
Facile emission color tuning and circularly polarized light generation of single luminogen in engineering robust forms†
 ab
Zikai
He
*ac
and
ab
Zikai
He
*ac
and
Abstract
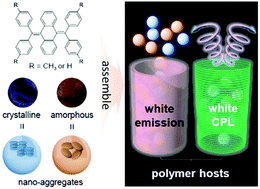
Controlling molecular assembled structures of a single luminogen through external manipulation is a general and powerful concept for designing emission-tunable materials. However, these reported powder-like compounds generally show limited emission color, high serendipitous-discovery dependence and poor mechanical properties. To overcome these limitations, we describe a simple yet powerful strategy that is adapted from methods used to control bulk commodity polymers, to modulate the molecular assembled architecture of dopant. We use luminogens with aggregation-induced emission characteristics and intrinsic blue and yellow emissions at their crystalline and amorphous states as demonstrations. When incorporating such luminogens (AIEgens) in the desired polymer matrix with tailored microstructure, white light and white circularly polarized light are generated, which are inaccessible from single and achiral AIEgen alone. The introduced strategy for realizing diverse light emission is an innovative way of developing luminogens and provides useful continuous emissive materials in the forms of macroscopic films and fibers for developing flexible devices and wearable systems.
 Please wait while we load your content...
Please wait while we load your content...

