaces is vital for understanding its physiological functions.7 Many studies using different techniques, such as NSOM,8 dSTORM,9 multiplexed exchange-PAINT10 (Point Accumulation for Imaging in Nanoscale Topography) and reversible cryo-arrest, have shown that EGFR forms clusters on cell surfaces. Among these techniques, single-molecule localization microscopy including (direct) stochastic optical reconstruction microscopy ((d)STORM)11–13 and photoactivated localization microscopy (PALM)14 has attracted our attention. It allows the resolution limit to be bypassed and has enabled unprecedented insights into the organization of subcellular components.15,16 Thus, it is very suitable to explore the distribution of proteins with nanometer precision at the single molecule level17,18 and decipher how the protein organization is linked to its functions.
To achieve a high-quality SMLM image, sufficient fluorophore labeling without being detrimental to biological targets is a crucial prerequisite.19,20 The most widely used labeling probes are organic fluorophore-conjugated antibodies and recombinant proteins that are fused to photoactivatable fluorescent proteins (FPs) or fluorogen-labeling enzymes, such as Halo- and SNAP-tags.21 However, the large size of antibodies results in steric hindrances and brings a linkage error of ∼10 to 20 nm by displacing the fluorophore from the target,22,23 and FPs and enzyme tags may affect the expression, cellular localization and functions of the target proteins. Therefore, elucidating the fine distribution and clustering of membrane proteins calls for new labeling strategies.
Fortunately, diverse kinds of labeling molecules have been provided and developed in recent years,19,23–25 one of which is aptamers. These synthetic single-stranded DNA or RNA oligonucleotides form specific three-dimensional conformations to bind to targets. They usually contain 20–50 bases,26 whose size is only a fifth of a full-length antibody.19,27 Not only does the base sequence-identified strategy endow aptamers with strong specificity and high affinity,28–30 but also the small size lowers the steric hindrance and linkage error. Additionally, aptamers can be produced via chemical synthesis in a large quantity with higher stability, less batch-to-batch variation, lower cost and shorter time than monoclonal antibodies.31,32 Meanwhile, they can be labeled and modified with a wide range of organic dyes that emit significantly more photons, thus obtaining better precision than FPs.33
Along this line, we here used a reported RNA aptamer6 to label the membrane EGFR on live cells for high-quality dSTORM imaging of EGFR distribution and clustering. Through comparison with the ligand EGF and the antibody cetuximab in both single- and dual-color imaging, we demonstrated the benefits of aptamers for sufficient fluorophore labeling and highly specific recognition, which elucidated more accurate and detailed spatial organization of EGFR. More importantly, the aptamer recognition method revealed the fine differences of clustering between active and resting EGFR on live cell membranes that have not been found by antibody labeling yet, suggesting aptamers to be promising tools for super-resolution imaging of membrane proteins.
Results and discussion
Super-resolution imaging of RNA aptamer-recognized EGFR
Since the size of the synthesized Cy3-conjugated RNA aptamer is so small, which is only about 2 nm × 3.5 nm and ∼13 kDa (the details are depicted in Fig. S1†), we first tested whether it would be internalized in live cells when used as a labeling probe. The live COS-7 cells were stained with the membrane fluorescent probe DiO and Cy3-conjugated EGFR aptamer at 4 °C for 10 min, and then detected under a confocal microscope. During the observation, the samples were always kept in a low-temperature environment. The fluorescent images showed that the Cy3-aptamer overlapped very well with the membrane and was not internalized under these experimental conditions (Fig. S2†). Thus, we proved that the aptamer probe could be used in live cells.Next, we used the aptamer in dSTORM imaging to investigate the spatial distribution of EGFR on live cell membranes (Fig. 1A). The process of staining and imaging was still maintained at the low temperature. We found that EGFR clusters distributed on the membranes. To characterize the detailed distribution pattern of EGFR, we employed Ripley's K function analysis,34,35 a widely used method to determine randomness or aggregation for spatial points in super-resolution data.36,37 The cell was first divided into four parts from the center, and a region of 3 × 3 μm2 in each part was stochastically selected for examination (Fig. 1D). The value of L(r) − r in the plot will be zero for molecules with a random distribution, and positive for clustering molecules. As shown in Fig. 1E, the aptamer labeled EGFR displayed a clustered distribution. The maximum of L(r) − r was 450, representing a high degree of clustering; the corresponding rmax was 216 nm, which was defined as the size of maximum clustering in this examined region; and the intercept of the plot on the horizontal axis was 740 nm, representing the maximal clustering range above the level for a random distribution. Secondly, a color-coded cluster map could be generated by interpolation with L(r) as the z-axis (Fig. 1F). Finally, a binary cluster image was produced by an appropriate threshold of L(r),9 from which the information of clustering could be extracted (Fig. 1G). Based on this method, we found that there were about 0.6 clusters per μm2 with an average area of 0.11 μm2 on the cell membrane (Fig. 1H and I).
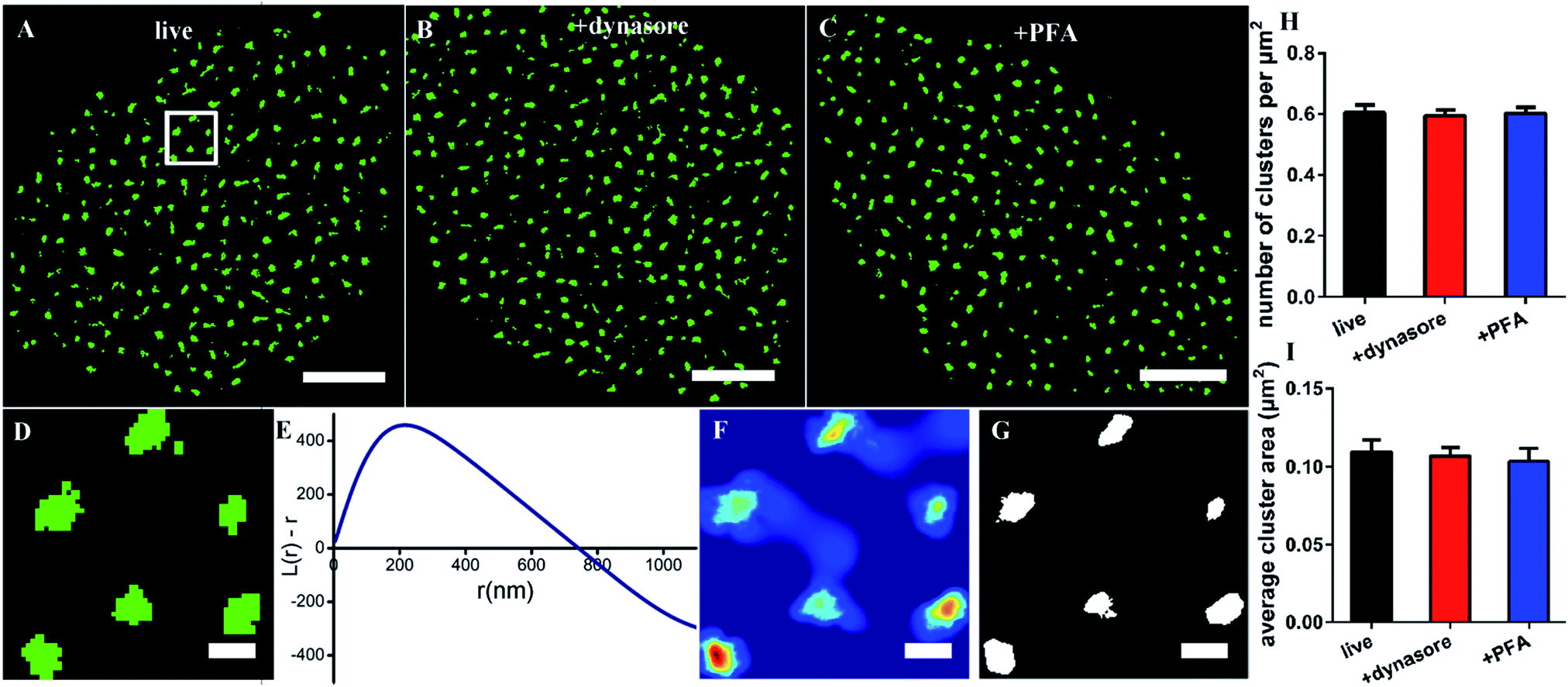 | ||
| Fig. 1 Spatial distribution and analysis of the aptamer labeled EGFR on COS-7 cells. (A–C) Reconstructed dSTORM images of EGFR labeled with the Cy3-conjugated aptamer on the membranes of the live cells (A), dynasore treated cells (B) and PFA fixed cells (C). (D) The expanded 3 × 3 μm2 region of the dSTORM image box in (A) showing the localization distribution of EGFR. (E) Ripley's K function plot representing that the peak clustering occurs at ∼216 nm and the degree of clustering scales up to 740 nm. (F) The interpolated cluster map through Ripley's K function analysis, indicating a highly clustered distribution. The clustering gradually deepens from blue to red color. (G) The binary cluster image generated from the color-map using a threshold, showing the shape, number and size of the clusters. Scale bars are 5 μm in A–C, and 500 nm in D, F and G. (H) The average number of clusters per unit area on the three kinds of cell membranes. (I) The average cluster area. Data were collected from 10 cells in three independent experiments (mean ± SD). | ||
Considering that it would take a relatively long time to capture the dSTORM frame sequence, we did not know whether the low temperature could ensure the aptamer against being internalized all the time. Hence, completely endocytosis-inhibited cells were taken as control groups. We pretreated the cells with dynasore, a cell-permeable dynamin inhibitor to block cell endocytosis,9,38 and then stained with the aptamer probe. Both the morphology of EGFR clusters and their clustering parameters were similar to those on live cells (Fig. 1B, H, and I). The results demonstrated that the aptamer could bind to EGFR but not enter the live cell at a low temperature. In addition, due to the fluidity of cell membranes,39,40 we also observed the EGFR on fixed cell membranes and found that there were no differences between live and fixed cells as well (Fig. 1C, H, and I), which verified again that the aptamer was suitable for live-cell labeling.
Collectively, the above results showed the clustered distribution of EGFR on live cell membranes, which was consistent with previous studies.9,19 The differences were the cluster size and shape, which might be caused by the different labeling probes, and we would address this problem in the following sections. Moreover, our data supported that the aptamer was highly desirable to label membrane proteins in live cells, because it neither affected a real distribution of proteins nor entered the cells by endocytosis.
Identifying aptamer's recognition efficiency
Some studies have stated that the smaller size of fluorescent probes made it possible for them to bind to more targets.19,23 As the size of aptamers is much smaller than that of common IgG antibodies, to test whether the RNA aptamer can actually recognize more EGFR molecules than a widely used antibody—cetuximab, we observed the membrane EGFR labeled with the Cy3-conjugated aptamer, EGF and cetuximab, respectively. Before imaging EGFR, we detected the resolution under the three labeling conditions (Fig. S3†). The FWHM of the aptamer probe was almost identical to that of EGF, which was ∼29 nm; whereas it was ∼32 nm for the antibody. Although the resolutions measured by these three probes have no significant differences due to the same dye they conjugated, aptamer and EGF probes showed a slightly smaller size than antibodies.By dSTORM imaging, we found that EGFR formed clusters on COS-7 cell membranes no matter which fluorescent probe was utilized (Fig. 2A–F), and the clustered distribution was confirmed by Ripley’s K-function analysis as well (Fig. S4†). Although clusters were observed in all states, the direct morphology of clusters provided by the enlarged images showed that aptamer and EGF labeled clusters were similar, whereas both of them were different from antibody labeled clusters. Quantitative circularity analysis indicated that the shape of the former two was more regular and more like a circle with a circularity of more than 0.7, while that of the latter was irregular and tended to a long ellipse with a circularity of only 0.5 (Fig. 2I). Meanwhile, large-area clusters (>0.16 μm2) were more under antibody labelling (Fig. 2J). However, both the number of localizations (Fig. 2G) and the cluster amount per unit (Fig. 2H) were nearly identical under the three labeling conditions. Hence, the above results hinted us that the molecular organization in clusters should be different when labeled with diverse probes. To further clarify this problem, we utilized semi-quantitative analysis to calculate the number of molecules within the clusters. It was estimated via dividing the localization number in one cluster by the average localization number of single dye-conjugated aptamer/EGF/cetuximab (see the Experimental section for details). The percentage of clusters containing different numbers of EGFR molecules (≥3) is displayed in Fig. 2K. Small clusters consisting of 3–5 molecules occupied a significantly large proportion in antibody labeled clusters, and for large clusters with more than 10 molecules, they were present in a much smaller proportion as antibody labeled clusters than as their EGF and aptamer counterparts. This result suggested that the aptamer and EGF can sufficiently recognize large and complex aggregates containing a number of molecules in crowded environments that antibodies cannot, which was also found in other studies.41 The reason might be that the bivalent structure and the large size of antibodies caused their cross-linking with each other and a relatively strong steric hindrance.19,23 As for the aptamer or EGF, they were greatly small (less than 10 nm) and flexible, and did not form cross-links, which might facilitate more efficient and correct recognition. Together, the results elucidated that the aptamer had a better and more efficient ability to recognize EGFR than the antibody, especially for large clusters, and its usage could largely reduce the labelling inaccuracy.
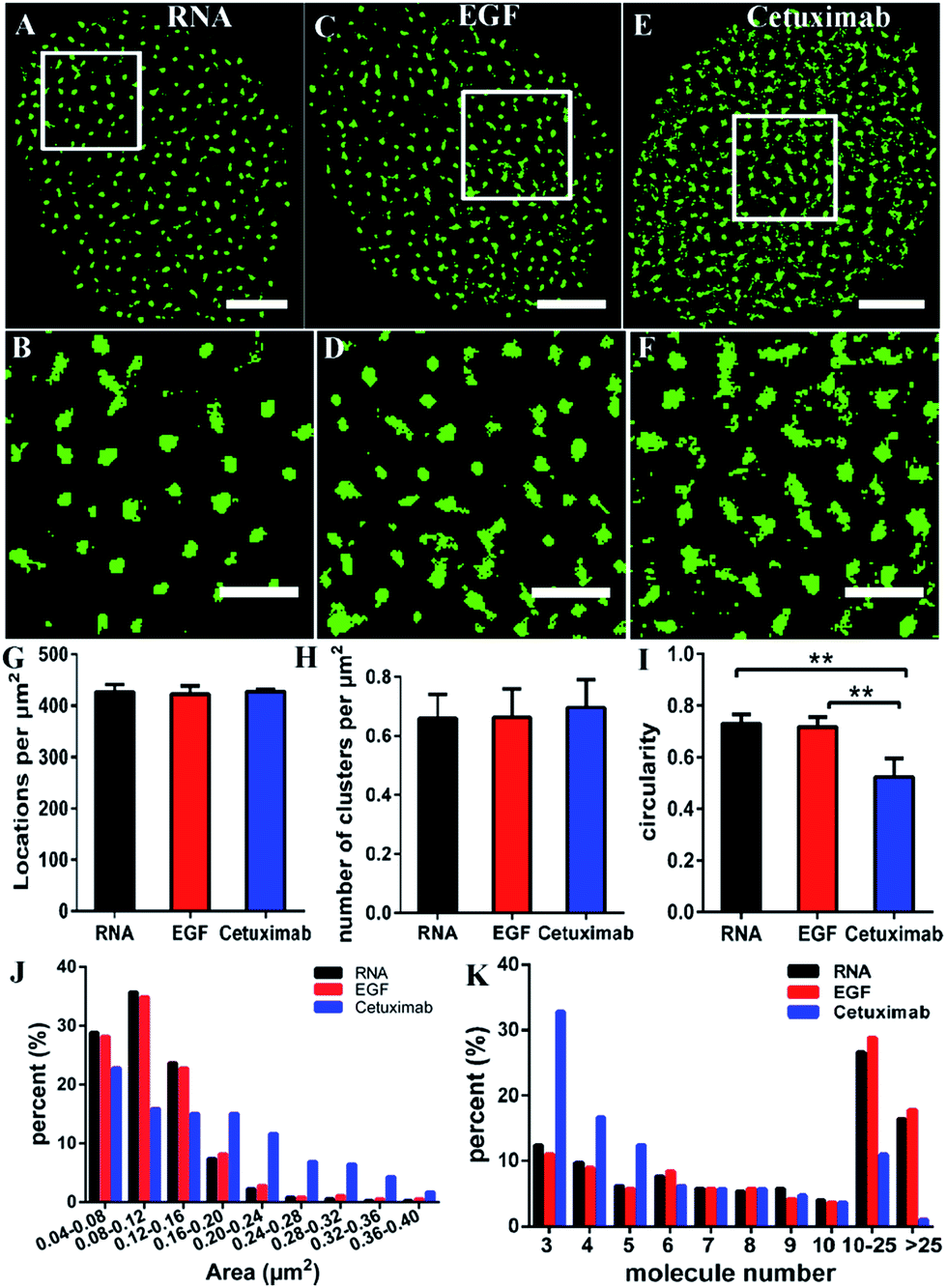 | ||
| Fig. 2 Comparison of different fluorescent probes in super-resolution imaging of EGFR. (A–F) Reconstructed dSTORM images of EGFR labeled with Cy3 conjugated RNA (A), EGF (C) and cetuximab (E), and the corresponding magnified images (B, D and F). Scale bars are 5 μm in A, C and E, and 2 μm in B, D and F. (G) The number of localizations per μm2 on COS-7 cells with different labeling probes. (H) The number of clusters per μm2. (I) The circularity of clusters. (J) The histogram of cluster areas. (K) The percentage of clusters containing different numbers of molecules. Data were collected from 10 cells in three independent experiments (mean ± SD). **p < 0.001 (two-tailed unpaired t-test). | ||
Verifying the high specificity of the RNA aptamer
To investigate the recognition specificity of the RNA aptamer, dual-color dSTORM imaging of EGFR labeled with EGF or cetuximab and the aptamer was performed. In these experiments, cells were first incubated with Alexa 647-conjugated EGF or cetuximab, and then stained with the Cy3-conjugated aptamer (see the Experimental section). We found that the majority of the aptamer labeled EGFR molecules were overlapped with EGF stained EGFR molecules, whereas the overlapping portion between the aptamer and cetuximab was not that high (Fig. 3A–H). To quantitatively analyze the degree of colocalization, the coordinate-based colocalization (CBC) algorithm42 was used, which is suited for single-molecule super-resolution data. A CBC value for each localization from species A, defined as CAi, indicates perfect colocalization for CAi = 1, colocalization for 0 < CAi < 1, no colocalization for CAi = 0 and segregated but near localizations for −1 < CAi < 0. For EGF and aptamer labeled EGFR, their individual localizations with 0 < CAi < 1 both occupied approximately 80 percent, and the percentage of maximal CAi was similar (Fig. 3I and J). This indicated that EGF and the aptamer colocalized well with each other, and both of them have high labeling efficiency and specificity. For cetuximab and aptamer labeled EGFR, the percentage of their individual CA that ranged from 0 to 1 was 0.6 and 0.8, respectively (Fig. 3K and L). Moreover, maximal CA, RNA–cetuximab occupied a higher percent than maximal CA, cetuximab–RNA, demonstrating that most aptamer recognized EGFR molecules were colocalized with antibody labeled EGFR molecules but EGFR stained with the antibody occupied more area. The reason was still the larger size of antibodies that increased the area of the labeled clusters. All the results indicated that the aptamer did not only have a high specificity but improved precision in recognizing membrane EGFR.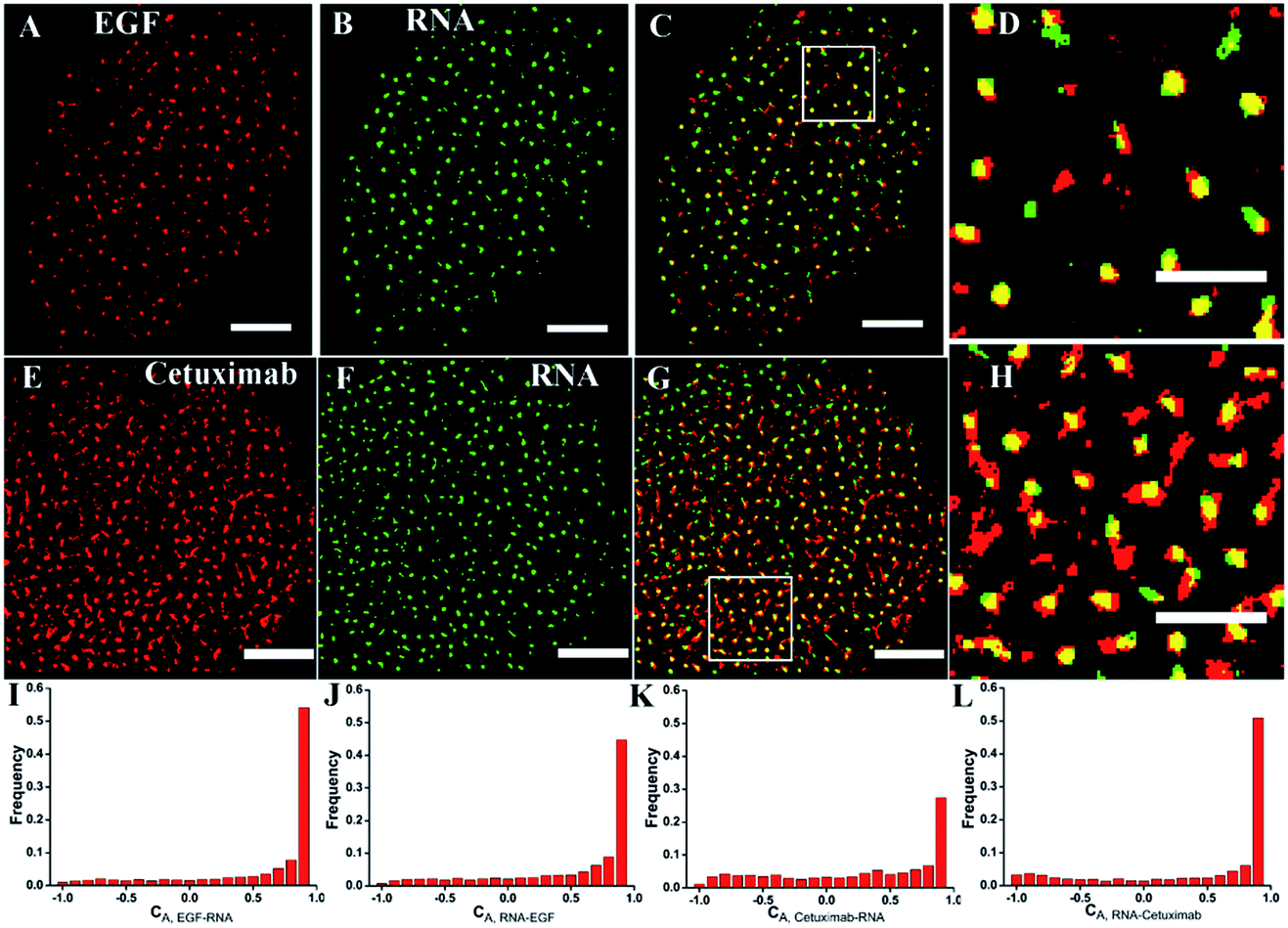 | ||
| Fig. 3 Dual-color dSTORM images revealing the high specificity of the EGFR aptamer. (A–D) Reconstructed dSTORM images of EGFR labeled with Alexa 647-conjugated EGF (A) and the Cy3-conjugated RNA aptamer (B) on the same cell membrane, and the merged image of the two channels (C) with a zoomed-in region (D). (E–H) dSTORM images of EGFR labeled with Alexa 647-conjugated cetuximab (E) and the Cy3-conjugated aptamer (F) on the same cell membrane, and the merged image (G) with a zoomed-in region (H). (I–L) The histograms show the distributions of the colocalization parameter CA for EGF and cetuximab labeled EGFR with respect to aptamer labeled EGFR. Scale bars are 5 μm in A–C, E–G, and 2 μm in D and H. | ||
It was noteworthy that if there was a competition between the two probes, the dual-color images could not show the accurate information of colocalization. A report has demonstrated that only at a high concentration (>1 μM) did EGF or cetuximab interfere with the aptamer binding.6 However, the concentration we used was 0.1 μM for EGF and 0.6 μM for cetuximab. Even so, to rule out the possibility of competitive labeling in experiments, we analyzed the number of total localizations, cluster amount and area under the single and double staining. There was nearly no difference of these parameters under the two conditions (Fig. 4). Almost the same staining efficiency of single and double labeling confirmed no competition between the aptamer and EGF or cetuximab at the experimental concentration. Moreover, the aptamer probes were added to samples afterwards in the dual-color imaging but localizations as many as with EGF or cetuximab were obtained (Fig. 4A and D). Taken together, we concluded that the aptamer can recognize membrane EGFR with a high specificity and accuracy even in the presence of natural ligands and antibodies.
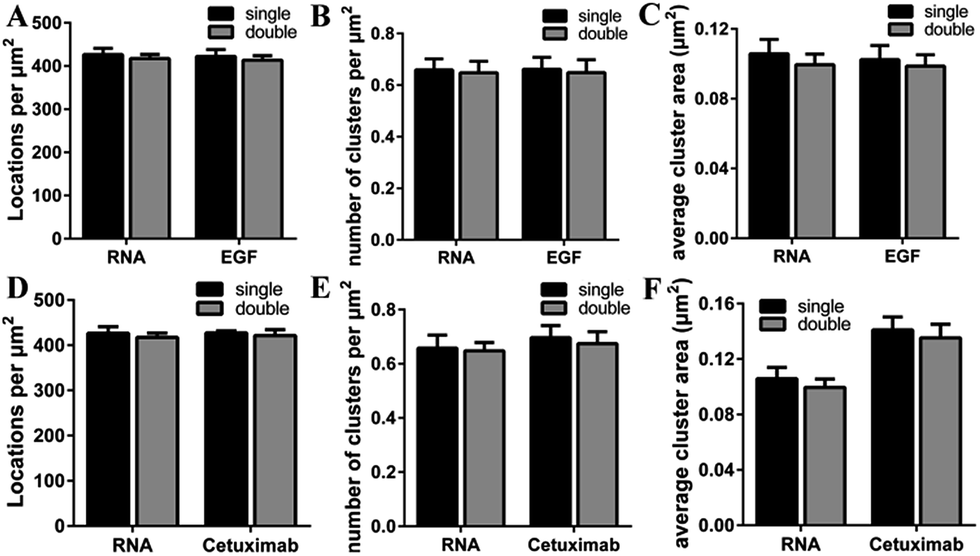 | ||
| Fig. 4 Analysis of single and double staining of EGFR on COS-7 cells with the aptamer and EGF or cetuximab. (A–C) The localization density (A), average cluster number per μm2 (B) and cluster area (C) under single labeling with the Cy3-aptamer and Cy3-EGF, and double labeling with Alexa 647–EGF and the Cy3-aptamer. (D–F) The same parameters as A–C under single labeling with the Cy3-aptamer and Cy3–cetuximab, and double labeling with Alexa 647–cetuximab and the Cy3-aptamer. Data were collected from 10 cells in 3 independent experiments (mean ± SD). | ||
Mapping the resting and stimulated EGFR with an RNA aptamer
EGFR is activated by ligand-induced dimerization,6 and the dynamic process of ligand binding has been studied by diverse methods, for example, FRET43 or universal PAINT.44 We wondered what changes the activation would bring to the EGFR distribution. Our previous study using the antibody labeling method found that the clustering characteristics of EGFR (such as size and number) were not changed after EGF stimulation.9 Nevertheless, considering the lower labeling efficiency of cetuximab which might cause the inaccuracy of clustering characteristics, we decided to employ aptamers to investigate the nanoscale organizations of activated EGFR on live COS-7 cell membranes. The serum-starved cells were first stimulated by EGF (100 ng mL−1) for 10 min, and then labeled with the aptamer or cetuximab, respectively. To guarantee the active EGFR was not internalized, we still carried out the experiment on ice.From the reconstructed dSTORM images, it was difficult to distinguish the clustering difference between active and resting EGFR either by aptamer or cetuximab labeling (Fig. 5A–D). The total localizations did not change after EGF stimulation as well (Fig. 5E), indicating a fixed number of EGFR molecules on live cell membranes either activated or not. However, the cluster number of active EGFR was a little more than that of inactive EGFR, especially when labeled with aptamers (Fig. 5F). As for the cluster area, we found that active EGFR formed larger clusters than resting ones (Fig. 5G and H), which was consistent with a previous study that used the reversible cryo-arrest method.45 The cluster size increased more significantly under aptamer labeling. Clusters with an area more than 0.12 μm2 all went up after activation. On the contrary, such clusters kept a downward tendency under cetuximab labeling except for those with an area of 0.2–0.24 μm2. Furthermore, by analyzing the molecular organization of clusters, we found that aptamer-labeled clusters with 2–3 molecules decreased and large clusters with more than 4 molecules increased after EGF stimulation, which was hardly shown in cetuximab-labeled clusters (Fig. 5I and J). Taken together, the aptamer labeling method revealed that active EGFR formed more and larger clusters containing more receptors on live cell membranes, which could not be clearly elucidated by antibody labeling. In addition, these results supported that the formation of EGFR clusters is associated with their activation. Molecules within the clusters have smaller distances, which might facilitate rapid dimerization and signal transduction.
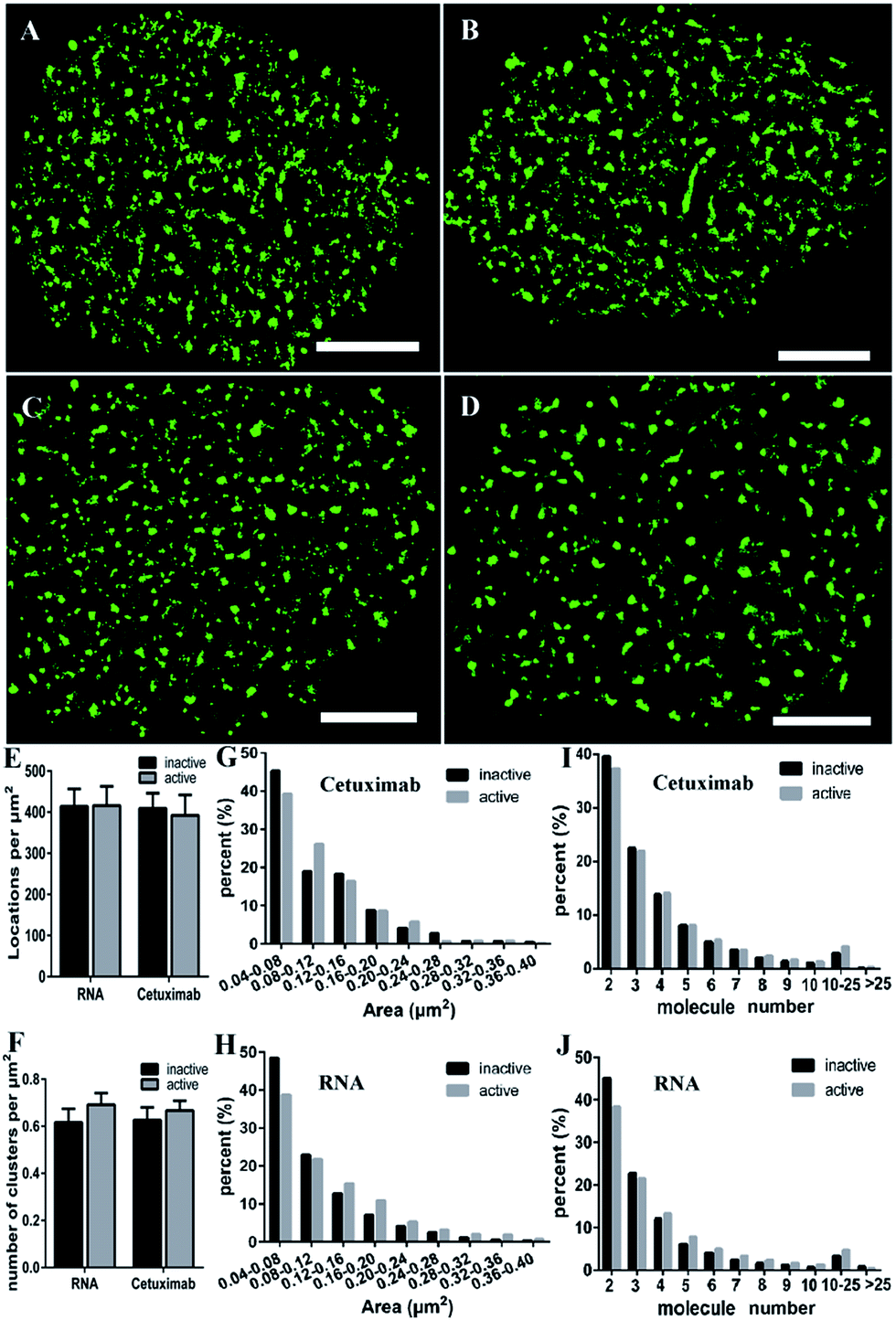 | ||
| Fig. 5 The distribution and clustering of active and resting EGFR on the serum-starved COS-7 cell membranes. (A–D) Reconstructed dSTORM images of Cy3-cetuximab labeled inactive (A) and active (B) EGFR, and Cy3-aptamer labeled inactive (C) and active (D) EGFR. Scale bars are 5 μm. (E) The number of localizations per μm2 of inactive and active EGFR with aptamer or antibody labeling. (F) The number of inactive and active EGFR clusters in unit area. (G and H) Histogram of inactive and active EGFR cluster areas labeled with cetuximab (G) or the aptamer (H). (I and J) The percentage of clusters containing different molecule numbers with cetuximab (I) or aptamer (J) labeling. Data were collected from 10 cells in three independent experiments (mean ± SD). | ||
Experimental section
Fluorescent probes
EGF (Sigma Aldrich) and cetuximab (Merck) were separately conjugated with Cy3 (Invitrogen) or Alexa 647 (Invitrogen) to act as probes for imaging as previously depicted.9,15 Briefly, different dyes were added to EGF or cetuximab solution at individual appropriate concentration, and then they were mixed and reacted at 20 °C for ∼2 hours with gentle vortexing. A PD Spin Trap G-25 filtration column (GE Healthcare) was used to collect the dye conjugated protein with a labeling ratio of ∼0.7 to 1 (dye/protein), and the excessive dye was removed.The 40-mer RNA aptamer was synthesized based on a reported sequence:6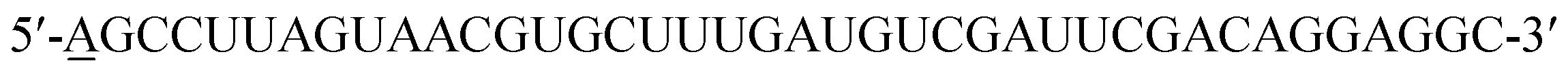 (Sangon Biotech). The 5′ end of the sequence was modified by adding one adenosine residue to which Cy3 was conjugated, and this simple modification would not affect the functional region of the aptamer. In addition, instead of 2′-F modified nucleic acids, only common nucleic acids were used for RNA synthesis.6 The 2-dimensional structure of the synthetic EGFR aptamer was predicted using the mfold web server (http://unafold.rna.albany.edu).
(Sangon Biotech). The 5′ end of the sequence was modified by adding one adenosine residue to which Cy3 was conjugated, and this simple modification would not affect the functional region of the aptamer. In addition, instead of 2′-F modified nucleic acids, only common nucleic acids were used for RNA synthesis.6 The 2-dimensional structure of the synthetic EGFR aptamer was predicted using the mfold web server (http://unafold.rna.albany.edu).
Cell culture
COS-7 cells, purchased from the Cell Bank of the Committee on Type Culture Collection of the Chinese Academy of Sciences, were cultured in Dulbecco's modified Eagle medium (DMEM, Hyclone) supplemented with 10% fetal bovine serum (FBS, Gibco) and antibiotics. Cells were maintained in a humidified environment with 5% CO2 at 37 °C. Before dSTORM imaging experiments, the cells were passaged into a dish where a cover slip was placed, and cultured for at least 24 hours.Confocal microscopy
COS-7 cells were cultured in glass bottom dishes for at least 24 hours. Then the cells were washed with warm PBS, and incubated with 5 μM DiO (Biotum) at 4 °C for 20 min. After removing the excess DiO and washing three times with PBS, cells were further stained with 0.2 μM EGFR aptamer at 4 °C for 10 min. Imaging was performed on a Leica TCS SP2 confocal microscope. Cy3-labeled EGFR was excited with a 532 nm He–Ne laser, and DiO-labeled cell membranes were excited with a 488 nm Ar–Kr laser. The fluorescence images were collected with a 100× 1.49 NA oil-immersion objective.Preparation of dSTORM samples
For imaging live cells (Fig. 1), cultured cells at ∼60% confluence were washed with warm PBS three times and stained directly with the Cy3-conjugated aptamer in 3% BSA solution at 4 °C for 10 min. The samples were kept on ice all the time during treatment and imaging. For dynasore treated cells, they were pretreated with 80 μM dynasore (Sigma-Aldrich) for 30 min to inhibit EGFR endocytosis, and then washed with PBS and stained with the EGFR aptamer as done in live cell imaging. For imaging fixed cells (Fig. 2), they were fixed with 4% PFA at room temperature for 10 min, blocked with 3% BSA for 30 min, and stained with the Cy3-conjugated aptamer, EGF or cetuximab at 4 °C for 10 min. For dual-color imaging (Fig. 3), the cells were fixed, blocked, first labeled with Alexa 647-conjugated EGF or cetuximab at 4 °C for 10 min, and subsequently stained with the Cy3-conjugated aptamer at 4 °C for 10 min. For imaging active EGFR (Fig. 5), cells were pre-cultured in serum-free DMEM medium for 24 h, and stimulated with EGF (100 ng mL−1) for 10 min. Immediately after that, they were washed three times with PBS and stained with the Cy3-conjugated aptamer or cetuximab (in 3% BSA) at 4 °C for 10 min.The labeling concentration of the Cy3-conjugated aptamer was 0.6 μM, which was determined by the concentration plot (Fig. S5†). Alexa 647 (Cy3)-conjugated EGF was 0.1 μM and cetuximab was 0.6 μM, according to a previous study.9
After staining, the cells were washed three times with PBS. Before sealing the sample, 25 μL imaging buffer containing 0.5 mg mL−1 of glucose oxidase (Sigma-Aldrich), 40 μg/mL catalase (Sigma-Aldrich) and 140 mM beta-mercaptoethanol (βME) were added.
Super-resolution imaging
dSTORM images were acquired using a Nikon Ti-E microscope (Nikon, Japan) equipped with a 100× 1.49 NA objective lens and TIRF illumination. Samples were illuminated in TIRF mode with a 532 nm (200 mW) and 640 nm laser (150 mW). For single-color imaging of EGFR, only a 532 nm laser was used to illuminate the samples. As for dual-color imaging, a 640 nm laser was first utilized to excite Alexa 647-conjugated EGF/cetuximab and then a 532 nm laser was switched on to excite the Cy3-conjugated aptamer. A dichroic mirror and an emission filter were installed in a beam splitter for illumination and an EMCCD camera (Photometrics, Cascade II) was equipped for capturing. To avoid the entering of aptamers into cells, samples were kept on ice during the whole imaging process. Cells were imaged at a 30 ms frame rate for 5000 frames. Thus, the capture time of one cell was usually less than 5 min. During this time, the sample was stabilized by a focus lock to eliminate the z-drift.The raw data were analyzed by a free available plug-in ThunderSTORM46 in Image J. First, the imaging stacks were processed with a series of feature-enhancing low-pass and band-pass filters, and the approximate molecular positions could be determined. Then, through post-processing routines to eliminate molecules with poor localization, merge molecules reappearing in subsequent frames and correct molecular positions for lateral drift, we finally obtained a reconstructed image of the precise localization of single fluorescent molecules.
Data analysis
To measure the resolution of the instrument under different labeling conditions, we recorded 5000 frames of different fluorescent probes on fixed cells at a very low concentration (∼5 nM) and analyzed the localization precision by measuring the FWHM of the Gaussian profile.47,48 As shown in Fig. S3,† the FWHM of the Cy3-conjugated aptamer, EGF and cetuximab was 29.4 ± 0.4 nm, 29.1 ± 0.2 nm and 32.2 ± 1.3 nm, respectively.The spatial distribution of EGFR on cell membranes was analyzed by Ripley's K function as in previous studies.15,36 A cell was divided into 4 parts, and a region of 3 × 3 μm2 in each part was stochastically selected. 10 cells from three independent experiments were examined. According to the function, a Ripley's K plot was first obtained. The positive values of L(r) − r in the plot indicated that molecules were significantly clustered rather than randomly distributed and the corresponding r value represented the size of the clusters in this region. Edge-effects were negated by weighting edge points and cropping image edges after calculation. Then an interpolated color map was generated by interpolating a surface plot with the L(r) value as the z-axis. Next, a binary cluster map was obtained through an appropriate L(r) threshold. For example, if the percentage of points which satisfy L(r) − r > 0 is 75%, the threshold is set as 25% of the maximum value of the L(r) from the plot. Finally, the information of cluster number, area and circularity can be extracted from the binary image using the tool of “Analyze particles” in Image J. To estimate the number of molecules in clusters, we divided the total number of localizations in the cluster by the average number of localizations in a single dye-conjugated aptamer (i.e., 33), EGF (i.e., 33) or cetuximab (i.e., 35).
For dual-color data analysis, the CBC approach of ThunderSTORM was applied to analyze the spatial association between cetuximab/EGF and aptamer recognized EGFR by calculating the colocalization values CA. CA can reach from −1 for anti-correlated, through 0 for non-correlated distributions (no colocalization), to +1 for perfectly correlated distribution (a high probability of colocalization).
Conclusions
In summary, we used an RNA aptamer to label EGFR on live cell membranes and obtained a superior dSTORM image of EGFR nanoscale clusters. Both single-labeling and dual-labeling with the aptamer, cetuximab and EGF demonstrated outstanding advantages of the aptamer in efficient recognition, high specificity and accurate information presentation. Moreover, we found that EGFR formed larger clusters with more molecules after being activated by EGF under aptamer labeling, implying that EGFR clustering may be linked to its activation. All the results suggested that aptamers can provide detailed morphological and clustering information of membrane proteins with minimal linkage error in super-resolution imaging, which is hard to find by the common antibody labeling method. Due to their small size and high affinity, we believe that aptamers will become a potential substitute for antibodies as super-resolution imaging probes in both live and fixed cells.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Key R&D Program of China (No. 2017YFA0505300 to HW) and NSFC (No. 21727816, 21525314, and 21721003 to HW; No. 21703231 to JG; No. 21503213 to MC; No. 31330082 to JJ).Notes and references
- M. Hamzeh-Mivehroud, A. Mahmoudpour and S. Dastmalchi, Chem. Biol. Drug Des., 2012, 79, 246–259 CrossRef CAS PubMed.
- A. Ahsan, D. Ray, S. G. Ramanand, A. Hegde, C. Whitehead, A. Rehemtulla, Y. Morishima, W. B. Pratt, Y. Osawa, T. S. Lawrence and M. K. Nyati, J. Biol. Chem., 2013, 288, 26879–26886 CrossRef CAS PubMed.
- E. G. Hofman, M. O. Ruonala, A. N. Bader, D. van den Heuvel, J. Voortman, R. C. Roovers, A. J. Verkleij, H. C. Gerritsen and P. M. van Bergen En Henegouwen, J. Cell Sci., 2008, 121, 2519–2528 CrossRef CAS PubMed.
- R. C. Roovers, M. J. Vosjan, T. Laeremans, R. el Khoulati, R. C. de Bruin, K. M. Ferguson, A. J. Verkleij, G. A. van Dongen and P. M. van Bergen en Henegouwen, Int. J. Cancer, 2011, 129, 2013–2024 CrossRef CAS PubMed.
- Y. Wan, D. Tamuly, P. B. Allen, Y. T. Kim, R. Bachoo, A. D. Ellington and S. M. Iqbal, Biomed. Microdevices, 2013, 15, 635–643 CrossRef CAS PubMed.
- C. L. Esposito, D. Passaro, I. Longobardo, G. Condorelli, P. Marotta, A. Affuso, V. de Franciscis and L. Cerchia, PLoS One, 2011, 6, e24071 CrossRef CAS PubMed.
- C. C. Valley, D. J. Arndt-Jovin, N. Karedla, M. P. Steinkamp, A. I. Chizhik, W. S. Hlavacek, B. S. Wilson, K. A. Lidke and D. S. Lidke, Mol. Biol. Cell, 2015, 26, 4087–4099 CrossRef CAS PubMed.
- A. Abulrob, Z. Lu, E. Baumann, D. Vobornik, R. Taylor, D. Stanimirovic and L. J. Johnston, J. Biol. Chem., 2010, 285, 3145–3156 CrossRef CAS PubMed.
- J. Gao, Y. Wang, M. Cai, Y. Pan, H. Xu, J. Jiang, H. Ji and H. Wang, Nanoscale, 2015, 7, 2511–2519 RSC.
- J. L. Werbin, M. S. Avendano, V. Becker, R. Jungmann, P. Yin, G. Danuser and P. K. Sorger, Sci. Rep., 2017, 7, 12150 CrossRef PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–795 CrossRef CAS PubMed.
- S. Nanguneri, B. Flottmann, F. Herrmannsdorfer, K. Thomas and M. Heilemann, Microsc. Res. Tech., 2014, 77, 510–516 CrossRef CAS PubMed.
- R. Henriques and M. M. Mhlanga, Biotechnol. J., 2009, 4, 846–857 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- J. Gao, F. Wang, Y. Liu, M. Cai, H. Xu, J. Jiang and H. Wang, Sci. Rep., 2015, 5, 9045 CrossRef CAS PubMed.
- J. Gao, J. Chen, M. Cai, H. Xu, J. Jiang, T. Tong and H. Wang, Methods Appl. Fluoresc., 2017, 5, 024004 CrossRef PubMed.
- S. van de Linde, S. Aufmkolk, C. Franke, T. Holm, T. Klein, A. Loschberger, S. Proppert, S. Wolter and M. Sauer, Chem. Biol., 2013, 20, 8–18 CrossRef CAS PubMed.
- E. Klotzsch, A. Smorodchenko, L. Lofler, R. Moldzio, E. Parkinson, G. J. Schutz and E. E. Pohl, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 130–135 CrossRef CAS PubMed.
- F. Opazo, M. Levy, M. Byrom, C. Schafer, C. Geisler, T. W. Groemer, A. D. Ellington and S. O. Rizzoli, Nat. Methods, 2012, 9, 938–939 CrossRef CAS PubMed.
- Y. Chen, M. Gu, P. W. Gunning and S. M. Russell, Histochem. Cell Biol., 2016, 146, 255–266 CrossRef CAS PubMed.
- G. Zhang, S. Zheng, H. Liu and P. R. Chen, Chem. Soc. Rev., 2015, 44, 3405–3417 RSC.
- M. Fernandez-Suarez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef CAS PubMed.
- J. Ries, C. Kaplan, E. Platonova, H. Eghlidi and H. Ewers, Nat. Methods, 2012, 9, 582–584 CrossRef CAS PubMed.
- T. Matsubara, R. Otani, M. Yamashita, H. Maeno, H. Nodono and T. Sato, Biomacromolecules, 2017, 18, 355–362 CrossRef CAS PubMed.
- C. Uttamapinant, J. D. Howe, K. Lang, V. Beranek, L. Davis, M. Mahesh, N. P. Barry and J. W. Chin, J. Am. Chem. Soc., 2015, 137, 4602–4605 CrossRef CAS PubMed.
- P. Zhang, N. Zhao, Z. Zeng, Y. Feng, C. H. Tung, C. C. Chang and Y. Zu, Lab. Invest., 2009, 89, 1423–1432 CrossRef CAS PubMed.
- M. Reth, Nat. Immunol., 2013, 14, 765–767 CrossRef CAS PubMed.
- S. Hui, Wensi Cui, Xiaoxiao He, Qiuping Guo, Kemin Wang, Xiaosheng Ye and Jinlu Tang, PLoS One, 2013, 8, 1–8 Search PubMed.
- T. Mairal, V. C. Ozalp, P. Lozano Sanchez, M. Mir, I. Katakis and C. K. O'Sullivan, Anal. Bioanal. Chem., 2008, 390, 989–1007 CrossRef CAS PubMed.
- A. Chen and S. Yang, Biosens. Bioelectron., 2015, 71, 230–242 CrossRef CAS PubMed.
- H. Ilkhani, M. Sarparast, A. Noori, S. Zahra Bathaie and M. F. Mousavi, Biosens. Bioelectron., 2015, 74, 491–497 CrossRef CAS PubMed.
- S. Y. Toh, M. Citartan, S. C. Gopinath and T. H. Tang, Biosens. Bioelectron., 2015, 64, 392–403 CrossRef CAS PubMed.
- H. Shi, Z. Tang, Y. Kim, H. Nie, Y. F. Huang, X. He, K. Deng, K. Wang and W. Tan, Chem. Asian J., 2010, 5, 2209–2213 CrossRef CAS PubMed.
- K. H. Roh, B. F. Lillemeier, F. Wang and M. M. Davis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1604–E1613 CrossRef CAS PubMed.
- D. M. Owen, C. Rentero, J. Rossy, A. Magenau, D. Williamson, M. Rodriguez and K. Gaus, J. Biophotonics, 2010, 3, 446–454 CrossRef CAS PubMed.
- J. Wu, J. Gao, M. Qi, J. Wang, M. Cai, S. Liu, X. Hao, J. Jiang and H. Wang, Nanoscale, 2013, 5, 11582–11586 RSC.
- Y. Wang, J. Gao, X. Guo, T. Tong, X. Shi, L. Li, M. Qi, Y. Wang, M. Cai, J. Jiang, C. Xu, H. Ji and H. Wang, Cell Res., 2014, 24, 959–976 CrossRef CAS PubMed.
- H. Al-Akhrass, T. Naves, F. Vincent, A. Magnaudeix, K. Durand, F. Bertin, B. Melloni, M. O. Jauberteau and F. Lalloue, Nat. Commun., 2017, 8, 1182 CrossRef PubMed.
- K. Montagne, H. Uchiyama, K. S. Furukawa and T. Ushida, J. Biomech., 2014, 47, 354–359 CrossRef PubMed.
- D. Oommen, N. J. Dodd, D. Yiannakis, R. Moyeed and A. N. Jha, Mutat. Res., 2016, 809, 43–49 CAS.
- M. A. Gomes de Castro, C. Hobartner and F. Opazo, PLoS One, 2017, 12, e0173050 CrossRef PubMed.
- S. Malkusch, U. Endesfelder, J. Mondry, M. Gelleri, P. J. Verveer and M. Heilemann, Histochem. Cell Biol., 2012, 137, 1–10 CrossRef CAS PubMed.
- H. Yamashita, Y. Yano, K. Kawano and K. Matsuzaki, Biochim. Biophys. Acta, 2015, 1848, 1359–1366 CrossRef CAS PubMed.
- P. Winckler, L. Lartigue, G. Giannone, F. De Giorgi, F. Ichas, J. B. Sibarita, B. Lounis and L. Cognet, Sci. Rep., 2013, 3, 2387 CrossRef PubMed.
- M. E. Masip, J. Huebinger, J. Christmann, O. Sabet, F. Wehner, A. Konitsiotis, G. R. Fuhr and P. I. H. Bastiaens, Nat. Methods, 2016, 13, 665–672 CrossRef CAS PubMed.
- M. Ovesny, P. Krizek, J. Borkovec, Z. Svindrych and G. M. Hagen, Bioinformatics, 2014, 30, 2389–2390 CrossRef CAS PubMed.
- B. F. Lillemeier, M. A. Mortelmaier, M. B. Forstner, J. B. Huppa, J. T. Groves and M. M. Davis, Nat. Immunol., 2010, 11, 90–96 CrossRef CAS PubMed.
- K. Notelaers, N. Smisdom, S. Rocha, D. Janssen, J. C. Meier, J. M. Rigo, J. Hofkens and M. Ameloot, Biochim. Biophys. Acta, 2012, 1818, 3131–3140 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00143j |
| This journal is © The Royal Society of Chemistry 2019 |