
Polymer networks based on photo-caged diene dimerization†
Tim
Krappitz‡
 a,
Florian
Feist‡
ab,
Iris
Lamparth
c,
Norbert
Moszner
c,
Hendrik
John
c,
James P.
Blinco
ad,
Tim R.
Dargaville
a and
Christopher
Barner-Kowollik
*ad
a,
Florian
Feist‡
ab,
Iris
Lamparth
c,
Norbert
Moszner
c,
Hendrik
John
c,
James P.
Blinco
ad,
Tim R.
Dargaville
a and
Christopher
Barner-Kowollik
*ad
aSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
bMax-Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
cIvoclar Vivadent AG, Research & Development, Bendererstrasse 2, FL-9494, Schaan, Liechtenstein
dMacromolecular Architectures, Institut für Technische Chemie und Polymerchemie, Karlsruhe Institute of Technology (KIT), Engesserstr. 18, 76131 Karlsruhe, Germany
First published on 24th September 2018
Abstract
A powerful, simple and efficient method for photochemical crosslinking, exploiting the self-dimerization of photo-caged dienes based on o-methylbenzaldehydes (o-MBAs) is introduced. Using a small molecule model system it is possible to identify the intermediate and final products of this light induced dimerization process. The bi-functional photocurable monomers contain o-MBA as well as vinylic units, which enables radical copolymerization with other monomers to tune the mechanical properties of the resulting networks. Crosslinked materials with reduced E-moduli ranging from 0.29 to 5.76 GPa and a hardness between 52 and 329 MPa were obtained, thus spanning a wide regime of different ‘soft’ to ‘hard’ material properties. The functional groups can be addressed through simultaneous reaction or in a λ-orthogonal fashion. This orthogonality can be achieved by inducing the free radical polymerization independently of the dimerization either thermally or via photoinitiation at a wavelength higher than the wavelength used for the non-radical dimerization process (>400 nm). Critically, this allows for a spatially defined adjustment of both the E-modulus and hardness within the respective material by controlling the irradiation parameters, such as wavelength, curing time and intensity. The introduced self-dimerizing resists thus represent a highly controllable combined radical/non-radical and λ-orthogonal curing system.
Conceptual insightsWe introduce a high performance yet chemically simple photoresist based on the dimerization of photo-caged dienes. In contrast to the majority of state-of-the-art photoresists, which are based on radical polymerization, the current resists allow curing via an efficient dimerization, allowing to λ-orthogonally execute photochemically induced radical curing and non-radical crosslinking. The study carefully maps the underpinning photochemical dimerization, which enables the λ-orthogonality to be selectively addressed, allowing to fine tune the resulting network properties. Critically, our study introduces highly effective photoresists that do not require an additional crosslinker that could potentially leach from the final material. The crosslinking itself proceeds without any additional catalyst. Thus, the herein exploited photodimerization concept represents a powerful technique enabling resist hardening in a simultaneous or λ-orthogonal consecutive fashion. |
Introduction
Crosslinked materials have a long standing history for the industrial processing of materials.1–3 Intermolecular crosslinking reactions are typically executed by utilizing multi-functional monomers for direct curing in situ or by post-crosslinking, i.e. combining a suitable multi-functional crosslinker with a functional polymer.4In contrast to many post-cross-linking approaches, methods utilizing o-quinodimethanes (o-QDMs) as crosslinking moiety exhibit key advantages, such as requiring no additional catalyst for activation.5o-Quinodimethanes are a class of highly reactive enes and can be obtained by the thermally or photoinduced cleavage of 1-functionalized benzocyclobutenes and o-methylcarbonyl compounds.5,6 In addition to their reactivity with electron deficient dienophiles7o-QDMs can undergo electrocyclisation, dimerization or oligomerization as previously shown for thermally generated o-QDMs.5 The product spectrum includes the formation of benzocyclobutanes, tetrahydrodibenzo[8]annulenes, hemiacetals, tetrahydro-anthracenes and spiro-compounds. Similar to the thermal transformation of benzocyclobutanes, o-QDMs can be obtained photochemically from photo-caged o-methylbenzaldehyde (o-MBA) moieties.7–9 While the potential of photochemically induced o-QDM formation and their subsequent Diels–Alder reaction for step-growth polymerization was initially shown by Meador,6 the mechanism of the photoinduced formation of o-QDM from o-MBA was primarily investigated by Porter and Tchir in 1970 and is summarized in Scheme 1.10
 | ||
| Scheme 1 Reaction mechanism for the reversible formation of an o-QDM from o-methylcarbonyl compounds (A1). | ||
Upon UV irradiation, o-MBAs, A1, initially form a short-lived excited singlet state, A2. Subsequent intersystem crossing and single bond rotation (A3 and A4) leads to (E)- and (Z)-enols (A6 and A7) via the biradicals A5a and A5b. However, the (Z)-enol A7 is the sole species active for reaction, as the less stable (E)-enol preferably undergoes a [1,5]-sigmatropic rearrangement.10–12 In the presence of an electron deficient dienophile (Z)-enol, A7 reacts to form a Diels–Alder cycloadduct or else slowly rearranges to the starting material.
The high reactivity of photoactivated o-QDMs towards dienophiles has been utilized for the generation of various polymer scaffolds, surfaces, and nanoparticles.13 The o-MBA driven photo-induced coupling (commonly termed ‘photoenol-ligation’) features several advantages, for instance reactivity at ambient temperatures, a broad functional group tolerance, a very high reaction rate with dienophiles and an excellent quantum yield, while exhibiting high stability under various reaction conditions.14
Photoinduced cross-linking reactions of polymers are essential for a broad range of applications exploiting their inherent advantages of spatial and temporal control in comparison to thermally induced cross-linking. Dual reactive structures become accessible via direct laser writing (DLW) based on photoenol-ligation, as well as spatially resolved surface patterning, which are among many examples where light-induced reactions involving o-QDMs are utilized.15,16 The concept of wavelength selective reactions for the synthesis of complex macromolecular architectures involving photoenol-ligation was introduced earlier by our group.17,18 Furthermore, we demonstrated that a radical polymerization and the photoenol-ligation can be performed consecutively.19 Macromolecular photocurable mixtures can be employed as photoresists and are of particular interest for soft matter materials design within the emerging field of 3D printing,20,21 yet most of these systems are based on an exclusive radical crosslinking mechanism. Radical systems are simple to execute and curing proceeds rapidly, however, a radical mediated curing system is then restricted to one chemical reaction type only, limiting the options for using disparate colours of light for curing control. There are notable exceptions in the literature that demonstrate the use of non-radical curing systems.22,23 For example, Bowman and co-workers recently described a sequential polymer network formation via wavelength selective irradiation.24 These authors utilized an irradiation wavelength between 400–500 nm for the initial curing step, leading to the cleavage of a selected photobase generator, thus, initiating an anion-mediated thiol-Michael polymerization. Subsequently, a second light source (λ = 365 nm) induced a radical polymerization with a photo-radical initiator. It thus appears highly attractive to combine different curing mechanisms into one resist system, enabling the addressing of the curing system with orthogonal triggers, for example different colours of light. The use of disparate wavelengths allows to control different stages of the curing process independently. Ideally, such multi-responsive resist systems should be chemically simple and the photoreactive precursors readily accessible with no need for added crosslinker.
Interestingly, to date, the reactivity of photochemically generated o-QDMs has exclusively been exploited for reactions between the o-QDM and an activated dienophile in the resist design, requiring additional crosslinker.22,23 In contrast, we introduce a spatially and temporally controlled crosslinking reaction with a simple resist design based on the o-QDM self-dimerization, exhibiting fast curing kinetics and the critical advantage of the absence of a small molecular crosslinker. Furthermore, we establish a simple yet powerful λ-orthogonal resist system by separating radical photopolymerization from non-radical o-QDMs self-dimerization with disparate colours of light, affording materials with widely adjustable transient properties (storage- and loss moduli, E-modulus and hardness).
Results and discussion
In the following, we will initially demonstrate the molecular basis of our o-QDM based crosslinking strategy and establish the exact chemistry of the dimer links. Subsequently, we progress to the network formation in different variants, including an attractive λ-orthogonal pathway. Finally, we demonstrate the versatility of our approach by introducing different co-monomers into the photocurable mixture and thus fine-tuning the mechanical properties of the resulting materials.o-QDM dimerization
We have previously observed the dimerization of o-QDMs during the synthesis of step growth polymers.25 In these processes, the precise analysis of the obtained structures and an in-depth understanding of the dimerization mechanism was inherently complex. However, in the present small-molecule study, we were able to identify reactive intermediates, as well as the final products of the photodimerization, which enables us to influence the reaction pathways (Scheme 2).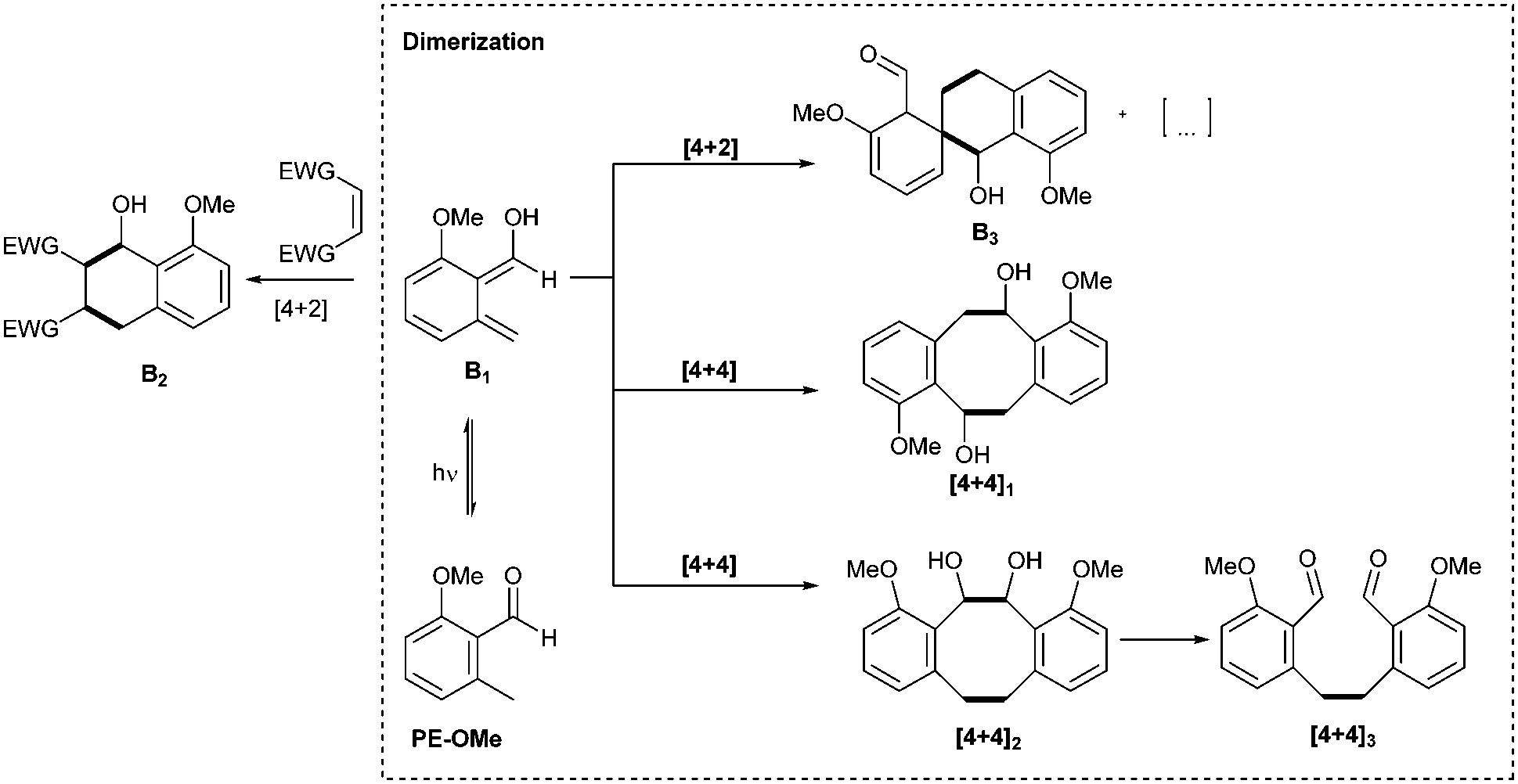 | ||
| Scheme 2 Reaction pathways within the PE-OMe model system via the photoinduced formation of diene B1 leading to the Diels–Alder product B2 in the presence of a suitable dienophile. In the absence of a dienophile, dimerization occurs, leading to the products B3, [4+4]1, [4+4]2 and [4+4]3, which we herein exploit for resist design. | ||
To assess the dimerization, we utilized 2-methoxy-6-methylbenzaldehyde (PE-OMe) as a model compound. A minimal amount of structural complexity in the PE-OMe molecule facilitated purification and characterization of the products.
To conduct photoreactions under continuous flow conditions offers various advantages compared to batch processes (improved irradiation homogeneity, reliable up-scaling, fast mixing and fast heat exchange).26 We performed photoflow reactions using PE-OMe to enable a high photon flux and temperature control during our model study.
The small molecule study allowed us to investigate and understand the dimerization reaction using standard organic analytical methods, such as nuclear magnetic resonance (NMR) spectroscopy (Section S5, ESI†) and liquid chromatography coupled to mass spectrometry (LC-MS, Section S6, ESI†). Noteworthy, the conditions of our small molecule study differed substantially from the conditions used for the crosslinking experiments. The photoflow experiments were conducted using low concentrations and low temperatures, whereas for the crosslinking, thin layers, a high photon flux, higher temperatures above the Tg of the polymers and considerably higher o-MBA concentrations were used. In the photoflow reactor high concentrations would lead to an inhomogeneous irradiation profile, resulting in impractical residence times.
We find that the rearrangement from B1 (Scheme 2) to form the starting material PE-OMe can be significantly slowed at low temperatures, increasing the lifetime of the o-QDM, thus enabling dimerization reactions even at low concentrations, leading to several products (Scheme 2). We anticipate that in the polymer crosslinking experiments the higher temperatures, the significantly higher concentration and photon cross-section will facilitate the dimerization, although these reaction conditions could not be applied in the small molecule photoflow study.
By screening various solvents (dichloromethane, ethyl acetate, toluene and dimethyl sulfoxide) for the model photoreaction, we observed a selectivity of approximately 80% for three main products ([4+4]1, [4+4]2, [4+4]3) in ethyl acetate. The percentage is based on integration of the crude liquid chromatography (LC) trace (refer to Fig. S29, ESI†). Finally, the dimerization was performed on the preparative scale using ethyl acetate as solvent in order to isolate the major components and fully characterize them by 1D and 2D NMR spectroscopy (Fig. 1 and Fig. S4–S13, ESI†) and LC-MS (Section S6, ESI†). Note that the [4+4]2 (the vicinal diol) was identified in the crude material (Fig. S29, ESI†) but could not be isolated, potentially due to decomposition at ambient temperature forming the bis-benzaldehyde [4+4]3 during the workup procedure. The aldehyde resonance 1 at δ = 10.65 ppm and the methyl resonance 2 at δ = 2.58 ppm of PE-OMe disappear upon irradiation to generate the resonances 1′, 1′′, 2′ and 2′′ (δ = 5.36 ppm, 10.66 ppm, 3.60/3.00 ppm, and 3.17 ppm, respectively) corresponding to the major products (Fig. 1). In addition to the major components, a variety of [4+2] cycloadducts were formed and identified by 1H-NMR spectroscopy in the crude material, although it was not possible to isolate these components due to their inherent reactivity and instability. In summary, at low temperatures (−20 to −40 °C) and low concentrations (5.00 mmol L−1), PE-OMe dimerizes in the absence of a dienophile to form [4+2] and [4+4] cycloadducts. The most abundant dimerization reaction (∼80%), however, is the [4+4] cycloaddition forming dibenzo[8]annulenes [4+4]1 and [4+4]2. The latter readily decomposes to form the bis-aldehyde [4+4]3.
 | ||
| Fig. 1 Left: 1H-NMR spectra from bottom to top: PE-OMe, the crude product after irradiation (ethyl acetate, λmax = 365 nm LED, 10 min retention time, −40 °C), and the two dominant products [4+4]1 (4,10-dimethoxy-5,6,11,12-tetrahydrodibenzo[a,e][8]annulene-5,11-diol) and [4+4]3 (6,6′-(ethane-1,2-diyl)bis(2-methoxybenzaldehyde)). Right: 1H-NMR spectrum and size exclusion chromatogram (SEC) of a typical copolymer recorded in deuterated chloroform (CDCl3) and dimethyl acetamide (DMAc), respectively. For the exact copolymer composition refer to Table 1, entry c. | ||
The inherent capability of o-QDMs to dimerize is exploited in the following formulation of photocurable mixtures, requiring no additional crosslinker. We expect a similar product spectrum for the crosslinked materials utilizing photoenol methacrylate (PEMA) compared to our observations in the small molecule study, utilizing PE-OMe as model compound. However, as the product selectivity of the dimerization largely varies depending on the reaction conditions, the product ratio in the photocrosslinking experiments might vary in comparison to the small molecule study.
Network formation
In the subsequent step, we install the o-MBA unit within a polymerizable monomer species, i.e. photoenol methacrylate (PEMA),27 enabling the formation of functional polymers that are the basis for the network formation driven by the photodimerization. Thus, PEMA features two reactive functionalities that can be selectively triggered. The o-MBA group is solely activated via irradiation between 300–400 nm, while the vinyl function can be activated by primary radicals derived from a variety of radical initiators that can be addressed by thermal or light triggers in the visible or UV light range. Herein, we follow three complementary pathways for the formation of the photocrosslinked polymeric networks, each featuring specific advantages (Scheme 3): | ||
| Scheme 3 Three approaches for the synthesis of photoinduced crosslinked polymeric materials in the absence of any additional small molecule as crosslinker, based on o-quinodimethane dimerization: (i) thermal initiation step leading to the formation of the linear polymeric precursors. (ii) All-in-one generation of the lateral chains and simultaneous crosslinking by o-QDM dimerization. (iii) λ-Orthogonal two step curing, initially affording the lateral chain architecture in the visible light regime, during which the o-MBA unit remains unaffected and subsequent UV light induced cross-linking. | ||
(i) Synthesis of PEMA containing polymeric precursors by thermally induced free radical polymerization, followed by their crosslinking with UV-A light.
(ii) Parallel photopolymerization and crosslinking in the UV-A spectral region.
(iii) λ-Orthogonal photopolymerization in the visible spectral region and subsequent crosslinking in the UV-A regime.
The formation of prepolymer resins is advantageous for applications where a potential leakage of low-molecular weight compounds must be prevented. Approach (i) thus allows for the isolation of the prepolymers from the polymerization mixture and, additionally, a detailed analysis of the intermediate material is possible. However, as the prepolymers are solids, additional heating is required before irradiation to process them as liquids, while approaches (ii and iii) enable facile processability due to low viscosities of the monomer-based resist mixtures. A minimization of required steps to obtain a cured material by simply using the same wavelength for both, the photopolymerization and the curing, is realized in the one-step approach (ii). This approach combines fast and simple processing with a uniform curing result. Most attractive, however, is approach (iii) as its λ-orthogonal nature allows for the generation of materials with widely different, spatially controllable hardness levels by directing the curing light source only at selected areas of the sample.
Precursor photocuring (approach i)
For precursor crosslinking, prepolymers with variable monomer feed ratios were synthesized by free radical polymerization at 65 °C in toluene utilizing 2,2′-azobis(2-methylpropionitrile) (AIBN) as thermal initiator and lauryl mercaptan as chain retarder. The chain retarder controlled the number average molecular weight, Mn, of the pre-polymers to well-below 10 kg mol−1 to facilitate the sample preparation for the material property testing. The synthesized polymers were fully characterized by 1H-NMR spectroscopy (Fig. S21–S25, ESI†), size exclusion chromatography (SEC, Fig. S32, ESI†), as well as differential scanning calorimetry (DSC, Fig. S33, ESI†) and thermo gravimetric analysis (TGA, Fig. S34, ESI†), confirming their structure and purity. The composition of the prepolymers was determined by 1H NMR spectroscopy and a typical example is shown in Fig. 1 on the right side. In all examined cases, the 1H-NMR spectrum clearly indicates the presence of the reactive aldehyde species within the o-MBA units after polymerization at δ = 10.67 ppm and 2.51 ppm, which are assigned to the aldehyde and the methyl protons, respectively. Furthermore, the methoxy protons of the MMA repeating unit appear at δ = 3.50 ppm and the characteristic O–CH2 resonance originating from the BMA repeating unit at δ = 3.86 ppm. The insert in Fig. 1 exemplarily shows the molecular weight distribution of the terpolymer comprised of butyl methacrylate (BMA), methyl methacrylate (MMA), and the photocrosslinker PEMA (entry c in Table 1). The number average molecular weight was determined based on a PMMA standard calibration to be 7000 g mol−1 (Đ = 1.7). By adjusting the feed ratio of the monomers and the chain retarder, the amount of photocrosslinkable PEMA units, the overall degree of polymerization, as well as the ratio of ‘hard’ building blocks, e.g. methyl methacrylate (MMA), isobornyl methacrylate (IBOMA), tetrahydrofurfuryl methacrylate (THFMA), and ‘soft’ building blocks, such as butyl methacrylate (BMA) can be controlled. Manipulation of the prepolymer composition allows for fine-tuning the viscoelastic properties of the photocurable mixture at elevated temperatures, which – in turn – will dictate the material properties of the crosslinked network. The material property dependence on the co-monomer composition can readily be followed by the respective glass transition temperature (Tg) for each polymer. Polymers containing a larger percentage of ‘soft’ monomer content exhibited a lower Tg (Table 1). For example, the Tg of a terpolymer consisting of various percentages of MMA, BMA and PEMA can be modified to range between 23 and 65 °C (Table 1, entry a–c).| Entry | BMA [%] | MMA [%] | PEMA [%] | THFMA [%] | IBOMA [%] | M n [g mol−1] | M w [g mol−1] | Đ | T g [°C] |
|---|---|---|---|---|---|---|---|---|---|
| a Feed ratio is stated; actual ratio could not be determined due to overlapping resonances. b Measured up to 140 °C. c Determined via size-exclusion chromatography in DMAc. The system was calibrated using narrow PMMA standards. d Determined via DSC. | |||||||||
| a | — | 77 | 23 | — | — | 6800 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
1.8 | 65 |
| b | 34 | 46 | 20 | — | — | 10000 |
16800 |
1.7 | 51 |
| c | 65 | 15 | 20 | — | — | 7000 | 11900 |
1.7 | 23 |
| d | — | — | 24 | 76 | — | 5700 | 11300 |
1.9 | 40b |
| e | — | — | 20a | — | 80a | 6600 | 9000 | 1.4 | 116b |
Thermal analysis was used to determine the temperature at which the first mass loss occurs and the sample begins to decompose, thus marking the upper limit for the temperature at which the polymers can still be safely processed. In all investigated cases, the temperature limit for processing is above 140 °C as determined by TGA (Fig. S34, ESI†). The glass transition temperatures, Tg, were determined using DSC. The highest Tg (116 °C) was found for the copolymer constituted of 20 mol% PEMA and 80 mol% isobornyl methacrylate, (Table 1 entry e and Fig. S33, ESI†). All other copolymers showed glass transition temperatures significantly below 100 °C (Table 1, entry a–d). A larger temperature gap between the Tg and the processing temperature (140 °C) significantly lowers the viscosity of the sample and facilitates their processability. In addition to the copolymer composition, the Tg is highly influenced by the degree of polymerization. We were able to follow the photodimerization process in situ by rheological experiments under periodic light irradiation (Fig. 2). The samples were loaded in a plate–plate measuring system of the rheometer by heating the prepolymers to 140 °C to minimize their viscosity and reach a state which is suitable for rheological experiments in the absence of any solvents. Furthermore, high temperatures facilitate chain mobility and result in an increased possibility of the crosslinking moieties to find a reaction partner. The bottom plate of the rheometer is constituted of a quartz glass slide, allowing for light irradiation at 365 nm with an intensity of 80 mW cm2. A narrow shear gap of 0.05 mm was selected to allow for full light penetration of the sample. After several minutes for temperature equilibration, the samples were investigated in oscillatory mode with a constant shear strain of 1% and an angular frequency of 10 rad s−1. The emission spectra of all light sources utilized herein are illustrated in Fig. S35 (ESI†). Initially, we conducted comparative curing experiments using a control copolymer without any photocrosslinkable units and our PEMA containing photocurable mixture (Fig. 2A).
 | ||
Fig. 2 Rheological data obtained at 140 °C in an oscillation experiment coupled with periodic UV irradiation centred at 365 nm. (A) Complex viscosity η*  p(MMA-co-BMA-co-PEMA) without crosslinker; p(MMA-co-BMA-co-PEMA) without crosslinker;  p(MMA-co-BMA-co-PEMA) with crosslinker; p(MMA-co-BMA-co-PEMA) with crosslinker;  p(MMA-co-BMA) reference copolymer. (B) p(MMA-co-BMA) reference copolymer. (B)  Storage modulus G′ p(MMA-co-BMA-co-PEMA) without crosslinker; Storage modulus G′ p(MMA-co-BMA-co-PEMA) without crosslinker;  loss modulus G′′ p(MMA-co-BMA-co-PEMA) without crosslinker. For the exact resist compositions, refer to Section S11 (ESI†). loss modulus G′′ p(MMA-co-BMA-co-PEMA) without crosslinker. For the exact resist compositions, refer to Section S11 (ESI†). | ||
In the absence of any photoreactive moiety, e.g. in case of poly(methyl methacrylate-co-butyl methacrylate), no increase in the complex viscosity was observed (Fig. 2A, blue  ) and hence no curing of the sample occurred during irradiation at 365 nm. Classically, o-QDM chemistry – including the one existing resist example22 – is employed with the addition of an electron deficient ene crosslinker. Comparative rheological experiments in the presence (Fig. 2A, red
) and hence no curing of the sample occurred during irradiation at 365 nm. Classically, o-QDM chemistry – including the one existing resist example22 – is employed with the addition of an electron deficient ene crosslinker. Comparative rheological experiments in the presence (Fig. 2A, red  ) and in the absence (Fig. 2A, grey
) and in the absence (Fig. 2A, grey  ) of a difumarate crosslinker (refer to the 1H-NMR spectra in Fig. S19 and S20, ESI†), resulted in an almost identical rheological behaviour. Critically, the temporal control over the reaction as evidenced by the ‘light-on/light-off’ experiments is retained in our dimerization driven photocurable system without additional crosslinker. Hence, as already shown in our small-molecule study (vide supra), there is no requirement for an additional crosslinker. The above finding is of key interest for applications requiring the absence of any low molecular weight compounds that could potentially leach from the material during processing or end-user application.
) of a difumarate crosslinker (refer to the 1H-NMR spectra in Fig. S19 and S20, ESI†), resulted in an almost identical rheological behaviour. Critically, the temporal control over the reaction as evidenced by the ‘light-on/light-off’ experiments is retained in our dimerization driven photocurable system without additional crosslinker. Hence, as already shown in our small-molecule study (vide supra), there is no requirement for an additional crosslinker. The above finding is of key interest for applications requiring the absence of any low molecular weight compounds that could potentially leach from the material during processing or end-user application.
The transition from a liquid to a solid is in general characterized by the intersection of the storage modulus G′ and the loss modulus G′′. In the present system, the transition time from a viscoelastic liquid to a photocured solid network is rapid with the material hardening in less than a minute under the given irradiation conditions. The loss- and storage moduli intersect after 17 s of irradiation (Fig. 2B) and the prepolymers can be post-cured, as evidenced by the increasing gap between storage modulus G′ and loss modulus G′′ after the intersection. A plateau of the storage modulus G′ is reached at approximately 106 Pa after 5 min curing time, marking the maximum degree of crosslinking.
Simultaneous curing by photopolymerization and o-QDM dimerization (approach ii)
The approach discussed above requires elevated temperatures to process and photocure the materials. Using mixtures of the monomers, which exhibit low viscosities, however, facilitates the processability at ambient temperature. Photoinduced radical polymerization and photoinduced non-radical curing by o-QDM dimerization can occur simultaneously, starting from mixtures of crosslinkable and non-crosslinkable monomers and a photoinitiator, under UV irradiation between 300–400 nm. The simultaneous polymerization and curing experiments were carried out for a variety of comonomer compositions in the presence of 50 mmol L−1 phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (MesBAPO-Ph, Irgacure® 819) as photoinitiator under UV-A irradiation centred at 350 nm. The curing times, tcure, were significantly increased (30 to 60 min) in comparison with the rheological experiments shown in Fig. 2 as the intensity of the utilized UV-A light source is significantly lower (5 mW cm−2), compared to 80 mW cm−2 utilized for the periodic irradiation experiments in the rheometer (vide supra). The curing temperatures were slightly elevated, depending on the solubility of PEMA in the respective monomer composition and the desired increase in the rate of dissolution. Rheological experiments could not be performed due to the volatile monomer mixture and the extremely low viscosity, preventing the use of the plate–plate measuring system used in the previous paragraph. However, the mechanical properties of the cured materials obtained via this combined approach (ii) were assessed by nanoindentation measurements providing information about the reduced E-modulus, Er, as well as the hardness of the cured samples. The reduced E-modulus Er was determined from the unloading curve of each indentation utilizing the Oliver and Pharr method (eqn (1)).28 | (1) |
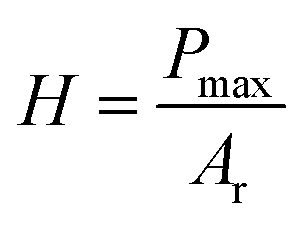 | (2) |
| Entry | BMA [%] | MMA [%] | MA [%] | PEMA [%] | T cure [°C] | t cure [min] | E r [GPa] | Hardness [MPa] |
|---|---|---|---|---|---|---|---|---|
| a | 34 | 61 | — | 5 | 45 | 60 | 3.85 ± 0.07 | 109.7 ± 1.7 |
| b | 32 | 58 | — | 10 | 45 | 60 | 5.43 ± 0.11 | 350 ± 19.8 |
| c | — | — | 97 | 3 | a.t. | 30 | 0.29 ± 0.04 | 52.0 ± 11.9 |
| d | — | — | 96 | 4 | a.t. | 30 | 0.89 ± 0.03 | 89.6 ± 7.4 |
| e | — | — | 95 | 5 | 50 | 30 | 1.26 ± 0.22 | 93.9 ± 17.7 |
| f | — | — | 90 | 10 | a.t. | 30 | 5.76 ± 0.06 | 329.3 ± 1.7 |
Thus, the reduced E-moduli determined for approach (ii) range from 0.29 GPa to 5.76 GPa and the hardness is adjustable between 52 MPa and 329 MPa. These values are in the typically observed range for uncured31 – e.g. polycarbonate 2.95 GPa, poly(methyl methacrylate) 3.75 GPa and chitosan 1.5 GPa – and cured32,33 – e.g. chitosan 4.7 GPa and multifunctional compositions up to 6.25 GPa – (bio)polymers. In order to achieve materials with increased flexibility, i.e. with a reduced E-modulus and hardness (Er ∼ 1 GPa and H < 100 MPa), the major component in the resist was changed from the methacrylic monomers MMA and BMA to methyl acrylate (MA, entry c–f). An increase of the PEMA content from 5 to 10 mol% leads to a significant increase of Er and H (entry e and f, Table 2). The combined curing and photopolymerization with a single colour of light is promising for applications that require a simple and fast formulation and curing process with a single hardness for the whole material.
λ-Orthogonal photocuring (approach iii)
An alternative, and even more powerful route, exploits the defined wavelength range in which the o-QDM dimerization can be addressed, to prepare crosslinked polymeric materials in a step-wise fashion. A previous investigation of the wavelength dependence of photoenol ligation revealed that the reaction does not occur upon irradiation above 400 nm and therefore the o-QDM intermediate is not formed.14 By careful selection of the photoinitiator, the initial photoinduced radical polymerization can be conducted in the visible light regime (>400 nm), while the subsequent non-radical crosslinking is addressed using a shorter wavelength (UV-A light centred at ∼350 nm). Critically, due to the separation of photopolymerization and curing, an additional dimension of control is introduced by the potential to solely cure, and thus influence the material properties, in defined regions of the material. We exemplify the approach on a reaction mixture comprising 55 mol% MMA, 30 mol% BMA and 15 mol% PEMA, as well as 50 mmol L−1 MesBAPO-Ph as the photoinitiator. The liquid monomer mixture was split into 0.3 mL per aluminium dish (∅ = 2.5 cm) in order to investigate the material properties of the same sample, but with a different irradiation history. Each sample was photopolymerized in a photoreactor chamber (refer to Fig. S36, ESI†) for 60 min at 45 °C at λ = 445–465 nm (7 mW cm−2). Subsequently, the photopolymerized samples were cured in the same photoreactor for various times with UV-A light centred at 350 nm (5 mW cm−2).The visible light photopolymerized product showed not only a decreased E-modulus and hardness (Fig. 3) compared to the cured samples, but was also completely soluble in suitable organic solvents, unambiguously demonstrating the λ-orthogonal nature of the resist as the o-MBA crosslinking is clearly not induced in the wavelength regime used for the photopolymerization.
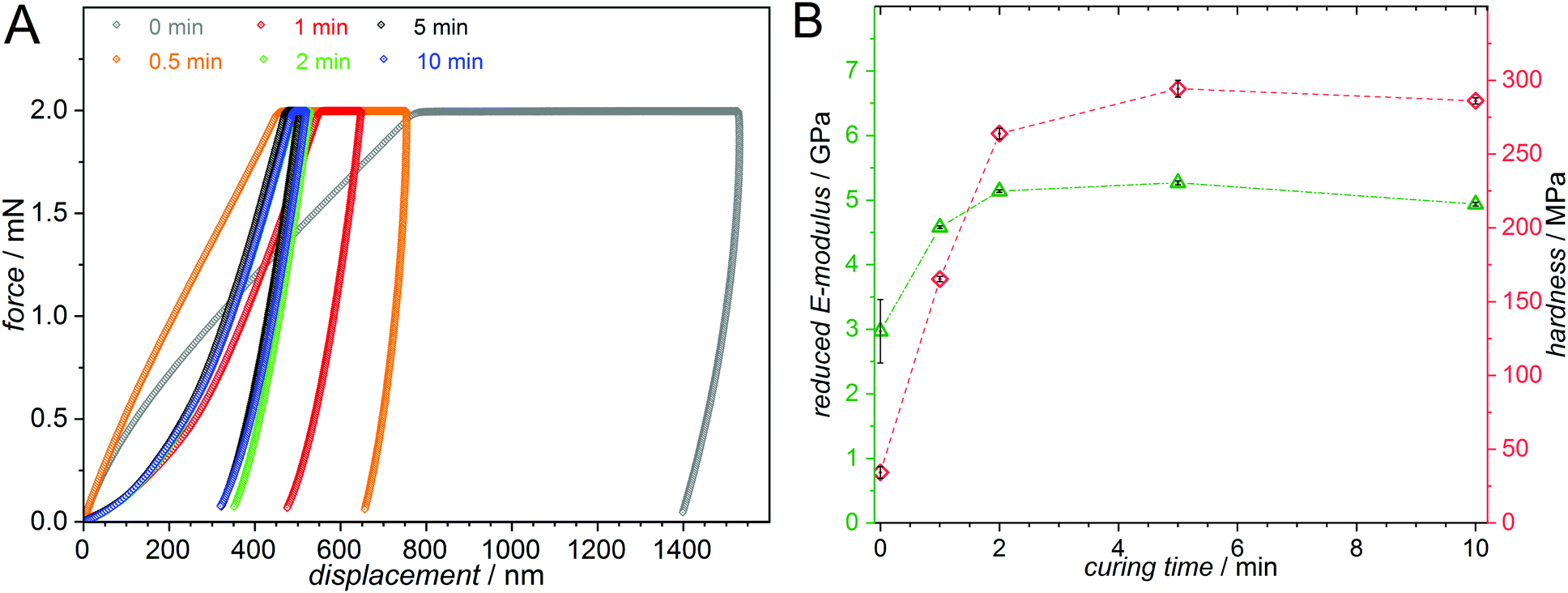 | ||
| Fig. 3 (A) Load–displacement curves from load controlled tests via nanoindentation. The measured samples consist of 55 mol% MMA, 30 mol% BMA and 15 mol% PEMA as well as 50 mmol L−1 MesBAPO-Ph as the photoinitiator and were photopolymerized with blue LEDs (λ = 445–465 nm, 7 mW cm−2, emission spectrum Fig. S35, ESI†) for 60 min and subsequently cured for different times using UV-A light centered at 350 nm (5 mW cm−2, emission spectrum Fig. S35, ESI†). The polymer creep significantly decreases with longer curing times. (B) The reduced E-moduli, Er, and hardnesses, H, obtained from the load-displacement curves exhibit a strong increase with increasing curing time. | ||
A clear trend for the polymer creep, i.e. the displacement over time at constant force is observed, drastically decreasing as a function of the curing time. As expected, both the reduced E-modulus and the hardness increase for longer curing times. The load-controlled indentations with 2000 μN peak load display Er values ranging from 3 to 5 GPa and hardness values between 30 and 300 MPa depending on the curing time for the respective sample. The simple formulation of the photocurable system and the λ-orthogonality shown in our experiments is expected to be highly valuable for applications in e.g. 3D printed structures with different hardness profiles obtained from a single photoresist by spatially and temporally controlling the crosslinking process.
Conclusions
In summary, we provide in-depth insights into the chemistry of the photoinduced dimerization of o-quinodimethanes – which forms the basis for a novel photoresist concept – via an elaborate small molecule study. The major composition of the product spectrum was found to consist of the [4+4]1, [4+4]2, [4+4]3 products. Further, we introduce three complementary approaches for the formulation and curing of photocurable mixtures without the requirement of additional crosslinker, solely based on the o-QDM dimerization. Specifically, the lateral chain architecture of the polymeric precursors can be obtained by thermally induced free radical polymerization enabling isolation and purification of the prepolymers by standard workup procedures prior to the crosslinking, which prevents any potential leakage of small molecules in the subsequent step. Alternatively, by exchanging the thermal radical initiator with a suitable photoinitiator, simultaneous photopolymerization and non-radical crosslinking in the resist mixture by irradiation with UV light can be realized. The use of monomeric resist mixtures with low viscosities facilitates their processability at ambient temperatures, whereas the thermally polymerized polymeric precursors require elevated temperatures to achieve suitable viscosities to process the resist. In addition, the material properties can be adjusted in a wide range, i.e. between 0.29 GPa and 5.76 GPa for the reduced E-modulus and between 52 and 329 MPa for the hardness, depending on the resist composition and selected irradiation parameters (e.g. wavelength, intensity and time).Critically, the photopolymerization can be conducted in the visible light regime (>400 nm) without inducing crosslinking, forming crosslinkable polymers in situ. Subsequently, the crosslinking is initiated photochemically via the formation and dimerization of o-QDMs in the UV spectral region (300–400 nm). Thus, our photocurable system provides the potential to process an o-QDM based resist mixture in a λ-orthogonal fashion, separating the photopolymerization from the curing process simply by utilizing two different colours of light. This enables the adjustment of different hardness profiles within the same material. The formulation of photoresists based on o-methylbenzaldehydes is a promising candidate for 3D printing or 3D laser lithography, especially for biomedical applications due to the absence of any additional crosslinker.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B.-K. acknowledges funding by Ivoclar Vivadent AG as well as the excellent collaboration and additional key support from the Australian Research (ARC) in the form of a Laureate Fellowship (FL170100014) enabling his photochemical research program. T. D. is supported by the ARC Future Fellowship scheme (FT150100408). F. F. is the recipient of an Australian Government funded International Post-Graduate Research Scholarship and gratefully acknowledges their support. This work was further enabled by use of the Central Analytical Research Facility hosted by the Institute for Future Environments at QUT. Kazuyuki Hosokawa (QUT) is gratefully acknowledged for his help with technical aspects. We thank Dr Christiane Lang (QUT) for proofreading and many helpful comments on the manuscript and Dr Anja Goldmann (QUT) for the graphical design of the ToC graphic.Notes and references
- K. Ueberreiter and G. Kanig, J. Chem. Phys., 1950, 18, 399 CrossRef CAS.
- T. G. Fox and S. Loshaek, J. Polym. Sci., 1955, 15, 371 CrossRef CAS.
- H. L. Fisher, Ind. Eng. Chem., 1939, 31, 1381 CrossRef CAS.
- S. Mane, S. Ponrathnam and N. Chavan, Can. Chem. Trans., 2016, 3, 473 Search PubMed.
- C. Pugh and A. R. Amrutkar, US2018030217A1, 2018.
- M. A. Meador, Macromolecules, 1996, 29, 8983 CrossRef CAS.
- J. L. Segura and N. Martín, Chem. Rev., 1999, 99, 3199 CrossRef CAS PubMed.
- P. Sammes, Tetrahedron, 1976, 32, 405 CrossRef CAS.
- J. L. Charlton and M. M. Alauddin, Tetrahedron, 1987, 43, 2873 CrossRef CAS.
- G. Porter and M. F. Tchir, J. Chem. Soc., 1970, 6, 1372 Search PubMed.
- G. Porter and M. F. Tchir, J. Chem. Soc., 1971, 0, 3772 RSC.
- W. Choy and H. Yang, J. Org. Chem., 1988, 53, 5796 CrossRef CAS.
- G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296 CrossRef CAS PubMed.
- J. P. Menzel, B. B. Noble, A. Lauer, M. L. Coote, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2017, 139, 15812 CrossRef CAS PubMed.
- B. Richter, V. Hahn, S. Bertels, T. K. Claus, M. Wegener, G. Delaittre, C. Barner-Kowollik and M. Bastmeyer, Adv. Mater., 2016, 29, 1604342 CrossRef PubMed.
- T. Pauloehrl, G. Delaittre, V. Winkler, A. Welle, M. Bruns, H. G. Börner, A. M. Greiner, M. Bastmeyer and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2012, 51, 1071 CrossRef CAS PubMed.
- K. Hiltebrandt, M. Kaupp, E. Molle, J. P. Menzel, J. P. Blinco and C. Barner-Kowollik, Chem. Commun., 2016, 52, 9426 RSC.
- M. Kaupp, K. Hiltebrandt, V. Trouillet, P. Mueller, A. S. Quick, M. Wegener and C. Barner-Kowollik, Chem. Commun., 2016, 52, 1975 RSC.
- K. N. R. Wuest, V. Trouillet, R. Köppe, P. W. Roesky, A. S. Goldmann, M. H. Stenzel and C. Barner-Kowollik, Polym. Chem., 2017, 8, 838 RSC.
- J. Zhang and P. Xiao, Polym. Chem., 2018, 9, 1530 RSC.
- C. Barner-Kowollik, M. Bastmeyer, E. Blasco, G. Delaittre, P. Müller, B. Richter and M. Wegener, Angew. Chem., Int. Ed., 2017, 56, 15828 CrossRef CAS PubMed.
- A. S. Quick, H. Rothfuss, A. Welle, B. Richter, J. Fischer, M. Wegener and C. Barner-Kowollik, Adv. Funct. Mater., 2014, 24, 3571 CrossRef CAS.
- M. M. Zieger, P. Mueller, A. S. Quick, M. Wegener and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2017, 56, 5625 CrossRef CAS PubMed.
- X. Zhang, W. Xi, S. Huang, K. Long and C. N. Bowman, Macromolecules, 2017, 50, 5652 CrossRef CAS PubMed.
- T. Gegenhuber, L. De Keer, A. S. Goldmann, P. H. M. Van Steenberge, J. O. Mueller, M.-F. Reyniers, J. P. Menzel, D. R. D’hooge and C. Barner-Kowollik, Macromolecules, 2017, 50, 6451 CrossRef CAS.
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276 CrossRef PubMed.
- T. K. Claus, B. Richter, V. Hahn, A. Welle, S. Kayser, M. Wegener, M. Bastmeyer, G. Delaittre and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016, 55, 3817 CrossRef CAS PubMed.
- W. C. Oliver and G. M. Pharr, J. Mater. Res., 1992, 7, 1564 CrossRef CAS.
- U. D. Cakmak, T. Schöberl and Z. Major, Meccanica, 2012, 47, 707 CrossRef.
- B. Sandmann, B. Happ, J. Vitz, R. M. Paulus, M. D. Hager, P. Burtscher, N. Moszner and U. S. Schubert, Macromol. Chem. Phys., 2014, 215, 1603 CrossRef CAS.
- C. Klapperich, K. Komvopoulos and L. Pruitt, J. Tribol., 2001, 123, 624 CrossRef CAS.
- B. Sandmann, B. Happ, M. D. Hager, J. Vitz, E. Rettler, P. Burtscher, N. Moszner and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 239 CrossRef CAS.
- D. A. Galloway, L. A. Laimins, B. Division and F. Hutchinson, J. Mech. Behav. Biomed. Mater., 2016, 5, 87 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization. See DOI: 10.1039/c8mh00951a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |