
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper relay path through the N-terminus of Wilson disease protein, ATP7B†
Kumaravel Ponnandai
Shanmugavel
and
Pernilla
Wittung-Stafshede
 *
*
Department of Biology and Biological Engineering, Chalmers University of Technology, 412 96 Gothenburg, Sweden. E-mail: Pernilla.wittung@chalmers.se
First published on 10th July 2019
Abstract
In human cells, copper (Cu) ions are transported by the cytoplasmic Cu chaperone Atox1 to the Wilson disease protein (ATP7B) in the Golgi for loading of Cu-dependent enzymes. ATP7B is a membrane-spanning protein which, in contrast to non-mammalian homologs, has six cytoplasmic metal-binding domains (MBDs). To address the reason for multiple MBDs, we introduced strategic mutations in which one, two or three MBDs had been blocked for Cu binding via cysteine-to-serine mutations (but all six MBDs are present in all) in a yeast system that probes Cu flow through Atox1 and ATP7B. The results, combined with earlier work, support a mechanistic model in which MBD1-3 forms a regulatory unit of ATP7B Cu transport. Cu delivery via Atox1 to this unit, followed by loading of Cu in MBD3, promotes release of inhibitory interactions. Whereas the Cu site in MBD4 can be mutated without a large effect, an intact Cu site in either MBD5 or MBD6 is required for Cu transport. All MBDs, expressed as single-domain proteins, can replace Atox1 and deliver Cu to full-length ATP7B. However, only MBD6 can deliver Cu to truncated ATP7B where all six MBDs are removed, suggesting a docking role for this structural unit.
Significance to metallomicsCopper is an essential metal ion facilitating biological function but can also promote human disease. To avoid toxicity of free copper ions, there are protein-based transport systems that deliver the metal to target proteins. Many questions around human copper transport proteins remain unanswered. We here used a yeast system that probes copper flow through the human copper transport proteins Atox1 and ATP7B to investigate how Cu is shuttled among ATP7B's unique set of six metal-binding domains. The yeast assay can be expanded to probe many mechanistic questions around copper transport and for investigations of human diseases involving Cu misbalance. |
Introduction
Copper (Cu) ions in oxidized and reduced forms are found in the active sites of many essential proteins that participate in key cellular reactions.1–3 However, free Cu ions are potentially toxic for cells since, due to their redox activity, they may produce reactive oxygen species.4 Therefore, the intracellular concentration of Cu is regulated via dedicated proteins that facilitate uptake, efflux and distribution of Cu (in its reduced form) to Cu-dependent proteins and enzymes.5–7 In the human cytoplasm, after the uptake of Cu by Ctr1,8 the Cu chaperone Atox1 transports the metal to ATP7A and ATP7B (also called Menke's and Wilson disease proteins, respectively), two homologous P1B-type ATPases located in the trans-Golgi network. Once transferred to ATP7A/B, the Cu ion is channeled through the protein to the lumen of the Golgi where it is loaded onto specific Cu-dependent proteins and enzymes. Cu distribution within cells is thought to be driven by gradients of increasing copper-binding affinity in combination with protein–protein specific recognition.9ATP7A/B are multi-domain proteins with several domains protruding in the cytoplasm as well as membrane-spanning parts that harbor a channel for Cu. Unique to ATP7/B are six cytoplasmic metal-binding domains (MBDs; named MBD1 to MBD6, with MBD1 being the first from the N-terminus and MBD6 the domain closest to the membrane-spanning part of ATP7A/B) connected by peptide linkers of various lengths constituting the N-terminal tail.10 Each MBD in ATP7A/B, as well as Atox1, has a ferredoxin-like α/β fold and a surface-exposed invariant CXXC motif (X = any residue) in which a single Cu can bind to the cysteine sulfurs. In contrast to the human ATP7A/B, the corresponding bacterial and yeast P1B-type ATPases have only one or two MBDs. The reason for the presence of multiple MBDs in ATP7A/B has been proposed to involve regulation of Cu transfer to the Golgi lumen and Cu-mediated protein trafficking between the Golgi and the plasma membrane.11–14 Since there is no high-resolution structural information on the arrangement of the six MBDs within full length ATP7A/B, it is unclear how these domains are arranged relative to each other at different stages of the catalytic cycle. Atox1 can deliver Cu to all the MBDs in vitro,15–21 through direct protein–protein interactions.22,23 It is unclear what MBD(s) receives Cu from Atox1 in vivo although recent data suggest MBD2 as the primary target.24 Using Cu transport assays in vitro, it has been shown that several MBDs can be deleted or mutated without loss of Cu transport activity, but the presence of at least one MBD appears to be required.14,25 We have identified various domain–domain interactions in ATP7B through in vitro and in silico analyses of multi-MBD constructs26–28 and recent NMR work beautifully showed that MBD1-3 (in the absence of Cu) adopted a compact unit that could interact (at least transiently) with other cytoplasmic ATP7B domains.13,24 Upon Cu delivery by Atox1 to MBD1-3, the dynamic restrictions on the unit were released, in accord with a role in Cu-dependent regulation.24,29
Here we used our previously described yeast system25 that probes Cu transport via Atox1 and ATP7B through growth on iron-limited media to address the Cu relay path among the MBDs in ATP7B. In contrast to our previous study, where MBDs were deleted from the N-terminus one by one, we here introduced Cu site Cys-to-Ser mutations to retain the domains but only remove the Cu-binding ability. We also tested the ability of the MBDs, when expressed as individual domains, to replace Atox1. Our current results, integrated with previous data,24,25 are used to propose a mechanistic model for how Cu is transported along the MBDs.
Results
The complementation assay builds on the fact that yeast requires Cu as a cofactor in Fet3p to survive in iron-limited conditions. Fet3p oxidizes ferrous to ferric iron ions, which are then taken up by the transmembrane permease Ftr1.14,30,31 Fet3p obtains Cu from the pathway involving the yeast Atox1 homolog, Atx1p, and the yeast ATP7A/B homolog, CCC2p. In the Δccc2Δatx1 double knockout yeast strain (without Atx1p and CCC2p) there is no functional Cu transport and, therefore, there is a lack of high-affinity iron uptake and the yeast cells grow poorly. By supplementing the Δccc2Δatx1 strain with the human homologs of Atx1p and CCC2p on high-copy plasmids, Cu transport can be restored, which facilitates iron uptake and improves cell growth.25 Earlier work has demonstrated that under these conditions, Cu transport limits yeast growth.14,31–33 Here, the Δccc2Δatx1 yeast strain supplemented with the human ATOX1 gene was used as the background in most experiments, to which high-copy plasmids with mutated ATP7B proteins were introduced. For all ATP7B variants tested, the yeast growth rate during the exponential phase was measured (data shown in Fig. S1, ESI†) and, like previously,25 used as an estimate of the Cu transport. Although the Cu transport efficiency relates directly to yeast growth, it is unclear which step(s) within the processes of Cu transfer from Atox1 to ATP7B, within ATP7B, and then to Fet3p, is rate limiting under the various conditions.The single, double, and triple Cu-site mutations introduced into the six MBDs of ATP7B are listed in Table 1. In all cases were the two cysteines in the CXXC motif exchanged to serines in order to block Cu binding. All mutated ATP7B variants were checked for the protein expression level (examples in Fig. S2, ESI†) to assure that alterations in yeast growth could be interpreted in terms of protein function.
| No | Mutant abbreviation | MBD1 | MBD2 | MBD3 | MBD4 | MBD5 | MBD6 |
|---|---|---|---|---|---|---|---|
| 1 | 1CS | X | |||||
| 2 | 2CS | X | |||||
| 3 | 3CS | X | |||||
| 4 | 4CS | X | |||||
| 5 | 5CS | X | |||||
| 6 | 6CS | X | |||||
| 7 | 123CS | X | X | X | |||
| 8 | 12CS | X | X | ||||
| 9 | 13CS | X | X | ||||
| 10 | 23CS | X | X | ||||
| 11 | 456CS | X | X | X | |||
| 12 | 56CS | X | X | ||||
| 13 | 46CS | X | X | ||||
| 14 | 45CS | X | X |
| No | Mutant abbreviation | MBD1 | MBD2 | MBD3 | MBD4 | MBD5 | MBD6 |
|---|---|---|---|---|---|---|---|
| 1 | ATP7B | ||||||
| 2 | DEL1-3 | X | X | X | |||
| 3 | DEL1-6 | X | X | X | X | X | X |
Single or triple Cu site mutated ATP7B variants
The data show that if the Cu sites in each domain are mutated individually, with the presence of the Cu sites in all other domains, the effect on the overall Cu transport is limited (Fig. 1A). The largest effects on Cu transport found for individual domain mutations of Cu sites were found for MBD3 and MBD6. In contrast, if all three Cu sites in MBD1-3 are mutated, full Cu transport activity remains, whereas mutation of all three Cu sites in MBD4-6 results in no Cu transport at all (Fig. 1B and C). The former results show that MBD1-3 are not needed for full activity but may instead regulate it (i.e., slow it down). The latter result shows that one or more of the MBDs closest to the membrane spanning part (MBD4-6) are essential for Cu transport, as proposed earlier by yeast complementation experiments.14 To elucidate further details, we divided the six domains into two sets, MBD1-3 and MBD4-6, and introduced all combinations of pairs of Cu site mutated domains within the two sets while retaining the Cu sites in the other three MBDs (Table 1).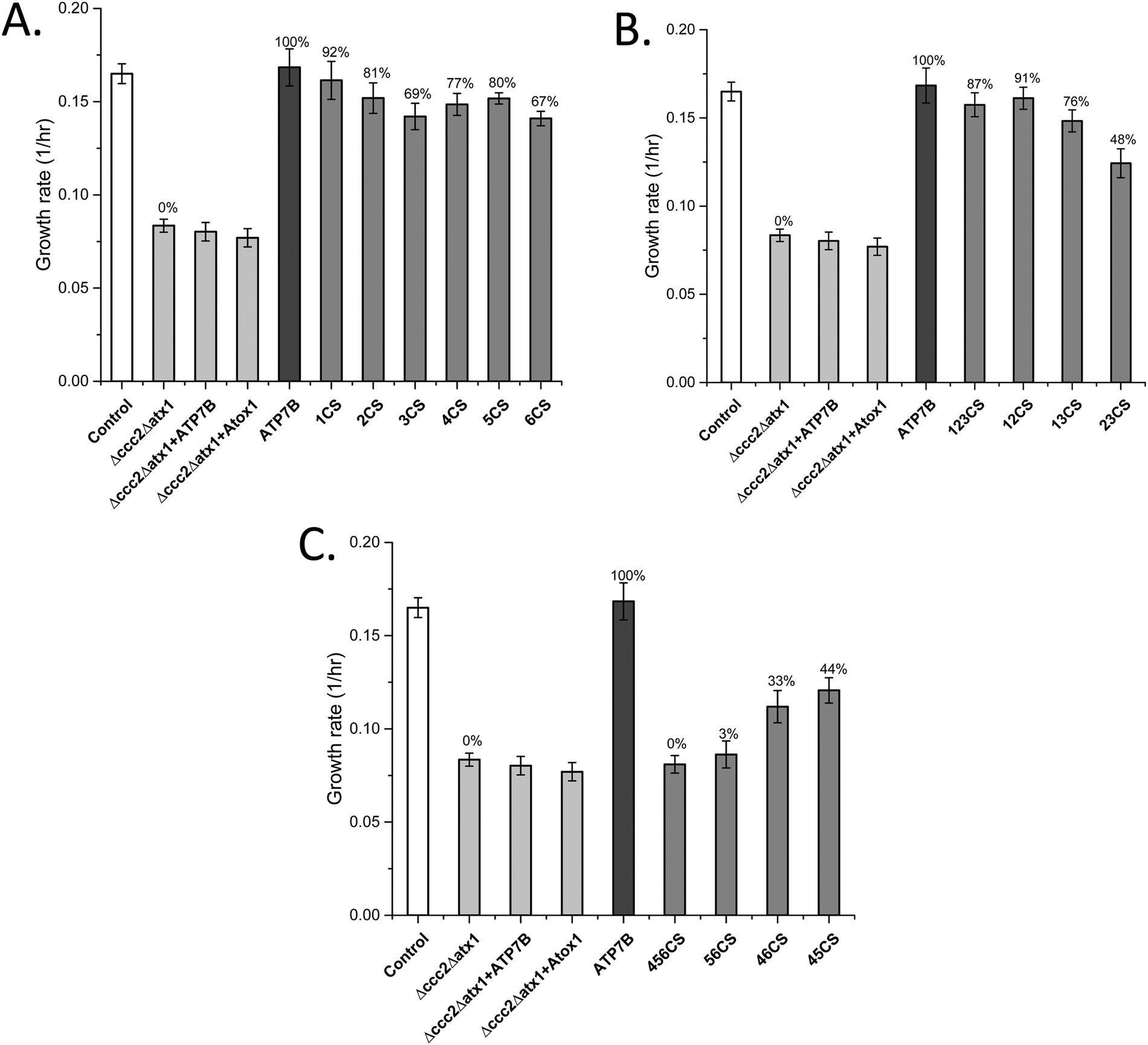 | ||
| Fig. 1 Growth rates of yeast expressing ATP7B with mutated MBDs (Table 1) as indicated in the Δccc2Δatx1 yeast strain complemented with Atox1 plasmid under iron-limited conditions. Error bars are based on standard deviations calculated for quadruplicate experiments in four biological replicas. (A) ATP7B variants with individual Cu sites mutated. (B) ATP7B variants with two or three Cu sites mutated among MBD1-3. (C) ATP7B variants with two or three Cu sites mutated among MBD1-3. Controls: S. cerevisiae Cen.pk 113.11C (positive control); Δccc2Δatx1, yeast with CCC2 and ATX1 knocked out, no added plasmids (negative control); ATP7B, Δccc2Δatx1 yeast with wild-type, full-length ATP7B and Atox1 plasmids (positive control); Δccc2Δatx1 + ATP7B, Δccc2Δatx1 yeast with ATP7B plasmid and without Atox1 plasmid; Δccc2Δatx1 + Atox1, Δccc2Δatx1 yeast with Atox1 plasmid and without ATP7B plasmid. The last two conditions also serve as negative controls as both proteins (chaperone and transporter) are needed to facilitate Cu transport.25 The Cu transport efficiency for the various variants (given as % of maximal effect, i.e., the difference between positive and negative controls) is given above each bar. | ||
MBD1-3 Cu site mutation combinations
If MBD12 cannot bind Cu, Cu transport is almost as efficient as with all Cu sites present in MBD1-3. If instead MBD13 or MBD23 cannot bind Cu, with MBD23 being most dramatic, the Cu transport is much reduced (Fig. 1B). Thus, when Cu can bind to MBD3, the Cu sites in MBD12 are not needed for efficient Cu transport. However, the domains themselves are needed as truncations of MBD1 or MBD12 resulted in much slower Cu transport.25 This implies that MBD3 is directly involved in inhibitory interactions with other ATP7B domains and that MBD12 must be present to release such interactions upon Cu loading. This also agrees with the NMR data that directly showed MBD1-3 to form a cooperative unit that was dissolved by Cu loading.24MBD4-6 Cu site mutation combinations
MBD4 alone (like MBD5 or MBD6 alone) can be blocked for Cu binding without a large reduction of the Cu transport (Fig. 1A). The decrease in the Cu transport upon mutation of the Cu site in MBD4 may be due to a direct role in the Cu relay from MBD3 to MBD56 or to a structural role (as previously proposed24) that, via conformational/dynamic changes, facilitates MBD3 to reach MBD56. In contrast, upon blockage of both the MBD5 and MBD6 Cu binding sites simultaneously, no Cu transport at all was detected, although the Cu sites in MBD1-4 are present (Fig. 1C). Blocking of Cu binding in MBD4 in combination with in MBD5 or with in MBD6 still results in some Cu transport activity (Fig. 1C). Thus, the data demonstrate that an intact Cu site in either MBD5 or MBD6 is absolutely required for Cu transport. Notably, the presence of a Cu site in MBD6 (without Cu sites in MBD45) results in higher Cu transport activity than a Cu site in MBD5 (without Cu sites in MBD46) (Fig. 1C), implying that the path through MBD6 dominates over the one through MBD5 for reaching the Cu site in the membrane spanning part of ATP7B.MBDs as cytoplasmic Cu chaperones
Both Atox1 and yeast Atx1 can deliver Cu efficiently to ATP7B in our yeast system.25 To reveal if Atox1 can be replaced with the individual MBDs in ATP7B, we changed the system and expressed ATP7B in combination with each MBD as a cytoplasmic single-domain protein (expressed on high-copy plasmids) instead of Atox1 (yeast growth curves shown in Fig. S1, ESI†). For full-length ATP7B, all MBDs can replace Atox1 and transfer Cu to ATP7B (Fig. 2A). MBD2, MBD4, MBD5 and MBD6 showed Atox1-like activity whereas MBD1 and MBD3 as chaperones resulted in somewhat lower Cu transfer activity. These results show that all MBDs are well expressed as individual domains and adopt folded states capable of binding Cu. For ATP7B with deletion of MBD1-3, i.e., with MBD4-6 remaining, again all MBDs can replace Atox1 and deliver Cu, although the activity was somewhat less than with Atox1 as the Cu chaperone (Fig. 2B). If all six MBDs are deleted in ATP7B, Atox1 cannot deliver Cu to ATP7B.25 However, with MBD1-6 deleted in ATP7B, we found that MBD6 as a cytoplasmic domain was able to deliver Cu to ATP7B whereas all other MBDs failed (Fig. 2C). This result implies that the MBD6 structural unit makes specific protein–protein interactions near the Cu entry site in the ATP7B membrane part.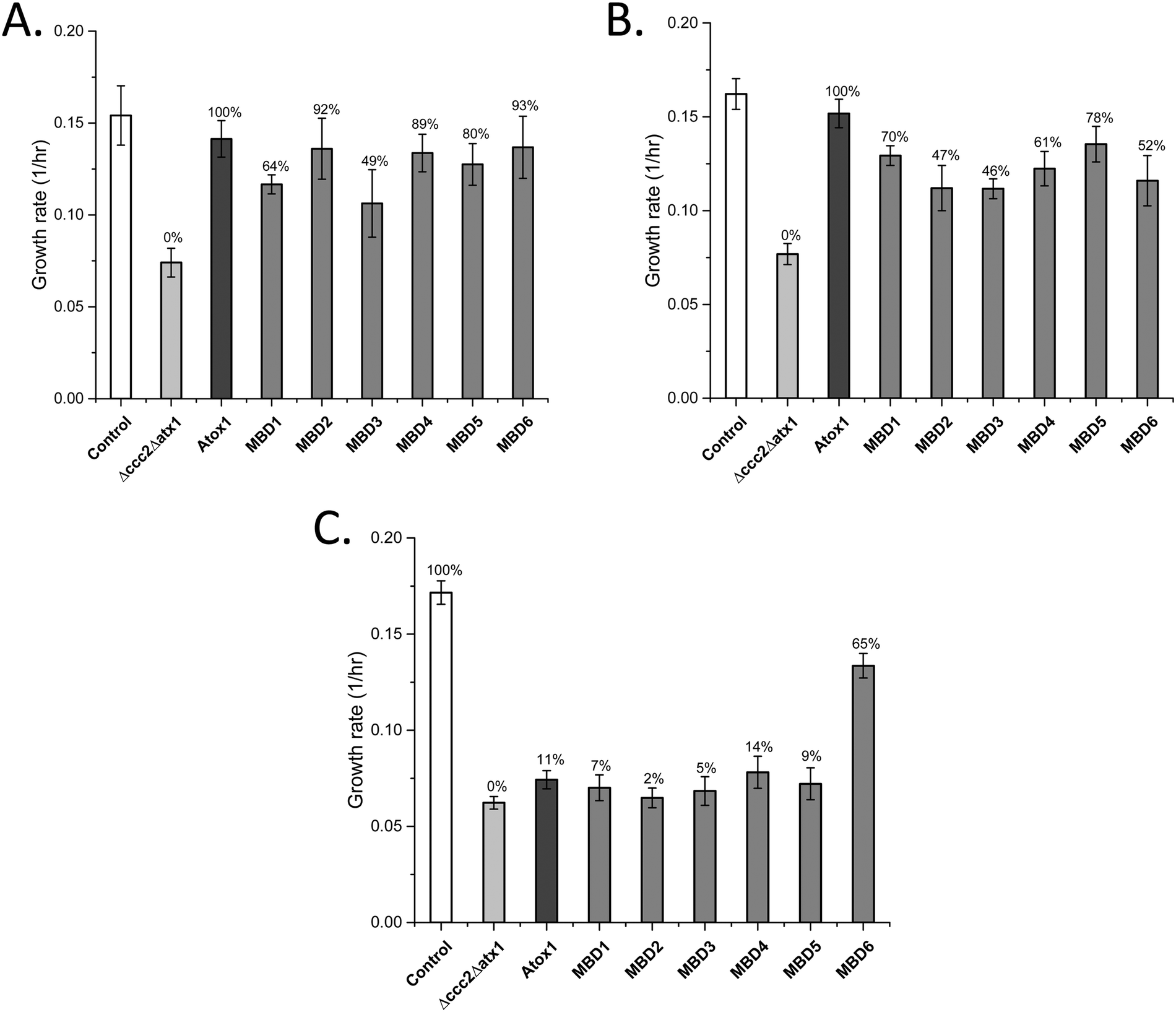 | ||
| Fig. 2 Growth rates of yeast expressing individual MBDs instead of Atox1, along with full-length or domain-truncated ATP7B variants (Table 2) as indicated in the Δccc2Δatx1 yeast strain under iron-limited conditions. Error bars are based on standard deviations calculated for quadruplicate experiments in four biological replicas. (A) Individual MBDs as chaperones and full-length ATP7B. (B) Individual MBDs as chaperones and ATP7B DEL1-3. (C) Individual MBDs as chaperones and ATP7B DEL1-6. Controls: S. cerevisiae Cen.pk 113.11C (positive control); Δccc2Δatx1, yeast with CCC2 and ATX1 knocked out, no added plasmids (negative control); Atox1, Δccc2Δatx1 yeast with ATP7B (full length in (A), DEL1-3 in (B), and DEL1-6 in (C)) and Atox1 plasmid. The control of Δccc2Δatx1 yeast with only ATP7B plasmid without Atox1 is shown in Fig. 1. Controls with only ATP7B DEL1-3 and ATP7B DEL1-6 plasmids in the absence of Atox1 in Δccc2Δatx1 yeast are reported in ref. 25 and match the data for Δccc2Δatx1 yeast without any supplemented plasmids. The Cu transport efficiency for the various combinations (given as % of maximal effect, i.e., the difference between positive and negative controls) is given above each bar. | ||
Discussion
The yeast S. cerevisiae provides an excellent model to characterize Cu transport to the secretory pathway because the proteins involved in Cu homeostasis are highly conserved from yeast to humans. Because specific Atox1 interactions appear to be involved in the Cu transport mechanism among the MBDs in ATP7B,29 our system, in contrast to previous reports,14,31–33 includes the exchange of the yeast Cu chaperone (Atx1) for the human one, i.e., Atox1, in addition to exchanging yeast CCC2 for human ATP7B. Although the MBDs have the same fold and Cu site, there are differences in biophysical properties among the six domains, such as electrostatic surface potentials, conformational dynamics and Cu-dependent structural changes, and variations in linker lengths between the domains,34–36 that all may contribute to a preferred Cu transport relay among them.Our finding that one of the Cu sites in MBD56 is essential for Cu transport is supported by our25 and others’ earlier work. It was shown that an intact Cu site in MBD6 was enough to restore Cu transport when the other MBDs were mutated31 and, in other studies, it was shown that a mutated Cu site in MBD6 but all other MBDs being functional,14 as well as with all MBDs present but only MBD5 with a functional Cu site,31 also promoted Cu transport. Because of our strategic combination of mutations, the current results (when merged with earlier findings) make possible a deepened analysis of how the six MBDs channel Cu in ATP7B, which is explained below (and illustrated in Fig. 3).
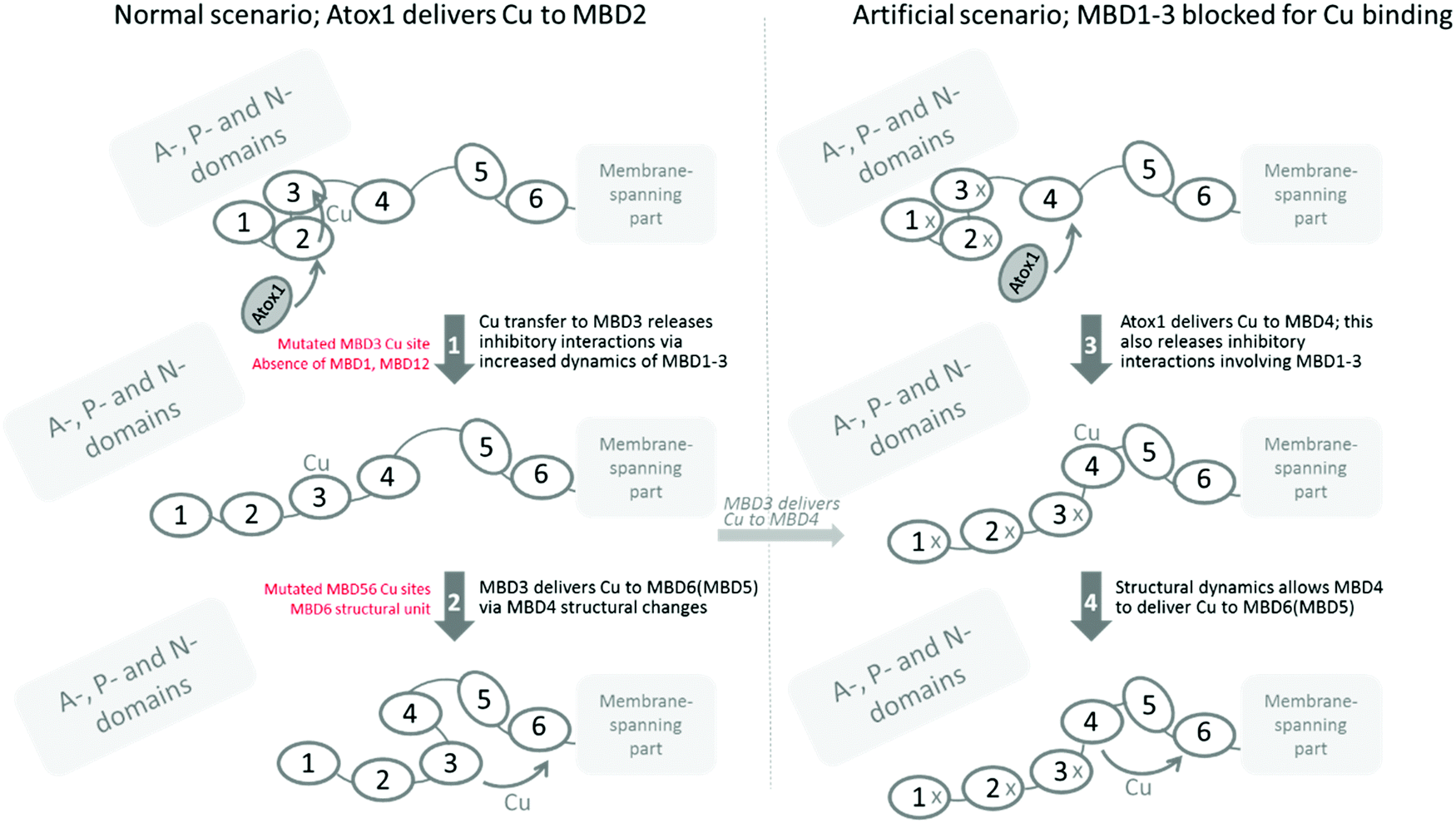 | ||
| Fig. 3 Scheme illustrating the major aspects of Cu transport among the MBDs in ATP7B deduced in this study, and taking into account others’ data. Left, normal scenario. Right, for the case where MBD1-3 have mutated Cu sites. The protein starts in an inhibitory form where MBD1-3 without Cu makes interactions with other (likely A-, P-, or N-) domains. Cu delivery by Atox1 to one of the MBD1-3 Cu sites, most likely the one in MBD2, results in further Cu transfer to MBD3. With Cu in MBD3, the domain interactions among MBD1-3 are released due to increased conformational dynamics; this relieves (inhibitory) interactions with other domains (1). This step can be blocked by mutation of Cu binding residues in MBD3 or by truncation of MBD1 or MBD12 domains. Next, conformational changes, possibly mediated by MBD4 as a spacer, allow MBD3 to deliver Cu to MBD5 or, more likely, MBD6, for Cu delivery to the membrane-spanning part (2). Mutation of the metal sites in MBD56 abolishes this step, and the MBD6 structural unit is required for correct docking at the membrane. In our artificial case, where Cu sites in MBD1-3 are mutated, we propose that Atox1 initially delivers Cu to MBD4, and Cu in MBD4 results in increased structural dynamics providing a pulling force on the MBD34 linker such that the inhibitory interactions between MBD1-3 and other domains are released (3). MBD4 would then deliver Cu directly to MBD5 or MBD6, for delivery to the Cu site in the membrane-spanning channel (4). | ||
Based on our data, when merged with the truncation data reported earlier,25 it appears that MBD1-3 forms a regulatory unit that controls Cu transport. In the absence of MBD1-3 (i.e., ATP7B with MBD1-3 deleted) there is normal Cu transport;25 in its presence, full Cu transport is only achieved if MBD3 can bind Cu. If the Cu site in MBD3 is mutated (Fig. 1B), or if the MBD1 or MBD12 domains are absent,25 the Cu transport activity becomes low. We imagine therefore that Cu is delivered to MBD2, as suggested by earlier data,24,37 and then is channeled to MBD3. Cu binding to MBD3 releases internal MBD1-3 interactions, which in turn will release MBD1-3 inhibitory interactions with other ATP7B cytoplasmic domains. This concept builds on what was proposed by Dmitriev et al.,24 but specifies that Cu must be able to reach MBD3 in order to release inhibitory interactions. Moreover, our earlier findings with truncated ATP7B (MBD1 or MBD12 absent) do not agree with the N-terminal peptide, residues 33–66 prior to MBD1,24 being responsible for the inhibitory interaction. Based on the current results, MBD3 must make physical interactions with other domains that can only be released by aid from MBD12. Still, both scenarios may be at play in vivo in the full length protein.
One puzzling piece of data is the fact that we propose an inhibitory unit of MBD1-3, but when we mutate all Cu sites in MBD1-3, we have an ATP7B protein that can transfer Cu rather well. This implies either that the Cys residues in the empty Cu sites are important for interactions within the MBD1-3 unit (e.g., transient disulfide bond formation) or, when no Cu can enter MBD1-3, instead Cu-Atox1 delivers Cu to MBD4 (which is the second most favored target after MBD217,38), and Cu placed in MBD4 also releases the MBD1-3 inhibitory interactions. For a construct with MBD4-6, it was shown by NMR that Atox1 preferred to deliver Cu to MBD4.38 That Cu-loading of MBD4 releases inhibitory interactions may be mediated by the MBD34 linker, pulling on MBD3 due to altered conformational dynamics when MBD4 becomes Cu-loaded. Notably, when Atox1 forms a Cu-dependent complex with MBD4, the entropy of the complex is increased.22 Also, with Cu in MBD4 in the full length protein as here, interactions with MBD56 may be favored, which may pull/strain the MBD34 linker. Since we can make Cys-to-Ser exchanges in two of the three domains in MBD1-3 (i.e., in MBD13 and MBD23), and still see inhibition of Cu transport, it does not appear likely that cysteine disulfides play a role in MBD1-3 internal/external interactions. The most likely scenario is thus that Atox1-mediated Cu loading of MBD4 can also release MBD1-3 inhibitory interactions. Notably, this (blockage of MBD1-3 Cu sites) would not be the normal scenario in vivo as, if possible, Atox1 would deliver Cu to MBD2 first.24,37 However, it could relate to regulation: at high Cu levels, i.e., many Cu-loaded Atox1, MBD4 may also get loaded and that could be a signal to release inhibition and begin to transfer Cu efficiently.
From MBD3, Cu may be transferred to MBD4 and then to one of MBD5 or MBD6, or MBD4 simply acts as a linker that allows MBD3 to reach MBD5 or MBD6, so Cu can be moved from one Cu site to the other via direct protein–protein interactions. It was speculated that MBD4 is a structural unit merely facilitating Cu transport between the other domains, as in the mouse and rat ATP7B homologs, MBD4 has no Cu site.29,39 Still in vitro data showed that MBD4 can deliver Cu to MBD56, with a preference for MBD6.17 Our results of somewhat lower Cu transport when the Cu site in MBD4 is mutated may be interpreted within both mechanisms. Importantly, our data clearly show that none of MBD1-4 can deliver Cu to the Cu entry site in the membrane-spanning part of ATP7B.
MBD5 or MBD6 may provide parallel routes to the Cu entry site in the membrane channel, as the Cu site in either of the two can be deleted without full loss of Cu transport. Nonetheless, for deletion of either MBD5 or MBD6 Cu sites, Cu transfer is significantly reduced, indicating that to obtain maximal Cu transport both Cu sites (MBD5 and MBD6) are needed. Also the intrinsic structural unit of MBD6 is essential, independent of the presence of the Cu site, as only MBD6 as a soluble domain (replacing Atox1) could deliver Cu to ATP7B when MBD1-6 were deleted (Fig. 2C). This implies that MBD6 makes compulsory protein–protein interactions with other parts of ATP7B that allow guiding of the Cu ion to the Cu entry site in the membrane-spanning channel. Based on an ATP7B structural model, it was proposed that attractive interactions between parts of MBD6 and the P-domain (involving residues conserved among homologs) may facilitate appropriate MBD6 positioning near the membrane Cu site.40Fig. 3 contains a schematic picture of how Cu transport among the ATP7B MBDs can be envisioned in the light of the current results.
In Archaeoglobus fulgidus, it was shown that when deleting the MBDs in the ATPase CopA the soluble Cu chaperone CopZ could deliver Cu directly to the Cu site in the membrane-spanning channel.41 This finding is similar to our results here for ATP7B, but in the human case in this study, the soluble chaperone (Atox1) and five out of the six MBDs could not do this job (with truncated ATP7B lacking the six MBDs), only MBD6. In contrast, none of the MBDs in A. fulgidus CopA, when expressed as soluble domains, could deliver Cu to CopA, either with or without MBDs present in CopA. Although here, and in the A. fulgidus study, the idea of creating soluble MBDs was to assess reaction mechanisms, it was recently shown that the CopA gene in E. coli can undergo programmed frameshifting to generate a soluble truncated protein that includes only the MBD.42 Thus, both the Cu transporter and the soluble Cu chaperone are made from the same gene in E coli. Notably, the authors suggested that similar programmed frameshifting may take place in ATP7B to generate a shorter version containing only MBD12.42
In conclusion, this study provides further support of MBD1-3 acting as a regulatory unit, in which all three domains cooperate and, upon Cu loading of MBD3, MBD1-3 interactions with each other and with other domains are formed, allowing for further Cu movement along the chain. We also show that a Cu site in either MBD5 or 6 is sufficient for Cu transport but the MBD6 structural unit is required to position the MBDs in such a way that allows Cu to move from MBD5 or MBD6 Cu sites to the metal entry site in the membrane-spanning part of ATP7B. It would be very interesting to assess how the here created variants function in ATP7B trafficking, which may be the major purpose of the MBD1-3 regulatory interactions. The six MBDs in ATP7B appear to be a system in which Nature has taken advantage of Cu-dependent variations of flexibility and conformational dynamics to fine-tune function.
Materials and methods
Yeast strains and plasmid construction
High-stress resistance yeast S. cerevisiae CEN.PK 113-11C (MATa SUC2 MAL2-8 URA3-52 HIS3-Δ1; provided by Dr P. Kötter, Institute of Microbiology, Johann Wolfgang Goethe-University, Frankfurt, German) was used as a reference strain. The yeast strains were cultivated in liquid YEPD/YPD (yeast extract peptone dextrose media) and grown at 30 °C. The yeast strain bearing CCC2 and ATX1 deletions was used in this study. For the expression of ATP7B and ATOX1, plasmids containing human ATP7B (p426GPD) and ATOX1 (p423GPD) were transformed to the yeast strain with CCC2 and ATX1 knockout (ΔCCC2ΔATX1). The detailed construction of the yeast strain and plasmids (p426GPD-ATP7B and p423GPD-ATOX1) was reported previously.25 The copper binding cysteines (CXXC) in the metal binding domains of ATP7B were mutated to serines (SXXS). Several combination of copper site mutations (CS) up to three metal binding domains were constructed. The mutations were introduced into the full-length ATP7B by a quick change site-directed mutagenesis kit using the previously constructed p426GPD-ATP7B plasmid25 as a template. The constructed ATP7B mutants are listed in Table 1. The sequences of the six ATP7B MBDs were amplified individually from the p426GPD-ATP7B plasmid with BamH1 and EcoR1 restriction sites, respectively. Next, the amplified MBD genes were inserted into the p423GPD vector (ATCC 87355; GDP, promoter for expression; His3 and ampR, markers). Construction of ATP7B DEL1-3 (deletion of MBDs 1-3) and DEL-6 (deletion of all six MBDs) plasmids was previously reported.25 All constructed plasmids were verified by sequencing (Eurofins). The DNA primers used for the plasmid construction are listed in Table S1 (Cu site mutated ATP7B variants) and Table S2 (individual MBDs as chaperones) (ESI†). All plasmids were transformed into yeast cells by the standard lithium acetate method.43Yeast complementation assay
The copper transport activity of the yeast strains was evaluated using growth curve analysis in iron limited medium. A single yeast colony from the plates was inoculated in iron limited medium (SD medium containing 1.7 g L−1 yeast nitrogen base without Fe and Cu), 50 mM MES buffer pH 6.1, 20 g L−1 glucose, 5 g L−1 ammonium sulfate, complete supplement mixture CSM–Ura–His, 1 mM ferrozine (Fe chelator), 1 μM CuSO4 and 100 μM FeSO4 and incubated overnight at 30 °C and 200 rpm.30 Yeast cells from this culture were washed with ice cold deionized water and cultivated in the fresh iron-limited medium at an initial cell density of OD600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm = 0.1. The growth of the cells was monitored using a growth profiler (Enzyscreen GP960), and growth rates were determined in the exponential phase. Growth was probed in 96-well plates (250 μL) every 30 min via green (from GP960) absorption, which was converted to OD600 equivalents. All yeast growth experiments were carried out under identical conditions and in four biological replicates. Error bars are based on standard deviations calculated for quadruplicate experiments. Statistical significance was determined by using two-tailed Student's t-test.
nm = 0.1. The growth of the cells was monitored using a growth profiler (Enzyscreen GP960), and growth rates were determined in the exponential phase. Growth was probed in 96-well plates (250 μL) every 30 min via green (from GP960) absorption, which was converted to OD600 equivalents. All yeast growth experiments were carried out under identical conditions and in four biological replicates. Error bars are based on standard deviations calculated for quadruplicate experiments. Statistical significance was determined by using two-tailed Student's t-test.
Protein extraction and Western blot
All yeast strains were grown in iron limited medium for 30 hours at 30 °C. Cells were spun down by centrifugation at 2000 × g at 4 °C for 10 min. Cells pellets were washed twice with ice-cold water and re-suspended in lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2.5 mM EDTA, 1% v/v Triton X100 and freshly added protease inhibitor). After disruption with glass beads, membranes were collected by centrifugation at 18000 × g (4 °C, 30 min). Samples were re-suspended in SDS loading buffer (0.5 M Tris–HCl, pH 6.8, 10% SDS, 0.5% (w/v) bromophenol blue, 87% glycerol, 100 mM DTT) and 50 mg of the membranes were loaded on a 4–12% Bis–Tris gel (Invitrogen) and blotted onto PVDF membranes. ATP7B and Atox1 were detected with monoclonal rabbit ATP7B and Atox1 antibodies, respectively (Abcam, 1:1000 dilution), upon incubation overnight at 4 °C. Next, blots were incubated with horse radish peroxidase conjugated anti-rabbit IgG reagent (Thermo Scientific Pierce) for 15 min, 4 °C. Bands were detected by Pierce™ fast western blot kits, SuperSignal™ West Femto, rabbit (ThermoScientific Pierce) and visualized with a BioRad ChemiDoc XRSimage analyzer.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Knut and Alice Wallenberg foundation, the Swedish Research Council and the Chalmers Foundation for funding.References
- D. L. Huffman and T. V. O’Halloran, Function, structure, and mechanism of intracellular copper trafficking proteins, Annu. Rev. Biochem., 2001, 70, 677–701 CrossRef CAS.
- S. Puig and D. J. Thiele, Molecular mechanisms of copper uptake and distribution, Curr. Opin. Chem. Biol., 2002, 6, 171–180 CrossRef CAS.
- E. D. Harris, Basic and clinical aspects of copper, Crit. Rev. Clin. Lab. Sci., 2003, 40, 547–586 CrossRef CAS.
- M. Valko, H. Morris and M. T. Cronin, Metals, toxicity and oxidative stress, Curr. Med. Chem., 2005, 12, 1161–1208 CrossRef CAS.
- T. V. O’Halloran and V. C. Culotta, Metallochaperones, an intracellular shuttle service for metal ions, J. Biol. Chem., 2000, 275, 25057–25060 CrossRef.
- R. A. Festa and D. J. Thiele, Copper: An essential metal in biology, Curr. Biol., 2011, 21, R877–R883 CrossRef CAS.
- N. J. Robinson and D. R. Winge, Copper metallochaperones, Annu. Rev. Biochem., 2010, 79, 537–562 CrossRef CAS.
- H. Ohrvik and D. J. Thiele, How copper traverses cellular membranes through the mammalian copper transporter 1, Ctr1, Ann. N. Y. Acad. Sci., 2014, 1314, 32–41 CrossRef CAS.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, T. Kozyreva, K. Zovo and P. Palumaa, Affinity gradients drive copper to cellular destinations, Nature, 2010, 465, 645–648 CrossRef CAS PubMed.
- S. Lutsenko, N. L. Barnes, M. Y. Bartee and O. Y. Dmitriev, Function and regulation of human copper-transporting ATPases, Physiol. Rev., 2007, 87, 1011–1046 CrossRef CAS.
- E. S. LeShane, U. Shinde, J. M. Walker, A. N. Barry, N. J. Blackburn, M. Ralle and S. Lutsenko, Interactions between copper-binding sites determine the redox status and conformation of the regulatory N-terminal domain of ATP7B, J. Biol. Chem., 2010, 285, 6327–6336 CrossRef CAS.
- N. M. Hasan, A. Gupta, E. Polishchuk, C. H. Yu, R. Polishchuk, O. Y. Dmitriev and S. Lutsenko, Molecular events initiating exit of a copper-transporting ATPase ATP7B from the trans-Golgi network, J. Biol. Chem., 2012, 287, 36041–36050 CrossRef CAS.
- Y. Huang, S. Nokhrin, G. Hassanzadeh-Ghassabeh, C. H. Yu, H. Yang, A. N. Barry, M. Tonelli, J. L. Markley, S. Muyldermans, O. Y. Dmitriev and S. Lutsenko, Interactions between metal-binding domains modulate intracellular targeting of Cu(I)-ATPase ATP7B, as revealed by nanobody binding, J. Biol. Chem., 2014, 289, 32682–32693 CrossRef CAS PubMed.
- J. R. Forbes, G. Hsi and D. W. Cox, Role of the copper-binding domain in the copper transport function of ATP7B, the P-type ATPase defective in Wilson disease, J. Biol. Chem., 1999, 274, 12408–12413 CrossRef CAS.
- L. Banci, I. Bertini, F. Cantini, C. Massagni, M. Migliardi and A. Rosato, An NMR study of the interaction of the N-terminal cytoplasmic tail of the Wilson disease protein with copper(I)-HAH1, J. Biol. Chem., 2009, 284, 9354–9360 CrossRef CAS.
- L. Banci, I. Bertini, F. Cantini, A. C. Rosenzweig and L. A. Yatsunyk, Metal binding domains 3 and 4 of the Wilson disease protein: solution structure and interaction with the copper(I) chaperone HAH1, Biochemistry, 2008, 47, 7423–7429 CrossRef CAS.
- D. Achila, L. Banci, I. Bertini, J. Bunce, S. Ciofi-Baffoni and D. L. Huffman, Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5729–5734 CrossRef CAS PubMed.
- A. K. Wernimont, D. L. Huffman, A. L. Lamb, T. V. O’Halloran and A. C. Rosenzweig, Structural basis for copper transfer by the metallochaperone for the Menkes/Wilson disease proteins, Nat. Struct. Biol., 2000, 7, 766–771 CrossRef CAS PubMed.
- R. A. Pufahl, C. P. Singer, K. L. Peariso, S. J. Lin, P. J. Schmidt, C. J. Fahrni, V. C. Culotta, J. E. Penner-Hahn and T. V. O’Halloran, Metal ion chaperone function of the soluble Cu(I) receptor Atx1, Science, 1997, 278, 853–856 CrossRef CAS.
- L. Banci, The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction, Nat. Chem. Biol., 2006, 2, 367–368 CrossRef CAS.
- L. Banci, I. Bertini, V. Calderone, N. Della-Malva, I. C. Felli, S. Neri, A. Pavelkova and A. Rosato, Copper(I)-mediated protein-protein interactions result from suboptimal interaction surfaces, Biochem. J., 2009, 422, 37–42 CrossRef CAS.
- M. S. Niemiec, A. P. Dingeldein and P. Wittung-Stafshede, Enthalpy-entropy compensation at play in human copper ion transfer, Sci. Rep., 2015, 5, 10518 CrossRef.
- M. S. Niemiec, C. F. Weise and P. Wittung-Stafshede, In vitro thermodynamic dissection of human copper transfer from chaperone to target protein, PLoS One, 2012, 7, e36102 CrossRef CAS.
- C. H. Yu, N. Yang, J. Bothe, M. Tonelli, S. Nokhrin, N. V. Dolgova, L. T. Braiterman, S. Lutsenko and O. Y. Dmitriev, The metal chaperone Atox1 regulates the activity of the human copper transporter ATP7B by modulating domain dynamics, J. Biol. Chem., 2017, 292(44), 18169–18177 CrossRef CAS.
- K. Ponnandai Shanmugavel, D. Petranovic and P. Wittung-Stafshede, Probing functional roles of Wilson disease protein (ATP7B) copper-binding domains in yeast, Metallomics, 2017, 9, 981–988 RSC.
- A. Rodriguez-Granillo, A. Crespo and P. Wittung-Stafshede, Interdomain interactions modulate collective dynamics of the metal-binding domains in the Wilson disease protein, J. Phys. Chem. B, 2010, 114, 1836–1848 CrossRef CAS.
- L. Nilsson, J. Aden, M. S. Niemiec, K. Nam and P. Wittung-Stafshede, Small pH and salt variations radically alter the thermal stability of metal-binding domains in the copper transporter, Wilson disease protein, J. Phys. Chem. B, 2013, 117, 13038–13050 CrossRef CAS.
- T. Mondol, J. Aden and P. Wittung-Stafshede, Copper binding triggers compaction in N-terminal tail of human copper pump ATP7B, Biochem. Biophys. Res. Commun., 2016, 470, 663–669 CrossRef CAS PubMed.
- C. H. Yu, N. V. Dolgova and O. Y. Dmitriev, Dynamics of the metal binding domains and regulation of the human copper transporters ATP7B and ATP7A, IUBMB Life, 2017, 69, 226–235 CrossRef CAS.
- I. Morin, S. Gudin, E. Mintz and M. Cuillel, Dissecting the role of the N-terminal metal-binding domains in activating the yeast copper ATPase in vivo, FEBS J., 2009, 276, 4483–4495 CrossRef CAS.
- M. A. Cater, J. Forbes, S. La Fontaine, D. Cox and J. F. Mercer, Intracellular trafficking of the human Wilson protein: the role of the six N-terminal metal-binding sites, Biochem. J., 2004, 380, 805–813 CrossRef CAS.
- M. Iida, K. Terada, Y. Sambongi, T. Wakabayashi, N. Miura, K. Koyama, M. Futai and T. Sugiyama, Analysis of functional domains of Wilson disease protein (ATP7B) in Saccharomyces cerevisiae, FEBS Lett., 1998, 428, 281–285 CrossRef CAS.
- G. Hsi, L. M. Cullen, G. Macintyre, M. M. Chen, D. M. Glerum and D. W. Cox, Sequence variation in the ATP-binding domain of the Wilson disease transporter, ATP7B, affects copper transport in a yeast model system, Hum. Mutat., 2008, 29, 491–501 CrossRef CAS.
- A. Rodriguez-Granillo, A. Crespo and P. Wittung-Stafshede, Conformational dynamics of metal-binding domains in Wilson disease protein: molecular insights into selective copper transfer, Biochemistry, 2009, 48, 5849–5863 CrossRef CAS.
- Y. Hatori and S. Lutsenko, An Expanding Range of Functions for the Copper Chaperone/Antioxidant Protein Atox1, Antioxid. Redox Signaling, 2013, 19(9), 945–957 CrossRef CAS.
- C. Arioz, Y. Li and P. Wittung-Stafshede, The six metal binding domains in human copper transporter, ATP7B: molecular biophysics and disease-causing mutations, Biometals, 2017, 30, 823–840 CrossRef.
- J. M. Walker, D. Huster, M. Ralle, C. T. Morgan, N. J. Blackburn and S. Lutsenko, The N-terminal metal-binding site 2 of the Wilson's Disease Protein plays a key role in the transfer of copper from Atox1, J. Biol. Chem., 2004, 279, 15376–15384 CrossRef CAS.
- N. Fatemi, D. M. Korzhnev, A. Velyvis, B. Sarkar and J. D. Forman-Kay, NMR characterization of copper-binding domains 4–6 of ATP7B, Biochemistry, 2010, 49, 8468–8477 CrossRef CAS.
- M. J. Tsay, N. Fatemi, S. Narindrasorasak, J. R. Forbes and B. Sarkar, Identification of the “missing domain” of the rat copper-transporting ATPase, atp7b: insight into the structural and metal binding characteristics of its N-terminal copper-binding domain, Biochim. Biophys. Acta, 2004, 1688, 78–85 CrossRef CAS.
- P. Gourdon, O. Sitsel, J. Lykkegaard Karlsen, L. Birk Moller and P. Nissen, Structural models of the human copper P-type ATPases ATP7A and ATP7B, Biol. Chem., 2012, 393, 205–216 CAS.
- M. Gonzalez-Guerrero and J. M. Arguello, Mechanism of Cu+ -transporting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5992–5997 CrossRef CAS PubMed.
- S. Meydan, D. Klepacki, S. Karthikeyan, T. Margus, P. Thomas, J. E. Jones, Y. Khan, J. Briggs, J. D. Dinman, N. Vázquez-Laslop and A. S. Mankin, Programmed Ribosomal Frameshifting Generates a Copper Transporter and a Copper Chaperone from the Same Gene, Mol. Cell, 2017, 65, 207–219 CrossRef CAS.
- R. D. Gietz and R. A. Woods, Yeast transformation by the LiAc/SS Carrier DNA/PEG method, Methods Mol. Biol., 2006, 313, 107–120 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 and S2. Fig. S1 and S2. See DOI: 10.1039/c9mt00147f |
| This journal is © The Royal Society of Chemistry 2019 |