
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurpassing 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) 000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy†
000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy†
Jan
Muntel‡
 ,
Tejas
Gandhi‡
,
Lynn
Verbeke
,
Oliver M.
Bernhardt
,
Tobias
Treiber
,
Roland
Bruderer
and
Lukas
Reiter
*
,
Tejas
Gandhi‡
,
Lynn
Verbeke
,
Oliver M.
Bernhardt
,
Tobias
Treiber
,
Roland
Bruderer
and
Lukas
Reiter
*
Biognosys AG, Wagistrasse 21, 8952 Schlieren, Switzerland. E-mail: lukas.reiter@biognosys.com
First published on 29th August 2019
Abstract
Comprehensive proteome quantification is crucial for a better understanding of underlying mechanisms of diseases. Liquid chromatography mass spectrometry (LC-MS) has become the method of choice for comprehensive proteome quantification due to its power and versatility. Even though great advances have been made in recent years, full proteome coverage for complex samples remains challenging due to the high dynamic range of protein expression. Additionally, when studying disease regulatory proteins, biomarkers or potential drug targets are often low abundant, such as for instance kinases and transcription factors. Here, we show that with improvements in chromatography and data analysis the single shot proteome coverage can go beyond 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 proteins in human tissue. In a testis cancer study, we quantified 11200 proteins using data independent acquisition (DIA). This depth was achieved with a false discovery rate of 1% which was experimentally validated using a two species test. We introduce the concept of hybrid libraries which combines the strength of direct searching of DIA data as well as the use of large project-specific or published DDA data sets. Remarkably deep proteome coverage is possible using hybrid libraries without the additional burden of creating a project-specific library. Within the testis cancer set, we found a large proportion of proteins in an altered expression (in total: 3351; 1453 increased in cancer). Many of these proteins could be linked to the hallmarks of cancer. For example, the complement system was downregulated which helps to evade the immune response and chromosomal replication was upregulated indicating a dysregulated cell cycle.
000 proteins in human tissue. In a testis cancer study, we quantified 11200 proteins using data independent acquisition (DIA). This depth was achieved with a false discovery rate of 1% which was experimentally validated using a two species test. We introduce the concept of hybrid libraries which combines the strength of direct searching of DIA data as well as the use of large project-specific or published DDA data sets. Remarkably deep proteome coverage is possible using hybrid libraries without the additional burden of creating a project-specific library. Within the testis cancer set, we found a large proportion of proteins in an altered expression (in total: 3351; 1453 increased in cancer). Many of these proteins could be linked to the hallmarks of cancer. For example, the complement system was downregulated which helps to evade the immune response and chromosomal replication was upregulated indicating a dysregulated cell cycle.
Introduction
Mass spectrometry-based proteomics has become a versatile tool for comprehensive proteome analysis.1 Up to now, a deep coverage was usually achieved by extensive fractionation. This enabled the quantification of more than 10000 proteins within one experiment2–4 and resulted in the first drafts of the human proteome in 2014.5,6
Fractionation-based approaches typically require a large amount of sample and MS time thereby limiting the throughput and complicating quantification. Therefore, methods for single-shot proteome analysis were developed further.7 A deep analysis of the yeast proteome in 2011 took 8 h8 and in 2014 it was possible to achieve an even greater depth in only about 1 h.9 This improvement in depth and analysis time were primarily driven by faster and more sensitive mass spectrometers.7 Nowadays, a comprehensive proteome coverage for less complex organisms, like yeast, can be routinely achieved in a single-shot analysis. However, it remains challenging for human tissue samples. Within the tissue, the proteome complexity is higher with an estimated expression of more than 10000 protein-coding genes and a higher dynamic range of protein expression.10,11 A milestone for mammalian tissues was achieved in 2018 in which >10000 proteins in a single-shot were detected using the BoxCar acquisition method and matching identifications to a previously acquired fractionated dataset.12 In the same year, a comprehensive single-shot proteome analysis was achieved using high-field asymmetric waveform ion mobility spectrometry (FAIMS) resulting in identification of >8000 proteins.13 A similar depth was also achieved using online parallel accumulation-serial fragmentation (PASEF) on the timsTOF Pro mass spectrometer.14
Data-independent acquisition (DIA) methods showed great potential for comprehensive single-shot proteome analysis. First introduced in the mid-2000s,15,16 various studies up to now showed that these methods enabled an accurate, precise and comprehensive proteome quantification.17–23 By fragmenting the interesting mass range in sequential, wide isolation windows (SWATH-type DIA approach24,25), it was possible to identify more peptides in short gradients than could be theoretically targeted in a sequential manner using data-dependent acquisition (DDA).26
Commonly, DIA data are analysed using a project-specific library. Such a library contains fragmentation information from previously acquired DDA data.24 For a project-specific library the DDA data are acquired using the same LC-MS setup and the same samples as used for the analysis of the samples by DIA. This generates an overhead to the DIA experiment. To avoid the overhead, publicly available resource libraries were utilized in the DIA data analysis.26,27 In an alternative approach, fasta-database search tools for DIA data were developed, e.g. DIA-Umpire,28 Pecan29 or DirectDIA in Spectronaut. Compared to library-based analysis of the DIA data, a database search resulted in less identified proteins, but an improved quantitative performance.30
A marriage of both of these approaches, i.e. combining data from a database search of DIA data with results of a search of DDA, holds the potential to the improve DIA data analysis in terms of quantitative precision and number of quantified proteins.31,32 Such a workflow has to address three key challenges: (1) degradation of indexed retention time (iRT) precision33 in the library as a result of potentially heterogenous chromatography, (2) homogeneous protein inference and false discovery rate (FDR) control when merging data from several sources to build the library, and (3) robust protein FDR control during DIA analysis. A solution to the RT precision proposed by MacCoss group32 utilized data from a narrow window DIA method to calibrate a protein database or library with chromatographic and MS-specific parameters. Whereas this approach greatly improved the number of quantified peptides and proteins in classical as well as resource libraries, the FDR estimate remained challenging and required additional tools. Furthermore, it required a new calibration of the library after each variation of the chromatography, e.g. column change.
Here, we propose a combined DIA data analysis strategy named hybrid library workflow which solves the three above mentioned challenges. The advantage of this approach is that it does not require any special calibration runs and the FDR control is robustly maintained throughout the entire pipeline at the library and DIA analysis level.34,35
The recent improvements in single-shot proteome analysis were mainly driven by development of better liquid chromatography, mass spectrometers, acquisition methods and data analysis strategies. On the liquid chromatography side, the last major progress was the introduction of commercial ultra-high pressure pumps which enabled the routine use of reversed phase sub-2 μm particles and long columns (>20 cm).7,36,37 Only recently, efforts have been made to improve the packing of the column by ultra-high pressure packing of columns38 and novel micro-pillar array columns have been introduced for proteomics applications.39
Today, sub-2 μm C18 solid phases are available with different chemistries. Interestingly, little work has been published to compare these particles in the context of single-shot proteomics. Hence, we decided to compare three different C18 solid phases that are commonly used in proteomics. We optimized the gradient shape and length to maximize the proteome coverage for single-shot experiments. In addition to the optimization of the chromatography, we introduce the afore-mentioned hybrid library approach for the analysis of the DIA data. Finally, we applied this optimized workflow in a small testis cancer study.
Testis tissue is one of the most complex human tissues.40–42 Due to the major role of the testis in the human reproduction system, it was studied mainly under the aspect of male fertility,43 and very little efforts have been made to study cancer in testis. In contrast to other cancers, the frequency of testis cancer peaks at an age of about 35 years similar to other germ cell cancers.44
The limited number of proteomic testis cancer studies and the high complexity of the tissue were the reasons, why we chose this tissue to demonstrate the large potential of our workflow for a comprehensive single-shot proteome analysis. Here we show the quantification of >10000 proteins at 1% FDR on precursor and protein level in testis tissue and how these data can serve for a better understanding of the molecular mechanisms of the cancer development.
Results and discussion
Optimization of chromatography (solid phases and gradient length) led to an increase of 67% in quantified precursors and of 36% in quantified proteins
First, we compared three sub-2 μm solid phases, widely used in proteomics, namely ReproSil Pur (1.9 μm), which is the standard phase in our facility and two solid phases from waters: BEH and CSH (both 1.7 μm). A 50 cm column was packed with each solid phase and tested using 2 μg of a HeLa digest. The MS data were collected with a Q Exactive HF-X mass spectrometer using a DIA method, which was adjusted to the peak width of the tested solid phases. The parallel nature of DIA has the advantage that the identification (ID) rate is not directly dependent on the scan speed of the mass spectrometer.26 Additionally, we investigated the influence of the flow rate on the number of quantified precursors, peptides and proteins. Three flow rates were tested on 2 h gradients: 200, 250 and 300 nl min−1. To avoid the overhead of library generation, we directly searched the DIA data using a fasta file (DirectDIA) during the optimization experiments. Using just a single library can introduce biases and the generation of a separate DDA based library for every optimization parameter is an unnecessary overhead.With all setups, we quantified between 77519 (ReproSil Pur, 300 nl min−1) and 82060 precursors (CSH, 250 nl min−1) (Fig. 1A and Table S2A, ESI†). Across all flow rates, we achieved the highest numbers of quantified precursors, peptides and proteins with the CSH solid phase; on average 5% more precursors were quantified in comparison to the ReproSil solid phase and 3% more peptides in comparison to the BEH solid phase. Similar trends were observed on peptide (+7% compared to ReproSil, +5% compared to BEH) and protein level (+8% compared to ReproSil and +5% compared to BEH). Interestingly, 250 nl min−1 was the optimal flow rate for all tested solid phases. For the CSH solid phase, we quantified 3% more precursors at 250 nl min−1 compared to 200 nl min−1 and 300 nl min−1 and observed similar trends on peptide and protein level. Additionally, these findings from the DirectDIA analysis were confirmed with a project-specific HeLa library (Fig. S1 and Table S2B, ESI†). It was possible to quantify in average 55% more precursors and peptides as well as 27% more proteins as compared to DirectDIA. Importantly, the qualitative differences between the solid phases and flow rates were consistent with the DirectDIA analysis. This demonstrated that the DirectDIA analysis is an effective alternative for experiments in which relative differences between workflows are under investigation and a high depth of the analysis is not required.
 | ||
| Fig. 1 Optimization of liquid chromatography. (A) Comparison of three solid phases using a DIA method, 2 h gradients and a HeLa sample. Gradient shape and DIA method were optimized for all solid phases. DIA data were analysed by DirectDIA. (B) Comparison of gradients lengths using the CSH solid phase, a DIA method and a HeLa sample. Gradient shape and DIA method were optimized for each length. Average run identifications and CVs <20% (orange bars) were calculated based on the data of the triplicate injection. | ||
Overall the differences in quantified precursors, peptides and proteins were rather small between the three tested solid phases and flow rates. Nevertheless, the results from the CSH phase showed a consistent better performance and 250 nl min−1 resulted in the highest number of quantified precursors, peptides and proteins. Therefore, the CSH phase and a flow rate of 250 nl min−1 was further used in this study.
Next, we incrementally increased the length of the gradient. We started with a 2 h gradient and ramped in 2 h steps up to 8 h. Again, the number of quantified precursors and proteins was compared using a HeLa sample and the DirectDIA analysis strategy. Each extension of the gradient increased the number of quantified precursors, peptides and proteins (Fig. 1B and Table S3A, ESI†). The largest improvement was observed by extending the gradient from 2 h to 4 h. The average number of quantified precursors in a triplicate measurement increased significantly by 38% from 86935 to 119992 (p = 3 × 10−7, based on two-sample t-test), on peptide level by 30% (p = 4 × 10−6) and on protein level by 17% to 6364 proteins (p = 1 × 10−4). Further, extension of the gradient to 6 and 8 h resulted in 7% more quantified precursors for each step, 9 to 6% more quantified peptides and 6 to 4% more quantified proteins. The highest number of quantified proteins was achieved by the 8 h gradient. In average 6970 proteins were quantified in a triplicate run. We noticed that for the 6 and 8 h gradients, the identification reproducibility decreased. The CV of the precursor identification was for the 2 h gradient 0.9% and increased to 5.4% for the 8 h gradient (compare error bars in Fig. 1B). This indicated that the shallow gradients with a very slow increase in percentage B became less reproducible. Because of this observation, we decided to analyse the number of precursors, peptides and proteins that were quantified with a CV below 20% based on an injection triplicate. This analysis allowed us to assess the quantitative precision of the measurements as well as the number of quantified precursors, peptides and proteins. As before, we observed the largest improvement by extending the gradient from 2 h to 4 h (+29% precursors, +26% peptides, +16% proteins). By further extending the gradient, we found a maximum of precursors with a quantitative CV below 20% for the 6 h gradient (95229 precursors). The same was true on peptide (61755) and protein level (4787). To exclude that the DirectDIA analysis might have introduced the lower quantitative precision for the 8 h gradient, the sample set was re-analysed with the HeLa project-specific library (Fig. S1B and S3B, ESI†). Again, this analysis revealed a higher sensitivity as shown by 54% more quantified precursors, 52% more peptides and 23% more proteins compared to the DirectDIA analysis. Importantly, the relative differences in quantified precursors, peptides and proteins between the gradient lengths as well as the respective numbers with a quantitative CV below 20% were consistent with the DirectDIA analysis. Due of the higher quantitative precision of the data generated with the 6 h gradient, we decided to use this gradient henceforth.
Overall with the chromatographic improvements using the CSH solid phase and extension of the gradient to 6 h, we were able increase the number of quantified precursors from 77184 to 128718 (+67%) and the number of quantified proteins from 4923 to 6712 (+36%).
Introduction of hybrid libraries increased the number of quantified proteins by 26% compared to DirectDIA analysis
The previous analyses showed that the DirectDIA approach is a valuable analysis method for workflow optimization, but it also showed limitations in sensitivity of the analysis compared to a project-specific library-based analysis. Typically, less precursors, peptides and proteins were quantified by DirectDIA. This was already shown previously, but on the other hand the DirectDIA analysis proved to provide a better quantitative performance than the library-based analysis.30 To combine the benefits of both approaches, we developed a hybrid library approach.There are three main challenges involved with this type of a workflow: (1) degradation of retention time precision from combining data with heterogenous chromatography, (2) loss of FDR control at the library level, and (3) robust protein FDR control during the DIA analysis. For DIA data analysis, we generally convert retention times into indexed retention times (iRTs),33 which are dimensionless and can be utilized for highly accurate retention time calibration.45
To solve the first challenge of degradation of retention time specificity, we approached it by allowing libraries with different iRT spaces instead of combining them into one. This means that each peptide in a library can have multiple associated empirical iRTs, if it was identified from multiple sources. This additional information in the library can be exploited during the DIA analysis in the form of source-specific RT to iRT calibration. This means that, if a peptide was identified both from DirectDIA and a DDA dataset, the retention time information from the best RT to iRT calibration will be used. The benefit of this approach is that the library moulds itself to best fit the DIA data without needing to recalibrate the library with each change in chromatography. The second challenge for this workflow is to maintain the FDR control at the library level. We solved this with our database search engine Pulsar and the introduction of search archives. Search archives are a collection of all PSMs identified without any FDR filtering (i.e. it contains the computationally expensive database search results). This enabled to combine previously searched DIA or DDA data with the target DIA runs in a manner that is both computationally efficient and FDR controlled. The average time to generate a library was reduced by >90% using search archives (Fig. S2, ESI†). Finally, we adapted the FDR calculation to account for the heterogeneity in the iRT space being targeted by building one separate precursor FDR model for each iRT source and normalizing the scores afterwards. We validated this approach by performing an empirical two species FDR test (Fig. 2A).
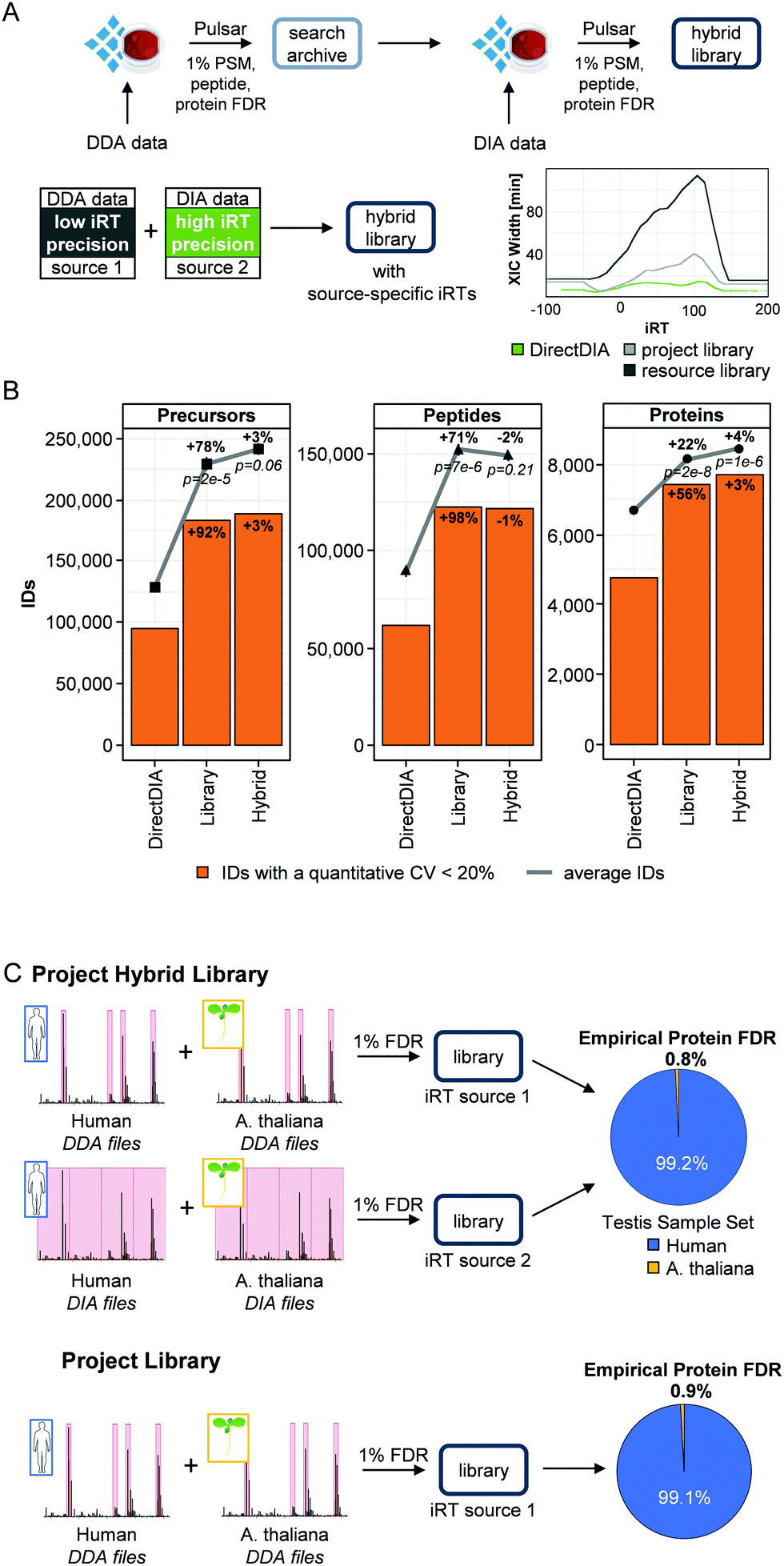 | ||
| Fig. 2 Hybrid library approach. (A) Workflow scheme. Database searches using Pulsar can be performed in Spectronaut or SpectroMine. XIC widths were extracted after analysis of a HeLa injection triplicate in Spectronaut. The HeLa project-specific library was generated based on high pH reverse-phase fractionation and the resource library was based on data published by Kim et al., 2014. (B) Comparison of DirectDIA, project-specific library and hybrid library for DIA data analysis of a triplicate injection of a HeLa sample. Average run identifications and CVs <20% (orange bars) were calculated based on the data of the triplicate injection. (C) Empirical Protein FDR Validation. The DDA files of the testis project library and of the A. thaliana project library were searched together to create the same iRT source for data analysis. The respective DIA files were also searched together. Both libraries were then applied for the analysis of the testis cancer set and the empirical protein FDR was calculated as the percentage of A. thaliana protein identifications within all identified proteins. | ||
To test the hybrid library approach, we combined the project-specific library and the DirectDIA data (hybrid library size: 415002 precursors, 231791 peptides and 10624 proteins, Fig. S3, ESI†). This hybrid library was applied to the injection triplicate of the 6 h HeLa runs. The performance was evaluated using the total number of quantified precursors, peptides and proteins as well as the respective quantifications with a CV below 20% (Fig. 2B and Table S4, ESI†).
As already shown in the previous analysis, the library-based analysis of the samples improved the number of quantified precursors (+78%, p = 2 × 10−5), peptides (+71%, p = 7 × 10−6) and proteins (+22%, p = 2 × 10−8) significantly compared to the DirectDIA analysis. Application of the hybrid library led only to a minor increase in quantified precursors (+3%) and proteins (+4%). The number of quantified peptides even slightly decreased in this analysis (−2%). The same was true for the quantifications with a CV below 20%. The overlap of quantified precursors, peptides and proteins was high between the three different analysis strategies (86% of the precursors and 88% of the peptides identified by DirectDIA were also identified by the other strategies, Fig. S3B, ESI†). The overlap on protein level was especially high; only <1% to 3% of the proteins were exclusively identified by only one approach.
It is noteworthy that we were able to quantify on average 8465 proteins and 7712 proteins with a quantitative CV below 20% within 6 h using the hybrid approach. This result represents a substantial improvement, especially in number of quantified proteins with a CV below 20%, to a recent comprehensive single-shot proteome study. In this publication more than 8000 proteins were identified in 5 h from which 6444 proteins were quantified with a CV below 20% in a human cell line sample.13
Analysis of human testis tissue resulted into the quantification of >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 000 proteins per run at 1% protein FDR
000 proteins per run at 1% protein FDR
After optimization of the liquid chromatography and development of the hybrid library approach, we were interested how well the single-shot proteome analysis would perform on a human tissue sample. We chose a small testis cancer sample set, consisting of three cancer samples and three near adjacent healthy tissue (NAT) samples. First, we generated a deep project-specific library using a pool of the NAT and a pool of the cancer samples. Both pooled samples were subjected to high-pH reverse phase fractionation and 20 fractions were generated. All fractions were analysed using DDA. The library comprised 436883 precursors, 255432 peptides and 13436 proteins representing the largest published proteomic dataset for a single tissue to our best knowledge (as compared to 11558 proteins in testis tissue reported by Sun et al.42). Additionally, we generated a hybrid library by combing the project library with a DirectDIA analysis of the samples (Fig. S4A, ESI†). We were also interested whether it would be possible to replace the project-specific library by a resource library. For this purpose, we used the raw files from the Kim et al. publication5 and generated a resource and a resource hybrid library. The sizes of these libraries were similar to our project-specific libraries (Fig. S4A, ESI†).
First, we analysed an injection triplicate of one of the testis cancer samples with the above-mentioned libraries. Using the project-specific hybrid library, we were able to quantify on average 10146 proteins per run (263791 precursors, 163825 peptides), which represented an improvement of 6% on protein level, 1% on peptide and 10% on precursor level compared to the project-specific library (Table S5A and B, ESI†). Out of the 10146 proteins, 8783 were quantified with a CV below 20% (Fig. S4B, ESI†). Notably, the largest increase in quantified precursors (+27%), peptides (+12%) and proteins (+10%), including the numbers with a CV below 20% (precursors: +16%, peptides: +14%, proteins: +10%), for the hybrid library was found by application of the hybrid library approach to the resource library (Fig. S4B, Tables S5C and D, ESI†). We noticed the largest overlap in quantified precursor and peptide between all four different analysis approaches (Fig. S4C, ESI†). The second largest overlap was found between project and project hybrid library, followed by the overlap of these two libraries to the resource hybrid library. This result was expected as these three libraries contained most of the sample-specific precursors and peptides. As for the HeLa dataset, the overlap on protein level was much higher compared to precursor and peptide level (76% of all quantified proteins were quantified by four libraries).
Based on these results and the results from the analysis of the HeLa using the hybrid library approach, we concluded that the benefit of the hybrid library depends on the size and quality of the initial library. For the HeLa dataset, we used a single HeLa digest for library generation and DIA analysis and observed minor differences in quantified precursors (+3%), peptides (−2%) and proteins (+4%) in the DIA experiment. The initial project-specific library provided already a very high coverage of the detectable precursors, peptides and proteins. A larger improvement was noticed for the testis sample (precursors: +10%, peptides: +1%, proteins: +6%). For the project-specific library, we created two condition pools each comprising three biologically different samples. In this commonly used strategy for DIA experiments, sample-specific proteins could be potentially missed, because of dilution of these proteins in the pooled sample. This effect has been described previously for a protein spike-in experiment in complex background30 and will likely get more prominent the more samples are pooled to generate the project-specific library. The largest improvement in quantified precursors (+27%), peptides (+12%) and proteins (+10%) using the hybrid library approach was found for the resource library which was based on DDA data from unrelated samples and on a different LC-MS setup. This finding showed that the hybrid library approach improved the usability of resource libraries by addition of sample-specific proteins, that were not part of the resource library.
During DIA data analysis, the FDR was calculated based on a target-decoy model. To experimentally cross validate the FDR estimates, we performed a two-species test using Arabidopsis thaliana as negative set and the testis cancer set (human) as positive set (Fig. 2C). For validation of the project hybrid library, we made two different libraries: (1) DDA library by searching human testis DDA runs together with A. thaliana DDA runs, and (2) DIA library by searching human testis DIA runs together with A. thaliana DIA runs. All the A. thaliana runs were acquired using the same setup as the testis data. In this manner, we had two different sources as before (DDA and DIA) but with a built-in negative set. Afterwards we used these libraries for the analysis of the DIA data from the testis cancer set. The empirical FDR was now calculated as the ratio of the identified human to A. thaliana proteins, which came out to be 0.8% (Table S7, ESI†). Additionally, we used the DDA data-based library only to validate the FDR of the project library, which was 0.9% (Fig. 2C). We concluded that the hybrid library approach did not inflate the protein FDR.
This was only the second time that more than 10000 quantified proteins in a single-shot were reported. In 2018 Meier et al. reported more than 10000 proteins detected in a single run by using the BoxCar acquisition method and an alignment strategy.12 Whereas an FDR control is difficult using an alignment strategy and generally not performed at all, FDR control on precursor and protein level is commonly applied in DIA experiments46 and was empirically validated for this study using an A. thaliana library as negative set.
Increased depth of analysis revealed deep insights into the cancer physiology
In the second part of the study, we analysed a testis cancer set (3 cancer and 3 NAT samples). All patients were diagnosed with seminoma cancer. Testis cancer is rare and has usually a good prognosis.47 Therefore, this kind of cancer has not been extensively studied by proteomics, so far.In total, we quantified 11197 proteins (including 715 one-peptide identifications) and 10554 proteins in average per sample (Fig. 3A and Table S6A, ESI†). The dataset covered 6 order of magnitude dynamic range of protein abundance (Fig. 3B). A median biological CV on protein level of 23% for the NAT and of 26% for the cancer cohorts indicated a good quantitative precision and we noticed only a minor dependency of the quantitative precision on the protein abundance. For the lowest abundant 2000 proteins, we determined a CV (including biological and technical variance) of 30% for the NAT cohort and 32% for the cancer cohort and for the 2000 highest abundant proteins 20% for the NAT and 23% for the cancer samples (Fig. 3B). Differential abundance between the NAT and cancer cohort was determined using a t-test including multiple testing correction with the method described by Storey48 (as implemented in Spectronaut; unfiltered candidate list: Table S8, ESI†). Proteins were considered differentially abundant with an absolute log2 fold-change larger than 1 and a Q value below 0.01. We found in total 3178 proteins in an altered amount between the cancer and NAT samples. Of these proteins, 1453 were found in an increased abundance in the cancer cohort and 1725 proteins in a lower abundance (Fig. 3C). This finding demonstrated a large impact on the proteome by the cancer, which was also reported already for cancer in other tissues.49,50
 | ||
| Fig. 3 Overview of testis cancer set. (A) Overview of identified precursors, peptides and proteins. Light coloured protein identifications indicate proteins identified with only one peptide. (B) Dynamic range of quantified proteins including biological quantitative CVs of the cancer cohort (red) and the NAT cohort (green) across all abundance ranges (CVs were calculated for bins of 2000 proteins). (C) Volcano plot of the statistical comparison of the NAT and cancer cohort (t-test). Proteins were considered candidates (red dots) with an absolute log2 fold change >1 and a Q value <0.01. | ||
As comparison we also analysed the testis cancer set with the 2 h and 4 h DIA method. Whereas with a 2 h gradient the number of identified proteins was still below the 10000 mark (on average 7610 and in total: 8488, Table S6B, ESI†), it was possible to identify in total 10404 proteins, but on average just below 10000 (9658, Fig. S5A and Table S6C, ESI†). Interestingly the number of differentially abundant proteins within the 4 h dataset (3351) was comparable to the 6 h dataset (3178, Fig. S5B, ESI†). Additionally, we also analysed the overlap of the candidate lists derived from the analysis of the data of the three different gradient lengths (Fig. S5C, ESI†). Overall, we found the largest overlap in candidates between the 4 h and 6 h dataset (∼80% of the candidates from both gradients). The overall overlap was large (1657 proteins) with only a low percentage of proteins quantified statistically significant in only one dataset (between 12 and 14%).
Unsupervised clustering showed a clear differentiation between the NAT and cancer samples. This finding indicated that the quantitative differences between the samples were driven by the underlying biological changes (Fig. 4A). For biological interpretation, the quantitative data were loaded into Ingenuity Pathway Analysis (IPA, Qiagen). The liver X receptor/retinoid X receptor (LXR/RXR) activation pathway (Fisher's exact test, p = 4.2 × 10−13), the complement system (p = 5.8 × 10−12), the acute phase response signalling (p = 2.2 × 10−11), the coagulation system (2.4 × 10−11) and the farnesoid X receptor/retinoid X receptor (FXR/RXR) activation pathway showed up as the top 5 pathways (top 10 pathways in Fig. 4B). In these pathways, the majority of the proteins were quantified in a lower amount in the cancer cohort. Only in the 6th most enriched pathway, cell cycle control chromosomal replication (p = 3.4 × 10−9), most of the proteins were found in a higher abundance in the cancer cohort.
 | ||
| Fig. 4 Biological Interpretation. (A) Empirical clustering of the data and heatmap visualization (based on intensities). (B) Top10 pathways of IPA analysis. Downregulated proteins were labelled blue (dark blue: Q value <0.01) and upregulated proteins were labelled orange (dark orange: Q value <0.01). White bars show the percentage of proteins in the pathway that were not identified. (C) Overview of complement system from the interpretation of the quantitative data in IPA (Qiagen). Bar chart depicts the quantification data for the proteins in this pathway. Stars indicate proteins that were quantified with a Q value <0.01. The shapes of the proteins in the pathway indicate different protein classes. (D) Overview of cell cycle control of chromosomal replication exported from IPA, including the quantitative data similar to Fig. 4C. | ||
The combined down-regulation of the LXR/RXR pathway and acute phase response signalling pathway was widely observed in proteomic studies for various cancer types, e.g. for colon adenocarcinomas,51 for triple-negative breast cancer (in combination with a downregulation of the FXR/RXR activation pathway,52 which was also found here) and in the urine of prostate cancer patients.53 LXR acts as sensor for cholesterol homeostasis and in normal cells the pathway is activated by high intracellular cholesterol concentrations to reduce synthesis and influx and enhance cholesterol efflux. Therefore, this pathway is typically downregulated in cancer cells to accumulate high intracellular cholesterol concentrations which are required to sustain a high growth rate.54 Interestingly, the cholesterol biosynthesis proteins were quantified in a lower amount in the cancer (average log2ratio = −2.1, Q value between 0.01 and 9.1 × 10−37). Additionally, we found the low-density lipoprotein receptor (LDLR), the major extracellular cholesterol capture protein, in a lower abundance in the cancer cohort (log2ratio = −3, Q value = 9 × 10−9). Cholesterol plays also an important role in apoptosis. Several death receptors are located in cholesterol-rich lipid rafts which trigger an apoptotic signal upon activation. Cancer cells can avoid apoptosis by modulating the composition of the lipid rafts leading to disruption of these receptors.55,56 Our data indicated by a lower expression level of two important classes of death receptors, the tumor necrosis factor receptor superfamily member 6 (FAS, log2ratio = −1.2, Q value = 2 × 10−9) and the tumor necrosis factor receptor superfamily member 10B (TNFRSF10B, log2ratio = −1.8, Q value = 0.03), that the testis cancer cells might use this strategy to avoid apoptosis.
It has also been shown that an activation of LXR leads to a cell-cycle arrest through downregulation of the S phase-associated kinase protein-2 (SKP2).57 In accordance with the downregulation of the LXR/RXR pathway in our dataset, SKP2 was quantified in an increased amount in the cancer samples (log2ratio = +1.4, Q value = 0.02). Because of the involvement of LXR in several cancer-related adaptations, an activation of the pathway is under investigation to facilitate cancer treatment in e.g. colon,58 prostate59 or gastric cancer.60
The second most significantly enriched pathway was the complement system (Fig. 4C). Almost all proteins of the pathway were quantified in decreased levels in the cancer samples (average log2ratio = −1.9, Q values between 0.2 and 4.6 × 10−173) with two exceptions: Integrin beta-2 (ITGB2, log2ratio = +2.1, Q value = 1 × 10−18) and the complement component 1 Q subcomponent-binding protein (C1QBP, log2ratio = +1, Q value = 2 × 10−5). Especially the finding of the elevated level of C1QBP was interesting because it acts as an inhibitor of the complement system and previous studies showed that cancer cells inhibit the complement system to escape immune response.61,62 Therefore, C1QBP was discussed as potential target for therapeutics.63
Cell cycle control chromosomal replication was the most enriched pathway (p = 3.4 × 10−9), in which most proteins were quantified in elevated levels in the cancer cohort (Fig. 4D, average log2ratio = +1.4, Q value between 0.03 and 3 × 10−52). This finding was to be expected as DNA replication is an important step in cell proliferation and a dysregulated cell cycle was described as one of the hallmarks of cancer.64 Therefore, cell cycle regulators are considered good targets in cancer therapy.65 Of special interest for therapy are the cyclin dependent kinases (CDKs). Promising results were achieved for CDK4 and CDK4 inhibitors,65 which both were not quantified in altered abundances in our study (CDK4: log2ratio = −0.5, Q value = 0.01; CDK6: log2ratio = 0, Q value = 0.08). Even though, our approach did not allow to measure the activity of these kinases, it indicated that the kinases would not be good targets in testis cancer. In contrast, CDK1 and CDK2 were quantified in significantly higher amounts in the cancer cohort (CDK1: log2ratio = +1.3, Q value = 3.3 × 10−12; CDK2: log2ratio = +1.1, Q value = 5 × 10−9). Thus, our data indicated that inhibition of CDK1 and CDK2 are potentially better targets for testis cancer treatment. Both kinases were already investigated as potential targets66,67 in cancer therapy. Additionally, several proteins of the cell cycle were linked to poor cancer prognosis, e.g. an increased expression of minichromosome maintenance proteins (MCMs, average log2ratio in our dataset = +1.8, Q value between 2.3 × 10−30 and 3 × 10−52) in breast cancer68 or colon cancer.69
Replication stress is regarded as one reason leading to genomic instability in cells and ultimately to the development of cancer. During replication stress the DNA replication fork progression slows down or stalls in S phase due to a high transcriptional activity leading to DNA double-strand breaks.70 Interestingly, we found besides an increase of the proteins involved in replication, suggesting an increased replication rate, that also the mismatch repair pathway was enriched in the IPA analysis (Fig. S6A, ESI,†p value = 6 × 10−7; average log2ratio = +1.3, Q values between 1.6 × 10−10 and 2.1 × 10−71). To support the hypothesis that this finding might be related to replication stress, we investigated the expression levels of the DNA-directed RNA polymerases, which are often dysregulated in cancer.71 The expression levels of the RNA polymerases were actually increased by an average log2ratio of +1; especially the proteins of the RNA polymerase I complex (average log2ratio = 1.2; Q values between 7.9 × 10−3 and 1.4 × 10−32; Fig. S6B, ESI†). A previous study linked the replication stress to an elevated expression of the general transcription factor TATA-box binding protein TBP.72 The expression level of TBP was also increased (log2ratio = +1, Q value = 1.2 × 10−4) in the cancer cohort indicating a potential role of TBP in replication stress in testis cancer.
Conclusions
The optimization of liquid chromatography and the introduction of the hybrid library data analysis strategy enabled us to identify and quantify more than 10000 proteins in a single DIA run. Our strategy allowed FDR controlled precursor and protein identification, which was also empirically validated for the dataset. We developed a new DIA analysis pipeline, namely hybrid library workflow, which combines the potential to generate libraries from the DIA runs with DDA based libraries. Our workflow is especially well-suited to exploit the depth of large resource data while keeping the iRT-precision of project-specific data.
The potential of the comprehensive single-shot proteome analysis workflow was exemplified on a small testis cancer study with a coverage of 11200 proteins. The high coverage of the testis cancer proteome enabled an in-depth analysis of several hallmarks of cancer like evasion of immune responses by downregulation of the complement system or a dysregulation of the cell cycle. These data showed how these pathways could be investigated down to the level of transcription factors (104 transcription factors were quantified based on protein description). This analysis demonstrates that deep proteome analysis paves the way to a better understanding of underlying molecular mechanisms of disease and helps to identify potential targets for disease treatment (339 of 516 described human kinases, according to https://www.uniprot.org/docs/pkinfam, were quantified as potential drug targets).
In the future, we imagine that additional ion separation devices like FAIMS13 or TIMS14 have the potential to further increase the depth of single-shot proteome analysis and/or to achieve such a high coverage with shorter gradients.
Material and methods
All chemicals were purchased from Sigma (St Louis, MO), otherwise the vendor is mentioned.Sample preparation
HeLa cells were purchased from Ipracell (Mons, Belgium). The fresh frozen testis cancer samples were obtained from Proteogenex (Los Angeles, CA). The sample set consisted of three seminoma cancer tissue samples plus three near adjacent tissue (NAT) samples as control. The tumour content of the cancer cohort was between 90 and 100%.Cells/tissue samples were lysed in lysis buffer (8 M urea, 0.1 M ammonium bicarbonate) using the TissueLyzer II (Qiagen, Heidelberg, Germany) with following settings: 3 cycles, 30 beats per s, 30 s. DNA was sheared using sonication in the Bioruptor (Diagenode, Seraing, Belgium) using following settings: 5 cycles, 30 s ON, 30 s OFF, 4 °C, high intensity. After clearing of the lysates by centrifugation (20 min, 16000 × g, room temperature), aliquots were reduced by parallel treatment with 10 mM tris(2-carboxyethyl)phosphine (TCEP) and 40 mM 2-chloroacetamide (CAA) for 1 h at 37 °C. Afterwards the urea concentration was lowered to 1.5 M by addition of 0.1 M ammonium bicarbonate buffer and digested by trypsin (1 to 100 ratio, Promega, Madison, WI) over night at 37 °C. Peptides were purified using MacroSpin clean-up columns (NEST group, Southborough, MA) following manufacturers protocol. Eluates were dried completely in a speed-vac (Savant SPD131DDA, Thermo Fisher Scientific, San Jose, CA). The samples were resuspended in buffer A (1% acetonitrile, 0.1% formic acid in water) containing iRT peptides (Biognosys, Schlieren, Switzerland). Peptide concentration were determined using nano-drop (Spectrostar Nano, BMG labtech, Ortenberg, Germany) and adjusted to 1 μg μl−1.
LC-MS analysis (data-independent acquisition, DIA)
For all optimization steps, 2 μg of the HeLa digest were injected (1 μg h−1). The different solid phases (C18 materials) were tested to optimize the chromatography, namely ReproSil Pur (1.9 μm, Dr Maisch, Ammerbuch, Germany), CSH (1.7 μm, Waters, Milford, MA) and BEH (1.7 μm, Waters, Milford, MA). The analytical columns were in-house packed into fritted tip emitters to a length of 50 cm (ID 75 μm, New Objective, Woburn, MA). The columns were operated using an Easy nLC 1200 (Thermo Fisher Scientific, San Jose, CA) coupled online to a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific). Peptides were eluted at three different flow rates (200 nl min−1, 250 nl min−1, 300 nl min−1) by a non-linear 2 h gradient from 1% buffer B (85% acetonitrile, 0.1% formic acid in water)/99% buffer A (0.1% formic acid in water) to 45% buffer B.Afterwards the non-linear gradient was ramped in steps of 2 h up to 8 h at a flow rate of 250 nl min−1 on the CSH column. For the chromatography optimization, the mass spectrometer was operated in data-independent acquisition (DIA) mode using following parameter for the MS1 scan: scan range: 350 to 1650 Th; AGC target: 3e6; max injection time: 20 ms; scan resolution: 120000. The MS1 was followed by DIA scan events with following settings: AGC target: 3e6; max injection time: 55 ms; scan resolution: 30000; first fixed mass: 200 Th; stepped normalized collision energy: 25.5, 27, 30. The number of DIA windows and the window widths were adjusted to the precursor density and to achieve 4–5 datapoints per peak for each experiment separately. The windows overlapped by 0.5 Th (window design: Table S1, ESI†). For the 6 h and 8 h method, the resolution of the DIA scan was increased to 60000. For a better calculation of the AGC target by the mass spectrometer two MS1 scans were acquired for the 6 h method (after half of the DIA scans) and three MS1 scans for the 8 h method (after a third of the DIA scans). The scan ranges of these additional scans were adjusted to the MS1 range covered by the subsequent DIA scans: for the 6 h method: 1st MS1 scan: 350 to 620 Th, 2nd MS1 scan: 600 to 1650 Th and for the 8 h method: 1st MS1 scan: 350 to 540 Th, 2nd MS1 scan: 530 to 720 Th, 3rd MS1 scan: 710 to 1650 Th.
For the testis dataset, 4 μg of the testis digests were injected (2/3 μg h−1), and the samples were analyzed by the 6 h gradient at a flow rate of 250 nl min−1 on the 50 cm CSH column using the above described 6 h DIA method.
LC-MS analysis (data-dependent acquisition, DDA)
A tissue specific library was generated for the analysis of the HeLa data as well as for the analysis of the testis samples. For the testis library, a pooled sample (in total 400 μg) of the NAT tissues as well as a pooled sample of the cancer cohort (400 μg) were fractionated using high pH reverse phase (HPRP) fractionation. For the HeLa library, 400 μg of peptides were subjected to HPRP fractionation. The fractionation was performed using a Dionex Ultimate 3000 LC (Thermo Scientific, Sunnyvale, CA) using an ACQUITY UPLC CSH1.7 μm C18 column (2.1 × 150 mm, Waters, Milford, MA). Peptides were separated by a 30 min non-linear gradient from 1% HPRP buffer B (100% acetonitrile)/99% HPRP buffer A (20 mM ammonium formiate, pH 10) to 40% buffer B. A micro fraction was taken every 45 s and pooled into 20 final fractions.73 Pooled fractions were dried completely by vacuum centrifugation and resuspended in 20 μl buffer A containing iRT peptides and peptide concentrations were determined by nano-drop.To generate the DDA data for the library, the same LC-MS setup as described for the DIA acquisition was operated in data-dependent Top20 mode. Peptides were separated by a non-linear 3 h gradient on the 50 cm CSH column. Following settings for the Q Exactive HF-X mass spectrometer were applied: MS1 scan resolution: 60000; MS1 AGC target: 3e6; MS1 maximum IT: 20 ms; MS1 scan range: 350–1650 Th; MS2 scan resolution: 30000; MS2 AGC target: 1e6; MS2 maximum IT: 55 ms; isolation window: 4 Th; first fixed mass: 200 Th; NCE: 27; minimum AGC target: 1e3; only charge states 2 to 4 considered; peptide match: preferred; exclude isotopes: on; dynamic exclusion: 30 s.
Data analysis – library generation
For the resource library, the raw files from the Kim et al.5 publication were downloaded. These raw files were searched with SpectroMine 1.0.21621.9.18427 (Biognosys) against the Human UniProt FASTA including isoforms (downloaded on Jul 1st, 2018) using following settings: fixed modification: carbamidomethyl (C); variable modifications: acetyl (protein N-term), oxidation (M); enzyme: trypsin/P with up to two missed cleavages. Mass tolerances were automatically determined by SpectroMine and other settings were set to default. Search results were filtered by a 1% FDR on precursor, peptide and protein level.74,75 The libraries (for HeLa as well as for testis) were generated using the default values in SpectroMine.For the project library, the DDA files from the fractionated samples were searched in SpectroMine with the same settings mentioned above. Additionally, the DIA runs from the testis cancer set were searched with SpectroMine in the same way. To generate the hybrid libraries, the “generate library from search archive” option with the default settings was used to create the project hybrid library (combination of project library and DIA search) and resource hybrid library (combination of resource library and DIA search). The generation of hybrid libraries from search archives in SpectroMine has three advantages: (1) it avoids having to re-search the raw data (2) it enables homogeneous protein inference (3) and it guarantees a peptide and protein FDR of 1% on the complete data set.74
The LC-MS data, libraries, results tables and Spectronaut projects of the testis analysis have been deposited to the ProteomeXchange Consortium76 (http://proteomecentral.proteomexchange.org) via the PRIDE77 partner repository with the dataset identifier PXD013658. The Spectronaut projects can be viewed using the free Spectronaut viewer (http://www.biognosys.com/technology/spectronaut-viewer).
DIA data analysis
Prior library-based analysis of the DIA data, the raw files were converted into htrms files using the htrms converter (Biognosys). MS1 and MS2 data were centroided during conversion. The other parameters were set to default.The htrms files were analyzed with Spectronaut X17 (version: 12.0.20491.18.30559, Biognosys) using the previously generated libraries and default settings.
For the analysis of the DIA data of the comparison of the different solid phases and the gradient ramp, the DirectDIA workflow was used (based on raw files). The workflow allowed a search of the DIA data against a FASTA file and quantification of the precursors, peptides and proteins. The principles were described by Tsou et al.28 The same search parameters as for the search of the DDA data in SpectroMine were applied and the results were filtered by a 1% FDR on precursor and protein level (Q value <0.01). These files were also searched using the generated HeLa library. To minimize effects of iRT precision on the identification result,45 the XIC extraction window was set to full for the comparison of the different solid phases. For all other analysis it was set to dynamic. The results of the DIA analysis were filtered within Spectronaut by 1% FDR on peptide and protein level using a target-decoy approach, which corresponds to a Q value ≤0.01.24,26,34,35 The decoy generation was done using a mutated decoys approach and protein FDR was calculated using an adapted version of Rosenberger and colleagues.35 Both strategies, as implemented in Spectronaut, were further described previously.26 For the testis cancer sample set, the quantification data were filtered with the Q value percentile filter set to 0.5. (outputs from Spectronaut can be found in the ESI;† Table S2: solid phase comparison, Table S3: gradient ramp, Table S4: hybrid library approach for HeLa sample, Table S5: library comparison for testis sample, Table S6: testis cancer set).
Empirical FDR validation
For the empirical FDR validation, we performed a two-species FDR test based on A. thaliana as negative control. All the A. thaliana samples were acquired using the same setup as for the testis data (20 fraction HPRP project library and six 6 h DIA runs). We curated the A. thaliana protein database (2019-04-08 uniprot A. thaliana) by removing all tryptic peptides which exist in any form in 2019-04-08 uniprot Human and our contaminants protein database. We made two different libraries: (1) DDA based library by searching human testis together with A. thaliana DDA runs, and (2) DIA library by searching human testis together with A. thaliana DIA runs. In this manner, we had two different sources as before (DDA and DIA) but with a built-in negative set. We searched the human testis and A. thaliana data together to use the human peptides for calibration in the DIA data analysis, because only very few A. thaliana peptides should be identified in the samples. The subsequent analysis was kept the same as with the main analysis (Table S7, ESI†).Biological interpretation of the data
To find the differentially abundant proteins between the NAT and cancer cohort, the statistical testing based on a paired samples t-test and multiple testing correction by the Storey method48 as integrated in Spectronaut was used. The heatmap was generated based on a hierarchical clustering in Spectronaut. For biological interpretation of the data, the unfiltered candidate list (Table S8, ESI†) from the post analysis view in Spectronaut was imported in to Ingenuity Pathway Analysis (IPA, Qiagen, Hilden, Germany). The complete human proteome was used as background set. Proteins were regarded as differentially abundant with an absolute log2 fold change of 1 and a Q value <0.01.
Conflicts of interest
All authors are employees of Biognosys AG. Spectronaut and SpectroMine are trademarks of Biognosys AG.Acknowledgements
We grateful to Ivana Karlovska for preparation of the HeLa sample.References
- R. Aebersold and M. Mann, Nature, 2016, 537, 347–355 CrossRef CAS PubMed.
- M. Beck, A. Schmidt, J. Malmstroem, M. Claassen, A. Ori, A. Szymborska, F. Herzog, O. Rinner, J. Ellenberg and R. Aebersold, Mol. Syst. Biol., 2011, 7, 549 CrossRef PubMed.
- N. Nagaraj, J. R. Wisniewski, T. Geiger, J. Cox, M. Kircher, J. Kelso, S. Pääbo and M. Mann, Mol. Syst. Biol., 2011, 7, 548 CrossRef PubMed.
- D. A. Wolters, M. P. Washburn and J. R. Yates, Anal. Chem., 2001, 73, 5683–5690 CrossRef CAS PubMed.
- M.-S. Kim, S. M. Pinto, D. Getnet, R. S. Nirujogi, S. S. Manda, R. Chaerkady, A. K. Madugundu, D. S. Kelkar, R. Isserlin, S. Jain, J. K. Thomas, B. Muthusamy, P. Leal-Rojas, P. Kumar, N. A. Sahasrabuddhe, L. Balakrishnan, J. Advani, B. George, S. Renuse, L. D. N. Selvan, A. H. Patil, V. Nanjappa, A. Radhakrishnan, S. Prasad, T. Subbannayya, R. Raju, M. Kumar, S. K. Sreenivasamurthy, A. Marimuthu, G. J. Sathe, S. Chavan, K. K. Datta, Y. Subbannayya, A. Sahu, S. D. Yelamanchi, S. Jayaram, P. Rajagopalan, J. Sharma, K. R. Murthy, N. Syed, R. Goel, A. A. Khan, S. Ahmad, G. Dey, K. Mudgal, A. Chatterjee, T.-C. Huang, J. Zhong, X. Wu, P. G. Shaw, D. Freed, M. S. Zahari, K. K. Mukherjee, S. Shankar, A. Mahadevan, H. Lam, C. J. Mitchell, S. K. Shankar, P. Satishchandra, J. T. Schroeder, R. Sirdeshmukh, A. Maitra, S. D. Leach, C. G. Drake, M. K. Halushka, T. S. K. Prasad, R. H. Hruban, C. L. Kerr, G. D. Bader, C. A. Iacobuzio-Donahue, H. Gowda and A. Pandey, Nature, 2014, 509, 575–581 CrossRef CAS PubMed.
- M. Wilhelm, J. Schlegl, H. Hahne, A. M. Gholami, M. Lieberenz, M. M. Savitski, E. Ziegler, L. Butzmann, S. Gessulat, H. Marx, T. Mathieson, S. Lemeer, K. Schnatbaum, U. Reimer, H. Wenschuh, M. Mollenhauer, J. Slotta-Huspenina, J.-H. Boese, M. Bantscheff, A. Gerstmair, F. Faerber and B. Kuster, Nature, 2014, 509, 582–587 CrossRef CAS PubMed.
- E. Shishkova, A. S. Hebert and J. J. Coon, Cell Syst., 2016, 3, 321–324 CrossRef CAS PubMed.
- S. S. Thakur, T. Geiger, B. Chatterjee, P. Bandilla, F. Fröhlich, J. Cox, M. Mann, F. Fröhlich, M. Mann, B. Chatterjee, P. Bandilla, S. S. Thakur, T. Geiger, B. Chatterjee, P. Bandilla, F. Fröhlich, J. Cox and M. Mann, Mol. Cell. Proteomics, 2011, 10, M110.003699 CrossRef PubMed.
- A. S. Hebert, A. L. Richards, D. J. Bailey, A. Ulbrich, E. E. Coughlin, M. S. Westphall and J. J. Coon, Mol. Cell. Proteomics, 2014, 13, 339–347 CrossRef CAS PubMed.
- J. W. Harper and E. J. Bennett, Nature, 2016, 537, 328–338 CrossRef CAS PubMed.
- B. Schwanhäusser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Nature, 2011, 473, 337–342 CrossRef PubMed.
- F. Meier, P. E. Geyer, S. Virreira Winter, J. Cox and M. Mann, Nat. Methods, 2018, 15, 440–448 CrossRef CAS PubMed.
- A. S. Hebert, S. Prasad, M. W. Belford, D. J. Bailey, G. C. McAlister, S. E. Abbatiello, R. Huguet, E. R. Wouters, J.-J. Dunyach, D. R. Brademan, M. S. Westphall and J. J. Coon, Anal. Chem., 2018, 90, 9529–9537 CrossRef CAS PubMed.
- A.-D. Brunner, S. Koch, N. Bache, N. Goedecke, M. A. Park, H. Koch, J. Decker, O. Räther, J. Cox, T. Kosinski, M. Krause, F. Meier, M. Mann, O. Hoerning and M. Lubeck, Mol. Cell. Proteomics, 2018, 17, 2534–2545 CrossRef PubMed.
- J. D. Venable, M. Q. Dong, J. Wohlschlegel, A. Dillin and J. R. Yates, Nat. Methods, 2004, 1, 39–45 CrossRef CAS PubMed.
- J. C. Silva, M. V. Gorenstein, G.-Z. Li, J. P. C. Vissers and S. J. Geromanos, Mol. Cell. Proteomics, 2006, 5, 144–156 CrossRef CAS PubMed.
- R. Bruderer, O. M. Bernhardt, T. Gandhi, S. M. Miladinović, L.-Y. Cheng, S. Messner, T. Ehrenberger, V. Zanotelli, Y. Butscheid, C. Escher, O. Vitek, O. Rinner and L. Reiter, Mol. Cell. Proteomics, 2015, 14, 1400–1410 CrossRef CAS PubMed.
- N. Selevsek, C.-Y. Chang, L. C. Gillet, P. Navarro, O. M. Bernhardt, L. Reiter, L.-Y. Cheng, O. Vitek and R. Aebersold, Mol. Cell. Proteomics, 2015, 14, 739–749 CrossRef CAS PubMed.
- U. Distler, J. Kuharev, P. Navarro, Y. Levin, H. Schild and S. Tenzer, Nat. Methods, 2014, 11, 167–170 CrossRef CAS PubMed.
- J. Vowinckel, F. Capuano, K. Campbell, M. J. Deery, K. S. Lilley and M. Ralser, F1000Research, 2014, 2, 272 Search PubMed.
- J. Muntel, Y. Xuan, S. T. Berger, L. Reiter, R. Bachur, A. Kentsis and H. Steen, J. Proteome Res., 2015, 14, 4752–4762 CrossRef CAS PubMed.
- C. D. Kelstrup, D. B. Bekker-Jensen, T. N. Arrey, A. Hogrebe, A. Harder, J. V. Olsen, T. Note and T. N. Nordisk, J. Proteome Res., 2017, 17, 727–738 CrossRef PubMed.
- R. Bruderer, J. Muntel, S. Müller, O. M. Bernhardt, T. Gandhi, O. Cominetti, C. Macron, J. Carayol, O. Rinner, A. Astrup, W. H. M. Saris, J. Hager, A. Valsesia, L. Dayon and L. Reiter, Mol. Cell. Proteomics, 2019, mcp.RA118.001288 Search PubMed.
- L. C. Gillet, P. Navarro, S. Tate, H. Röst, N. Selevsek, L. Reiter, R. Bonner and R. Aebersold, Mol. Cell. Proteomics, 2012, 11, O111.016717 CrossRef PubMed.
- B. C. Collins, C. L. Hunter, Y. Liu, B. Schilling, G. Rosenberger, S. L. Bader, D. W. Chan, B. W. Gibson, A.-C. Gingras, J. M. Held, M. Hirayama-Kurogi, G. Hou, C. Krisp, B. Larsen, L. Lin, S. Liu, M. P. Molloy, R. L. Moritz, S. Ohtsuki, R. Schlapbach, N. Selevsek, S. N. Thomas, S.-C. Tzeng, H. Zhang and R. Aebersold, Nat. Commun., 2017, 8, 291 CrossRef PubMed.
- R. Bruderer, O. M. Bernhardt, T. Gandhi, Y. Xuan, J. Sondermann, M. Schmidt, D. Gomez-Varela and L. Reiter, Mol. Cell. Proteomics, 2017, mcp.RA117.000314 Search PubMed.
- G. Rosenberger, C. C. Koh, T. Guo, H. L. Röst, P. Kouvonen, B. C. Collins, M. Heusel, Y. Liu, E. Caron, A. Vichalkovski, M. Faini, O. T. Schubert, P. Faridi, H. A. Ebhardt, M. Matondo, H. Lam, S. L. Bader, D. S. Campbell, E. W. Deutsch, R. L. Moritz, S. Tate and R. Aebersold, Sci. Data, 2014, 1, 140031 CrossRef CAS PubMed.
- C.-C. Tsou, D. Avtonomov, B. Larsen, M. Tucholska, H. Choi, A.-C. Gingras and A. I. Nesvizhskii, Nat. Methods, 2015, 12, 258–264 CrossRef CAS PubMed.
- Y. S. Ting, J. D. Egertson, J. G. Bollinger, B. C. Searle, S. H. Payne, W. S. Noble and M. J. MacCoss, Nat. Methods, 2017, 14, 903–908 CrossRef CAS PubMed.
- J. Muntel, J. Kirkpatrick, R. Bruderer, T. Huang, O. Vitek, A. Ori and L. Reiter, J. Proteome Res., 2019, 18, 1340–1351 CrossRef CAS PubMed.
- T. Gandhi, L. Verbeke, O. M. Bernhardt, J. Muntel, S. Müller, R. Bruderer, Y. Xuan and L. Reiter, ASMS Conference, San Diego, 2018.
- B. C. Searle, L. K. Pino, J. D. Egertson, Y. S. Ting, R. T. Lawrence, B. X. MacLean, J. Villén and M. J. MacCoss, Nat. Commun., 2018, 9, 5128 CrossRef PubMed.
- C. Escher, L. Reiter, B. Maclean, R. Ossola, F. Herzog, J. Chilton, M. J. Maccoss and O. Rinner, Proteomics, 2012, 12, 1111–1121 CrossRef CAS PubMed.
- L. Reiter, O. Rinner, P. Picotti, R. Hüttenhain, M. Beck, M.-Y. Brusniak, M. O. Hengartner and R. Aebersold, Nat. Methods, 2011, 8, 430–435 CrossRef CAS PubMed.
- G. Rosenberger, I. Bludau, U. Schmitt, M. Heusel, C. L. Hunter, Y. Liu, M. J. MacCoss, B. X. MacLean, A. I. Nesvizhskii, P. G. A. Pedrioli, L. Reiter, H. L. Röst, S. Tate, Y. S. Ting, B. C. Collins and R. Aebersold, Nat. Methods, 2017, 14, 921–927 CrossRef CAS PubMed.
- J. W. Jorgenson, Annu. Rev. Anal. Chem., 2010, 3, 129–150 CrossRef CAS PubMed.
- F. Gritti and G. Guiochon, J. Chromatogr. A, 2012, 1228, 2–19 CrossRef CAS PubMed.
- E. Shishkova, A. S. Hebert, M. S. Westphall and J. J. Coon, Anal. Chem., 2018, 90, 11503–11508 CrossRef CAS PubMed.
- J. Op De Beeck, J. Pauwels, A. Staes, N. Van Landuyt, D. Van Haver, W. De Malsche, G. Desmet, A. Argentini, L. Martens, P. Jacobs, F. Impens and K. Gevaert, bioRxiv, 2018, 472134 Search PubMed.
- M. Uhlen, L. Fagerberg, B. M. Hallstrom, C. Lindskog, P. Oksvold, A. Mardinoglu, A. Sivertsson, C. Kampf, E. Sjostedt, A. Asplund, I. Olsson, K. Edlund, E. Lundberg, S. Navani, C. A.-K. Szigyarto, J. Odeberg, D. Djureinovic, J. O. Takanen, S. Hober, T. Alm, P.-H. Edqvist, H. Berling, H. Tegel, J. Mulder, J. Rockberg, P. Nilsson, J. M. Schwenk, M. Hamsten, K. von Feilitzen, M. Forsberg, L. Persson, F. Johansson, M. Zwahlen, G. von Heijne, J. Nielsen and F. Ponten, Science, 2015, 347, 1260419 CrossRef PubMed.
- D. Wang, B. Eraslan, T. Wieland, B. Hallström, T. Hopf, D. P. Zolg, J. Zecha, A. Asplund, L. Li, C. Meng, M. Frejno, T. Schmidt, K. Schnatbaum, M. Wilhelm, F. Ponten, M. Uhlen, J. Gagneur, H. Hahne and B. Kuster, Mol. Syst. Biol., 2019, 15, e8503 CrossRef PubMed.
- J. Sun, J. Shi, Y. Wang, Y. Chen, Y. Li, D. Kong, L. Chang, F. Liu, Z. Lv, Y. Zhou, F. He, Y. Zhang and P. Xu, J. Proteome Res., 2018, 17, 4171–4177 CrossRef CAS PubMed.
- M. Alikhani, M. Mirzaei, M. Sabbaghian, P. Parsamatin, R. Karamzadeh, S. Adib, N. Sodeifi, M. A. S. Gilani, M. Zabet-Moghaddam, L. Parker, Y. Wu, V. Gupta, P. A. Haynes, H. Gourabi, H. Baharvand and G. H. Salekdeh, J. Proteomics, 2017, 162, 141–154 CrossRef CAS PubMed.
- A. Vassilev and M. L. DePamphilis, Genes, 2017, 8, 45 CrossRef PubMed.
- R. Bruderer, O. Bernhardt, T. Gandhi and L. Reiter, Proteomics, 2016, 1–20 Search PubMed.
- G. Rosenberger, I. Bludau, U. Schmitt, M. Heusel, C. L. Hunter, Y. Liu, M. J. MacCoss, B. X. MacLean, A. I. Nesvizhskii, P. G. A. Pedrioli, L. Reiter, H. L. Röst, S. Tate, Y. S. Ting, B. C. Collins and R. Aebersold, Nat. Methods, 2017, 14, 921–927 CrossRef CAS PubMed.
- R. D. Neal, N. Stuart and C. Wilkinson, BMJ Clin. Evid., 2007, 2007, 1807 Search PubMed.
- J. D. Storey, A direct approach to false discovery rates, 2002, vol. 64 Search PubMed.
- S. Tenzer, P. Leidinger, C. Backes, H. Huwer, A. Hildebrandt, H.-P. Lenhof, T. Wesse, A. Franke, E. Meese and A. Keller, Oncotarget, 2016, 7, 14857–14870 CrossRef PubMed.
- B. Zhang, J. Wang, X. Wang, J. Zhu, Q. Liu, Z. Shi, M. C. Chambers, L. J. Zimmerman, K. F. Shaddox, S. Kim, S. R. Davies, S. Wang, P. Wang, C. R. Kinsinger, R. C. Rivers, H. Rodriguez, R. R. Townsend, M. J. C. Ellis, S. A. Carr, D. L. Tabb, R. J. Coffey, R. J. C. Slebos, D. C. Liebler and NCI CPTAC, Nature, 2014, 513, 382–387 CrossRef CAS PubMed.
- H. Tang, S. Mirshahidi, M. Senthil, K. Kazanjian, C.-S. Chen and K. Zhang, Cancer Biomarkers, 2014, 14, 313–324 CAS.
- O. Torres-Luquis, K. Madden, N. M. N’dri, R. Berg, O. F. Olopade, W. Ngwa, D. Abuidris, S. Mittal, B. Lyn-Cook and S. I. Mohammed, Breast Cancer, 2019, 11, 1–12 Search PubMed.
- I. Maleva Kostovska, O. Stankov, G. Petrusevska, S. Stavridis, S. Kiprijanovska, M. Polenakovic, K. Davalieva and S. Komina, Proteomes, 2017, 6, 1 CrossRef PubMed.
- F. Bovenga, C. Sabbà and A. Moschetta, Cell Metab., 2015, 21, 517–526 CrossRef CAS PubMed.
- Y. C. Li, M. J. Park, S.-K. Ye, C.-W. Kim and Y.-N. Kim, Am. J. Pathol., 2006, 168, 1107–1118 CrossRef CAS PubMed ; quiz 1404–1405.
- J. H. Song, M. C. L. Tse, A. Bellail, S. Phuphanich, F. Khuri, N. M. Kneteman and C. Hao, Cancer Res., 2007, 67, 6946–6955 CrossRef CAS PubMed.
- J. Fukuchi, J. M. Kokontis, R. A. Hiipakka, C. Chuu and S. Liao, Cancer Res., 2004, 64, 7686–7689 CrossRef CAS PubMed.
- L.-L. Vedin, J.-Å. Gustafsson and K. R. Steffensen, Mol. Carcinog., 2013, 52, 835–844 CrossRef CAS PubMed.
- W. Fu, J. Yao, Y. Huang, Q. Li, W. Li, Z. Chen, F. He, Z. Zhou and J. Yan, Cell. Physiol. Biochem., 2014, 33, 195–204 CrossRef CAS PubMed.
- Q. Wang, F. Feng, J. Wang, M. Ren, Z. Shi, X. Mao, H. Zhang and X. Ju, J. Cell. Mol. Med., 2019, 23, 789–797 CrossRef CAS PubMed.
- V. Afshar-Kharghan, J. Clin. Invest., 2017, 127, 780–789 CrossRef PubMed.
- R. Pio, L. Corrales and J. D. Lambris, Adv. Exp. Med. Biol., 2014, 772, 229–262 CrossRef CAS PubMed.
- A. M. McGee, D. L. Douglas, Y. Liang, S. M. Hyder and C. P. Baines, Cell Cycle, 2011, 10, 4119–4127 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- T. Otto and P. Sicinski, Nat. Rev. Cancer, 2017, 17, 93–115 CrossRef CAS PubMed.
- S. Costa-Cabral, R. Brough, A. Konde, M. Aarts, J. Campbell, E. Marinari, J. Riffell, A. Bardelli, C. Torrance, C. J. Lord and A. Ashworth, PLoS One, 2016, 11, e0149099 CrossRef PubMed.
- S. Campaner, M. Doni, P. Hydbring, A. Verrecchia, L. Bianchi, D. Sardella, T. Schleker, D. Perna, S. Tronnersjö, M. Murga, O. Fernandez-Capetillo, M. Barbacid, L.-G. Larsson and B. Amati, Nat. Cell Biol., 2010, 12, 54–59 CrossRef CAS PubMed.
- H. F. Kwok, S. D. Zhang, C. M. McCrudden, H. F. Yuen, K. P. Ting, Q. Wen, U. S. Khoo and K. Y. K. Chan, Am. J. Cancer Res., 2015, 5, 52–71 CAS.
- C. Giaginis, M. Georgiadou, K. Dimakopoulou, G. Tsourouflis, E. Gatzidou, G. Kouraklis and S. Theocharis, Dig. Dis. Sci., 2009, 54, 282–291 CrossRef CAS PubMed.
- P. Kotsantis, R. M. Jones, M. R. Higgs and E. Petermann, Adv. Clin. Chem., 2015, 69, 91–138 CAS.
- M. J. Bywater, R. B. Pearson, G. A. McArthur and R. D. Hannan, Nat. Rev. Cancer, 2013, 13, 299–314 CrossRef CAS PubMed.
- P. Kotsantis, L. M. Silva, S. Irmscher, R. M. Jones, L. Folkes, N. Gromak and E. Petermann, Nat. Commun., 2016, 7, 13087 CrossRef CAS PubMed.
- F. Yang, Y. Shen, D. G. Camp and R. D. Smith, Expert Rev. Proteomics, 2012, 9, 129–134 CrossRef CAS PubMed.
- L. Reiter, M. Claassen, S. P. Schrimpf, M. Jovanovic, A. Schmidt, J. M. Buhmann, M. O. Hengartner and R. Aebersold, Mol. Cell. Proteomics, 2009, 8, 2405–2417 CrossRef CAS PubMed.
- M. M. Savitski, M. Wilhelm, H. Hahne, B. Kuster and M. Bantscheff, Mol. Cell. Proteomics, 2015, 14, 2394–2404 CrossRef CAS PubMed.
- E. W. Deutsch, A. Csordas, Z. Sun, A. Jarnuczak, Y. Perez-Riverol, T. Ternent, D. S. Campbell, M. Bernal-Llinares, S. Okuda, S. Kawano, R. L. Moritz, J. J. Carver, M. Wang, Y. Ishihama, N. Bandeira, H. Hermjakob and J. A. Vizcaíno, Nucleic Acids Res., 2017, 45, D1100–D1106 CrossRef CAS PubMed.
- J. A. Vizcaíno, R. G. Côté, A. Csordas, J. A. Dianes, A. Fabregat, J. M. Foster, J. Griss, E. Alpi, M. Birim, J. Contell, G. O’Kelly, A. Schoenegger, D. Ovelleiro, Y. Pérez-Riverol, F. Reisinger, D. Ríos, R. Wang and H. Hermjakob, Nucleic Acids Res., 2013, 41, D1063–D1069 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mo00082h |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |