
Lipid mechanisms in hallmarks of cancer
J.
Molendijk
 ab,
H.
Robinson
ab,
Z.
Djuric
c and
M. M.
Hill
*ab
ab,
H.
Robinson
ab,
Z.
Djuric
c and
M. M.
Hill
*ab
aQIMR Berghofer Medical Research Institute, Herston, Brisbane, 4006, Australia. E-mail: michelle.hill@qimrberghofer.edu.au
bUniversity of Queensland Diamantina Institute, Faculty of Medicine, The University of Queensland, Translational Research Institute, Woolloongabba, Brisbane, 4102, Australia
cUniversity of Michigan, Ann Arbor, Michigan 48109-5930, USA
First published on 14th November 2019
Abstract
Excess adiposity is a risk factor for several cancer types. This is likely due to complex mechanisms including alterations in the lipid milieu that plays a pivotal role in multiple aspects of carcinogenesis. Here we consider the direct role of lipids in regulating well-known hallmarks of cancer. Furthermore, we suggest that obesity-associated remodelling of membranes and organelles drives cancer cell proliferation and invasion. Identification of cancer-related lipid-mediated mechanisms amongst the broad metabolic disturbances due to excess adiposity is central to the identification of novel and more efficacious prevention and intervention strategies.
 J. Molendijk | Jeffrey Molendijk obtained a PhD in biochemistry from the University of Queensland (Australia) and a Master's degree in toxicology and environmental health from the Utrecht University (the Netherlands). As a research officer at the QIMR Berghofer Medical Research Institute, Jeff investigates the roles of metabolites in diseases such as esophageal adenocarcinoma, hepatic encephalopathy and cystic fibrosis. He gained extensive experience in developing and applying mass spectrometry techniques to discover disease-related markers. Jeff is focused on developing and implementing novel bioinformatics approaches to improve or automate analytical workflows, recently publishing the lipidr R package for lipidomics data analyses. |
 H. Robinson | Harley Robinson received a Bachelor of Biomedical Science with Honors from The University of Queensland (Australia) in 2016. After experience as a research assistant, Harley commenced a PhD degree in Biomedical Science in 2017 at QIMR Berghofer Medical Research Institute (enrolled at the University of Queensland). Her current work involves the development subcellular proteomics and lipidomics methods to assess membrane remodeling underpinning oncogenic function and extracellular vesicle cargo loading. |
 Z. Djuric | Zora Djuric, PhD is a research professor at the University of Michigan. The goal of her research program is to develop dietary approaches for reducing cancer risk. This work has included both clinical trials and animal studies. She is interested in developing methodology for achieving dietary changes that target key mechanisms in the carcinogenic processes. Her current work evaluates the interplay of excess adiposity and background diet in modulating the efficacy of preventive agents. She is also working to apply the methodologies developed to elicit dietary changes in research settings to community and clinical settings. |
 M. M. Hill | Michelle Hill, PhD leads the Precision and Systems Biomedicine Laboratory at QIMR Berghofer Medical Research Institute, Brisbane, Australia. Through innovative use of mass spectrometry-based omics technologies, including the development of novel biomarker pipelines and subcellular profiling methods, her laboratory his working towards blood biomarkers for early detection of oesophageal adenocarcinoma, and discovery of novel lipid-mediated mechanisms for extracellular vesicle cargo loading. |
As early as 2002, the International Agency for Research on Cancer (IARC) recommended avoidance of weight gain, based on the evidence linking obesity to cancers of the colon, esophagus, kidney, and corpus uteri, and postmenopausal breast cancer.1 An additional eight cancer types were added to the list in 2016, with over 1000 studies considered.2 These findings are alarming in view of the obesity epidemic that has spread throughout the world,3,4 and it is imperative to understand the molecular mechanisms linking obesity to carcinogenesis.
Obesity is characterised by excess body fat that stems from excess energy intake over energy expenditure. Body fat accumulates as metabolic pathways switch from fat oxidation to provide energy to fat synthesis for storage. Epidemiological studies have evaluated the role of lipids in obesity-related cancers using dietary surveys, and blood lipid profiles to correlate specific lipid classes with cancer incidence and risk. These studies provide strong evidence for the association of obesity-linked cancers with higher dietary cholesterol5–9 and saturated fat,9–14 elevated blood LDL-cholesterol15,16 and triacylglycerol,15,17–23 and decreased HDL-cholesterol.17,22,24,25 Lipids are comprised of highly diverse molecular structures, which are now able to be measured with advanced mass spectrometry-based lipidomics techniques. Recent applications of lipidomics have shown potential to reveal novel roles of lipid species in the development and progression of many diseases and highlight the need to evaluate the roles of specific lipid species in molecular mechanisms.26–29
Here, we evaluate the current data on the multiple roles of lipids in mediating the effects of obesity on cancer, linking specific molecular mechanisms to key cancer hallmarks (Fig. 1). We propose obesity-associated membrane remodelling as a potentially modifiable mechanism in cancer.
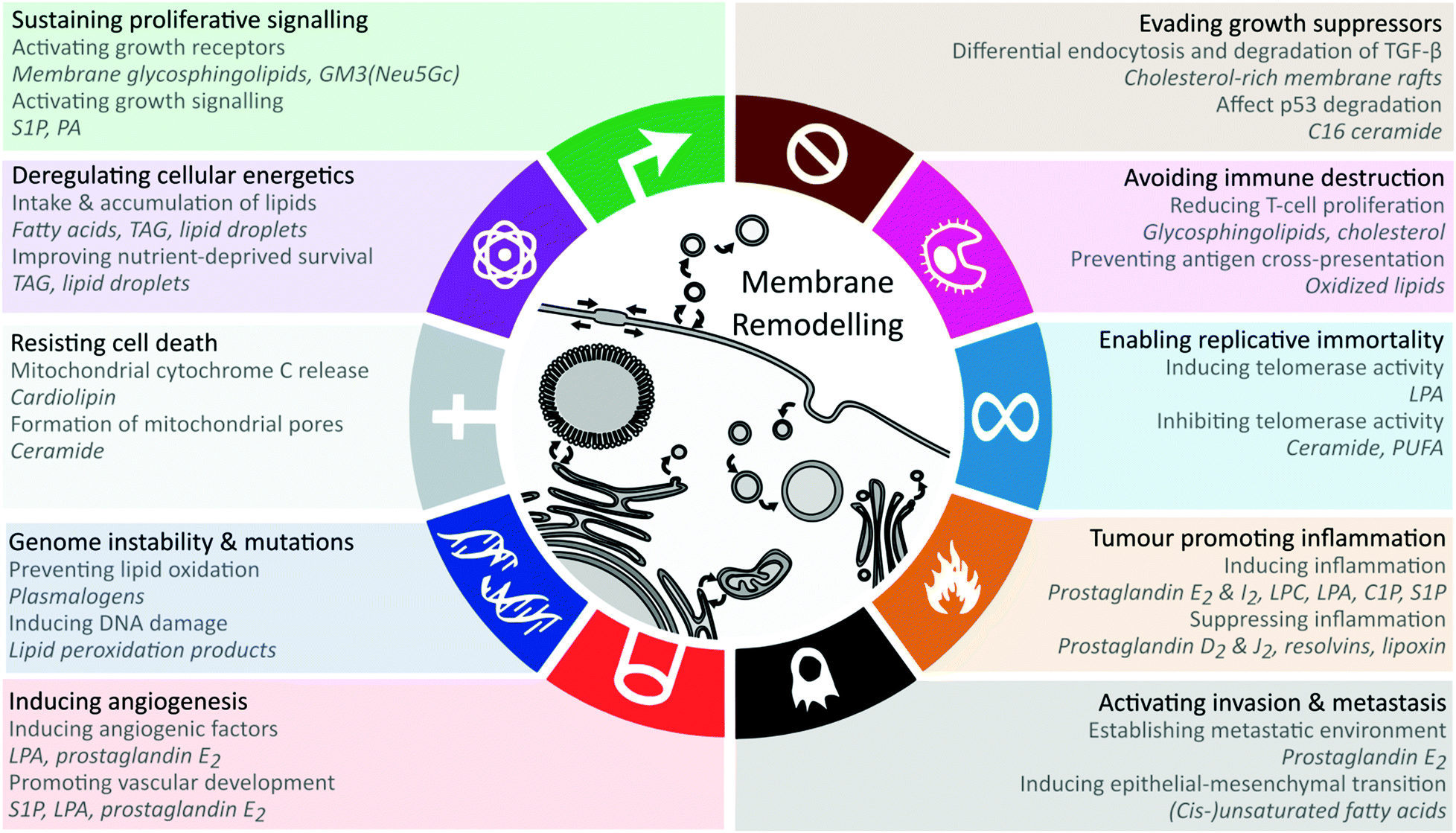 | ||
| Fig. 1 Lipid-mediated mechanisms that promote hallmarks and enabling characteristics of cancer. For each hallmark the lipid-related mechanisms are displayed in addition to the relevant lipids displayed in italic. See text for details. At the centre, membrane remodelling is depicted for the subcellular compartments, where altered membrane composition through metabolism and trafficking enables many of the hallmarks. Abbreviations: GM3 – monosialodihexosylganglioside, LPA – lysophosphatidic acid, PA – phosphatidic acid, PI – phosphatidylinositol, PIP2 – phosphatidylinositol biphosphate, PIP3 – phosphatidylinositol trisphosphate, S1P – sphingosine-1-phosphate, TAG – triacylglycerol. | ||
Derailed cellular lipidome in obesity and cancer
The cellular lipidome comprises a diverse range of compounds that are classified based on their chemical structures (Fig. 2). The LIPID MAPS structure database currently contains >43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 biologically relevant lipid structures.30 Recent advances in mass spectrometry-based lipidomics enabled the high throughput measurement of >1500 unique lipids in a single patient derived sample.31 However, even these methods do not capture the full complexity of lipids in biological samples, and novel lipids are continuing to be characterised with new methods. Broad cellular functions are known for the major lipid classes, underpinned by their biophysical properties. Glycerophospholipids are major constituents of cellular membranes, that together with sphingolipids and cholesterol modulate membrane fluidity. Cardiolipins are a structurally distinct group of glycerophospholipids with two phosphatidylglyceride backbones and a total of four acyl chains. Cardiolipins are considered signature lipids for the mitochondria, and are essential for mitochondrial functions, including apoptosis signalling.32 Sphingolipids and fatty acids also function in signal transduction, while glycerolipids primarily serve as central intermediates in glycerophospholipid synthesis or as lipid storage molecules.
000 biologically relevant lipid structures.30 Recent advances in mass spectrometry-based lipidomics enabled the high throughput measurement of >1500 unique lipids in a single patient derived sample.31 However, even these methods do not capture the full complexity of lipids in biological samples, and novel lipids are continuing to be characterised with new methods. Broad cellular functions are known for the major lipid classes, underpinned by their biophysical properties. Glycerophospholipids are major constituents of cellular membranes, that together with sphingolipids and cholesterol modulate membrane fluidity. Cardiolipins are a structurally distinct group of glycerophospholipids with two phosphatidylglyceride backbones and a total of four acyl chains. Cardiolipins are considered signature lipids for the mitochondria, and are essential for mitochondrial functions, including apoptosis signalling.32 Sphingolipids and fatty acids also function in signal transduction, while glycerolipids primarily serve as central intermediates in glycerophospholipid synthesis or as lipid storage molecules.
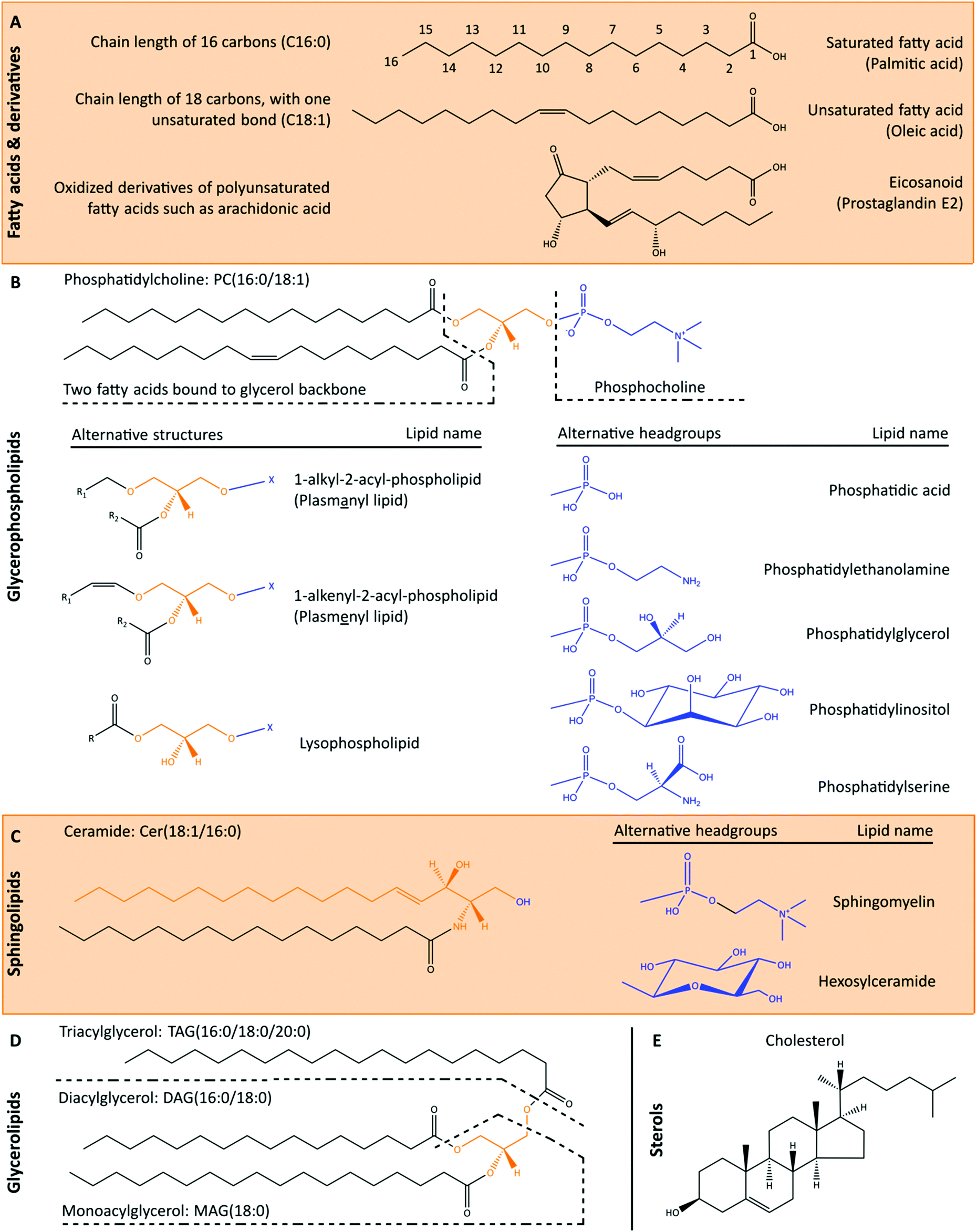 | ||
| Fig. 2 Cellular lipids can be broadly divided into fatty acids, glycerophospholipids, sphingolipids, glycerolipids and sterols. (A) Fatty acids and derivatives consist of a carboxyl group and hydrocarbon chain of varying lengths and number of unsaturated bonds which determines the three-dimensional structure of lipids and subsequently its functions and interactions. The nomenclature of lipids reflects the number of carbons and unsaturated bonds in the fatty acid chains of the molecule. (B) Glycerophospholipids contain a glycerol backbone (orange), lipid headgroup (blue) and at least one fatty acid or fatty alcohol chain. For the alternative structures, R refers to a fatty acid or fatty alcohol group, whilst X refers to the headgroup. Glycerophospholipids are major constituents of cellular membranes. (C) Sphingolipids contain a sphingoid backbone (orange), such as sphingosine or sphinganine, instead of a glycerol backbone. Sphingolipids play important roles in membrane fluidity and cell signalling. Ceramides contain a sphingoid backbone and a fatty acid chain but lack a polar headgroup, and function as diffusible signalling lipids. Sphingomyelins have a similar structure to ceramides but also contain a phosphocholine headgroup. A structurally distinct group of sphingolipids are the glycosphingolipids, which consists of a ceramide molecule bound to a sugar residue. A glycosphingolipid containing a single hexose such as glucose or galactose can be referred to as glucosylceramide or galactosylceramide, respectively. Glycosphingolipids containing more complex sugar structures and a sialic acid residue are called gangliosides, of which GM2, GM3, GD2 and GD3 are well-known examples. (D) Glycerolipids contain a glycerol backbone with at least one fatty acid chain but lack a headgroup, and generally function as central intermediates in glycerophospholipid synthesis or as lipid storage molecules. (E) Sterols are classified as a subgroup of the steroid molecules bound to a hydroxyl group. The unique rigid steroid structure of cholesterol disrupts the interaction between fatty acid chains to permit many biophysical roles, rather than relying on head group or fatty acid chains for function. Furthermore, the hydroxyl group of sterols can be esterified, forming sterol esters such as cholesteryl ester, which is commonly stored in lipid droplets. | ||
While the broad functions of lipid classes are known, minor differences in lipid structures can lead to changes in biophysical and functional properties. For example, ceramides with long-chain fatty acids are reported to have pro-apoptotic properties, whilst very long-chain fatty acid-containing ceramides are anti-apoptotic.33 The presence of double bonds (unsaturation) in fatty acid side chains alters the three-dimensional structure of lipids (Fig. 2A), and consequently their functions. With advancements in mass spectrometry-based lipidomics in the past decade, the ability to measure individual lipid species has increased our knowledge on the quantitative changes of specific lipids. These bulk lipidomics measurements have been further evaluated in the cellular context, revealing that obesity leads to broad derangements in the organelle composition and membrane dynamics of many cell types.
Organelle function
Altered lipid homeostasis leads to changes in organelle lipid composition which can impact their size, shape, subcellular distribution and function. While the functions of several organelles have been reported to be de-regulated in obesity, further studies on other organelles are required.Lipid droplets are dynamic cellular organelles primarily comprised of a neutral lipid core of triacylglycerols and cholesteryl esters within a phospholipid monolayer. As lipid storage organelles, it is no surprise that obesity leads to the increase in size and number of lipid droplets in multiple tissues.34 Lipid droplet composition is modified by high-fat diets, with significant enrichment of sphingomyelin and diacylglycerol, as well as alterations in unsaturated lysophosphatidylcholines (LPC 20:4, 22:5 and 22:6 increase while LPC 16:0 and 18:0 decrease) and unsaturated phosphatidylcholines (PC 18:0/20:4, 40:5, 18:0_22:6 and 38:5 increase, while PC 16:0/18:2, PC 32:0 and PC 32:1 decrease).35,36 However, lipid droplets are also an active site for the production of eicosanoids that act as lipid messengers with roles in chronic inflammation and immunosuppression.37,38 Diet-induced increases in certain diacylgylcerols containing arachidonic acid (20:4, omega-6) are precursors for eicosanoid production, and thereby may stimulate greater eicosanoid production.35,36 Hence this is a mechanism by which obesity-induced changes in lipid droplets disrupt systemic inflammation and cellular health.
Remodelling of cardiolipin profiles in mitochondria was reported in an obese mouse model, correlating with increased oxygen consumption and reactive oxygen species (ROS) production, altered calcium signalling and energetics.39 Cholesterol concentrations in mitochondrial membranes were decreased in obese placental tissue when compared to healthy weight.40 Mitochondrial lipid changes also influence other organelle composition and function through cholesterol-rich mitochondrial-associated membranes (MAMs), which function as lipid transfer sites between mitochondria and other membranes including the endoplasmic reticulum.41 Mitochondria and peroxisomes are sites of very long-chain fatty oxidation during stress states. Interestingly, adipose tissues from overweight patients with cancer-related lymphedema show a reduction in nervonic acid (C24:1) and caprinic acid (C10:0), which are thought to trigger increased peroxisome oxidation and stress.42 This process, requiring cooperation with mitochondria, leads to changes in peroxisome morphology, ROS accumulation and activation of peroxisome proliferator-activator receptor (PPAR) pathways with well-known effects on inhibiting apoptosis, and regulating cell cycle.42,43
Endoplasmic reticulum membrane lipid composition changes observed in obesity include enrichment of saturated phosphatidylcholine, diacylglycerol species and monounsaturated fatty acids and reduction of phosphatidylethanolamine species (including PE 18:2, 18:3 and 16:0) when compared to lean subjects.44 These obesity-associated alterations lead to impaired endoplasmic reticulum function and chronic endoplasmic reticulum stress, as observed by increased transcription of ER stress markers, HERPUD1 and Derlin-2.44 Conversely, correcting the endoplasmic reticulum phospholipid abnormalities in vivo improves glucose homeostasis and reduces chronic endoplasmic reticulum stress.44
Lastly, lysosomal membranes are altered in obesity, impacting autophagy, contributing to obesity-associated inflammation,45 and deregulating cellular energetics due to autophagy enabling bioavailability of macromolecules.46 Lysosomal membranes tend to accumulate long-chain saturated phosphatidylcholine, long-chain lysobisphosphatidic acid, and ceramides in high-fat diets,46,47 leading to impaired acidification.
Lateral membrane organisation and secretory function
Membrane lipids laterally segregate into functional domains based on biophysical properties of the lipid species. Cholesterol- and sphingolipid-rich liquid-ordered membrane microdomains (termed rafts) regulate signal transduction through nanoscale spatial regulation of membrane receptors, cytoskeleton anchoring, adhesion contacts, endocytosis and membrane trafficking.48 Obesity modulates the size, abundance and composition of membrane rafts in diverse tissue types.49 Lipidomics studies indicate that excess body fat resulted in remodelling of adipose plasma membrane and raft-mediated lipid metabolism, cell adhesion and pro-inflammatory processes.50,51 Specifically, enrichment of plasmanyl phosphatidylcholine and sphingomyelins, was observed, and a decrease in PUFA-containing ether lipids likely enhanced formation of membrane rafts at the plasma membrane in obese models.50 Real-time Cell Analyzer experiments confirmed changes in cell adhesion under different dietary stimuli (high-fat versus normal) and an increase in FAT/CD36 transcription activity infers heightened inflammation in obese tissue.51 As part of the immune system, T-cell differentiation via raft-dependent pathways is altered in overweight and obese individuals compared to lean individuals.52 Increases in unsaturated phosphatidylcholines and decreases in unsaturated phosphatidylethanolamines were also observed in these T-cell membranes in obese mice, which may drive the altered function of membrane rafts.52 Similarly, B-cell activity in obese human and mouse studies is reduced and can be rectified through exposure to polyunsaturated fatty acid, docosahexaenoic acid.49 As polyunsaturated fatty acids tend to affect the interaction between fatty acid chains due to structural rigidity, and thus limit membrane raft formation,53 it likely membrane raft alterations are at the heart of these functional changes.Altered membrane rafts also influences secretory functions. Of note, obesity is associated with altered composition and action of extracellular vesicles (EVs), small vesicles which mediate long-range intercellular communications including in cancer.54 Similar to membrane rafts, EVs are enriched in cholesterol and sphingolipids. Circulating EVs from obese patients or mouse models tend to effect pathways involving Wnt/β-catenin, TGF-β, insulin signalling, monocyte activation and ROS generation, often relating to inflammatory responses.54 Lipidomics analyses of obesity-derived EVs are scarce. However the process of adipogenesis (fat cell maturation) tends to enrich EVs with fatty acids, including oleic acid, phosphatidylethanolamine, sphingomyelin and phosphatidylinositol.55 In a recent melanoma study, obese patients and mice tended to produce more adipose-derived EVs which contained fatty acid oxidation proteins, leading to an increase in fatty acid oxidation-dependent cell migration.56
A low-grade, systemic inflammatory state is associated with obesity.37,38 In adipose tissue and blood, excess energy intake shifts fatty acid metabolism towards endogenous fatty acid synthesis and storage, increasing fatty acid flux, and altering fatty acid profiles to increase saturated fatty acids and decrease the proportion of omega-3 fatty acids.57–62 The products of endogenous fatty acid synthesis are saturated fatty acids and monounsaturated fatty acids, allosteric activators of inducible cyclooxygenase-2.63,64 Obesity also increases intestinal permeability to increase exposures to LPS via trans-cellular and para-cellular routes.65,66 This sets up a local pro-inflammatory state via activation of the immune system resulting in increased production of prostaglandin E2 and other pro-inflammatory metabolites from the oxygenation of the ω-6 fatty acid arachidonic acid.67–71 For example, the production of prostaglandin E2 is significantly increased with increasing BMI in the rectal mucosa.72
Contribution of lipids to cancer hallmarks
The cellular lipid remodelling induced by obesity impacts multiple facets of physiology relevant to carcinogenesis. To distil the potential contribution of lipid remodelling in carcinogenesis, we outline the known lipid mechanisms for each of the cancer hallmarks (Fig. 1).Sustaining proliferative signalling
Hanahan & Weinberg refer to sustaining proliferative signalling as the most fundamental trait of cancer cells,73 and recent studies demonstrate the regulatory roles of lipids in proliferative signalling by modulating growth factor receptor activation or interacting with tumour suppressors.74,75 Lipids promote proliferative signalling through lateral membrane microdomain re-organisation via membrane rafts or binding and regulating protein activity. Membrane glycosphingolipids such as the ganglioside GM3 regulate lateral membrane movements via cholesterol and sphingolipid-rich membrane rafts. The lipid GM3(Neu5Gc) regulates epidermal growth factor receptor signalling, thereby directly influencing proliferative signalling.74 The signalling lipid sphingosine-1-phosphate binds to the G-protein-coupled-receptor S1PR1 to promote cell proliferation through PI3K, AKT and MAPK pathways,75 while phosphatidic acid directly interacts with the proteins Large tumour suppressor homolog 1/2 (LATS1/2) and Neurofibromin 2 (NF2) to interrupt their downstream activities and leads to the activation of the oncoprotein YAP.76 LATS, NF2 and YAP are part of the Hippo signalling pathway which regulates cell growth and tumour suppression.76 The (lysophosphatidic acid-mediated) activation of YAP has been associated with aggressive tumour phenotypes and leads to cancer progression and metastasis.77 Obesity-mediated endoplasmic reticulum remodelling can play a role in proliferative signalling as well.78Enabling replicative immortality
In contrast to normal cells, cancer cells gain the ability of unlimited replication due to telomeres protecting the ends of chromosomes.73 Lysophosphatidic acid and ceramides have been reported to regulate telomerase or human-telomerase reverse transcriptase (hTERT), thereby influencing telomere lengths and replicative immortality. Yang et al. reported that lysophosphatidic acid exposure induces the expression of hTERT and telomerase activity in ovarian cancer cells through the HIF-1α and PI3K pathways.79 However, further studies are required, as the specific lysophosphatidic acid fatty acid composition was not reported.79 Opposite effects were found for ceramides (C6 and C18-ceramides80,81) and several polyunsaturated fatty acids (eicosapentaenoic acid, C20:5 and docosahexaenoic acid, C22:6) which downregulate hTERT and inhibit telomerase activity.82 Inhibiting telomerase activity is beneficial to cancer cells, since telomere shortening induces senescence. Unfortunately, no consistent ceramide alterations are found between cancer types.83–86 Detailed reporting of the exact species (including chain compositions) or groups (patterns in chain length or unsaturation) of ceramides that are altered in a study could potentially elucidate the contrasting findings.Evading growth suppressors
The major cancer growth evasion pathways, TGF-β and p53, have been reported to be regulated by cholesterol and ceramides.87–90 Membrane rafts in bone marrow stem cells can be altered by obesity, leading to rapid differentiation and proliferation by altering raft-mediated TGF-β signalling.87 Cholesterol-rich membrane rafts regulate TGF-β signalling through differential endocytosis which either leads to TGF-β degradation or signalling.88 Conversely, cholesterol-lowering statin drugs prevent the cholesterol-mediated TGF-β degradation.88 Furthermore, cholesterol decreases TGF-β binding to TGF-β receptors, whereas statins reverse this effect.89 Since the intake of dietary cholesterol is consistently associated with obesity-associated cancers,5–9 this hallmark may be of particular relevance in prevention and intervention measures. C16 ceramide has been shown to bind to p53 and prevent its ubiquitination and degradation,90 having the potential to sustain tumour growth suppression. While C16 ceramide was reported to be elevated in a colorectal cancer study,83 more data are required for other cancers and ceramide chain lengths to establish the broader relevance of this mechanism.Deregulating cellular energetics
Although cancer metabolism research has focused on glycolysis, cancer cells also accumulate energy-rich lipids including triacylglycerol and cholesteryl esters.91 Intracellular lipid droplets serve as a source of nutrients for cancer cells and improve cell survival under normoxia and hypoxic conditions.91 Furthermore, cancer-associated adipocytes provide energy and lipids to cancer cells, contributing to cancer progression and metastasis.92–94 Co-culture of adipocytes and breast cancer cells leads to decreased PPARγ expression in adipocytes, suggesting that cancer cells promotes of adipocyte catabolism.95Cancer cells up-regulate the expression of cell surface lipases, such as lipoprotein lipase (LPL) and endothelial lipase (LIPG) to hydrolyze triglycerides from lipoproteins, releasing free fatty acids which can be taken up through fatty acid transport proteins.96,97 CD36 is the major fatty acid importer that is upregulated in cancer cells and is required to support lipid synthesis and cell proliferation.92,98–100 Although other fatty acid transport proteins share the same function, CD36 was shown to be over 30-fold more effective than FATP4 and ACSL1.101 Cells expressing high concentrations of CD36 are shown to make use of dietary lipids, whereas inhibition of CD36 impairs metastasis and is linked to improved patient survival rates.99 Zhao et al. found that CD36 expression promotes the growth of breast cancer cells through the uptake of exogenous lipids.98
The altered lipid metabolism can also contribute to cancer cachexia, in which patients rapidly lose skeletal muscle and body fat mass, accounting for up to 20% of cancer deaths.102 Increases in lipolytic activities in the white adipose tissue of cancer patients leads to the release of fatty acids and glycerol.102 Additionally, the activities of lipoprotein LPL are decreased in white adipose tissue, leading to a decreased uptake of nutrients.102 Altogether, these findings highlight a complex cancer mechanism in which adipose tissue functions as an energy source to support the rapid energy expenditure of proliferating cancer cells.
Inducing angiogenesis
Obesity-induced changes to endoplasmic reticulum function are also known to regulate angiogenic activity.78 Membrane abnormalities in the endoplasmic reticulum trigger ATF4 activation due to stimulating the unfolded protein response (UPR), which directly binds to the VEGF protomor resulting in its overexpression. Subsequent release of VEGF from the tumour or tumour-associated adipocyte will promote angiogenesis by binding to vascular endothelial growth factor (VEGF) receptor on vascular epithelial cells.78 Interestingly, VEGFR activation can trigger UPR in the recipient cells, resulting in amplification of the angiogenic signals.103Several lipids regulate the development of vasculature during embryonic development.104 Since angiogenesis plays a key role in tumour growth by providing access to nutrients and oxygen circulating in the blood, obesity-induced lipids may modulate tumour angiogenesis although supporting experimental evidence is required.
Sphingosine-1-phosphate and lysophosphatidic acid are thought to act as bioactive messenger molecules involved in angiogenesis through several mechanisms. Sphingosine-1-phosphate acts by binding to S1P receptors (S1PR1-5), with S1PR1 required for promoting vascular barrier functions and suppressing vascular sprouting.104 Similarly, lysophosphatidic acid acts a ligand for a plethora of receptors involved in angiogenesis.104 Loss of the enpp2 gene involved in the synthesis of lysophosphatidic acid leads to a lethal embryonic phenotype in mice and zebrafish as a result of vascular defects. Lysophosphatidic acid induces the production of the angiogenic factor IL-8 through interactions with lysophosphatidic acid receptors 1, 2 and 3.104 Specific LPA species were not examined in these studies, but should be evaluated in the future.
Prostaglandin E2 is shown to be an angiogenic factor in cancer which induces the production of vascular endothelial growth factor (VEGF) through a positive feedback loop, by which VEGF also induces the COX-2 mediated prostaglandin E2 production.104 Other lipid mediators such as lipoxin A4, Resolvin D1 and Resolvin E1 are anti-angiogenic factors which are produced from omega-3 fatty acids, competing with production of prostaglandin E2 from omega-6 fatty acids, inhibiting cytokine action or downregulating VEGF receptor signalling.104
Resisting cell death
Cancer cells gain the ability to resist cell death by subverting apoptosis73 through alterations of mitochondrial membrane composition and reducing pro-apoptotic lipid signalling. In addition to promoting angiogenesis as outlined above, obesity-induced endoplasmic reticulum stress may play a role in apoptosis resistance through activation of UPR-related transcription factors that modulate apoptotic proteins such as XBP1, ATF6f and ATF4.44,78The release of cytochrome c from the mitochondria leads to cell death by activating caspase proteins. Cytochrome c is retained at the mitochondrial membrane through interactions with lipid fatty acid chains of cardiolipins.105 Therefore, altering the mitochondrial membrane composition of cardiolipins can directly lead to cytochrome c mediated apoptosis.32 Increased cardiolipin concentrations were associated with aggressive prostate cancer,106,107 potentially by preventing cytochrome c-mediated apoptosis. However, not only the concentrations of mitochondrial cardiolipins are important, but also the degree of unsaturation in the fatty acid chains of cardiolipins, which affect its (three-dimensional) structure. Stearoyl-CoA desaturase-1 (SCD1) inhibition leads to a mitochondrial cardiolipin composition higher in saturated fatty acids, which subsequently causes cytochrome c release from the mitochondria.108 Furthermore, treating SCD inhibited cells with oleic acid (C18:1) prevents cancer cell death.98,108 These results suggest that increased lipid desaturation, not only through metabolic enzymes but also dietary uptake, is a mechanism of increased cancer risk by preventing cardiolipin-mediated apoptosis in cancer cells.
Sphingolipids are involved in regulating cell death through the “sphingolipid rheostat”, in which opposing sphingolipid-mediated signalling pathways determine the fate of the cell.109 Specifically, the sphingolipid profile rather than ceramide concentration per se determines the fate of the cell, with ceramides as pro-apoptotic and sphingosine-1-phosphate as pro-survival molecules.110 It is speculated that in cancer cells sphingosine kinase 1 (SPH1) activation by the RAS pathway promotes pro-survival signalling and drug resistance by indirectly depleting ceramide concentrations.111 Chemotherapy treatments commonly lead to the accumulation of ceramides, though activation of ceramide synthases or sphingomyelinase enzymes, leading to cancer cell death.111
Multiple modes of action for ceramide-induced apoptosis have been discovered.33,112–114 It has been reported that C16-ceramides create pores in the outer mitochondrial membrane, releasing pro-apoptotic proteins, whereas very long-chain (containing a C20–26 fatty acid) ceramides interfere with this mechanism.33 Activation of neutral sphingomyelinase 1 induces apoptosis by breaking down sphingomyelins into ceramides.115 Additionally, cancer cells could prevent apoptosis by metabolising ceramides into other lipids that are not involved in apoptosis.
AKT, protein kinase C (PKC) and survivin are other ceramide-mediated regulators of cell survival and apoptosis.116 For example, the activation of p38 MAPK by ceramides leads to AKT downregulation and subsequent translocation of the apoptosis regulator BAX to the mitochondria.116 Furthermore, ceramides induce the translocation of PKC to the mitochondrial membrane and promote cytochrome c release.116
Avoiding immune destruction
Immune evasion, or the ability of cancer cells to avoid immunosurveillance, has emerged as a key cancer hallmark.73 Excess glycosphingolipids and cholesterol impairs the function of several immune cell subsets, and may be a mechanism of obesity-associated carcinogenesis.117The glycosphingolipid GM3(Neu5Gc) is shown to have immune suppressive effects and induces the down-modulation of CD4 in CD4+ T lymphocytes.74 Similarly, the presence of the complex glycosphingolipids GD3 and GD2 on cancer cell membranes suppresses cytotoxic T cells.118 Furthermore, an unidentified lipid tumour antigen was found to be released by ovarian cancer cells to inhibit T cell proliferation.119 More recently, it has been reported that hypercholesterolemia reduces the production of natural killer T (NKT) cells by limiting differentiation of hematopoietic stem cells.117 Finally, accumulation of intracellular lipids and oxidized lipids in dendritic cells of cancer patients was correlated with impaired antigen-presentation and immunity.120,121 Not surprisingly, (immune) cell-type specific lipid droplet content affects the amount of eicosanoids stored in the cell121 affecting antigen presentation, chemotaxis and phagocytosis.
Tumour-promoting inflammation
Tumour-promoting inflammation is described as an enabling characteristic of cancer, affecting multiple other cancer hallmarks.73 Bioactive lipids play a central role in chronic inflammation, and are perpetuated in obesity as the lipid droplet is a major site of eicosanoid synthesis.122 Classic eicosanoids, including prostaglandin E2 and prostaglandin I2 play roles in the development of chronic inflammation by acting as cytokine amplifiers.123 Resolvins formed from omega-3 fatty acids (Resolvins D1–4 and E1–3) and also eicosanoids such as prostaglandin D2 and prostaglandin J2 act as anti-inflammatory molecules.124 The cellular actions depend on both the concentrations of pro- and anti-inflammatory molecules as well as the presence of specific receptors. In cancer cells, the pro-inflammatory pathways prevail.125 The altered eicosanoid profile in obesity further contributes to the cellular inflammatory state favourable for cancer development and growth.37,38Several membrane lipid classes are implicated in inflammation through membrane raft and/or signal transduction in obesity-associated cancers.123 Specifically, lysophosphatidic acid and lysophosphatidylcholine were shown to influence the activation, distribution and trafficking of immune cells.123 Unsaturated (C18:1, C20:4) but not saturated (C16:0, C18:0) lysophosphatidic acid were shown to induce chemotaxis of immature dendritic cells though a LPA3 receptor-mediated response.126 It has been reported that lysophosphatidylcholine is frequently upregulated at inflammation sites, where multiple lysophosphatidylcholine species are shown to induce chemotaxis of immune cells (C16:0, C18:0, C18:1).127 Additionally, ceramide-1-phosphate and sphingosine-1-phosphate regulate cytokine production, and the latter also promotes eicosanoid storm.123
Activating invasion & metastasis
Invasion and metastasis characterise cancer spread, with induction of the epithelial–mesenchymal transition (EMT) as a central mechanism. In addition to the inflammatory regulation described above, specific lipids play key roles. Prostaglandin E2 binds to the prostaglandin E2 receptor EP2 and induces a COX2 driven epithelial–mesenchymal transition (EMT) to promote invasion and metastasis.128 Glycosphingolipids, including GM2, were shown to be associated to tumour invasiveness by increasing N-cadherin expression and inducing EMT.129 Endoplasmic reticulum stress can also alter N-cadherin expression via activation of the PERK pathway.78 High ceramide concentrations induce the proteasome-mediated degradation of β-catenin, reducing cell adhesion and inducing invasion.130The induction of EMT through altering the Wnt pathway is common in many cancers, leading to the maintenance of cancer stem cells and metastasis.131 Lipids play an important role in the Wnt signalling pathway, as lipid-modified Wnt is required for the interaction with the cysteine-rich domain of frizzled (FZD) receptors.132 FZD receptors specifically recognize cis-unsaturated fatty acids, due to the U-shaped geometry of these lipids, allowing the bond between the ligand and receptor.132 The interaction of FZD8 and Wnt11 promotes TFG-β-mediated EMT in cancer cells.133
Genome instability & mutation
As one of the cancer characteristics described by Hanahan & Weinberg in 2011, genome instability & mutation is an enabling characteristic which is directly related to several of the cancer hallmarks.73 The best example is the mutation of TP53, which impairs its tumour suppressive function, leading to cancer development. Reactive oxygen species lead to lipid peroxidation, generating reactive lipid peroxide species. The primary products of lipid peroxidation, malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), contribute to cancer by forming adducts with proteins, lipids, and DNA, thereby modulating the functions of these molecules including gene mutations.134,135At least two molecular mechanisms regulate lipid-based anti-oxidant action, namely lipid desaturation and formation of anti-oxidant lipids. Lipid peroxidation is initiated at the double bonds of fatty acid chains, and polyunsaturated lipids are therefore more prone to lipid peroxidation. The rate limiting enzyme in fatty acid desaturation, SCD1, creates monounsaturated lipids from saturated lipids, and is highly expressed in several cancer types, such as colorectal and endometrial cancers.136–138 In addition to SCD1, desaturation of lipids beyond the first double bond is regulated by FADS1 and FADS2, which produce polyunsaturated fatty acids. A direct relation between FADS gene mutations and the production of lipid peroxides has been discovered.139 Correlative lipidomic and proteomic studies are required to decipher the relationship between total fatty saturation and the formation of lipid peroxide species.
As a counterpoint to lipid peroxidation, the class of plasmenyl lipids are known scavenging lipids with antioxidant properties since the alkenyl-bond is preferentially oxidized, thereby protecting nearby polyunsaturated lipids from oxidation.140 In line with an antioxidant function, high concentrations of plasmenyl lipids are observed in tissues and organs which are prone to oxidative stress and require protection against free radicals.141 Plasmenyl-phosphatidylethanolamine has been shown to have anti-tumour properties such as inhibiting proliferation and migration,142 and has been reported to be downregulated in breast cancer tissues.143
Next-generation lipidomics
While the maturing lipidomics technologies have already revealed unprecedented molecular details of lipid species, and enabled assignment of lipid-mediated mechanisms to cancer hallmarks, further improvements in molecular, subcellular and functional lipidomics techniques will be required to fully realise the potential of lipidomics as a research tool. Novel methods are also being developed to better define the high risk lipidome, which may exist independent of classical measures of obesity. For elucidating lipid structures, this includes ozone-induced dissociation (OzID)144 and electron-impact excitation of ions from organics (EIEIO)145 mass spectrometry. In a human plasma dataset, Marshall et al. reported 22 unique phosphatidylcholine lipids at the sum composition level (e.g. PC 34:1), 41 unique lipids at the molecular lipid level (e.g. PC 16:0_18:1) and 76 unique lipids at the defined sn-position level (e.g. PC 16:0/18:1).144 The next, more detailed step involves distinguishing isomeric lipids at the structurally defined level (e.g. PC 16:0/18:1(n-9)) based on the unsaturation bond position on lipid hydrocarbon chains144 using OzID or EIEIO.Other advances include imaging mass spectrometry that has enabled spatial lipidomics at the tissue level.146 The impact of obesity on organelle lipidome, proteome and function remain to be fully investigated. For example, while the Golgi apparatus plays an important role in lipid metabolism, membrane raft formation, lipid droplet formation and lipid trafficking,147,148 to our knowledge no study has assessed changes in lipid composition of the Golgi apparatus in cancer or obesity. The lipid profile of the nucleus also appears to be dynamic and altered with food intake,149 influencing chromosomal segregation and nucleus morphology.150 However, it is unknown whether obesity alters the molecular composition and function of the nuclear proteome and lipidome, and if there are any direct roles in cancers. While current techniques focus on individual organelles in separate studies, development of high throughput organelle molecular profiling and computational biology methods to interrogate multiple/all organelles in one experiment will enable more rapid advances in this area.
The fundamental mechanisms of membrane lateral organisation and its impact on signal transduction, vesicular transport and secretory pathway remain an intense area of study. While the plasma membrane was the starting point and major focus of raft biologists, it is clear that lateral organisation exist in other organelle membranes.151,152 The detailed investigations of dynamic membrane lateral organisation at the subcellular, nanometer scale will require innovations and collaborations in biophysics, cell biology, imaging and lipidomics.
Finally, adoption of harmonized lipidomics guidelines153 will be required for acceptance of lipidomics as a reproducible omics technology and facilitate its progress into clinical diagnostics as scientific findings are translated to the clinic.
Conclusions
Based on the plethora of reported lipid-mediated mechanisms, we propose that obesity-associated lipid derangement is a potential driver fuelling multiple cancer hallmarks. From this, it follows that lipid-related treatments and diagnoses have the potential to improve patient outcomes. Indeed, the effectiveness of lipid altering drugs in the treatment and prevention of cancer has previously been demonstrated.154–156 Recently, exercise has been suggested to be beneficial in improving the quality of life and cancer-related outcomes, mainly based on observational studies.157,158 Whether or not this is due to a more favourable energy balance imparted by engaging in physical activity or by an increase in fitness per se is not known. Dietary changes, especially those that result in a small weight loss or prevent weight gain, have been shown to reduce cancer risk.159–161 The further development of analytical methods, cross-disciplinary research, and adoption of guidelines are expected to contribute to full understanding of lipid-mediated carcinogenesis mechanisms, providing the scientific basis to design lifestyle-based prevention and intervention strategies to target specific pathways involved in the aetiology of obesity-associated cancers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
HR and JM were supported by Australian Government Research Training Program Scholarships. ZD was supported by a U.S. Fulbright Global Scholar Award.Notes and references
- International Agency for Research on Cancer, IARC handbooks of cancer prevention: weight control and physical activity, Lyon, France, 2002.
- International Agency for Research on Cancer, IARC Handbooks of Cancer Prevention: Body Fatness, Lyon, France, 2016.
- M. Bluher, Nat. Rev. Endocrinol., 2019, 15, 288–298 CrossRef PubMed.
- L. M. Jaacks, S. Vandevijvere, A. Pan, C. J. McGowan, C. Wallace, F. Imamura, D. Mozaffarian, B. Swinburn and M. Ezzati, Lancet Diabetes Endocrinol., 2019, 7, 231–240 CrossRef PubMed.
- T. T. Gong, D. Li, Q. J. Wu and Y. Z. Wang, Oncotarget, 2016, 7, 16996–17008 Search PubMed.
- J. Hu, C. La Vecchia, M. de Groh, E. Negri, H. Morrison, L. Mery and Canadian Cancer Registries Epidemiology Research Group, Ann. Oncol., 2012, 23, 491–500 CrossRef CAS PubMed.
- C. Li, L. Yang, D. Zhang and W. Jiang, Nutr. Res., 2016, 36, 627–635 CrossRef CAS PubMed.
- S. T. Mayne, H. A. Risch, R. Dubrow, W. H. Chow, M. D. Gammon, T. L. Vaughan, D. C. Farrow, J. B. Schoenberg, J. L. Stanford, H. Ahsan, A. B. West, H. Rotterdam, W. J. Blot and J. F. Fraumeni Jr, Cancer Epidemiol., Biomarkers Prev., 2001, 10, 1055–1062 CAS.
- E. H. Allott, L. Arab, L. J. Su, L. Farnan, E. T. Fontham, J. L. Mohler, J. T. Bensen and S. E. Steck, Prostate Cancer Prostatic Dis., 2017, 20, 48–54 CrossRef CAS PubMed.
- J. Zhao, C. Lyu, J. Gao, L. Du, B. Shan, H. Zhang, H. Y. Wang and Y. Gao, Medicine, 2016, 95, e4121 CrossRef CAS PubMed.
- K. E. Brock, G. Gridley, B. C. Chiu, A. G. Ershow, C. F. Lynch and K. P. Cantor, Br. J. Nutr., 2009, 101, 1228–1238 CrossRef CAS PubMed.
- J. Hu, C. La Vecchia, M. DesMeules, E. Negri, L. Mery and Canadian Cancer Registries Epidemiology Research Group, Nutr. Cancer, 2008, 60, 720–728 CrossRef CAS PubMed.
- J. Hu, C. La Vecchia, E. Negri, M. de Groh, H. Morrison, L. Mery and Canadian Cancer Registries Epidemiology Research Group, Cancer Causes Control, 2015, 26, 839–847 CrossRef PubMed.
- D. He, X. Huang, Z. P. Wang, D. Chen, J. Chen and C. Y. Duan, Oncotarget, 2017, 8, 99049–99056 Search PubMed.
- Y. Tian, K. Wang, J. Li, J. Wang, Z. Wang, Y. Fan, Y. Ye, G. Ji and Y. Li, Public Health Nutr., 2015, 18, 3355–3370 CrossRef PubMed.
- G. M. Zhang, Y. Zhu, L. Luo, H. L. Zhang, C. Y. Gu, L. J. Sun and D. W. Ye, BJU Int., 2014, 113, E75–E81 CrossRef CAS PubMed.
- P. D. Chandler, Y. Song, J. Lin, S. Zhang, H. D. Sesso, S. Mora, E. L. Giovannucci, K. E. Rexrode, M. V. Moorthy, C. Li, P. M. Ridker, I. M. Lee, J. E. Manson, J. E. Buring and L. Wang, Am. J. Clin. Nutr., 2016, 103, 1397–1407 CrossRef CAS PubMed.
- T. Stocks, A. Lukanova, T. Bjorge, H. Ulmer, J. Manjer, M. Almquist, H. Concin, A. Engeland, G. Hallmans, G. Nagel, S. Tretli, M. B. Veierod, H. Jonsson, P. Stattin and Metabolic Syndrome Cancer Project Me-Can Group, Cancer, 2011, 117, 2398–2407 CrossRef PubMed.
- H. Ulmer, W. Borena, K. Rapp, J. Klenk, A. Strasak, G. Diem, H. Concin and G. Nagel, Br. J. Cancer, 2009, 101, 1202–1206 CrossRef CAS PubMed.
- K. Lindemann, L. J. Vatten, M. Ellstrom-Engh and A. Eskild, Int. J. Cancer, 2009, 124, 2938–2941 CrossRef CAS PubMed.
- C. Haggstrom, K. Rapp, T. Stocks, J. Manjer, T. Bjorge, H. Ulmer, A. Engeland, M. Almqvist, H. Concin, R. Selmer, B. Ljungberg, S. Tretli, G. Nagel, G. Hallmans, H. Jonsson and P. Stattin, PLoS One, 2013, 8, e57475 CrossRef PubMed.
- V. Michalaki, G. Koutroulis, K. Syrigos, C. Piperi and A. Kalofoutis, Mol. Cell. Biochem., 2005, 268, 19–24 CrossRef CAS PubMed.
- R. Arthur, H. Moller, H. Garmo, L. Holmberg, P. Stattin, H. Malmstrom, M. Lambe, N. Hammar, G. Walldius, D. Robinson, I. Jungner and M. V. Hemelrijck, Cancer Med., 2016, 5, 1307–1318 CrossRef CAS PubMed.
- G. Andreotti, J. Chen, Y. T. Gao, A. Rashid, S. C. Chang, M. C. Shen, B. S. Wang, T. Q. Han, B. H. Zhang, K. N. Danforth, M. D. Althuis and A. W. Hsing, Int. J. Cancer, 2008, 122, 2322–2329 CrossRef CAS PubMed.
- M. Touvier, P. Fassier, M. His, T. Norat, D. S. Chan, J. Blacher, S. Hercberg, P. Galan, N. Druesne-Pecollo and P. Latino-Martel, Br. J. Nutr., 2015, 114, 347–357 CrossRef CAS PubMed.
- F. Baenke, B. Peck, H. Miess and A. Schulze, Dis. Models Mech., 2013, 6, 1353–1363 CrossRef CAS PubMed.
- S. Beloribi-Djefaflia, S. Vasseur and F. Guillaumond, Oncogenesis, 2016, 5, e189 CrossRef CAS PubMed.
- M. L. Doria, C. Z. Cotrim, C. Simoes, B. Macedo, P. Domingues, M. R. Domingues and L. A. Helguero, J. Cell. Physiol., 2013, 228, 457–468 CrossRef CAS PubMed.
- I. C. Kim, J. H. Lee, G. Bang, S. H. Choi, Y. H. Kim, K. P. Kim, H. K. Kim and J. Ro, Anticancer Res., 2013, 33, 2467–2472 CAS.
- M. Sud, E. Fahy, D. Cotter, A. Brown, E. A. Dennis, C. K. Glass, A. H. Merrill Jr., R. C. Murphy, C. R. Raetz, D. W. Russell and S. Subramaniam, Nucleic Acids Res., 2007, 35, D527–D532 CrossRef CAS PubMed.
- R. t'Kindt, E. D. Telenga, L. Jorge, A. J. Van Oosterhout, P. Sandra, N. H. Ten Hacken and K. Sandra, Anal. Chem., 2015, 87, 4957–4964 CrossRef PubMed.
- Z. T. Schug and E. Gottlieb, Biochim. Biophys. Acta, 2009, 1788, 2022–2031 CrossRef CAS PubMed.
- J. Stiban and M. Perera, Biochim. Biophys. Acta, 2015, 1848, 561–567 CrossRef CAS PubMed.
- T. D’Aquila, A. S. Zembroski and K. K. Buhman, Front. Physiol., 2019, 10, 180 CrossRef PubMed.
- C. Chitraju, M. Trötzmüller, J. Hartler, H. Wolinski, G. G. Thallinger, A. Lass, R. Zechner, R. Zimmermann, H. C. Köfeler and F. Spener, J. Lipid Res., 2012, 53, 2141–2152 CrossRef CAS PubMed.
- A. Seyer, M. Cantiello, J. Bertrand-Michel, V. Roques, M. Nauze, V. Bézirard, X. Collet, D. Touboul, A. Brunelle and C. Coméra, PLoS One, 2013, 8, e58224 CrossRef CAS PubMed.
- S. E. Shoelson, L. Herrero and A. Naaz, Gastroenterology, 2007, 132, 2169–2180 CrossRef CAS PubMed.
- E. Yehuda-Shnaidman and B. Schwartz, Obes. Rev., 2012, 13, 1083–1095 CrossRef CAS PubMed.
- J. Li, C. Romestaing, X. Han, Y. Li, X. Hao, Y. Wu, C. Sun, X. Liu, L. S. Jefferson, J. Xiong, K. F. Lanoue, Z. Chang, C. J. Lynch, H. Wang and Y. Shi, Cell Metab., 2010, 12, 154–165 CrossRef CAS PubMed.
- L. Lassance, M. Haghiac, J. Minium, P. Catalano and S. Hauguel-deMouzon, J. Clin. Endocrinol. Metab., 2015, 100, E11–E18 CrossRef PubMed.
- P. Theurey and J. Rieusset, Trends Endocrinol. Metab., 2017, 28, 32–45 CrossRef CAS PubMed.
- L. M. Sedger, D. L. Tull, M. J. McConville, D. P. De Souza, T. W. T. Rupasinghe, S. J. Williams, S. Dayalan, D. Lanzer, H. Mackie, T. C. Lam and J. Boyages, PLoS One, 2016, 11, e0154650 CrossRef PubMed.
- F. J. Gonzalez, J. M. Peters and R. C. Cattley, JNCI, J. Natl. Cancer Inst., 1998, 90, 1702–1709 CrossRef CAS PubMed.
- S. Fu, L. Yang, P. Li, O. Hofmann, L. Dicker, W. Hide, X. Lin, S. M. Watkins, A. R. Ivanov and G. S. Hotamisligil, Nature, 2011, 473, 528 CrossRef CAS PubMed.
- L. Ju, J. Han, X. Zhang, Y. Deng, H. Yan, C. Wang, X. Li, S. Chen, M. Alimujiang, X. Li, Q. Fang, Y. Yang and W. Jia, Cell Death Dis., 2019, 10, 121 CrossRef PubMed.
- T. Yamamoto, Y. Takabatake, A. Takahashi, T. Kimura, T. Namba, J. Matsuda, S. Minami, J.-Y. Kaimori, I. Matsui, T. Matsusaka, F. Niimura, M. Yanagita and Y. Isaka, J. Am. Soc. Nephrol., 2017, 28, 1534 CrossRef CAS PubMed.
- J. A. Rodriguez-Navarro, S. Kaushik, H. Koga, C. Dall'Armi, G. Shui, M. R. Wenk, G. Di Paolo and A. M. Cuervo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E705–E714 CrossRef CAS PubMed.
- B. P. Head, H. H. Patel and P. A. Insel, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 532–545 CrossRef CAS PubMed.
- R. Kosaraju, W. Guesdon, M. J. Crouch, H. L. Teague, E. M. Sullivan, E. A. Karlsson, S. Schultz-Cherry, K. Gowdy, L. C. Bridges, L. R. Reese, P. D. Neufer, M. Armstrong, N. Reisdorph, J. J. Milner, M. Beck and S. R. Shaikh, J. Immunol., 2017, 198, 4738 CrossRef CAS PubMed.
- K. H. Pietiläinen, T. Róg, T. Seppänen-Laakso, S. Virtue, P. Gopalacharyulu, J. Tang, S. Rodriguez-Cuenca, A. Maciejewski, J. Naukkarinen, A.-L. Ruskeepää, P. S. Niemelä, L. Yetukuri, C. Y. Tan, V. Velagapudi, S. Castillo, H. Nygren, T. Hyötyläinen, A. Rissanen, J. Kaprio, H. Yki-Järvinen, I. Vattulainen, A. Vidal-Puig and M. Orešič, PLoS Biol., 2011, 9, e1000623 CrossRef PubMed.
- E. Berger, S. Héraud, A. Mojallal, C. Lequeux, M. Weiss-Gayet, O. Damour and A. Géloën, Adipocyte, 2015, 4, 161–180 CrossRef CAS PubMed.
- C. Mauro, J. Smith, D. Cucchi, D. Coe, H. Fu, F. Bonacina, A. Baragetti, G. Cermenati, D. Caruso, N. Mitro, A. L. Catapano, E. Ammirati, M. P. Longhi, K. Okkenhaug, G. D. Norata and F. M. Marelli-Berg, Cell Metab., 2017, 25, 593–609 CrossRef CAS PubMed.
- H. F. Turk and R. S. Chapkin, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2013, 88, 43–47 CrossRef CAS PubMed.
- F. Pardo, R. Villalobos-Labra, B. Sobrevia, F. Toledo and L. Sobrevia, Mol. Aspects Med., 2018, 60, 81–91 CrossRef CAS PubMed.
- K. D. Connolly, I. A. Guschina, V. Yeung, A. Clayton, M. S. Draman, C. Von Ruhland, M. Ludgate, P. E. James and D. A. Rees, J. Extracell. Vesicles, 2015, 4, 29159 CrossRef PubMed.
- I. Lazar, E. Clement, S. Dauvillier, D. Milhas, M. Ducoux-Petit, S. LeGonidec, C. Moro, V. Soldan, S. Dalle, S. Balor, M. Golzio, O. Burlet-Schiltz, P. Valet, C. Muller and L. Nieto, Cancer Res., 2016, 76, 4051–4057 CrossRef CAS PubMed.
- M. Micallef, I. Munro, M. Phang and M. Garg, Br. J. Nutr., 2009, 102, 1370–1374 CrossRef CAS PubMed.
- J. Y. Kim, J. Y. Park, O. Y. Kim, B. M. Ham, H. J. Kim, D. Y. Kwon, Y. Jang and J. H. Lee, J. Proteome Res., 2010, 9, 4368–4375 CrossRef CAS PubMed.
- M. Karlsson, S. Marild, J. Brandberg, L. Lonn, P. Friberg and B. Strandvik, Obesity, 2006, 14, 1931–1939 CrossRef CAS PubMed.
- C. A. Pickens, L. M. Sordillo, S. S. Comstock, W. S. Harris, K. Hortos, B. Kovan and J. I. Fenton, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2015, 95, 31–40 CrossRef CAS PubMed.
- S. Caspar-Bauguil, A. Fioroni, A. Galinier, S. Allenbach, M. C. Pujol, R. Salvayre, A. Cartier, I. Lemieux, D. Richard, S. Biron, P. Marceau, L. Casteilla, L. Penicaud and P. Mauriege, Obes. Surg., 2012, 22, 935–944 CrossRef CAS PubMed.
- G. M. Corbi, S. Carbone, P. Ziccardi, G. Giugliano, R. Marfella, F. Nappo, G. Paolisso, K. Esposito and D. Giugliano, J. Clin. Endocrinol. Metab., 2002, 87, 2080–2083 CrossRef CAS PubMed.
- L. Dong, C. Yuan, B. J. Orlando, M. G. Malkowski and W. L. Smith, J. Biol. Chem., 2016, 291, 25641–25655 CrossRef CAS PubMed.
- L. Dong, H. Zou, C. Yuan, Y. H. Hong, D. V. Kuklev and W. L. Smith, J. Biol. Chem., 2016, 291, 4069–4078 CrossRef CAS PubMed.
- M. Guerville and G. Boudry, Am. J. Physiol.: Gastrointest. Liver Physiol., 2016, 311, G1–G15 CrossRef PubMed.
- J. Amar, R. Burcelin, J. B. Ruidavets, P. D. Cani, J. Fauvel, M. C. Alessi, B. Chamontin and J. Ferrieres, Am. J. Clin. Nutr., 2008, 87, 1219–1223 CrossRef CAS PubMed.
- J. H. Gao, S. L. Wen, H. Tong, C. H. Wang, W. J. Yang, S. H. Tang, Z. P. Yan, Y. Tai, C. Ye, R. Liu, Z. Y. Huang, Y. M. Tang, J. H. Yang and C. W. Tang, Am. J. Physiol.: Gastrointest. Liver Physiol., 2016, 310, G962–G972 CrossRef PubMed.
- S. S. Short, J. Wang, S. L. Castle, G. E. Fernandez, N. Smiley, M. Zobel, E. M. Pontarelli, S. C. Papillon, A. V. Grishin and H. R. Ford, Lab. Invest., 2013, 93, 1265–1275 CrossRef CAS PubMed.
- D. Wang and R. N. Dubois, Oncogene, 2010, 29, 781–788 CrossRef CAS PubMed.
- S. Frosali, D. Pagliari, G. Gambassi, R. Landolfi, F. Pandolfi and R. Cianci, J. Immunol. Res., 2015, 2015, 489821 Search PubMed.
- H. Oshima and M. Oshima, J. Gastroenterol., 2012, 47, 97–106 CrossRef CAS PubMed.
- M. E. Martinez, D. Heddens, D. L. Earnest, C. L. Bogert, D. Roe, J. Einspahr, J. R. Marshall and D. S. Alberts, J. Natl. Cancer Inst., 1999, 91, 950–953 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- M. Labrada, D. Dorvignit, G. Hevia, N. Rodriguez-Zhurbenko, A. M. Hernandez, A. M. Vazquez and L. E. Fernandez, Semin. Oncol., 2018, 45, 41–51 CrossRef CAS PubMed.
- V. Garcia-Gonzalez, J. F. Diaz-Villanueva, O. Galindo-Hernandez, I. Martinez-Navarro, G. Hurtado-Ureta and A. A. Perez-Arias, Int. J. Mol. Sci., 2018, 19, 2527 CrossRef PubMed.
- H. Han, R. Vargas, G. Seo and W. Wang, Mol. Cell. Oncol., 2019, 6, 1558683 CrossRef PubMed.
- J. S. A. Warren, Y. Xiao and J. M. Lamar, Cancers, 2018, 10, 115 CrossRef PubMed.
- H. Urra, E. Dufey, T. Avril, E. Chevet and C. Hetz, Trends Cancer, 2016, 2, 252–262 CrossRef PubMed.
- K. Yang, D. Zheng, X. Deng, L. Bai, Y. Xu and Y. S. Cong, J. Cell. Biochem., 2008, 105, 1194–1201 CrossRef CAS PubMed.
- B. Ogretmen, D. Schady, J. Usta, R. Wood, J. M. Kraveka, C. Luberto, H. Birbes, Y. A. Hannun and L. M. Obeid, J. Biol. Chem., 2001, 276, 24901–24910 CrossRef CAS PubMed.
- L. G. Wooten-Blanks, P. Song, C. E. Senkal and B. Ogretmen, FASEB J., 2007, 21, 3386–3397 CrossRef CAS PubMed.
- T. Eitsuka, K. Nakagawa, S. Kato, J. Ito, Y. Otoki, S. Takasu, N. Shimizu, T. Takahashi and T. Miyazawa, Int. J. Mol. Sci., 2018, 19, 478 CrossRef PubMed.
- L. Chen, H. Chen, Y. Li, L. Li, Y. Qiu and J. Ren, Oncol. Rep., 2015, 34, 447–454 CrossRef CAS PubMed.
- M. Nagahashi, J. Tsuchida, K. Moro, M. Hasegawa, K. Tatsuda, I. A. Woelfel, K. Takabe and T. Wakai, J. Surg. Res., 2016, 204, 435–444 CrossRef CAS PubMed.
- Z. Li, M. Guan, Y. Lin, X. Cui, Y. Zhang, Z. Zhao and J. Zhu, Int. J. Mol. Sci., 2017, 18, 2550 CrossRef PubMed.
- P. Knapp, L. Bodnar, A. Blachnio-Zabielska, M. Swiderska and A. Chabowski, Gynecol. Oncol., 2017, 147, 139–144 CrossRef CAS PubMed.
- F. Hermetet, A. Buffière, A. Aznague, J.-P. Pais de Barros, J.-N. Bastie, L. Delva and R. Quéré, Nat. Commun., 2019, 10, 523 CrossRef PubMed.
- C. L. Chen, I. H. Liu, S. J. Fliesler, X. Han, S. S. Huang and J. S. Huang, J. Cell Sci., 2007, 120, 3509–3521 CrossRef CAS PubMed.
- C. L. Chen, S. S. Huang and J. S. Huang, J. Cell. Physiol., 2008, 215, 223–233 CrossRef CAS PubMed.
- B. Fekry, K. A. Jeffries, A. Esmaeilniakooshkghazi, Z. M. Szulc, K. J. Knagge, D. R. Kirchner, D. A. Horita, S. A. Krupenko and N. I. Krupenko, Nat. Commun., 2018, 9, 4149 CrossRef PubMed.
- S. Koizume and Y. Miyagi, Int. J. Mol. Sci., 2016, 17, 1430 CrossRef PubMed.
- A. Ladanyi, A. Mukherjee, H. A. Kenny, A. Johnson, A. K. Mitra, S. Sundaresan, K. M. Nieman, G. Pascual, S. A. Benitah, A. Montag, S. D. Yamada, N. A. Abumrad and E. Lengyel, Oncogene, 2018, 37, 2285–2301 CrossRef CAS PubMed.
- M. N. Duong, A. Geneste, F. Fallone, X. Li, C. Dumontet and C. Muller, Oncotarget, 2017, 8, 57622–57641 CrossRef PubMed.
- K. M. Nieman, H. A. Kenny, C. V. Penicka, A. Ladanyi, R. Buell-Gutbrod, M. R. Zillhardt, I. L. Romero, M. S. Carey, G. B. Mills, G. S. Hotamisligil, S. D. Yamada, M. E. Peter, K. Gwin and E. Lengyel, Nat. Med., 2011, 17, 1498–1503 CrossRef CAS PubMed.
- Q. Wu, S. Sun, Z. Li, Q. Yang, B. Li, S. Zhu, L. Wang, J. Wu, J. Yuan, C. Yang, J. Li and S. Sun, Mol. Cancer, 2018, 17, 155 CrossRef PubMed.
- I. J. Goldberg, R. H. Eckel and N. A. Abumrad, J. Lipid Res., 2009, 50(Suppl), S86–S90 CrossRef PubMed.
- F. Slebe, F. Rojo, M. Vinaixa, M. Garcia-Rocha, G. Testoni, M. Guiu, E. Planet, S. Samino, E. J. Arenas, A. Beltran, A. Rovira, A. Lluch, X. Salvatella, O. Yanes, J. Albanell, J. J. Guinovart and R. R. Gomis, Nat. Commun., 2016, 7, 11199 CrossRef CAS PubMed.
- J. Zhao, Z. Zhi, C. Wang, H. Xing, G. Song, X. Yu, Y. Zhu, X. Wang, X. Zhang and Y. Di, Oncol. Rep., 2017, 38, 2105–2115 CrossRef CAS PubMed.
- G. Pascual, A. Avgustinova, S. Mejetta, M. Martin, A. Castellanos, C. S. Attolini, A. Berenguer, N. Prats, A. Toll, J. A. Hueto, C. Bescos, L. Di Croce and S. A. Benitah, Nature, 2017, 541, 41–45 CrossRef CAS PubMed.
- J. S. Hale, B. Otvos, M. Sinyuk, A. G. Alvarado, M. Hitomi, K. Stoltz, Q. Wu, W. Flavahan, B. Levison, M. L. Johansen, D. Schmitt, J. M. Neltner, P. Huang, B. Ren, A. E. Sloan, R. L. Silverstein, C. L. Gladson, J. A. DiDonato, J. M. Brown, T. McIntyre, S. L. Hazen, C. Horbinski, J. N. Rich and J. D. Lathia, Stem Cells, 2014, 32, 1746–1758 CrossRef CAS PubMed.
- H. Schneider, S. Staudacher, M. Poppelreuther, W. Stremmel, R. Ehehalt and J. Fullekrug, Arch. Biochem. Biophys., 2014, 546, 8–18 CrossRef CAS PubMed.
- J. M. Argiles, S. Busquets, B. Stemmler and F. J. Lopez-Soriano, Nat. Rev. Cancer, 2014, 14, 754–762 CrossRef CAS PubMed.
- M. J. Pagliassotti, P. Y. Kim, A. L. Estrada, C. M. Stewart and C. L. Gentile, Metabolism, 2016, 65, 1238–1246 CrossRef CAS PubMed.
- Y. Hisano and T. Hla, Pharmacol. Ther., 2019, 193, 91–98 CrossRef CAS PubMed.
- E. K. Tuominen, C. J. Wallace and P. K. Kinnunen, J. Biol. Chem., 2002, 277, 8822–8826 CrossRef CAS PubMed.
- E. C. Randall, G. Zadra, P. Chetta, B. G. C. Lopez, S. Syamala, S. S. Basu, J. N. Agar, M. Loda, C. M. Tempany, F. M. Fennessy and N. Y. R. Agar, Mol. Cancer Res., 2019, 17, 1155–1165 CrossRef CAS PubMed.
- A. Sapandowski, M. Stope, K. Evert, M. Evert, U. Zimmermann, D. Peter, I. Page, M. Burchardt and L. Schild, Mol. Cell. Biochem., 2015, 410, 175–185 CrossRef CAS PubMed.
- B. Peck, Z. T. Schug, Q. Zhang, B. Dankworth, D. T. Jones, E. Smethurst, R. Patel, S. Mason, M. Jiang, R. Saunders, M. Howell, R. Mitter, B. Spencer-Dene, G. Stamp, L. McGarry, D. James, E. Shanks, E. O. Aboagye, S. E. Critchlow, H. Y. Leung, A. L. Harris, M. J. O. Wakelam, E. Gottlieb and A. Schulze, Cancer Metab., 2016, 4, 6 CrossRef PubMed.
- J. Newton, S. Lima, M. Maceyka and S. Spiegel, Exp. Cell Res., 2015, 333, 195–200 CrossRef CAS PubMed.
- M. P. Wymann and R. Schneiter, Nat. Rev. Mol. Cell Biol., 2008, 9, 162–176 CrossRef CAS PubMed.
- A. C. Lewis, C. T. Wallington-Beddoe, J. A. Powell and S. M. Pitson, Cell Death Discovery, 2018, 4, 4 CrossRef PubMed.
- G. Seumois, M. Fillet, L. Gillet, C. Faccinetto, C. Desmet, C. Francois, B. Dewals, C. Oury, A. Vanderplasschen, P. Lekeux and F. Bureau, J. Leukocyte Biol., 2007, 81, 1477–1486 CrossRef CAS PubMed.
- L. Samsel, G. Zaidel, H. M. Drumgoole, D. Jelovac, C. Drachenberg, J. G. Rhee, A. M. Brodie, A. Bielawska and M. J. Smyth, Prostate, 2004, 58, 382–393 CrossRef CAS PubMed.
- M. Park, V. Kaddai, J. Ching, K. T. Fridianto, R. J. Sieli, S. Sugii and S. A. Summers, J. Biol. Chem., 2016, 291, 23978–23988 CrossRef CAS PubMed.
- T. Yabu, H. Shiba, Y. Shibasaki, T. Nakanishi, S. Imamura, K. Touhata and M. Yamashita, Cell Death Differ., 2015, 22, 258–273 CrossRef CAS PubMed.
- S. A. Morad and M. C. Cabot, Nat. Rev. Cancer, 2013, 13, 51–65 CrossRef CAS PubMed.
- G. Tie, J. Yan, L. Khair, J. A. Messina, A. Deng, J. Kang, T. Fazzio and L. M. Messina, Cancer Res., 2017, 77, 2351–2362 CrossRef CAS PubMed.
- J. Liu, X. Zheng, X. Pang, L. Li, J. Wang, C. Yang and G. Du, Acta Pharm. Sin. B, 2018, 8, 713–720 CrossRef PubMed.
- C. Wefers, T. Duiveman-de Boer, P. L. M. Zusterzeel, L. Massuger, D. Fuchs, R. Torensma, C. E. Wheelock and I. J. M. de Vries, Int. J. Mol. Sci., 2018, 19, 273 CrossRef PubMed.
- R. Arai, S. Soda, T. Okutomi, H. Morita, F. Ohmi, T. Funakoshi, A. Takemasa and Y. Ishii, J. Immunol. Res., 2018, 2018, 5708239 Search PubMed.
- M. H. den Brok, T. K. Raaijmakers, E. Collado-Camps and G. J. Adema, Trends Immunol., 2018, 39, 380–392 CrossRef CAS PubMed.
- M. T. Accioly, P. Pacheco, C. M. Maya-Monteiro, N. Carrossini, B. K. Robbs, S. S. Oliveira, C. Kaufmann, J. A. Morgado-Diaz, P. T. Bozza and J. P. Viola, Cancer Res., 2008, 68, 1732–1740 CrossRef CAS PubMed.
- V. Chiurchiu, A. Leuti and M. Maccarrone, Front. Immunol., 2018, 9, 38 CrossRef PubMed.
- C. Zhang, K. Wang, L. Yang, R. Liu, Y. Chu, X. Qin, P. Yang and H. Yu, Analyst, 2018, 143, 4526–4536 RSC.
- J. Korbecki, I. Baranowska-Bosiacka, I. Gutowska and D. Chlubek, Acta Biochim. Pol., 2014, 61, 639–649 CrossRef PubMed.
- L. C. Chan, W. Peters, Y. Xu, J. Chun, R. V. Farese Jr. and S. Cases, J. Leukocyte Biol., 2007, 82, 1193–1200 CrossRef CAS PubMed.
- I. Sevastou, E. Kaffe, M. A. Mouratis and V. Aidinis, Biochim. Biophys. Acta, 2013, 1831, 42–60 CrossRef CAS PubMed.
- J. Qiu, Q. Li, K. A. Bell, X. Yao, Y. Du, E. Zhang, J. J. Yu, Y. Yu, Z. Shi and J. Jiang, Br. J. Pharmacol., 2019, 176, 1680–1699 CrossRef CAS PubMed.
- S. Ishihara, K. Aoki, T. Mizutani, M. Amano, S. I. Nishimura and H. Haga, Cell Struct. Funct., 2018, 43, 177–185 CrossRef CAS PubMed.
- M. Garcia-Barros, N. Coant, J. P. Truman, A. J. Snider and Y. A. Hannun, Biochim. Biophys. Acta, 2014, 1841, 773–782 CrossRef CAS PubMed.
- T. Zhan, N. Rindtorff and M. Boutros, Oncogene, 2017, 36, 1461–1473 CrossRef CAS PubMed.
- A. H. Nile, S. Mukund, K. Stanger, W. Wang and R. N. Hannoush, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4147–4152 CrossRef CAS PubMed.
- V. Murillo-Garzon, I. Gorrono-Etxebarria, M. Akerfelt, M. C. Puustinen, L. Sistonen, M. Nees, J. Carton, J. Waxman and R. M. Kypta, Nat. Commun., 2018, 9, 1747 CrossRef PubMed.
- L. J. Marnett, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1999, 424, 83–95 CrossRef CAS PubMed.
- H. Zhong and H. Yin, Redox Biol., 2015, 4, 193–199 CrossRef CAS PubMed.
- W. Li, H. Bai, S. Liu, D. Cao, H. Wu, K. Shen, Y. Tai and J. Yang, Oncotarget, 2018, 9, 12064–12078 Search PubMed.
- H. Ran, Y. Zhu, R. Deng, Q. Zhang, X. Liu, M. Feng, J. Zhong, S. Lin, X. Tong and Q. Su, J. Exp. Clin. Cancer Res., 2018, 37, 54 CrossRef PubMed.
- U. V. Roongta, J. G. Pabalan, X. Wang, R. P. Ryseck, J. Fargnoli, B. J. Henley, W. P. Yang, J. Zhu, M. T. Madireddi, R. M. Lawrence, T. W. Wong and B. A. Rupnow, Mol. Cancer Res., 2011, 9, 1551–1561 CrossRef CAS PubMed.
- J. H. Kwak, J. K. Paik, O. Y. Kim, Y. Jang, S. H. Lee, J. M. Ordovas and J. H. Lee, Atherosclerosis, 2011, 214, 94–100 CrossRef CAS PubMed.
- B. Engelmann, Biochem. Soc. Trans., 2004, 32, 147–150 CrossRef CAS PubMed.
- P. J. Sindelar, Z. Guan, G. Dallner and L. Ernster, Free Radical Biol. Med., 1999, 26, 318–324 CrossRef CAS PubMed.
- M. C. F. Messias, G. C. Mecatti, D. G. Priolli and P. de Oliveira Carvalho, Lipids Health Dis., 2018, 17, 41 CrossRef PubMed.
- E. Cifkova, M. Holcapek, M. Lisa, D. Vrana, J. Gatek and B. Melichar, Anal. Bioanal. Chem., 2015, 407, 991–1002 CrossRef CAS PubMed.
- D. L. Marshall, A. Criscuolo, R. S. E. Young, B. L. J. Poad, M. Zeller, G. E. Reid, T. W. Mitchell and S. J. Blanksby, J. Am. Soc. Mass Spectrom., 2019, 30, 1621–1630 CrossRef CAS PubMed.
- T. Baba, J. L. Campbell, J. C. Y. Le Blanc, P. R. S. Baker and K. Ikeda, J. Lipid Res., 2018, 59, 910–919 CrossRef CAS PubMed.
- S. R. Ellis, M. R. L. Paine, G. B. Eijkel, J. K. Pauling, P. Husen, M. W. Jervelund, M. Hermansson, C. S. Ejsing and R. M. A. Heeren, Nat. Methods, 2018, 15, 515–518 CrossRef CAS PubMed.
- U. Loizides-Mangold, FEBS J., 2013, 280, 2817–2829 CrossRef CAS PubMed.
- J. C. Holthuis, G. van Meer and K. Huitema, Mol. Membr. Biol., 2003, 20, 231–241 CrossRef CAS PubMed.
- R. Aviram, G. Manella, N. Kopelman, A. Neufeld-Cohen, Z. Zwighaft, M. Elimelech, Y. Adamovich, M. Golik, C. Wang, X. Han and G. Asher, Mol. Cell, 2016, 62, 636–648 CrossRef CAS PubMed.
- M. W. Hetzer, Cold Spring Harbor Perspect. Biol., 2010, 2, a000539 Search PubMed.
- A. Mohamed, H. Robinson, P. J. Erramouspe and M. M. Hill, Expert Rev. Proteomics, 2018, 15, 1053–1063 CrossRef CAS PubMed.
- T. Nishimura and C. J. Stefan, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2019 DOI:10.1016/j.bbalip.2019.07.001.
- B. Burla, M. Arita, M. Arita, A. K. Bendt, A. Cazenave-Gassiot, E. A. Dennis, K. Ekroos, X. Han, K. Ikeda, G. Liebisch, M. K. Lin, T. P. Loh, P. J. Meikle, M. Oresic, O. Quehenberger, A. Shevchenko, F. Torta, M. J. O. Wakelam, C. E. Wheelock and M. R. Wenk, J. Lipid Res., 2018, 59, 2001–2017 CrossRef CAS PubMed.
- R. Mamtani, J. D. Lewis, F. I. Scott, T. Ahmad, D. S. Goldberg, J. Datta, Y. X. Yang and B. Boursi, PLoS Med., 2016, 13, e1002007 CrossRef PubMed.
- M. Mansourian, S. Haghjooy-Javanmard, A. Eshraghi, G. Vaseghi, A. Hayatshahi and J. Thomas, J. Pharm. Pharm. Sci., 2016, 19, 72–81 Search PubMed.
- H. Yokomichi, A. Nagai, M. Hirata, A. Tamakoshi, Y. Kiyohara, Y. Kamatani, K. Muto, T. Ninomiya, K. Matsuda, M. Kubo, Y. Nakamura, BioBank Japan Cooperative Hospital Group and Z. Yamagata, J. Epidemiol., 2017, 27, S84–S91 CrossRef PubMed.
- N. M. Iyengar and L. W. Jones, JAMA Oncol., 2019, 5, 1620–1627 CrossRef PubMed.
- D. Lugo, A. L. Pulido, C. G. Mihos, O. Issa, M. Cusnir, S. A. Horvath, J. Lin and O. Santana, Complement. Ther. Med., 2019, 44, 9–13 CrossRef PubMed.
- R. T. Chlebowski, A. K. Aragaki, G. L. Anderson, C. A. Thomson, J. E. Manson, M. S. Simon, B. V. Howard, T. E. Rohan, L. Snetselar, D. Lane, W. Barrington, M. Z. Vitolins, C. Womack, L. Qi, L. Hou, F. Thomas and R. L. Prentice, J. Clin. Oncol., 2017, 35, 2919–2926 CrossRef CAS PubMed.
- M. Azrad, C. K. Blair, C. L. Rock, R. L. Sedjo, K. Y. Wolin and W. Demark-Wahnefried, Breast Cancer Res. Treat., 2019, 176, 649–656 CrossRef PubMed.
- C. M. Ulrich, C. Himbert, A. N. Holowatyj and S. D. Hursting, Nat. Rev. Gastroenterol. Hepatol., 2018, 15, 683–698 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |