
Aggregation-induced emission: fundamental understanding and future developments
Yuncong
Chen†
 abc,
Jacky W. Y.
Lam†
ac,
Ryan T. K.
Kwok
ac,
Bin
Liu
*d and
Ben Zhong
Tang
*ace
abc,
Jacky W. Y.
Lam†
ac,
Ryan T. K.
Kwok
ac,
Bin
Liu
*d and
Ben Zhong
Tang
*ace
aHKUST Shenzhen Research Institute, No. 9 Yuexing 1st RD, South Area, Hi-tech Park, Nanshan, Shenzhen 518057, China. E-mail: tangbenz@ust.hk
bState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China
cDepartment of Chemistry, The Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction and Institute for Advanced Study, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
dDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: cheliub@nus.edu.sg
eCenter for Aggregation-Induced Emission, SCUT-HKUST Joint Research Laboratory, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
First published on 15th November 2018
Abstract
Since the introduction of the concept of aggregation-induced emission (AIE) in 2001, many research groups have become involved in AIE research. Aggregation-induced emission luminogens (AIEgens) have emerged as a novel type of advanced material with excellent performance in various fields. Much effort has been devoted to determining the AIE mechanism(s) by theoreticians and experimentalists. Restriction of intramolecular motion has been recognized as the general working mechanism of AIE, but the mechanims of some AIE systems still remain unclear. In this focus article, the progress of the fundamental understanding of the AIE mechanism is reviewed and the future developments in AIE research are discussed. The goal is to provide a brief yet insightful introduction and interpretation of the subject to both new and experienced AIE researchers.
Luminescent materials have been extensively studied for decades because of their application in various fields such as light emitting devices, chemical sensing and especially bio-imaging.1–4 However, because of the strong intermolecular π–π stacking, most conventional organic dyes exhibit aggregation-caused quenching (ACQ) when their molecules are aggregated (Fig. 1, left).5 The ACQ effect greatly limits their use in fields such as organic light emitting diodes (OLEDs) and organic nano-dots for bio-imaging, because the emission is often quenched when the organic dyes are used as solid films or in the aggregate state. Because conventional organic luminophores usually possess planar aromatic cores which favour the occurrence of π–π stacking when aggregated, the ACQ effect is very common and becomes the accepted belief.
 | ||
| Fig. 1 Photographs of perylene (left) and hexaphenylsilole (HPS) solutions (right) with different poor solvent fractions taken under ultraviolet (UV) light irradiation. | ||
In 2001, a new concept called “aggregation-induced emission (AIE)” was first proposed by Tang et al. Unlike conventional ACQ dyes, AIE luminogens (AIEgens) show weak or negligible emission in dilute solution but emit intensely in the aggregate or solid state (Fig. 1, right). AIE is thus the exact opposite of ACQ.6 Using AIEgens is a powerful approach for addressing the ACQ problem. In fact, a few systems had been reported to show stronger photoluminescence (PL) in the solid state than in dilute solution, one of which can be dated back to 1853 by Stokes.7,8 However, little attention was given to the significance of this phenomenon and its potential for practical implications. After the AIE concept was proposed, great effort has been devoted to deciphering its mechanism(s) and exploring its high technology applications in various fields.
AIE research has now gained extensive attention and AIEgens have been recognized as an important type of advanced functional material for a wide variety of applications. Deciphering its working mechanism is not only of fundamental importance but also beneficial for providing design guidelines for the development of new AIE systems for new practical applications. Although some debate still exists, restriction of intramolecular motions (RIM) has been generally accepted as central to the AIE working mechanism. According to the RIM mechanism, it can be concluded that structural rigidification in high viscosity media, low temperature, and doping of chromophores in rigid matrices can also lead to strong emission. Thus, aggregation is not necessary for strong emission on some occasions. As AIEgens often show unique advantages such as high solid quantum yield, high photostability and no ACQ effect, they have been successfully applied in various fields such as OLEDs, stimuli responsive sensing, bioimaging and theranostics.9–12 In this brief focus article, an overview of the current fundamental understanding of the AIE working mechanism, clarification of the existing controversies and disputes, and discussion of the future developments in AIE research are provided.
To investigate the AIE working mechanism, first the structural difference between typical AIEgens and ACQ dyes should be understood. Distinctly different from the large planar structures of conventional ACQ dyes, most of the AIEgens show highly twisted propeller-like structures. This suggests that the twisted structure plays a critical role for the AIE phenomenon. It is reasonable to postulate that the intramolecular motions (rotation and vibration) of the AIEgens such as tetraphenylethene (TPE) and silole are rapid in dilute solution and the emission is quenched because of the high nonradiative decay rate (knr). However, because of the highly twisted conformation of AIEgens, both intermolecular π–π stacking and intramolecular motions are restricted in the aggregate or solid state which results in a suppressed knr. Therefore, the radiative decay rate (kr) can now effectively compete with the knr, leading to an enhanced emission quantum yield. The RIM mechanism has now been recognized as the general mechanism of AIE, supported by the experimental data and theoretical simulation results that show that the knr of an AIEgen is suppressed by up to four orders of magnitude, whereas the kr shows little change from a solution to an aggregate or solid state.13,14
It is not difficult to understand that the active intramolecular motions in solution will be restricted in aggregates or the solid state because of the physical constraint, and this is supported by many experimental results based on the suppression of molecular motions by incorporation of chromophores into a high viscosity medium, lowering the temperature, doping them in a rigid polymer matrix or embedding them into metal organic frameworks.15–19 Therefore, the key issue to understanding the AIE phenomenon is to determine why the knr in solution is so large, i.e., what types of intramolecular motions lead to a such large knr. Numerous studies have been made by both theoreticians and experimentalists to investigate how an excited AIEgen decays to the ground state. Several probable mechanisms were proposed for different AIE systems, including E/Z isomerization, photo-cyclization (PC), rotation of phenyl rings, rotation of double bonds, easy access of conical intersection (CI), and so on.9,20–24 In this paper, TPE, one of the most classic AIEgens, has been chosen as an example to discuss its excited-state decay pathways.
As illustrated in Fig. 2(a), a TPE molecule is largely twisted in the ground state because of the steric hindrance of the peripheral phenyl rings. Upon excitation, the π bond is destroyed and a diradical-like species is generated. This species can be described using several resonance structures as the radicals are conjugated with the neighbouring phenyl rings (only one representative resonance structure is drawn for clarity). According to the Frank–Condon (FC) principle, the conformation of the molecule does not change upon light absorption because of the fast (fs range) excitation process.20 Therefore, the FC excited state is very unstable and tends to relax to a geometry with lower energy. The following structural relaxations are among those that could occur from the FC excited state: (1) extension of the central quasi-single bond, (2) rotation around the central single bond axis, (3) rotation of the phenyl rings towards a lower twist angle, and (4) shortening of the peripheral single bonds. All these motions will help in stabilizing the excited state and have been predicted from theoretical calculations.25,26
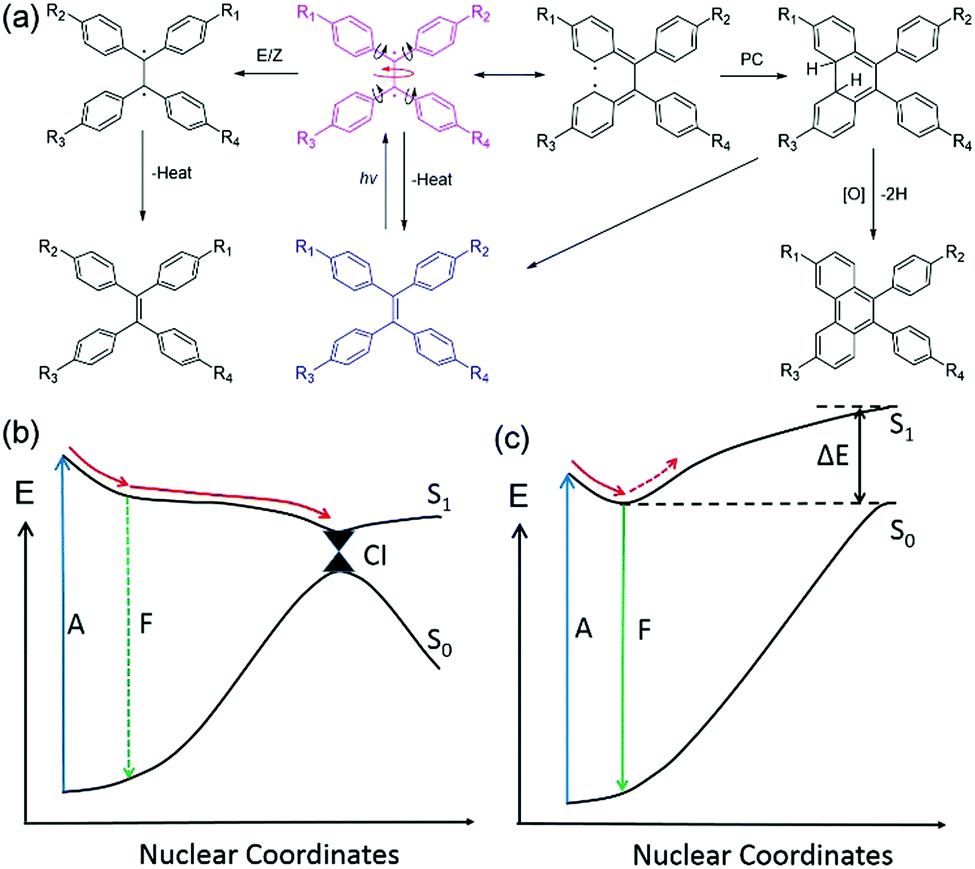 | ||
| Fig. 2 (a) Simplified schematic illustration of the possible photophysical and photochemical processes of TPE derivatives. (b and c) Proposed decay pathways along the potential energy curves of a typical AIE molecule in (b) solution with low viscosity and (c) solid or crystal state. Abbreviations: A = absorption, CI = conical intersection, F = fluorescence. | ||
In dilute solution with a low viscosity, the energy of the excited state (S1) is stabilized by structural relaxation whereas that of the ground state (S0) is distinctly elevated, leading to a dramatic decrease of the energy gap between the two states [Fig. 2(b)]. Therefore, it is easy for the relaxation to reach the vicinity of the CI, where the potential energy surfaces (curves) are degenerate (intersect) and non-adiabatic coupling takes place. This leads to a very large knr and the molecule decays back to the ground state and sometimes an E/Z isomerization occurs.27 A small oscillator strength (f) at near 90° conformation will lead to a low kr and the emission of this conformation is often too weak to be detected. Meanwhile, PC can also take place near the CI in a proper conformation with a double bond rotation angle smaller than 90°.28,29 The PC product is usually unstable and tends to transform to the original structure once the molecule decays to the ground state. In the presence of oxidant, the product could be isolated after dehydrogenization. The potential energy curve (PEC) of S1 is shallow with a small or no energy barrier to reach the CI region. Thus, the CI is easy to access in both cases. The kr (107–108 s−1) cannot compete with the fast vibrational relaxation rate (up to 1011 s−1). As a result, the fluorescence of TPE derivatives was quenched in dilute solutions of low viscosity. It is worth noting that the effect of twisted intramolecular charge transfer (TICT) needs to be taken into consideration when the substituents are strong electron donors and acceptors. TICT fluorophores often show strong short-wavelength emission in low polarity solvents but weak red shifted-emission in highly polar solvents.30
However, in the solid or crystal states, the PEC of S1 becomes steep and the energy of S1 rises dramatically after the minimum point because of steric hindrance of the neighbouring molecules. The intramolecular motions were restricted because of the large energy barrier [Fig. 2(c)]. Therefore, the kr can now compete with the suppressed vibrational decay rate and the kIC. Consequently, the fluorescence is recovered. To gain deeper insight into the AIE mechanism and to guide future design of AIE systems, it is important to understand how the excited molecules decay to the ground state and which intramolecular motions cause the large non-radiative decay rate in solution. To do this, quantum chemical calculations need to be performed to simulate the excited-state decay pathways of different AIE cores as well as TPE and silole.
It is worth noting that the intermolecular interactions in the crystal state are usually stronger than those in the amorphous solid state, leading to further restriction of the intramolecular motions and a smaller reorganization energy.31 This is in agreement with the fact that the crystals of most AIEgens show stronger and blue-shifted PL than their amorphous counterparts.32 In some systems, the solid powders are still weakly emissive or completely non-luminescent because of the loose aggregation and thus, there is more freedom for intramolecular motions. However, intense emission was observed in their crystals, which displayed a phenomenon of crystallization-induced emission (CIE).33,34 This phenomenon is more commonly observed in phosphorescent materials because their triplet states more readily undergo non-radiative decay than their singlet counterparts. As phosphorescence of most organic molecules was only observed in the crystal state at room temperature, this phenomenon is normally termed as crystallization-induced phosphorescence or room-temperature phosphorescence (RTP).35,36 The RTP systems are promising for time-gated imaging and have excellent signal-to-noise ratios because of their long emission lifetime.37,38
According to Kasha's rule, the emission of a luminophore originates almost exclusively from the lowest excited state (S1 or T1).39 However, some recently reported AIE or RTP systems show emission from the higher excited states, which offers new insights into the AIE mechanism and novel design principles for AIE systems (Fig. 3). Aprahamian et al. developed a series of boron difluoride (BF2)-hydrazine-based dyes or BODIHY derivatives with viscosity dependent emission and AIE property. They claimed that the emission is not from the S1 state but from higher energy states (>S1).40 In solution, the excited BODIHY derivatives are flexible and the dark S1 state is easily accessible, resulting in weak emission. However, in the solid state or a highly viscous environment, the intramolecular motions of the excited BODIHYs are restricted and therefore radiative decay to S0 could compete with vibrational relaxation to S1. Thus, the observed emission is assumed to be from the higher excited states. Peng and Tang reported a series of pure organic RTP molecules with dual phosphorescence from both T1 and T2.41 Theoretical calculations suggested that T2 arose mainly from n–π* transition whereas T1 showed more π–π* transition. According to El-Sayed's rule, T2 is expected to exhibit a much faster radiative decay rate and a shorter lifetime than T1. Because of the small experimental energy gap (ΔEST; 0.19 eV) between T2 and T1, T2 could also be populated at room temperature. Thanks to the restricted nonradiative decay by crystallization, both phosphorescence from T1 (yellow) and T2 (blue) are observed to generate a white light emission.
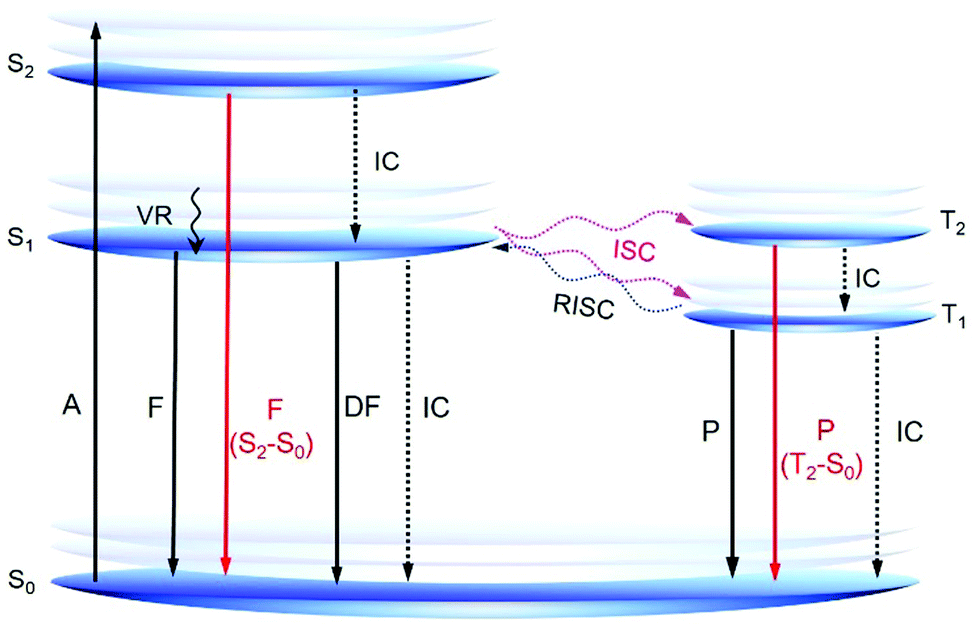 | ||
| Fig. 3 Simplified Jablonski diagram of the photophysical processes in singlet (left) and triplet (right) states. Abbreviations: A = absorption, DF = delayed fluorescence, F = fluorescence, IC = internal conversion, ISC = intersystem crossing, P = phosphorescence, RISC = reverse intersystem crossing, VR = vibrational relaxation. | ||
The observations of novel AIE systems with emission from higher excited states are very interesting and are an exciting progression in AIE research, offering a much wider utilization of light emission. However, such kind of reports are still limited and those that do exist lack principles of design. Constructing new AIE systems with fluorescence or phosphorescence from higher singlet or triplet states and investigation of the structure–property relationship of these systems are fundamentally important. In-depth theoretical investigations are also highly required for a better understanding of these interesting systems.
Traditional luminophores usually contain large aromatic rings or π-conjugated systems as chromophores. However, a growing number of nonconventional systems such as non-conjugated polymers and natural products with no conventional conjugated building blocks have recently been reported to show AIE characteristics (Fig. 4).42–47 Some oligomers or polymers with electron-rich atoms (N, O, S, P) or subunits (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –CN–, –COOH) are non-emissive in solution but become emissive upon aggregation. The clusteration-triggered emission (CTE) was proposed as the mechanism of such phenomenon. Overlapping of the electron clouds of the electron-rich atoms or groups forms an intermolecular through-space interaction upon clustering or aggregation [Fig. 4(a)].9 The resulting “clusteroluminogens” exhibit higher electronic conjugation than the isolated atoms or groups. Meanwhile, the intramolecular motions are restricted upon aggregation. All these collective factors contribute to the observable emission. Polyacrylonitrile (PAN) is a cyano-containing polymer showing CIE. The PAN polymer emits blue light in the solid powder state but shows virtually no emission in dilute dimethylformamide solution [Fig. 4(b)].48 Intramolecular through-space conjugation can also serve as a possible working mechanism of nonconventional AIE systems. Tetraphenylethane (s-TPE) with isolated phenyl rings showed prominent emission in the solid state but was non-emissive in dilute solution [Fig. 4(c)].49 Xie et al. reported that gold(I)–thiolate [Au(I)–thiolate] was an interesting nonconventional AIE system [Fig. 4(d)].50 The emission of the Au(I)–thiolate complex became distinctly stronger upon aggregation. However, the AIE mechanism in terms of the luminescent chromophore is still not clear.
O, –CN–, –COOH) are non-emissive in solution but become emissive upon aggregation. The clusteration-triggered emission (CTE) was proposed as the mechanism of such phenomenon. Overlapping of the electron clouds of the electron-rich atoms or groups forms an intermolecular through-space interaction upon clustering or aggregation [Fig. 4(a)].9 The resulting “clusteroluminogens” exhibit higher electronic conjugation than the isolated atoms or groups. Meanwhile, the intramolecular motions are restricted upon aggregation. All these collective factors contribute to the observable emission. Polyacrylonitrile (PAN) is a cyano-containing polymer showing CIE. The PAN polymer emits blue light in the solid powder state but shows virtually no emission in dilute dimethylformamide solution [Fig. 4(b)].48 Intramolecular through-space conjugation can also serve as a possible working mechanism of nonconventional AIE systems. Tetraphenylethane (s-TPE) with isolated phenyl rings showed prominent emission in the solid state but was non-emissive in dilute solution [Fig. 4(c)].49 Xie et al. reported that gold(I)–thiolate [Au(I)–thiolate] was an interesting nonconventional AIE system [Fig. 4(d)].50 The emission of the Au(I)–thiolate complex became distinctly stronger upon aggregation. However, the AIE mechanism in terms of the luminescent chromophore is still not clear.
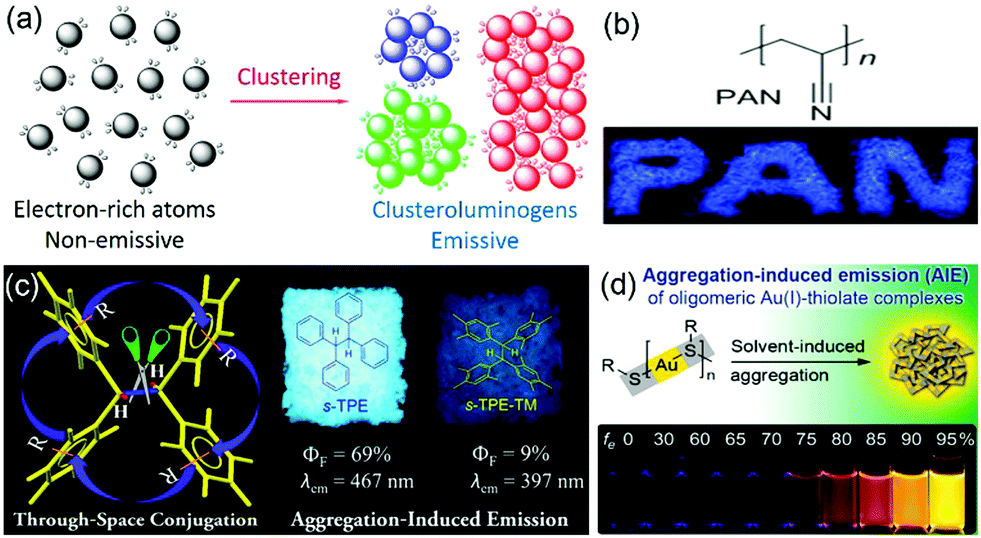 | ||
| Fig. 4 (a) Diagram of the proposed mechanism of the nonconventional AIE systems. (b) An example of a nonconventional AIE system with no phenyl ring in the chemical structure and its solid emission under UV light irradiation. (c) An example of a nonconventional AIE system with through-space conjugation as the working mechanism. (d) An interesting illustration of Au(i)–thiolate complexes showing AIE property. | ||
Many nonconventional AIE systems have been reported to date, which is very interesting and greatly broadens the research scope of AIE. Auto-fluorescence in biological samples has long been thought to originate from proteins containing tryptophan, tyrosine and phenylalanine or the biomolecules such as NADPH and flavins. However, the studies of nonconventional AIE systems may suggest other probable origins. Novel concepts of CTE and “clusteroluminogens” have been proposed to describe these phenomena and materials. However, the mechanisms behind different nonconventional AIE systems are still not clear. Although the formation of new chromophores by through-space conjugation after clustering provides a promising hypothesis to explain some of the systems, in-depth theoretical and experimental investigations are still required to gain an in-depth understanding of these phenomena.
Developing high performance OLEDs is one of the most promising applications of AIEgens.51 Recently it was reported that non-doped OLEDs based on aggregation-induced delayed fluorescence (AIDF) materials showed excellent performance.52–55 Their external quantum efficiencies are comparable to those of traditional doped OLEDs and showed significantly reduced efficiency roll-off, indicating that AIDF materials have potential for use in large-scale commercial OLEDs. The scope of the applications of AIEgens in sensing and imaging is huge, probably only limited by the endeavors and imaginations of the researchers.56–61 Some AIEgens have shown great potential for super resolution imaging.62,63 AIEgens have also demonstrated excellent performance in theranostics.64–67 AIEgen or AIE nanoparticles linked with antibodies have shown capabilities for specific cancer cell imaging.68,69 The non-radiative processes of AIEgens could also be utilized for imaging and therapy. AIEgen-based imaging guided photodynamic therapy, photoacoustic (PA) imaging and photothermal therapy have recently gained much attention.70,71 Developing AIEgens with long excitation wavelength, large mole absorbance and excellent reactive oxygen species generation rates is still challenging but is greatly needed. Furthermore, combining fluorescence imaging and PA imaging with other clinical imaging methods such as computed tomography, MRI and ultrasonography to achieve multimodal clinical diagnosis is also a promising topic. In addition, multifunctional AIEgens acting as both imaging agents and drugs could be used for imaging guided chemotherapy. The biocompatibility, circulation, distribution, retention time and clearing dynamics of these AIEgens need to be systematically evaluated before they can be used in clinic applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Science Foundation of China (21788102), the Research Grants Council of Hong Kong (16305518, 16308016, 16305015, C6009-17G and A-HKUST605/15), the International Science & Technology Cooperation Program of Guangzhou (No. 201704030069), the Innovation and Technology Commission (ITC-CNERC14S01 and ITS/254/17) and the Science and Technology Plan of Shenzhen (JCYJ20170818113851132, JCYJ20170818113840164 and JCYJ20170307173739739).Notes and references
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973 CrossRef CAS PubMed.
- T. Ueno and T. Nagano, Nat. Methods, 2011, 8, 642 CrossRef CAS PubMed.
- Y. Chen, Y. Bai, Z. Han, W. He and Z. Guo, Chem. Soc. Rev., 2015, 44, 4517 RSC.
- M. Vendrell, D. Zhai, J. C. Er and Y.-T. Chang, Chem. Rev., 2012, 112, 4391 CrossRef CAS PubMed.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- G. G. Stokes, Philos. Trans. R. Soc. London, 1853, 143, 385 CrossRef.
- S. Wang, W. J. Oldham, R. A. Hudack and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 5695 CrossRef CAS.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174 RSC.
- Q. Peng, Y. Yi, Z. Shuai and J. Shao, J. Am. Chem. Soc., 2007, 129, 9333 CrossRef CAS PubMed.
- T. Zhang, Y. Jiang, Y. Niu, D. Wang, Q. Peng and Z. Shuai, J. Phys. Chem. A, 2014, 118, 9094 CrossRef CAS PubMed.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535 CrossRef CAS.
- N. B. Shustova, B. D. McCarthy and M. Dincă, J. Am. Chem. Soc., 2011, 133, 20126 CrossRef CAS PubMed.
- N. B. Shustova, A. F. Cozzolino and M. Dincă, J. Am. Chem. Soc., 2012, 134, 19596 CrossRef CAS PubMed.
- N. B. Shustova, T. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dincă, J. Am. Chem. Soc., 2012, 134, 15061 CrossRef CAS PubMed.
- B. He, S. Ye, Y. Guo, B. Chen, X. Xu, H. Qiu and Z. Zhao, Sci. China: Chem., 2013, 56, 1221 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- J. B. Xiong, Y. X. Yuan, L. Wang, J. P. Sun, W. G. Qiao, H. C. Zhang, M. Duan, H. Han, S. Zhang and Y. S. Zheng, Org. Lett., 2018, 20, 373 CrossRef CAS PubMed.
- D. A. Shultz and M. A. Fox, J. Am. Chem. Soc., 1989, 111, 6311 CrossRef CAS.
- Q. Li and L. Blancafort, Chem. Commun., 2013, 49, 5966 RSC.
- S. Sasaki, S. Suzuki, W. M. C. Sameera, K. Igawa, K. Morokuma and G. Konishi, J. Am. Chem. Soc., 2016, 138, 8194 CrossRef CAS PubMed.
- K. Kokado, T. Machida, T. Iwasa, T. Taketsugu and K. Sada, J. Phys. Chem. C, 2018, 122, 245 CrossRef CAS.
- G.-J. Zhao and K.-L. Han, J. Chem. Phys., 2007, 127, 094307 CrossRef PubMed.
- Y.-J. Gao, X.-P. Chang, X.-Y. Liu, Q.-S. Li, G. Cui and W. Thiel, J. Phys. Chem. A, 2017, 121, 2572 CrossRef CAS PubMed.
- A. Prlj, N. Doslic and C. Corminboeuf, Phys. Chem. Chem. Phys., 2016, 18, 11606 RSC.
- Y. Cai, L. Du, K. Samedov, X. Gu, F. Qi, H. H. Y. Sung, B. O. Patrick, Z. Yan, X. Jiang, H. Zhang, J. W. Y. Lam, I. D. Williams, D. L. Phillips, A. Qin and B. Z. Tang, Chem. Sci., 2018, 9, 4662 RSC.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845 CrossRef CAS.
- X. Zheng, Q. Peng, L. Zhu, Y. Xie, X. Huang and Z. Shuai, Nanoscale, 2016, 8, 15173 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Sun, J. Liu, Z. Li, J. Sun, H. H. Y. Sung, I. D. Williams, H. S. Kwok and B. Z. Tang, Chem. Commun., 2007, 3255 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, Z. Li, J. Sun, H. H.-Y. Sung, I. D. Williams and B. Z. Tang, Chem. Commun., 2007, 40 RSC.
- X. Cheng, D. Li, Z. Zhang, H. Zhang and Y. Wang, Org. Lett., 2014, 16, 880 CrossRef CAS PubMed.
- Y. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090 CrossRef.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 207 Search PubMed.
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- M. Kasha, Discuss. Faraday Soc., 1950, 14 RSC.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83 CrossRef CAS PubMed.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- R. Ye, Y. Liu, H. Zhang, H. Su, Y. Zhang, L. Xu, R. Hu, R. T. K. Kwok, K. S. Wong, J. W. Y. Lam, W. A. Goddard and B. Z. Tang, Polym. Chem., 2017, 8, 1722 RSC.
- S. Zhu, Y. Song, J. Shao, X. Zhao and B. Yang, Angew. Chem., Int. Ed., 2015, 54, 14626 CrossRef CAS PubMed.
- Y. Ruff and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 3556 CrossRef CAS PubMed.
- M. Sun, C.-Y. Hong and C.-Y. Pan, J. Am. Chem. Soc., 2012, 134, 20581 CrossRef CAS PubMed.
- J.-J. Yan, Z.-K. Wang, X.-S. Lin, C.-Y. Hong, H.-J. Liang, C.-Y. Pan and Y.-Z. You, Adv. Mater., 2012, 24, 5617 CrossRef CAS PubMed.
- Y. Ruff, E. Buhler, S.-J. Candau, E. Kesselman, Y. Talmon and J.-M. Lehn, J. Am. Chem. Soc., 2010, 132, 2573 CrossRef CAS PubMed.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586 CrossRef CAS PubMed.
- H. Zhang, X. Zheng, N. Xie, Z. He, J. Liu, N. L. C. Leung, Y. Niu, X. Huang, K. S. Wong, R. T. K. Kwok, H. H. Y. Sung, I. D. Williams, A. Qin, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16264 CrossRef CAS PubMed.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662 CrossRef CAS PubMed.
- Z. Zhao, H. Nie, C. Ge, Y. Cai, Y. Xiong, J. Qi, W. Wu, R. T. K. Kwok, X. Gao, A. Qin, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2017, 4, 1700005 CrossRef PubMed.
- R. Furue, T. Nishimoto, I. S. Park, J. Lee and T. Yasuda, Angew. Chem., Int. Ed., 2016, 55, 7171 CrossRef CAS PubMed.
- J. Huang, H. Nie, J. Zeng, Z. Zhuang, S. Gan, Y. Cai, J. Guo, S. J. Su, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 12971 CrossRef CAS PubMed.
- J. Guo, X. L. Li, H. Nie, W. Luo, S. Gan, S. Hu, R. Hu, A. Qin, Z. Zhao, S. J. Su and B. Z. Tang, Adv. Funct. Mater., 2017, 27, 1606458 CrossRef.
- J. Guo, X. L. Li, H. Nie, W. Luo, R. Hu, A. Qin, Z. Zhao, S. J. Su and B. Z. Tang, Chem. Mater., 2017, 29, 3623 CrossRef CAS.
- C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62 CrossRef CAS PubMed.
- M. Gao, Q. Hu, G. Feng, B. Z. Tang and B. Liu, J. Mater. Chem. B, 2014, 2, 3438 RSC.
- E. Wang, E. Zhao, Y. Hong, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem. B, 2014, 2, 2013 RSC.
- Y. Chen, W. Zhang, Z. Zhao, Y. Cai, J. Gong, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 5011 CrossRef CAS PubMed.
- D. Wang, H. Su, R. T. K. Kwok, X. Hu, H. Zou, Q. Luo, M. M. S. Lee, W. Xu, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2018, 9, 3685 RSC.
- Y.-C. Chen, W.-J. Zhang, Y.-J. Cai, R. T. K. Kwok, Y.-B. Hu, J. W. Y. Lam, X.-G. Gu, Z.-K. He, Z. Zhao, X.-Y. Zheng, B. Chen, C. Gui and B.-Z. Tang, Chem. Sci., 2017, 8, 2047 RSC.
- J. Yu, X. Sun, F. Cai, Z. Zhu, A. Qin, J. Qian, B. Tang and S. He, Opt. Lett., 2015, 40, 2313 CrossRef CAS PubMed.
- X. Gu, E. Zhao, T. Zhao, M. Kang, C. Gui, J. W. Y. Lam, S. Du, M. M. T. Loy and B. Z. Tang, Adv. Mater., 2016, 28, 5064 CrossRef CAS PubMed.
- J. Qi, C. Chen, D. Ding and B. Z. Tang, Adv. Healthcare Mater., 2018, 7, 1800477 CrossRef PubMed.
- J. Qian and B. Z. Tang, Chem, 2017, 3, 56 CAS.
- G. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404 CrossRef CAS PubMed.
- W. Wu, D. Mao, F. Hu, S. Xu, C. Chen, C. J. Zhang, X. Cheng, Y. Yuan, D. Ding, D. Kong and B. Liu, Adv. Mater., 2017, 29, 1700548 CrossRef PubMed.
- X. Shi, C. Yu, H. Su, R. Kwok, M. Jiang, Z. He, J. Lam and B. Tang, Chem. Sci., 2017, 8, 7014 RSC.
- M. Gao, H. Su, G. Lin, S. Li, X. Yu, A. Qin, Z. Zhao, Z. Zhang and B. Z. Tang, Nanoscale, 2016, 8, 15027 RSC.
- J. Qi, Y. Fang, R. T. K. Kwok, X. Zhang, X. Hu, J. W. Y. Lam, D. Ding and B. Z. Tang, ACS Nano, 2017, 11, 7177 CrossRef CAS PubMed.
- X. Cai, J. Liu, W. H. Liew, Y. Duan, J. Geng, N. Thakor, K. Yao, L.-D. Liao and B. Liu, Mater. Chem. Front., 2017, 1, 1556 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |