
Design strategies for developing non-precious metal based bi-functional catalysts for alkaline electrolyte based zinc–air batteries

Abstract
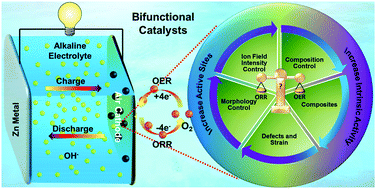
Compared with the current dominant energy storage system (lithium-ion batteries (LIBs)), rechargeable zinc–air batteries (ZABs) with alkaline electrolyte are safer and less expensive, have much higher theoretical volumetric energy density, can be manufactured in ambient air rather than a dry room, and have much higher tolerance to moisture and air during operation. A mature aqueous alkaline electrolyte could also significantly improve safety while minimizing the fabrication cost. Hence, ZABs have great potential to challenge the dominant position of LIBs in the future. Nevertheless, the widespread application of this energy storage system is seriously hindered by the sluggish kinetics of the oxygen reduction (ORR) and evolution reactions (OER) at the liquid–gas–solid phase cathode interface. Therefore, to further promote the development of this technology, the development of low-cost, high-activity catalysts for the OER/ORR has long been recognized as a crucial measure. This paper summarizes the existing strategies that could be used to develop non-precious-metal based, high activity bifunctional OER/ORR catalysts for the alkaline electrolyte based zinc–air system.
- This article is part of the themed collection: Horizons Community Board Collection – Advanced Energy Storage Technologies
 Please wait while we load your content...
Please wait while we load your content...

