
Ultralong tumor retention of theranostic nanoparticles with short peptide-enabled active tumor homing†

Abstract
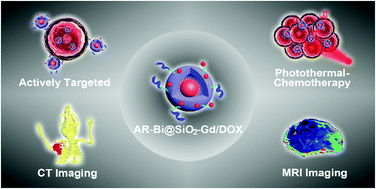
Computer tomography (CT) and magnetic resonance imaging (MRI) are noninvasive cancer imaging methods in clinics. Hence, a material that enables MRI/CT dual-modal imaging-guided therapy is in high demand. Currently, the available materials lack active tumor targeting, deep tumor penetration, and ultralong tumor retention and may lose their imaging elements. To overcome these drawbacks, herein, nanoparticles (NPs) were developed by integrating an MRI contrast-enhancing chelated gadolinium (Gd) complex within a doxorubicin (DOX)-loaded protective silica shell as well as a CT imaging/photothermal biocompatible bismuth (Bi) nano-core, which surface-displayed an MCF-7 breast tumor-homing peptide (AREYGTRFSLIGGYR, termed AR); we found that the resultant NPs AR-Bi@SiO2-Gd/DOX could home to and penetrate deep into the tumors with the unexpected ultralong retention of at least 14 days (as determined by CT/MRI imaging) and the tumor retention half-life of 104.5 h (as determined by ICP-MS analysis) under the guidance of the AR peptide. These NPs can be further used to image tumors with significantly increased sharp contrasts via both CT and MRI, which are much better than the commercial standard contrast agents; moreover, they significantly inhibit tumor growth via the synergistic action of both Bi-enabled photothermal therapy and DOX-induced chemotherapy. The NPs are cleared by the spleen, liver and kidney and then excreted from the body along with faeces and urine. The precise tumor targeting and ultralong tumor retention of these unique NPs would enable both precise tumor detection for early diagnosis and signal-persistent tumor tracking for monitoring the treatment with only a single injection of these NPs.
 Please wait while we load your content...
Please wait while we load your content...

