
A centimeter scale self-standing two-dimensional ultra-thin mesoporous platinum nanosheet†

Abstract
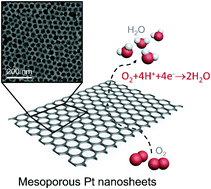
Here, we report the unprecedented synthesis of centimeter scale self-standing mesoporous Pt nanosheets. Due to solvent evaporation, spherical micelles made of poly(styrene-b-2-vinyl pyridine-b-ethylene oxide) (PS-P2VP-PEO) self-assemble in a well-ordered fashion, over the entire surface of the substrate, and confined within an ultra-thin layer. Metal deposition around the micelles is carried out through a two-step reduction process using two different reducing agents. After removing the micelles and substrate, a continuous self-standing ultra-thin mesoporous Pt nanosheet can be obtained (∼15 nm thick). Due to several unique porous structural features, including abundant active sites, high surface area, and long-range orthogonally ordered pores, the obtained material is expected to deliver high performance in a wide range of electrochemical applications. This novel synthetic strategy opens a new route to design and control novel 2D mesoporous metal nanosheets with various functions.
 Please wait while we load your content...
Please wait while we load your content...

