
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hole delocalization as a driving force for charge pair dissociation in organic photovoltaics†
Andrew B.
Matheson
 ,
Arvydas
Ruseckas
*,
Scott J.
Pearson
and
Ifor D. W.
Samuel
*
,
Arvydas
Ruseckas
*,
Scott J.
Pearson
and
Ifor D. W.
Samuel
*
Organic Semiconductor Centre, SUPA, School of Physics and Astronomy, University of St. Andrews, St. Andrews, Fife, KY16 9SS, UK. E-mail: ar30@st-andrews.ac.uk; idws@st-andrews.ac.uk
First published on 27th February 2019
Abstract
Charge carrier photogeneration is studied in photovoltaic blends of the conjugated polymer PTB7 with the electron acceptor PC71BM at low excitation densities using broadband transient absorption spectroscopy. In the optimized blend we observe a rise of hole polaron absorption on a 500 ps time scale which implies an increase of hole delocalization when photo-generated charge pairs dissociate into free charges. This concept is supported by the observed saturation of polaron absorption with electrochemical oxidation of polymer films, and the significant differences in transient absorption spectra observed in an inefficient blend due to bound charge transfer (CT) states. Our results suggest that hole polaron delocalization on polymer chains and entropy provide driving force for charge separation by lowering the free energy of the spatially separated charge pair. A potential barrier to the reformation of CT states appears as a result of carrier delocalization which also helps to reduce non-geminate carrier recombination.
Conceptual insightsOrganic solar cells have made substantial progress in the last decade in both efficiency and stability. Yet the precise mechanism and driving forces for dissociation of inter-molecular charge transfer (CT) states into free carriers are not fully understood. Here we show that the delocalization of holes on conjugated polymer chains increases upon charge separation in a polymer–fullerene blend, something that has not been observed before. The dynamic increase of hole delocalization not only reduces the Coulomb attraction in a charge pair and the effective mass of carriers as suggested previously, but also provides driving force for charge pair dissociation by lowering the Gibbs free energy. Because the free energy of the spatially separated pair is lower than that of the bound CT state, a potential barrier is formed for non-geminate charge recombination through the CT state. Our results give a new perspective on charge separation in organic photovoltaic materials. |
1. Introduction
The field of organic photovoltaics (OPV) has developed steadily in the last decade and single junction solar cells with power conversion efficiencies up to 14% have been reported.1–6 Charge pairs in OPV devices are generated by electron or hole transfer at heterojunctions between electron donor and acceptor materials. Because charge transfer is a short-range process and the dielectric constants of organic semiconductors are low (typically around 3), the generated charge pairs are expected to be bound by Coulomb attraction. Yet in optimized active layers the majority of pairs (including the directly excited low-energy charge transfer (CT) states) dissociate into free carriers in the weak built-in electric field.7–9 The mechanism and driving forces for dissociation are not fully understood. Several studies have suggested that the Gibbs free energy decreases upon pair dissociation because of increased entropy10–12 and also because of decreased site energy when charges are transported in bulk heterojunctions from a mixed donor–acceptor phase to pure donor and acceptor domains.13–15 Both electron and hole have to leave the interface between donor and acceptor to avoid geminate recombination and it has been shown that low mobility of slower carriers often limits the device efficiency.16 Heterojunctions with fullerene derivatives as electron acceptors usually show high initial electron mobility which makes the dissociation kinetically feasible.11,17–22 Electron delocalization over several fullerene molecules has been suggested to explain high initial electron mobility and efficient pair dissociation.23–26 In contrast, the role of hole delocalization and mobility in charge separation is much less understood.27–29In this paper we show that hole delocalization increases upon the dissociation of CT states in the efficient photovoltaic blend PTB7:PC71BM. This provides a significant driving force for charge pair dissociation by lowering the Gibbs free energy. The role of hole delocalization gives a new perspective on the charge separation mechanism and shows advantages of conjugated polymers for OPV applications. It also provides new insight for understanding charge photo-generation in non-fullerene solar cells and reduced non-geminate recombination.
2. Experimental
PTB7 with a molecular weight of 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da and a polydispersity of 2.6 was obtained from 1-Material Inc., and PC71BM of 99% purity was obtained from Solenne. PTB7 and fullerene were dissolved in chlorobenzene (HPLC grade from Sigma Aldrich) at a 40:60 ratio by weight and stirred at 50 °C for five hours. Blends with optimized morphology for photovoltaic action were prepared by adding 3% by volume of 1,8-diodooctane (DIO) from Sigma-Aldrich to the solution which was then stirred for other 5 min. Blended films for transient absorption (TA) spectroscopy were prepared by spin-coating on clean fused silica substrates at 1000 rpm in the nitrogen-filled glove box. The samples were excited with 180 fs pulses from an optical parametric amplifier pumped by the second harmonic of a Pharos regenerative amplifier at the pulse repetition rate of 2.5 kHz. The excitation energy density was kept low at 1.5 μJ cm−2 to give the excitation density of 2.6 × 1017 cm−3. This is low enough to minimize non-geminate recombination on a picosecond time scale. A white light continuum probe was generated in a YAG crystal using ∼1 μJ pulses of Pharos output at 1.2 eV which were delayed optically with respect to the pump pulses. The probe light was dispersed after the sample with a prism and detected by an InGaAs photodiode array assembled by Entwicklungsbüro Stresing.
000 Da and a polydispersity of 2.6 was obtained from 1-Material Inc., and PC71BM of 99% purity was obtained from Solenne. PTB7 and fullerene were dissolved in chlorobenzene (HPLC grade from Sigma Aldrich) at a 40:60 ratio by weight and stirred at 50 °C for five hours. Blends with optimized morphology for photovoltaic action were prepared by adding 3% by volume of 1,8-diodooctane (DIO) from Sigma-Aldrich to the solution which was then stirred for other 5 min. Blended films for transient absorption (TA) spectroscopy were prepared by spin-coating on clean fused silica substrates at 1000 rpm in the nitrogen-filled glove box. The samples were excited with 180 fs pulses from an optical parametric amplifier pumped by the second harmonic of a Pharos regenerative amplifier at the pulse repetition rate of 2.5 kHz. The excitation energy density was kept low at 1.5 μJ cm−2 to give the excitation density of 2.6 × 1017 cm−3. This is low enough to minimize non-geminate recombination on a picosecond time scale. A white light continuum probe was generated in a YAG crystal using ∼1 μJ pulses of Pharos output at 1.2 eV which were delayed optically with respect to the pump pulses. The probe light was dispersed after the sample with a prism and detected by an InGaAs photodiode array assembled by Entwicklungsbüro Stresing.
PTB7 films for spectro-electrochemical (SEC) measurements were drop-cast from toluene onto the indium tin oxide (ITO) working electrode and immersed in an electrochemical cell with an electrolyte of tetrabutylammonium hexafluorophosphate (Bu4NPF6) dissolved in acetonitrile. The cell had a silver reference electrode, a platinum counter electrode and a hose to supply nitrogen gas. The hose was used to purge the electrolyte solution for 10 minutes prior to measurements and then was placed above the surface of the solution during measurements. A CH Instruments electrochemical analyser connected to a computer was used to control measurements. A Perkin-Elmer Lambda 950 spectrometer was used to measure electrochemically induced absorption. The voltage across the electrodes was applied for 1 minute before each absorption measurement was taken to allow the doping level to equilibrate. The PC71BM film dissolved in the electrolyte when electrochemically reduced, hence the PC71BM− anion absorption was measured in di-chlorobenzene solution mixed with the electrolyte at a 4:1 volume ratio.
3. Results
Optimized PTB7:PC71BM blends have been used as active layers to make solar cells with up to 10% efficiency.3 The highest efficiencies are achieved with the use of the high boiling point additive 1,8-diiodooctane (DIO) to prepare the blends. Fig. 1 shows the TA spectra of the 40:60 PTB7:PC71BM optimized blend prepared with DIO and of the 90:10 PTB7:PC71BM blend. The excitation photon energies were carefully selected to give similar ground state absorbance of 0.25 and hence similar and nearly uniform excitation density of ∼2.6 × 1017 cm−3 in both bends (see Fig. S1 in ESI†). Both blends display ground state bleach at photon energies E > 1.8 eV but it is stronger in the optimized blend. Both blends show two photo-induced absorption bands, one at 1.1 eV and the other at ∼1.65 eV but the intensities of these bands differ substantially. The optimized blend shows strong absorption at 1.1 eV which grows with time from ΔA ∼ 0.45 × 10−3 at 3 ps to ΔA ∼ 0.63 × 10−3 at 1 ns and also shifts to a lower energy. The 90:10 PTB7:PC71BM blend shows stronger induced absorption at 1.65 eV and very little rise of the 1.1 eV band.
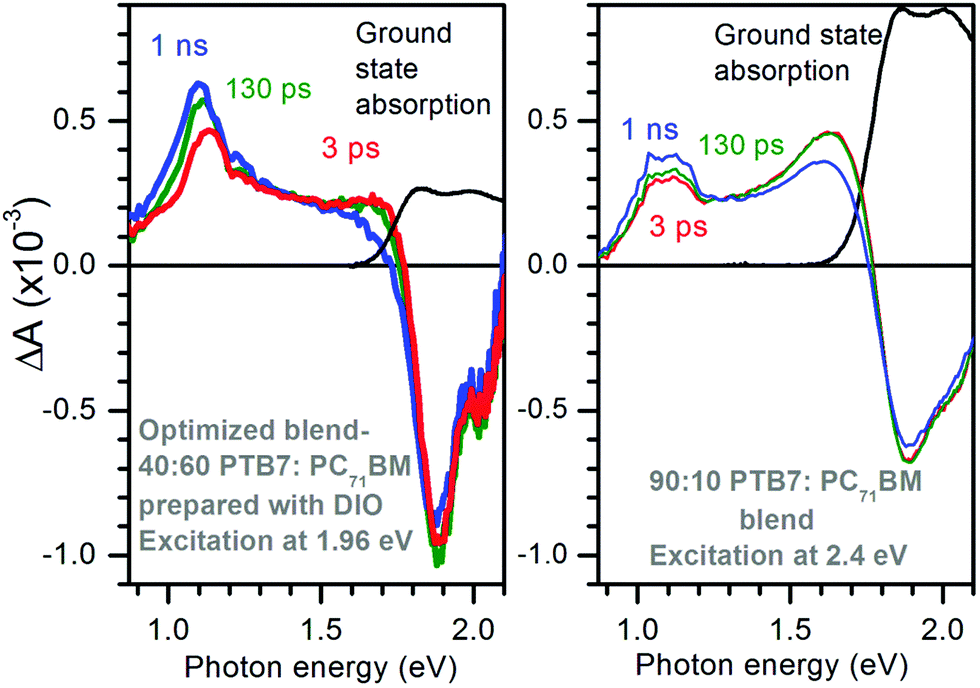 | ||
| Fig. 1 Transient absorption spectra of the optimized PTB7:PC71BM blend with 40:60 ratio prepared from chlorobenzene solution with DIO additive (left) and of the 90:10 PTB7:PC71BM blend (right) at selected time delays after excitation with 180 fs optical pulses. The excitation photon energies were selected to give the similar initial excitation density of ∼2.6 × 1017 cm−3 in both blends. Ground state absorption spectra are shown by black lines for reference. | ||
We wish to assign the spectral features observed in Fig. 1, so we compare a normalized TA spectrum of the optimized blend to that of a neat PTB7 film in Fig. 2. The TA spectrum of the neat PTB7 film at 0.1 ns shows a broad peak at around 1 eV which shifts to about 1.2 eV on a nanosecond scale. This can be attributed to conversion of primary excitons into PTB7 triplet state. The TA spectra of the blends are very different from the spectra of the neat PTB7 film implying that the singlet and triplet excitons present in the neat film cannot account for the spectra observed in the blends. Instead the majority of primary excitons must split into electron–hole pairs within 3 ps in both blends. This observation is supported by streak camera measurements which show a fast decay of PTB7 fluorescence in the optimized blend and in the 90:10 blend, predominantly within 3 ps (Fig. S2 in ESI†), suggesting that singlet excitons are quenched by electron transfer to PC71BM in <3 ps. It is also consistent with previous measurements of ultrafast fluorescence12,30 and TA.30,31 It should be noted, however, that previous TA studies of PTB7:PC71BM blends have not observed any rise in the strength of the 1.1 eV band. Judging from the five times stronger ΔA signals as compared to our measurements, we may assume these previous studies used significantly higher excitation densities, and thus the rise at 1.1 eV was masked by non-geminate recombination.
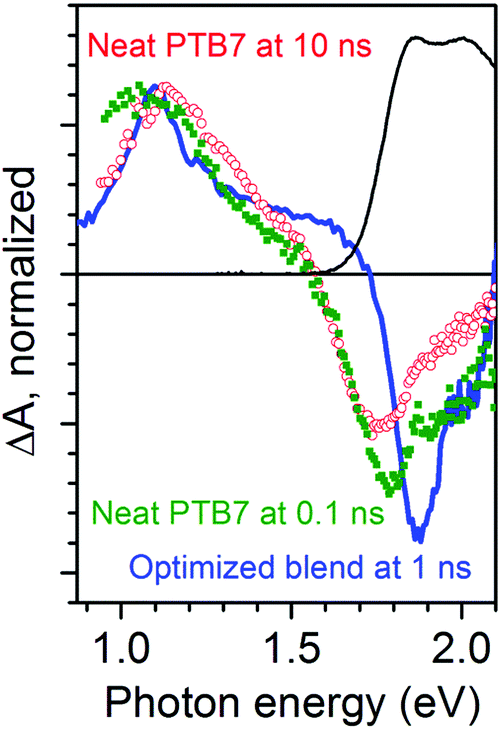 | ||
| Fig. 2 Transient absorption spectra of neat PTB7 film after excitation at 2.3 eV and of optimized PTB7:PC71BM blend normalized at the photo-induced absorption maximum around 1.1 eV. Black line shows ground state absorption spectrum of neat PTB7 film. | ||
Having shown that the spectral features and dynamics observed in Fig. 1 cannot be assigned to species present in optically excited PTB7 films, to better understand the physical significance of these spectral features and their dynamics we look at the electrochemically induced absorption of both PTB7 and PC71BM. Fig. 3 shows the results for a PTB7 film. In the cyclic voltammogram (CV) shown in Fig. 3b we observe a strong positive current for V > 0.7 V in the forward scan direction, and therefore we assume this to be the voltage range in which oxidation is taking place. We did not observe a peak in current over this 0–1.1 V range when scanning in the forward direction, with the current rising over this full range until we reverse scanning direction. We did observe a peak in the negative current at ∼0.6 V when ramping down from 1.1 V down to 0 V. That we did not seen a current peak in the forward scan direction, or multiple current peaks when scanning in the reverse direction is a good indication that we are still safely in the region of first PTB7 oxidation over the full range of the experiment, rather than accessing a higher oxidized state. Ideally, we would have scanned the voltage in the positive direction far enough to also observe a humped peak in the current in the forward scan too (i.e. something looking similar to that we observe for PC71BM in Fig. 4), but we did not want to exceed 1.1 V and risk irreversible electrochemical degradation of the film. Following oxidation, we observe the bleaching of ground state absorption at photon energies above 1.75 eV because the number of neutral chromophores has decreased (Fig. 3a). Simultaneously, an electrochemically induced absorption appears with a peak at approximately 1.1 eV and a smaller peak at approximately 1.67 eV. The electrochemically induced absorption spectra for the voltage up to 1 V closely resemble TA spectra observed in the optimized blend (vide supra). They are also very similar to the induced absorption observed by exposure of PTB7 film to iodine vapour and can be attributed to P2 transition in hole polaron whilst P1 transition previously observed at 0.4 eV is beyond our measurement range.32 With increasing voltage to 1.1 V the peak broadens to a higher energy side as absorption increases more in the region of 1.2–1.6 eV than at 1.1 eV. In contrast, the position of the 1.67 eV peak shows a slight apparent shift to lower energy as doping is increased, perhaps due to an increasing contribution from the low energy band. In Fig. 3c the induced absorption peak values for the two bands are plotted as a function of the fraction of chromophores oxidized which is calculated using the proportion of the ground state absorption which has been bleached
 | (1) |
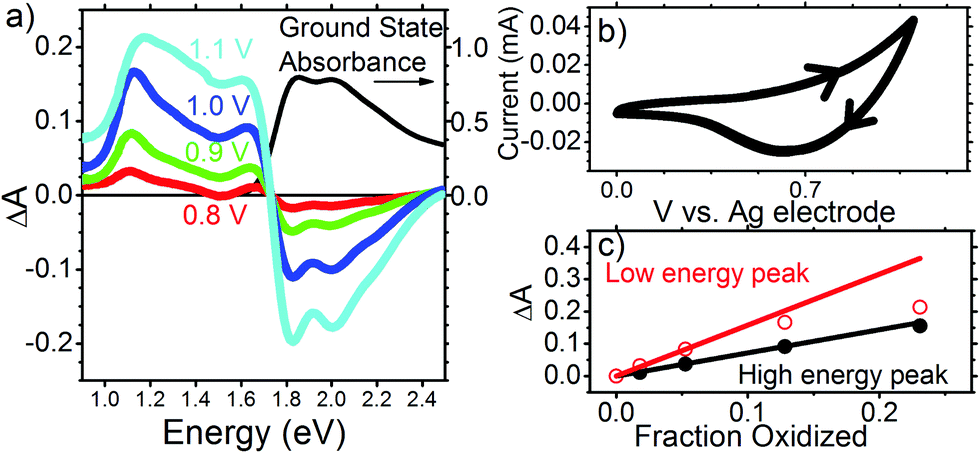 | ||
| Fig. 3 (a) Electrochemically induced change of absorbance in PTB7 film at different voltages applied to the ITO electrode (left axis) and its ground state absorbance (right axis). (b) Cyclic voltammogram for PTB7 with direction of scan shown with arrow heads. (c) Induced absorbance at the low energy peak (around 1.1 eV) and the high energy peak (around 1.65 eV) vs. the fraction of chromophores oxidized, which was obtained from eqn (1). | ||
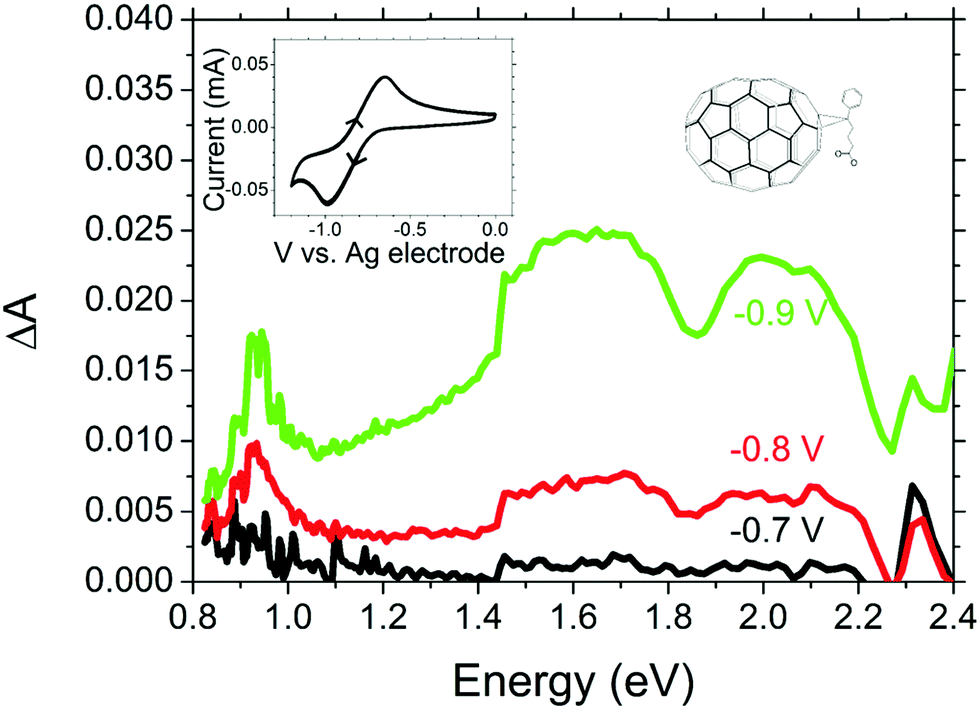 | ||
| Fig. 4 Electrochemically induced absorbance in PC71BM with cyclic voltammogram and chemical structure shown in the inset. | ||
The induced absorption in both bands shows a linear dependence on the fraction oxidised, up to 0.07. However, while the high energy band follows the linear relationship even when 25% of the polymer chains are oxidized, when the fraction oxidised exceeds 0.1, the low energy peak shows partial saturation. A similar shift of hole polaron absorption to higher energy at high doping has previously been observed in polyfluorenes,33,34 and explained by the distribution of conjugation lengths in the polymer. The energy of the hole polaron is reduced by hole delocalization, and thus polarons will initially tend to occupy long conjugated segments of the polymer backbone. At very high doping levels all long polymer segments are oxidized and hole polarons begin to occupy shorter conjugated segments, on which they are less delocalized. Even if two hole polarons were to reside side-by-side on long segments, they would repel each other and become more compressed at either end of their segment, and thus more localized. Either way, absorption saturation at 1.1 eV and the shift to higher energy indicate that hole polarons are delocalized along conjugated segments and they start interacting at high doping levels. Based on these observations we assign the 1.1 eV absorption band to delocalized hole polarons.
Given the position of the 1.65 eV band relative to the ground state absorption, it might be thought that this peak is due to a Stark shift. However, measurements of Stark shift have shown absorption changes ΔA < 10−3 even for electric fields many orders of magnitude stronger than the field in our electrochemical cell.35 Hence the electrochemically induced absorption of ΔA ∼ 0.1 we observe at 1.65 eV is far too strong to come from a Stark shift of the ground state absorption and must be due to a species produced through electrochemical doping, hence, hole polarons. Because this absorption is not sensitive to Coulomb repulsion between holes even at very high doping concentration, we attribute it to a different optical transition which is more localized than the transition at 1.1 eV. Similar observation was reported by Lane et al. when comparing photo-induced absorption spectra in the oligomer sexithiophene and polythiophene.36 They observed that whilst the spectral position of the lower energy absorption band depended on conjugation length, the higher energy band did not shift when conjugation length was reduced and concluded that the latter must be much more localized polaronic transitions such that changes in conjugation had minimal effect on their energy.
Optical excitation of PTB7:PC71BM blends will also result in the generation of electrons in the PC71BM domain, thus, we also investigate the spectrum of reduced PC71BM. Fig. 4 shows the electrochemically induced absorption upon reduction of PC71BM with corresponding CV curve shown in the inset. From the shape of the CV curve, which shows a single peak in the forward and reverse scans, we may safely assume that only the first PC71BM reduction is occurring between 0 V and −1 V relative to our reference electrode. Having identified this range of potentials as being the region in which the first reduction occurs, we measured the absorption spectra.
Electrochemically induced absorbance shows a narrow band at ∼0.93 eV and a broad band between 1.5 eV and 2.3 eV. A similar absorption spectrum but with a peak at 0.9 eV has been reported for the bare C70− anion,37 and a similar 0.93 eV peak has been observed previously in photo-induced absorption spectra of photovoltaic blends with PC71BM as an electron acceptor.38 However, to the best of our knowledge, this is the first time the electrochemically induced anion absorption for PC71BM− is observed “directly” without interpretation of photo-induced absorption spectra where multiple species may be present. Due to the solubility of reduced fullerenes being significantly higher in polar solvents than that of the neutral state, it was not possible to perform these measurements in film, as the PC71BM molecules dissolved in the electrolyte when reduced and then precipitated out of solution once reduction was reversed. Previous work suggested minimal differences between the C60 anion absorption spectra in film and solution.39 On this basis we expect that the PC71BM− anion absorption spectrum in film would be similar to that in solution.
4. Discussion
Now we can interpret the TA results shown in Fig. 1 and 2 using findings from electrochemically induced absorption spectra. The overall shape of TA spectra in the optimized blend is very similar to that of oxidized PTB7 shown in Fig. 3a and does not show a narrow PC71BM− anion peak at 0.93 eV. Each absorbed photon generates an electrically neutral exciton which may form an electron–hole pair, hence, the numbers of photo-generated radical cations and anions are equal. The fact the TA spectra so closely resemble those of doped PTB7 rather than doped PC71BM implies that absorption of the PC71BM− anion is much weaker than that of the PTB7+ cation and the TA spectra in the optimized blend must be dominated by photo-generated hole polarons.We previously assigned the TA spectrum of the neat PTB7 film at 10 ns shown in Fig. 2 as being dominated by triplet excitons. It has a maximum at 1.2 eV and does not show characteristic polaron absorption in the region of 1.5–1.7 eV we observe in SEC which confirms this further. We have considered a possibility that the 30% rise of the 1.1 eV band in the optimized blend may arise from charge pair recombination to the PTB7 triplet state. We can rule out such a scenario based on the following arguments. First, we constructed a composite TA spectrum by taking the spectrum at 3 ps and adding the PTB7 triplet exciton contribution assuming that the 30% rise of the signal at 1.1 eV comes from triplet excitons and compared it with the measured spectrum at 1 ns (Fig. S3 in ESI†). The measured spectrum at 1 ns shows a narrower peak compared to the composite spectrum and also there is a 0.03 eV difference in the peak position. This shows that the observed red-shift is incompatible with recombination to the triplet state. Second, geminate recombination of charge pairs to the polymer triplet state would require a spin flip and is unlikely to occur on a picosecond time scale whilst non-geminate recombination would show the excitation density dependence. We found that the rise time of the induced absorption at 1.1 eV did not change when the excitation density was decreased by a factor of three (Fig. S4 in ESI†). Based on these observations we conclude that triplet excitons cannot be responsible for the rise and dynamic redshift of the 1.1 eV peak observed in the optimized blend. Instead, the spectral shift is more similar to the difference in shape between the low voltage and high voltage SEC traces shown in Fig. 3a, which we assigned to compressed polarons when they interact at high doping levels. We thus consider it more likely that the rise and shift observed in Fig. 1 are due to the increasing delocalization of the hole polaron with time as the polarons relax to lower energy and occupy extended polymer chains with a greater conjugation length. The initial excitation density in our TA experiments was in the range of (0.8–2.6) × 1017 cm−3. This is slightly higher than the charge densities in organic solar cells of up to 1 × 1017 cm−3 under AM1.5 conditions. At these excitation densities the carrier–carrier interactions will be weak and so our findings are highly relevant to explain charge generation under standard solar cell illumination.
The strength of any ΔA signal is proportional to the oscillator strength of the probed optical transition which for a conjugated chromophore can be evaluated using the particle in the box model. In this model the total oscillator strength of a conjugated chromophore is proportional to the number of π electrons in the box. This scaling approximately works for the strongest optical transition in conjugated oligomers in all-trans configuration.40,41 Thus, the 30% rise of ΔA we observe in the 1–1.1 eV region for the optimized blend on a picosecond time scale suggests a 30% increase in hole delocalization (i.e. the box length).
The TA spectra measured in a 90:10 PTB7:PC71BM blend show weaker absorption at 1.1 eV relative to ∼1.65 eV band implying that majority of holes are more localized in the 90:10 blend. This blend does not show any red-shift of the 1.1 eV peak with time. Previous ultrafast carrier drift measurements showed that CT states are strongly bound in the 90:10 PTB7:PC71BM blend and can dissociate only in very strong electric field.22 The low concentration of fullerene molecules in the 90:10 blend inhibits electron transport and bound pairs do not dissociate. Therefore, weaker induced absorption at 1.1 eV is consistent with holes localized in the strongly bound CT state. Hole localization is expected in the bound CT state because of the strong Coulomb attraction, as has been reported recently in CdSe nanorods following electron transfer to an acceptor.42 In contrast, the much stronger polaron absorption observed in the optimized PTB7:PC71BM blend, the rise in signal, and shift to lower energies imply that the hole is delocalized immediately after exciton splitting and delocalization increases further with time.
A delocalized hole is less strongly bound to its sibling electron and also has lower effective mass, which was invoked to explain the dependence of photocurrent on electric field in planar heterojunctions using electron donors with different conjugation length.28 The field strength at which photocurrent saturation is observed was found to decrease as the conjugation length of the polymer used as an electron donor increased. This implies that materials which promote carrier delocalization have lower Coulomb barriers for pair dissociation. The dynamic increase of hole delocalization which we observe in this work can thus provide an additional driving force for charge pair dissociation and potentially can explain field-independent barrier-less dissociation of photo-generated charge pairs observed in optimized PTB7:PC71BM blends.22,43 We have also studied a 40:60 blend prepared without DIO and observed very similar TA spectra to the optimized blend but the signal at 1.1 eV was ∼30% smaller without DIO whilst the signal at 1.7 eV had similar strength in both blends (Fig. S5, ESI†). This suggests that the holes in the blend prepared without DIO are on average less delocalized than in the optimized blend but more delocalized than in the 90:10 blend. The 40:60 blend without DIO shows an ∼20% rise of the induced absorption at 1.1 eV on the same time scale as the optimized blend indicating an increase in hole delocalization with charge separation. The smaller signal and less rise at 1.1 eV without DIO correlates with previous observations that the charge pair dissociation efficiency is halved in samples prepared without DIO.22,43 This can be explained by a more heterogeneous morphology of the blend prepared without DIO where large fullerene-rich domains give efficient charge separation whilst the polymer-rich phases have insufficient fullerene aggregation to support charge pair dissociation, which is similar to the 90:10 blend.12,44
In order to understand the role of holes and electrons in charge separation we discuss our findings in the light of previously reported results for this material. A hole delocalization of over 4 nm was estimated for photogenerated radical pairs in PTB7:PC71BM blends at 50 K using studies of hyperfine interactions of electron spin with protons by electron paramagnetic resonance spectroscopy.45 This experiment probes spatially separated pairs with lifetimes >100 ns. Electron transfer to the fullerene and hole polaron formation is complete by 10 ps in the optimized blend. As discussed earlier, our results demonstrate a 30% increase in hole delocalization between 3 ps and 1 ns. If the dissociated hole is delocalized over 4 nm whilst free of Coulombic interactions at ∼1 ns, then the 30% increase in the strength of the 1.1 eV absorption band implies that at 3 ps its delocalization was over about 3 nm. The TA spectra measured in a blend with 10 wt% PC71BM show about twice weaker absorption at 1.1 eV than the optimized blend at 1 ns suggesting that hole delocalization length in the bound CT state is about 2 nm. Ultrafast carrier drift study showed that electrons are more mobile than holes in optimized PTB7:PC71BM blends and they drive the initial charge separation to about 2 nm in 10 ps.46 This initial charge separation is very important for OPVs because it prevents hole localization by Coulomb attraction which occurs in the blend with 90 wt% PTB7. The hole is thus merely a stationary spectator in the initial charge separation process but its ability to delocalize is very important because it reduces the Coulomb attraction. This provides a driving force for dissociation of the loosely bound charge pair by delocalizing further, as we shall discuss further in the next paragraph.
The Coulomb binding energy of the charge pair separated by 1 nm is about 0.4 eV. With entropy taken into account in an energetically ordered model at a planar donor–acceptor interface the free energy barrier of ΔG‡ = 0.18 eV was calculated for dissociation of the charge pair separated by 1 nm.47 However, the experimentally observed activation energy for CT state dissociation, Ea, is in the range of 0.035–0.09 eV in optimized PTB7:PC71BM blends.48,49 The fact Ea < ΔG‡ implies that other factors in addition to entropy influence charge separation. We can estimate the effect of hole and electron delocalization on the free energy of the system by taking the difference between the relaxed CT state energy (ECT), which is observed as the electroluminescence peak at 1.27 eV in PTB7:PC71BM blends,50 and of the photovoltaic gap EGap = 1.10 eV in this heterojunction, which was determined using a combination of ultraviolet and inverse photoelectron spectroscopies (UPS and IPES).51 UPS removes an electron to infinity and leaves a delocalized hole on the polymer, whereas IPES adds an electron to fullerene which can delocalize and hence reduce its energy. The estimated energy gain from charge delocalization is ECT − EGap = 0.17 ± 0.09 eV, with the uncertainty limited by the accuracy of UPS and IPES measurements. This figure is very similar to the calculated ΔG‡ = 0.18 eV suggesting that carrier delocalization in combination with entropy gain may be the major driving force in making CT state dissociation favourable. Our proposed energy landscape for CT state and separated charge pairs is illustrated in Fig. 5. Lower energy of separated pairs will also create an energy barrier to the reformation of the CT state, hence, it also helps to reduce non-geminate charge recombination.
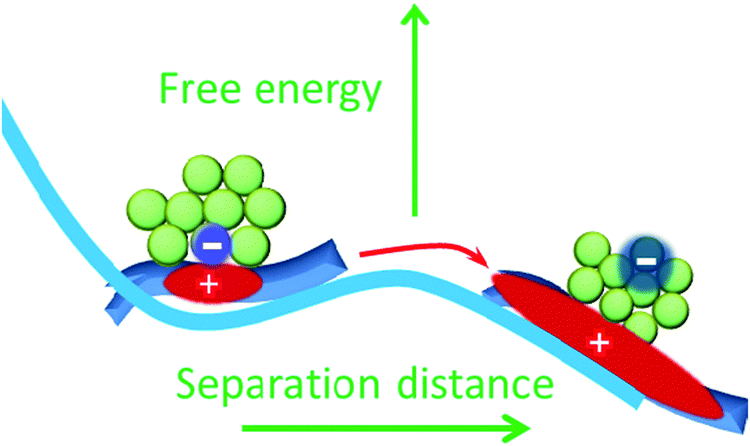 | ||
| Fig. 5 Proposed Gibbs free energy surface along the charge separation coordinate. Hole delocalization provides driving force for dissociation of CT state by lowering the free energy of spatially separated charge pair. | ||
5. Conclusions
By combining broadband TA spectroscopy with electrochemically induced charge absorption measurements we have shown that the hole delocalization length in the donor polymer PTB7 increases upon charge pair dissociation. We estimate that the free energy gained by hole and electron delocalization effectively lowers the Coulomb barrier and allows the electron to escape the Coulomb potential of the hole. Although the hole is merely a spectator in the charge separation process, its ability to delocalize is important for efficiency of OPVs. Our results show advantages of conjugated molecules which support charge delocalization for applications in organic photovoltaics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (grants EP/L017008/1, EP/G03673X/1 and EP/J009016/1) and the European Research Council (grant 321305). I. D. W. S. acknowledges a Royal Society Wolfson Research Merit Award. We are grateful to Dr Francisco Montilla for his help in setting up the apparatus for electrochemically induced absorption. The research data supporting this publication can be accessed at DOI: 10.17630/ca6397c2-16f4-4329-a1e5-5cd7832f68ceReferences
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- X. Ouyang, R. Peng, L. Ai, X. Zhang and Z. Ge, Nat. Photonics, 2015, 9, 520–524 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28, 4734–4739 CrossRef CAS PubMed.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2016, 16, 363 CrossRef PubMed.
- J. Lee, K. Vandewal, S. R. Yost, M. E. Bahlke, L. Goris, M. A. Baldo, J. V. Manca and T. V. Voorhis, J. Am. Chem. Soc., 2010, 132, 11878–11880 CrossRef CAS PubMed.
- K. Vandewal, S. Albrecht, E. T. Hoke, K. R. Graham, J. Widmer, J. D. Douglas, M. Schubert, W. R. Mateker, J. T. Bloking, G. F. Burkhard, A. Sellinger, J. M. J. Fréchet, A. Amassian, M. K. Riede, M. D. McGehee, D. Neher and A. Salleo, Nat. Mater., 2014, 13, 63–68 CrossRef CAS PubMed.
- T. G. J. van der Hofstad, D. Di Nuzzo, M. van den Berg, R. A. J. Janssen and S. C. J. Meskers, Adv. Energy Mater., 2012, 2, 1095–1099 CrossRef CAS.
- B. A. Gregg, J. Phys., Lett., 2011, 2, 3013–3015 CAS.
- D. A. Vithanage, A. Devižis, V. Abramavičius, Y. Infahsaeng, D. Abramavičius, R. C. I. MacKenzie, P. E. Keivanidis, A. Yartsev, D. Hertel, J. Nelson, V. Sundström and V. Gulbinas, Nat. Commun., 2013, 4, 2334 CrossRef PubMed.
- G. J. Hedley, A. J. Ward, A. Alekseev, C. T. Howells, E. R. Martins, L. A. Serrano, G. Cooke, A. Ruseckas and I. D. W. Samuel, Nat. Commun., 2013, 4, 2867 CrossRef PubMed.
- F. C. Jamieson, E. B. Domingo, T. McCarthy-Ward, M. Heeney, N. Stingelin and J. R. Durrant, Chem. Sci., 2012, 3, 485–492 RSC.
- C. Groves, Energy Environ. Sci., 2013, 6, 1546–1551 RSC.
- T. M. Burke and M. D. McGehee, Adv. Mater., 2014, 26, 1923–1928 CrossRef CAS PubMed.
- M. Stolterfoht, A. Armin, S. Shoaee, I. Kassal, P. Burn and P. Meredith, Nat. Commun., 2016, 7, 11944 CrossRef CAS PubMed.
- V. Abramavicius, D. Amarasinghe Vithanage, A. Devizis, Y. Infahsaeng, A. Bruno, S. Foster, P. E. Keivanidis, D. Abramavicius, J. Nelson, A. Yartsev, V. Sundstrom and V. Gulbinas, Phys. Chem. Chem. Phys., 2014, 16, 2686–2692 RSC.
- V. Pranculis, Y. Infahsaeng, Z. Tang, A. Devižis, D. A. Vithanage, C. S. Ponseca, O. Inganäs, A. P. Yartsev, V. Gulbinas and V. Sundström, J. Am. Chem. Soc., 2014, 136, 11331–11338 CrossRef CAS PubMed.
- A. Devižis, D. Hertel, K. Meerholz, V. Gulbinas and J. E. Moser, Org. Electron., 2014, 15, 3729–3734 CrossRef.
- R. Augulis, A. Devižis, D. Peckus, V. Gulbinas, D. Hertel and K. Meerholz, J. Phys. Chem. C, 2015, 119, 5761–5770 CrossRef CAS.
- A. Devižis, J. De Jonghe-Risse, R. Hany, F. Nüesch, S. Jenatsch, V. Gulbinas and J.-E. Moser, J. Am. Chem. Soc., 2015, 137, 8192–8198 CrossRef PubMed.
- V. Pranculis, A. Ruseckas, D. A. Vithanage, G. J. Hedley, I. D. W. Samuel and V. Gulbinas, J. Phys. Chem. C, 2016, 120, 9588–9594 CrossRef CAS PubMed.
- B. Bernardo, D. Cheyns, B. Verreet, R. D. Schaller, B. P. Rand and N. C. Giebink, Nat. Commun., 2014, 5 Search PubMed.
- V. Abramavicius, V. Pranculis, A. Melianas, O. Inganäs, V. Gulbinas and D. Abramavicius, Sci. Rep., 2016, 6, 32914 CrossRef CAS PubMed.
- S. Gélinas, A. Rao, A. Kumar, S. L. Smith, A. W. Chin, J. Clark, T. S. van der Poll, G. C. Bazan and R. H. Friend, Science, 2014, 343, 512–516 CrossRef PubMed.
- B. M. Savoie, A. Rao, A. A. Bakulin, S. Gelinas, B. Movaghar, R. H. Friend, T. J. Marks and M. A. Ratner, J. Am. Chem. Soc., 2014, 136, 2876–2884 CrossRef CAS PubMed.
- C. Deibel, T. Strobel and V. Dyakonov, Phys. Rev. Lett., 2009, 103, 036402 CrossRef PubMed.
- C. Schwarz, S. Tscheuschner, J. Frisch, S. Winkler, N. Koch, H. Bässler and A. Köhler, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 155205 CrossRef.
- A. B. Matheson, S. J. Pearson, A. Ruseckas and I. D. W. Samuel, J. Phys., Lett., 2013, 4, 4166–4171 CAS.
- Y. Kouhei, K. Hayato, Y. Takeshi, H. Liyuan and M. Yutaka, Jpn. J. Appl. Phys., 2013, 52, 062405 CrossRef.
- J. M. Szarko, B. S. Rolczynski, S. J. Lou, T. Xu, J. Strzalka, T. J. Marks, L. Yu and L. X. Chen, Adv. Funct. Mater., 2014, 24, 10–26 CrossRef CAS.
- T. Basel, U. Huynh, T. Zheng, T. Xu, L. Yu and Z. V. Vardeny, Adv. Funct. Mater., 2015, 25, 1895–1902 CrossRef CAS.
- N. Takeda, S. Asaoka and J. R. Miller, J. Am. Chem. Soc., 2006, 128, 16073–16082 CrossRef CAS PubMed.
- F. Montilla, A. Ruseckas and I. D. W. Samuel, Chem. Phys. Lett., 2013, 585, 133–137 CrossRef CAS.
- L. Sebastian, G. Weiser and H. Bässler, Chem. Phys., 1981, 61, 125–135 CrossRef CAS.
- P. A. Lane, X. Wei and Z. V. Vardeny, Primary Photoexcitations in Conjugated Polymers: Molecular Exciton Versus Semiconductor Band Model, 1998, pp. 292–317 DOI:10.1142/9789814447201_0011.
- D. R. Lawson, D. L. Feldhiem, C. A. Foss, P. K. Dorhout, C. M. Elliott, C. R. Martin and B. Parkinson, J. Phys. Chem., 1992, 96, 7175–7177 CrossRef CAS.
- A. Sperlich, M. Liedtke, J. Kern, H. Kraus, C. Deibel, S. Filippone, J. L. Delgado, N. Martín and V. Dyakonov, Phys. Status Solidi RRL, 2011, 5, 128–130 CrossRef CAS.
- K. Kaneto, T. Abe and W. Takashima, Solid State Commun., 1995, 96, 259–264 CrossRef CAS.
- S. Schumacher, A. Ruseckas, N. A. Montgomery, P. J. Skabara, A. L. Kanibolotsky, M. J. Paterson, I. Galbraith, G. A. Turnbull and I. D. W. Samuel, J. Chem. Phys., 2009, 131, 154906 CrossRef PubMed.
- M. S. Vezie, S. Few, I. Meager, G. Pieridou, B. Dörling, R. S. Ashraf, A. R. Goñi, H. Bronstein, I. McCulloch, S. C. Hayes, M. Campoy-Quiles and J. Nelson, Nat. Mater., 2016, 15, 746 CrossRef CAS PubMed.
- Y. Yang, K. Wu, A. Shabaev, A. L. Efros, T. Lian and M. C. Beard, ACS Energy Lett., 2016, 1, 76–81 CrossRef CAS.
- A. Foertig, J. Kniepert, M. Gluecker, T. Brenner, V. Dyakonov, D. Neher and C. Deibel, Adv. Funct. Mater., 2014, 24, 1306–1311 CrossRef CAS.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65–74 CrossRef CAS.
- J. Niklas, K. L. Mardis, B. P. Banks, G. M. Grooms, A. Sperlich, V. Dyakonov, S. Beaupre, M. Leclerc, T. Xu, L. Yu and O. G. Poluektov, Phys. Chem. Chem. Phys., 2013, 15, 9562–9574 RSC.
- D. A. Vithanage, A. B. Matheson, V. Pranculis, G. J. Hedley, S. J. Pearson, V. Gulbinas, I. D. W. Samuel and A. Ruseckas, J. Phys. Chem. C, 2017, 121, 14060–14065 CrossRef CAS.
- S. N. Hood and I. Kassal, J. Phys. Chem. Lett., 2016, 7, 4495–4500 CrossRef CAS PubMed.
- M. Gerhard, A. P. Arndt, I. A. Howard, A. Rahimi-Iman, U. Lemmer and M. Koch, J. Phys. Chem. C, 2015, 119, 28309–28318 CrossRef CAS.
- J. y. Tsutsumi, H. Matsuzaki, N. Kanai, T. Yamada and T. Hasegawa, J. Phys. Chem. C, 2013, 117, 16769–16773 CrossRef CAS.
- M. K. Etherington, J. Wang, P. C. Y. Chow and N. C. Greenham, Appl. Phys. Lett., 2014, 104, 063304 CrossRef.
- S. Park, J. Jeong, G. Hyun, M. Kim, H. Lee and Y. Yi, Sci. Rep., 2016, 6, 35262 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mh01204k |
| This journal is © The Royal Society of Chemistry 2019 |