
Identifying glioblastoma margins using dual-targeted organic nanoparticles for efficient in vivo fluorescence image-guided photothermal therapy†
Xiaolei
Cai‡
 a,
Aishwarya
Bandla‡
b,
Chan Kim
Chuan
b,
Gayathiri
Magarajah
b,
Lun-De
Liao
bc,
Daniel Boon Loong
Teh
d,
Brian K.
Kennedy
d,
Nitish V.
Thakor
*b and
Bin
Liu
*a
a,
Aishwarya
Bandla‡
b,
Chan Kim
Chuan
b,
Gayathiri
Magarajah
b,
Lun-De
Liao
bc,
Daniel Boon Loong
Teh
d,
Brian K.
Kennedy
d,
Nitish V.
Thakor
*b and
Bin
Liu
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: cheliub@nus.edu.sg
bSingapore Institute for Neurotechnology (SINAPSE), National University of Singapore, 28 Medical Drive, Singapore 117456, Singapore. E-mail: eletnv@nus.edu.sg
cInstitute of Biomedical Engineering and Nanomedicine, National Health Research Institutes, 35 Keyan Rd., Zhunan Town, Miaoli County 35053, Taiwan
dDepartments of Biochemistry and Physiology, Yong Loo Lin School of Medicine, National University of Singapore, 8 Medical Drive, Singapore 117456, Singapore
First published on 13th October 2018
Abstract
Current therapeutics for glioblastoma multiforme (GBM) treatment are unsatisfactory due to their limited ability to control the progression from tumour margins. In this work, organic nanoparticles (NPs) are synthesized by co-encapsulating a fluorogen with aggregation-induced emission to generate a bright red emission for imaging and a semiconducting polymer to offer NIR absorption for photothermal therapy. The NPs are further modified with different ratios of two targeting ligands, folate and cRGD peptide. The best ratio that performs specific and efficient GBM targeting is screened out through in vitro and ex vivo fluorescence imaging analysis. The NPs with an FA to cRGD ratio of 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75 exhibit superior ability to target GBM cells in vitro and also show efficient accumulation at the GBM margin and in the tumour interior after in vivo administration. The progression of GBM can be greatly suppressed through photothermal therapy, which provides a simple but promising strategy for GBM treatment.
:75 exhibit superior ability to target GBM cells in vitro and also show efficient accumulation at the GBM margin and in the tumour interior after in vivo administration. The progression of GBM can be greatly suppressed through photothermal therapy, which provides a simple but promising strategy for GBM treatment.
Conceptual insightsGlioblastoma multiforme (GBM) is the most aggressive brain tumour which has extremely poor median survival. Current treatment including surgery, radiotherapy and chemotherapy could not effectively act on the GBM margins, resulting in tumour progression and recurrence. One of the problems is that the GBM margin could not be accurately demarcated by the current imaging techniques. On the other hand, the existence of the blood–brain barrier inhibits the delivery of therapeutic agents or contrast agents to the GBM. To solve these two problems, we report a novel design of organic nanoparticles with dual-targeting ligands to realize efficient delivery of nanoparticles towards the GBM margin through the blood–brain tumour barrier. The intrinsic bright red fluorescence from the nanoparticles showed that promising GBM marginal targeting was achieved. Meanwhile, as the nanoparticles also possess high NIR absorption, the implementation of NIR laser irradiation also realized precise photothermal treatment of GBM, which successfully controlled the growth of GBM. This work demonstrates a simple but efficient strategy to overcome the two obstacles faced by current GBM treatment, which provides promising opportunities for future clinical applications. |
Glioblastoma multiforme (GBM), the deadliest form of brain tumour in adults, is responsible for 15% of all brain-tumour diagnoses worldwide.1–3 The median survival of patients diagnosed with GBM is only 16–19 months.4,5 Current clinical therapy for GBM involves a three-pronged approach, consisting of surgical resection followed by adjuvant radiotherapy and chemotherapy. While these could prolong patient survival by several months, there is an increased chance of healthy tissues suffering adverse side effects of these adjuvant therapies.6,7 Despite the fact that recent advances in radiotherapy and chemotherapy have led to enhanced therapeutic efficacy, the curative effect for GBM is still limited.8,9 Further, recurrence and progression of the GBM due to incomplete resection, insufficient treatment at the tumour margin, and infiltration of tumour cells into the surrounding healthy brain are always the causes of death.6,10
The current brain imaging techniques, such as computed tomography (CT) and magnetic resonance imaging (MRI), are not able to accurately determine the margin of GBM. Biopsy and post-mortem studies have shown that gliomas extend further than what could be determined using these imaging techniques. Studies involving CT imaging have indicated that tumour cells extend beyond the area of CT enhancement and are frequently seen in regions of peritumoural edema.11 Tumour cells may also be found in areas that appeared normal on CT in 20% of serial stereotactic biopsy specimens.12 Moreover, MRI with improved soft-tissue resolution has also failed to improve the identification of the tumour margin. Biopsy studies have shown that the tumour extends beyond the margin of T2 signal change in most GBM. In some cases, tumours extended up to 2.5 cm beyond the area of T2 signal change.13 The inefficient imaging of GBM margin, on one hand, always leads to incomplete resection; on the other hand, limits the evaluation of delivery efficiency and therapeutic performance of new therapeutics at the GBM margin, hampering the study of innovative therapeutic approaches.
Another emerging focus is the function of the blood–brain barrier (BBB) and blood–brain tumour barrier (BBTB) in the tumour microenvironment. Although the BBB could become permeable to a certain degree with the progression of GBM,14,15 the BBTB formed by abnormal neovasculature is considered to be the main barrier that prevents the crossing of therapeutics since it is not as leaky as the blood–tumour barrier in other cancer types.16 In particular, at the marginal area of GBM where the infiltrative GBM cells exist, the delivery of therapeutics is highly restricted by the less permeable BBTB, leading to infiltration and metastasis of GBM.17,18 Although various delivery strategies with the aid of targeting ligands to cross the BBB and BBTB have been developed,19–21 their capabilities to cross the BBB and BBTB lack detailed evaluation and analysis, and whether they have the ability to precisely detect tumour margins is not clear.22,23 As a result, the lack of sufficient therapeutics at the marginal area may still lead to recurrence and unstoppable progression of GBM.
Therefore, it is extremely important to develop a novel platform that is indeed helpful in effectively crossing the BBTB to target the GBM marginal area and provide sufficient therapeutic effect to inhibit the progression of GBM. In this work, we present a simple but efficient strategy to address these issues by synthesizing organic nanoparticles (NPs) with a red fluorescence and photothermal effect. We further modified the NPs with different targeting ligands for GBM marginal targeting. To precisely assess the GBM margin targeting efficiency of the NPs and screen out the best formulation through fluorescence imaging, an organic fluorogen TPETPAFN (Fig. 1A), possessing a typical aggregation-induced emission (AIE) property, that is non-emissive in organic solvents but highly emissive once encapsulated into organic NPs, was chosen as the fluorescence emitter to be encapsulated into NPs. Due to the unique AIE property, the NPs exhibit high brightness, good photostability and good biocompatibility. Meanwhile, in the paradigm of cancer therapeutics, a semiconducting polymer, SP (Fig. 1A), possessing high molar absorptivity in the near infrared (NIR) region, was co-encapsulated into the NPs to act as the photothermal therapy (PTT) agent for GBM treatment with high specificity, minimal invasiveness and precise spatial-temporal selectivity.24,25 The surface of the synthesized TPETPAFN–SP (TNSP) NPs was further decorated with a single targeting ligand, folate (FA), or dual targeting ligands, FA and cyclic arginine–glycine–aspartic acid (cRGD) peptide. In vitro fluorescence imaging of GBM cells and ex vivo fluorescence imaging of brain tissues revealed the good targeting effect of NPs with optimized targeting ligands. Photothermal therapy was further carried out in both in vitro and in vivo experiments by irradiating the cells or animals with a 808 nm laser after the administration of the NPs with dual targeting ligands, which demonstrated an efficient photothermal effect to ablate GBM cells in vitro and suppress the growth of GBM in vivo. Overall, this study reveals that the strategy of using dual targeting ligands and PTT is promising in the paradigm of precise targeting and treatment of GBM. It also provides valuable information regarding the BBB and BBTB function with the formation and progression of GBM.
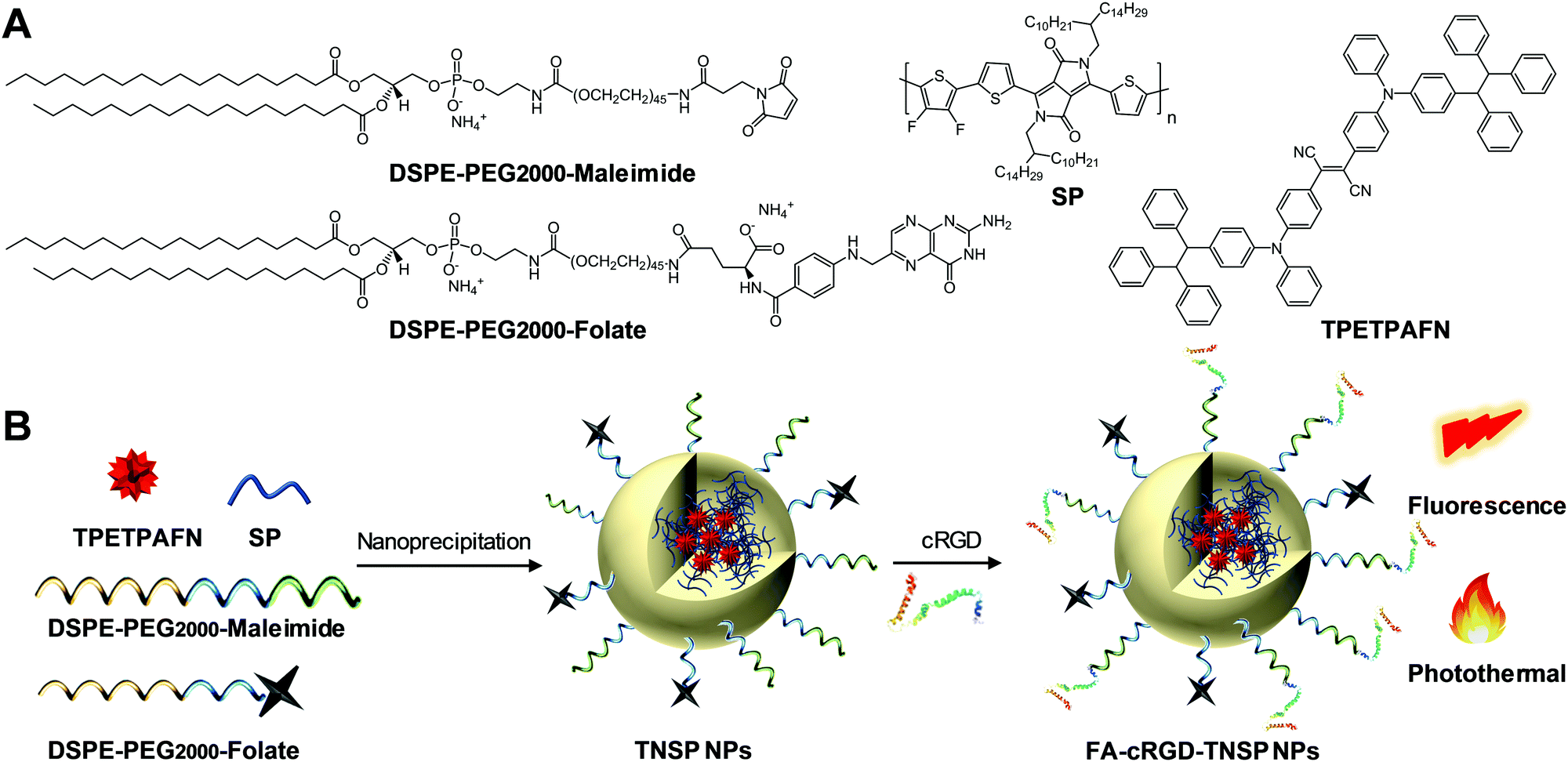 | ||
| Fig. 1 (A) Chemical structures of DSPE-PEG2000-maleimide, DSPE-PEG2000-folate, SP, and TPETPAFN. (B) Schematic illustration of NP preparation. | ||
As shown in Fig. 1, an AIE fluorogen, TPETPAFN, and a semiconducting polymer (SP) with a mass ratio of 1:1 were co-encapsulated into 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(methoxy(polyethylene glycol)-2000) (DSPE-PEG2000) through a modified nanoprecipitation method to form TNSP NPs with a PEG surface. TPETPAFN was selected as the fluorescence emitter as it exhibits a bright red fluorescence from 550 to 900 nm with an emission peak at ∼655 nm in the aggregate state (Fig. 2A).26 The SP was chosen to serve as the PTT agent as it has long wavelength absorption, high NIR absorptivity at 808 nm with almost no emission, which could generate an effective photothermal effect to realize efficient PTT. Furthermore, as compared to conventional PTT agents, such as gold nanomaterials27,28 and carbon nanomaterials,29,30 semiconducting polymers exhibit better biocompatibility, which is more favorable for biological applications. As shown in Fig. 2B, the obtained TNSP NPs show two absorption peaks located at 510 nm and 750 nm, arising from TPETPAFN and SP, respectively. The mass absorptivity of TNSP NPs is 91.6 cm−1 mg−1 mL at 808 nm. The absorption of TPETPAFN ends at 650 nm, which does not interfere with the absorption and photothermal effect of SP when excited with a 808 nm NIR laser. Despite the partial overlap between the emission spectrum of TPETPAFN and the absorption of SP, which partially quenches the TPETPAFN fluorescence, TNSP NPs emit a bright red fluorescence with a peak at 655 nm and a quantum yield of ∼16.5% upon excitation at 510 nm. Laser light scattering (LLS) results and Field-emission transmission electron microscopy (FE-TEM) images indicate that the NPs have a diameter of ∼20 nm (Fig. 2C). Elemental analysis (Fig. S3, ESI†) shows that the actual values of C, H, N, and S in the NPs are consistent with the calculated values. Moreover, thermogravimetric analysis (Fig. S4, ESI†) indicates that the NPs are thermally stable up to a temperature of 200 °C.
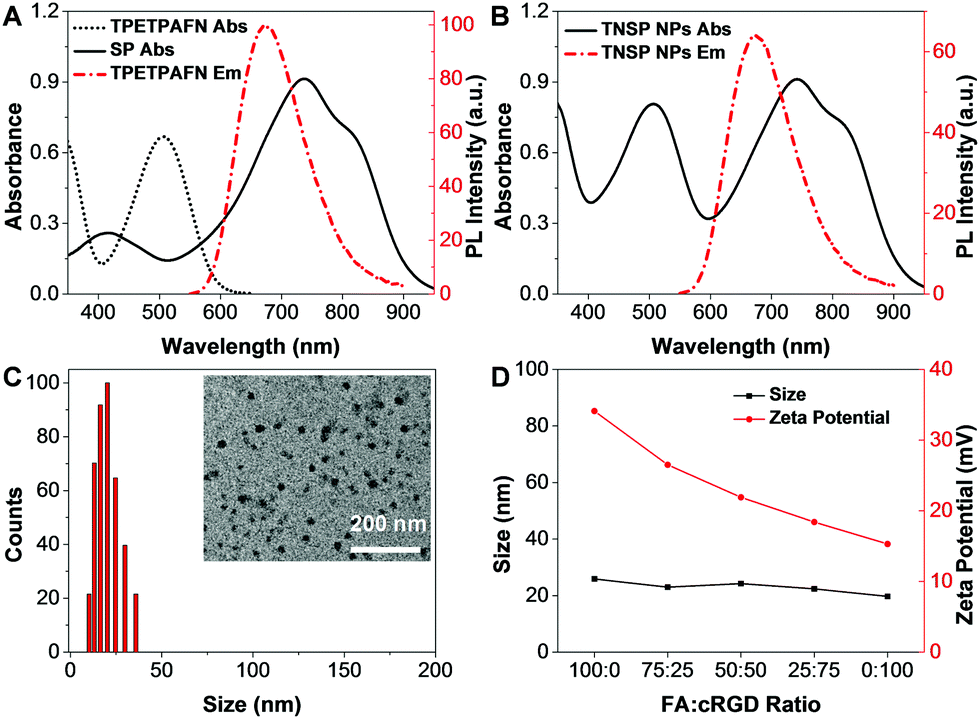 | ||
| Fig. 2 (A) UV-vis absorption spectra of TPETPAFN and SP NPs. PL spectrum of TPETPAFN NPs. (B) UV-vis absorption and PL spectra of TNSP NPs. (C) LLS size distribution of TNSP NPs. Inset: FE-TEM images of NPs. (D) LLS size and zeta potential of TNSP NPs with different surface density of FA and cRGD. | ||
To improve the targeting ability to GBM cells and tumour margins, TNSP NPs with PEG surfaces (PEG-TNSP NPs) were further modified with a single targeting ligand, FA, to obtain FA-TNSP NPs which could help target folate receptor overexpressed GBM cells.31 In addition, TNSP NPs were also modified with dual targeting ligands, FA and cRGD, as cRGD could not only target αvβ3 integrin receptor overexpressed GBM cells,32 but also help penetrate through the αvβ3 integrin receptor overexpressed BBTB.33 To explore how the ratio of the two targeting ligands could affect the cell targeting efficiency, DSPE-PEG2000-folate and DSPE-PEG2000-maleimide with different feeding ratios of 100:0, 75:25, 50:50, 25:75 and 0:100 were used as the encapsulation matrices to fine-tune the surface density of the two targeting ligands. After NP formation, cRGD was conjugated to DSPE-PEG2000-maleimide through a thiol–maleimide coupling reaction. As shown in Fig. 2D, LLS results indicate that there is almost no change in NP size after modifying the surface density of the two targeting ligands. Meanwhile, the zeta potentials of the NPs become less negative with the increase of cRGD density. All the NPs show good colloidal stability without any aggregation after 30-day storage at 4 °C.
After preparation of TNSP NPs with different surface functionalization, the capability of these NPs for GBM targeting was firstly tested through in vitro cell imaging. After 4 h of incubation with the PEG-TNSP NPs, as indicated by Fig. S7 (ESI†), the cellular uptake into U87MG GBM cells and NIH/3T3 normal cells is quite poor. Meanwhile, as shown in Fig. 3A, the internalization of surface functionalized NPs into the U87MG GBM cells and NIH/3T3 normal cells varies with the change of surface targeting ligands. When the surface density of FA decreases and cRGD increases, the cellular uptake of TNSP NPs into U87MG cells shows obvious enhancement while the selectivity to NIH/3T3 cells is reduced. A possible reason is that FA is more negatively charged than cRGD, which could reduce the surface zeta potential of the NPs and is less favourable for uptake into cells through the negatively-charged cell membrane. On the other hand, the dual targeting ligands could produce a synergistic effect to improve the targeting efficiency to tumour cells and the selectivity over normal cells as compared to the NPs with the single targeting ligand. Considering both the U87MG cellular uptake and selectivity over NIH/3T3 cells, TNSP NPs with a surface density of 75% cRGD and 25% FA (FA-cRGD-TNSP NPs) exhibit the best performance for in vitro targeted imaging of GBM. Subsequently, FA-cRGD-TNSP NPs were tested in neuronal glia mixed culture, which contains neurons, astrocytes, and oligodendrocytes to mimic the real brain microenvironment. As indicated by the fluorescence images in Fig. 3B, compared to the control group without NP incubation, almost no fluorescence could be observed from the cells after incubation with FA-cRGD-TNSP NPs, indicating that the NPs could barely enter neurons or glia cells. These results indicate that the synthesized NPs are potentially inert in the real brain microenvironment. Moreover, NIH/3T3 normal cells and neuronal glia mixed culture showed excellent viability upon incubation with 0 to 100 μg mL−1 (mass concentration based on TPETPAFN and SP) of FA-cRGD-TNSP NPs (Fig. 3C). These promising results prove that FA-cRGD-TNSP NPs have high biocompatibility. Together with their good stability in a simulated in vivo environment (1× PBS buffer with human serum, 37 °C, 24 h incubation) as shown in Fig. S8 (ESI†), FA-cRGD-TNSP NPs are suitable for in vivo studies.
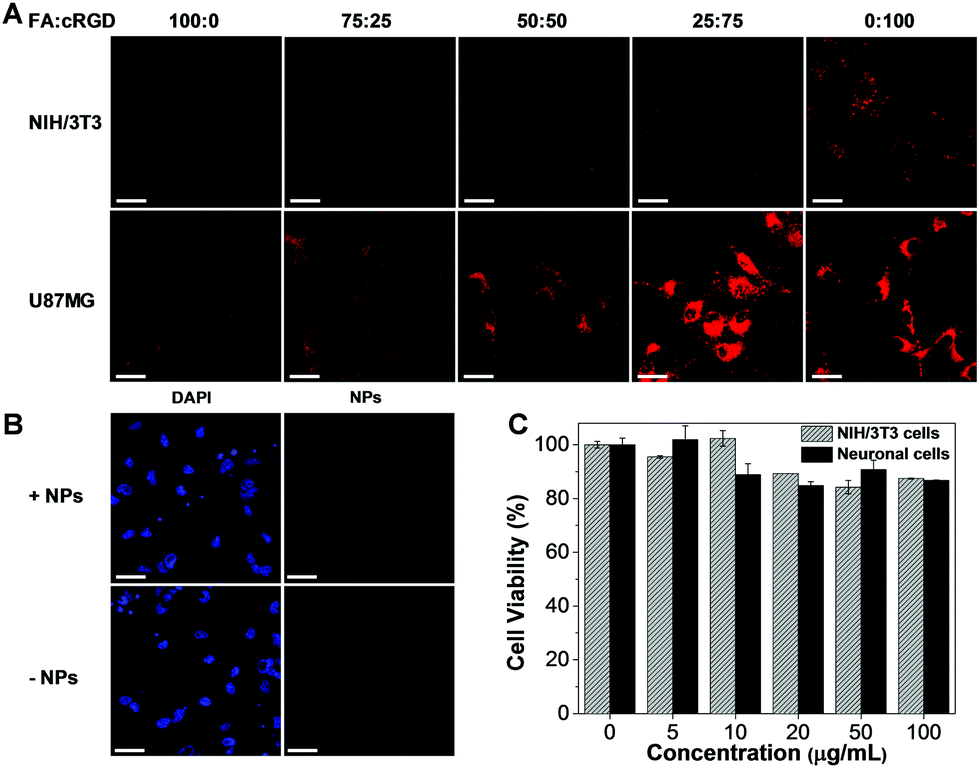 | ||
| Fig. 3 (A) Confocal images of NIH/3T3 normal cells and U87MG GBM cells after incubation with TNSP NPs with different surface functionalization for 4 h at 37 °C. Concentration: 10 μg mL−1. Excitation: 488 nm. Emission: 620–750 nm. Scale bars: 50 μm. (B) Confocal images of neuronal glia mixed culture after incubation with FA-cRGD-TNSP NPs (25% FA and 75% cRGD) for 4 h at 37 °C. Concentration: 10 μg mL−1. Excitation: 405 nm (DAPI) and 488 nm (NPs). Emission: 420–470 nm (DAPI) and 620–750 nm (NPs). Scale bars: 50 μm. (C) Cell viabilities of NIH/3T3 normal cells and neuronal glia mixed culture after incubation with different concentrations of FA-cRGD-TNSP NPs (25% FA and 75% cRGD) for 24 hours. | ||
Using PEG-TNSP NPs and FA-TNSP NPs as controls, FA-cRGD-TNSP NPs were further studied in an in vivo U87MG-cell based murine glioblastoma model to assess the feasibility of the dual targeting design for in vivo BBTB penetration and GBM margin targeting. The three kinds of NPs were administrated into mice through IV injection from the tail vein on day 7 post tumour implantation. Brain tissues were harvested at a time point of 4 h after administration of NPs and investigated by ex vivo fluorescence imaging. Fig. 4A indicates that the PEG-TNSP NPs could marginally target the tumour as compared to the non-tumour region. However, as indicated by the weak red fluorescence, only a limited quantity of NPs accumulate at the tumour margins and in the interior of the tumour. Following this we observed that the FA-TNSP NPs facilitate better accumulation of NPs in the tumour cells at the margin and the core part of the tumour, though the quantity is still not sufficient enough. As both PEG-TNSP NPs and FA-TNSP NPs do not have any BBB or BBTB penetration capability, their appearance at tumour margins and interior reveals the increase of BBB permeability after GBM formation. FA-cRGD-NPs achieve the best performance in accurately demarcating the tumour margins and also realizing sufficient accumulation of NPs in the tumour core as indicated by the bright red fluorescence, which indicates that the synergistic effect of FA and the BBTB-penetrating peptide, cRGD, could extensively enhance the extravasation of NPs through the BBTB and the targeting efficiency towards GBM in this animal model. To further confirm the GBM margin and interior, the GBM overexpressed EGFR receptors were immunostained by primary anti-EGFR antibody and secondary antibody conjugated with Alexa Fluor® 488. As shown in Fig. S9 (ESI†), the tumour margin and interior could be clearly differentiated by the bright green fluorescence. The fluorescence overlaps between the green and red fluorescence are consistent with Fig. 4A. Moreover, in vivo fluorescence imaging of GBM (Fig. S10, ESI†) further validated the promising accumulation of FA-cRGD-NPs at the GBM region post injection.
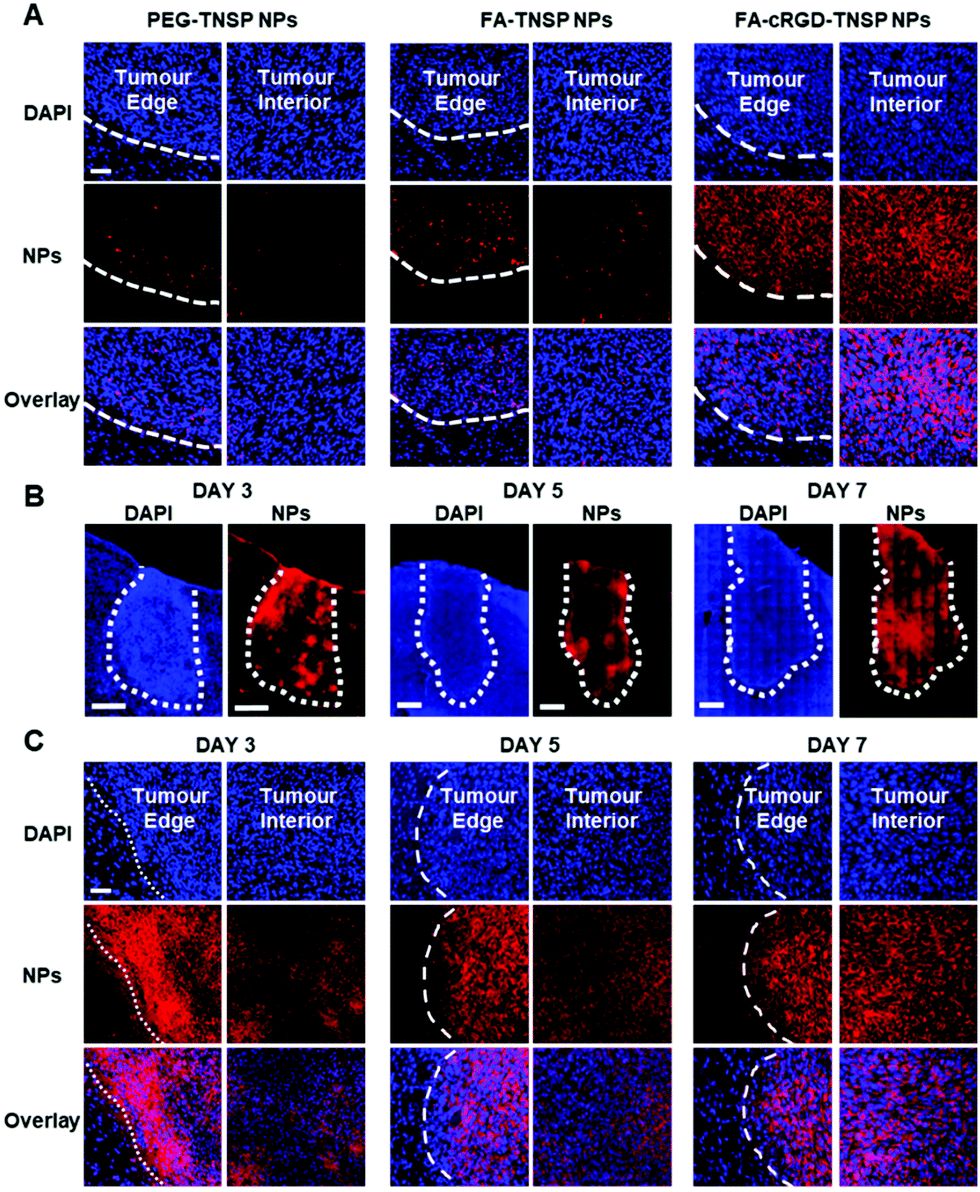 | ||
| Fig. 4 (A) Confocal images of brain slices at 7 days post tumour implantation after administration of TNSP NPs with different surface functionalization for 4 h. Excitation: 405 nm (DAPI) and 488 nm (NPs). Emission: 420–470 nm (DAPI) and 620–750 nm (NPs). Scale bar: 100 μm. (B) Confocal tile scans of brain slices at 3 days, 5 days and 7 days post tumour implantation after administration of FA-cRGD-TNSP NPs for 4 h. Excitation: 405 nm (DAPI) and 488 nm (NPs). Emission: 420–470 nm (DAPI) and 620–750 nm (NPs). Scale bars: 500 μm. (C) Confocal images of brain slices at 3 days, 5 days and 7 days post tumour implantation after administration of FA-cRGD-TNSP NPs for 4 h. Excitation: 405 nm (DAPI) and 488 nm (NPs). Emission: 420–470 nm (DAPI) and 620–750 nm (NPs). Scale bar: 100 μm. | ||
Subsequently, FA-cRGD-TNSP NPs were used to study their capability to target GBM at different stages of tumour growth. The tile scan images of whole brain slices containing the tumour (Fig. 4B) at 3 days, 5 days and 7 days post tumour implantation show that the entire tumours exhibit a bright red fluorescence. This indicates the strong capability of FA-cRGD-TNSP NPs in targeting and accumulating in brain tumours. As shown in Fig. 4C (the enlarged images of Fig. 4B), the FA-cRGD-TNSP NPs can clearly classify the tumour margins while the interior of tumours are also labeled with a considerable number of NPs with tumour progression. These observations demonstrate that FA-cRGD-TNSP NPs could offer highly specific and targeted nanodiagnostics for GBM at different tumour stages in the U87MG-cell-based murine model. Meanwhile, the adequate delivery of NPs into the GBM margin and interior also provides promising opportunities to implement photothermal therapy of GBM. In addition, as no BBB penetration ligand was used in the design of FA-cRGD-TNSP NPs, the non-specific BBB penetration at the non-tumour region was avoided and the subsequent potential adverse effect on neuronal cells was minimized.
The temperature evolution of TNSP NP suspensions with different concentrations (SP mass concentration) upon 808 nm laser irradiation from 1 to 10 min is shown in Fig. 5A. The TNSP NP suspension with a concentration of 10 μg mL−1 exhibits a rapid temperature increase of ∼65 °C after 10 min 808 nm laser irradiation. Even at a lower concentration of 2.5 μg mL−1, the TNSP NP suspension could still be heated up to ∼60 °C, attributing to the high photothermal conversion efficiency (59%, Fig. S11, ESI†). Under the same condition, TPETPAFN NPs, which were prepared by the same method using DSPE-PEG2000 as the encapsulation matrix, showed a temperature increase of only ∼3 °C in aqueous media (Fig. S12, ESI†). Furthermore, the photothermal stability of the TNSP NPs was tested by repeating the laser irradiation ON/OFF cycle five times, during which the sample exhibits stable temperature increase and decrease (Fig. S13, ESI†), indicating the good photothermal stability of the NPs.
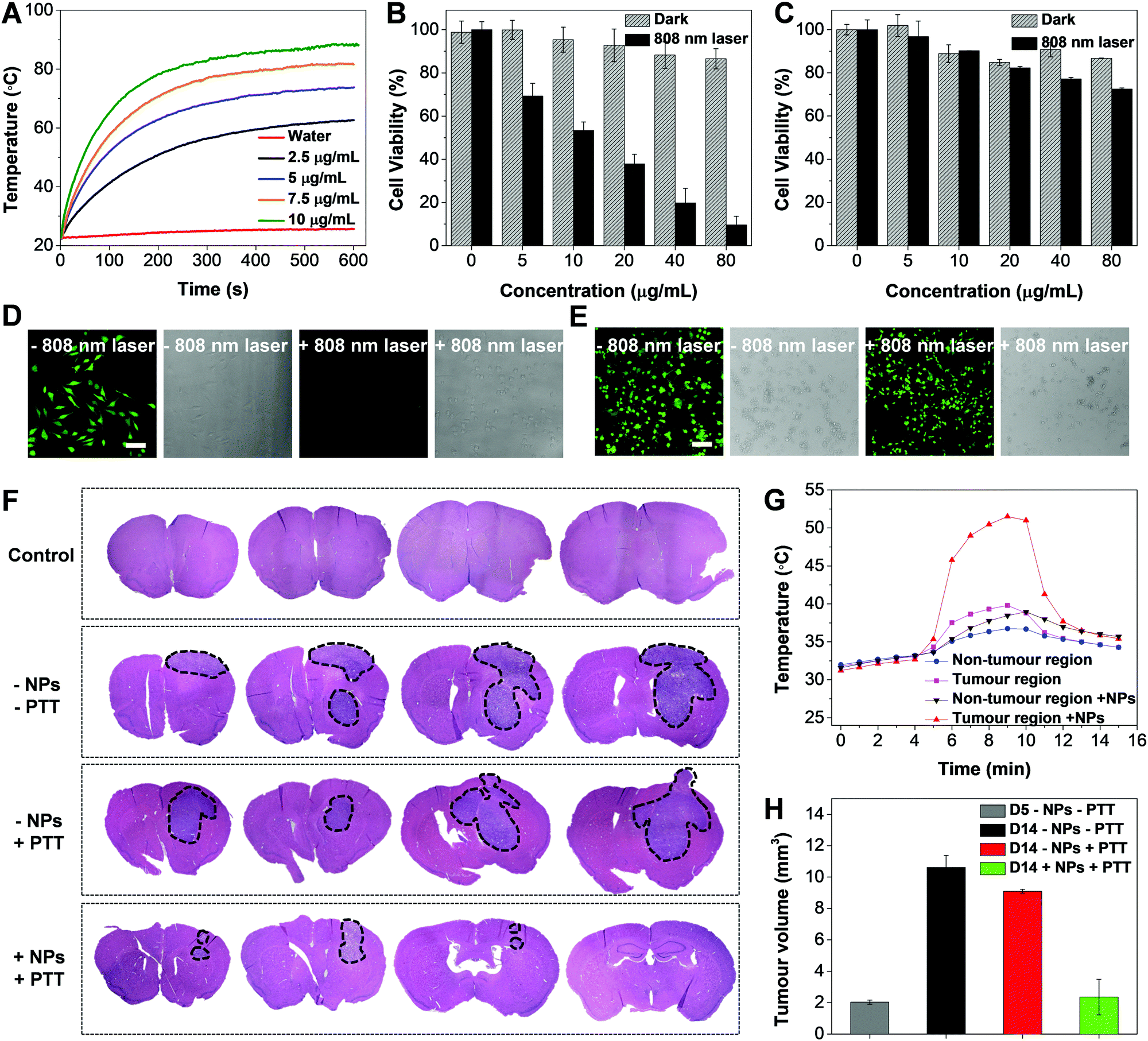 | ||
| Fig. 5 (A) Temperature evolution of water and different concentrations of FA-cRGD-TNSP NP suspensions in water after 808 nm laser irradiation at a laser power of 0.8 W from 1 to 10 min. Cell viabilities of U87MG cells (B) and neuronal glia mixed culture (C) after incubation with different concentrations of FA-cRGD-TNSP NPs and kept in the dark environment or irradiated with a 808 nm laser (0.8 W, 5 min). Confocal images of U87MG cells (D) and neuronal glia mixed culture (E) incubated with FA-cRGD-TNSP NPs before and after 808 nm laser irradiation (0.8 W, 5 min) and stained by FDA. (F) Representative microscopic images of H&E stained coronal brain sections from the control and tumour-bearing animals. The brain slices were serially sectioned from the frontal lobe to the parietal lobe, shown in sequence from left to right. The black dotted lines indicate the tumour region. The FA-cRGD-TNSP NPs were injected into the mice at day 5 after tumour implantation. PTT treatment was conducted at 4 h after NP injection at day 5. All animals were sacrificed at the same time point at day 14 post tumour implantation. (G) Cortical temperature evolution after 808 nm laser irradiation. (H) Tumour volume change of treated and untreated mice at different days post tumour implantation. | ||
In vitro evaluation of the PTT effect was conducted by incubation of U87MG GBM cells and neuronal cells with FA-cRGD-TNSP NPs for 4 h and subsequent irradiation with the 808 nm CW laser for 5 min at a power of 0.8 W, using cells kept in the dark as control groups. The MTT studies of cell viability indicate that the U87MG cells can be efficiently killed by 808 nm laser irradiation due to the photothermal effect while the cells kept in the dark environment remain highly viable (Fig. 5B). Meanwhile, as the cellular uptake of NPs into neurons and glia cells is negligible, they could maintain high viability with and without 808 nm laser irradiation (Fig. 5C). This reveals the negligible influence of 808 nm laser treatment towards the neuronal population and the high safety of using PTT for GBM treatment. The MTT results were further validated by confocal images (Fig. 5D and E). The 808 nm laser treated U87MG cells exhibit unhealthy spherical morphologies and cannot be stained by fluorescein diacetate (FDA), which could selectively label live cells and generate a green fluorescence, while the untreated U87MG cells and the two groups of neuronal cells with or without 808 nm laser treatment remain healthy and generate a bright green fluorescence after FDA staining.
Finally, in vivo photothermal treatment of GBM was carried out after intravenous administration of FA-cRGD-TNSP NPs to tumour-implanted mice at day 5 post tumour implantation using a 808 nm CW laser, which can penetrate through the mouse skull as reported previously.34 As indicated by the H&E-stained brain slices in Fig. 5F, a single laser exposure (808 nm CW laser, 5 min, 0.8 W) after 4 h of NP administration at day 5 post tumour implantation could successfully suppress tumour growth, leading to an obviously smaller tumour when observed at day 14 post tumour implantation, as compared to the group of untreated mice and the group of mice with only laser irradiation. The cortical temperature evolution was monitored by a microprobe thermometer before and after 808 nm laser irradiation. A rapid increase in temperature to ∼50 °C at the tumour site of NP treated mice was observed after laser irradiation (Fig. 5G), while the non-tumour site and control group without NP administration exhibit negligible temperature increase, which further validates the selective NP accumulation at GBM and the good photothermal effect of the NPs. Moreover, as revealed by Fig. 5H, obvious GBM growth in the untreated mice could be observed from day 5 to day 14 post implantation, while in the mice treated with both FA-cRGD-TNSP NPs and 808 nm laser irradiation greatly controlled progression of GBM could be observed. These observations prove the feasibility of applying the dual targeting NPs for efficient photothermal treatment of GBM attributing their good targeting efficiency to the margin of GBM and the efficient photothermal effect. Meanwhile, after NP administration, no obvious body weight loss of the animals could be observed (Fig. S14, ESI†) while the H&E stained slices of different organs assessed by three independent pathologists suggest that the NPs do not lead to any significant lesions (Fig. S15, ESI†). The biodistribution and metabolism of the NPs were studied by ex vivo fluorescence imaging of important organs. As shown in Fig. S16 (ESI†), the NPs mainly accumulated at the liver and the spleen post injection due to the uptake by the reticuloendothelial system. At day 10 post injection, almost no fluorescence could be observed from the organs, indicating the rapid clearance of the NPs from the animal's body. The half-life of FA-cRGD-TNSP NPs was measured to be ∼2.8 h (Fig. S17, ESI†). These results proves the good biocompatibility and safety of the FA-cRGD-TNSP NPs for in vivo applications.
Conclusions
In summary, we developed a simple but promising strategy for identifying the margin of GBM and achieving efficient photothermal therapy to supress the growth of GBM by organic nanoparticles with dual-targeting ligands. The co-encapsulation of the AIE fluorogen, TPETPAFN, and the semiconducting polymer, SP, generates NPs with a bright red emission and high molar absorptivity in the NIR region, which exhibit promising optical properties, small size and uniform morphology. After surface functionalization with different ratios of the two targeting ligands, the NPs with a FA to cRGD ratio of 25:75 demonstrate the best performance for in vitro GBM cell targeting. Meanwhile, the NPs with dual targeting ligands also show superior capability in targeting tumour margins as well as the tumour interior as compared to the NPs without targeting ligands or with one targeting ligand. Furthermore, the NPs with dual targeting ligands show promising delivery efficiency to the tumour margin at different tumour stages from day 3, day 5 to day 7 post tumour implantation due to the synergistic targeting effect of the dual targeting ligands, which could help vividly analyse the tumour size at various stages of GBM by ex vivo fluorescence imaging and provide promising opportunities for the effective treatment of GBM. The PTT treatment of GBM demonstrates sufficient killing efficiency of GBM cells in vitro and promising suppression of GBM growth in vivo with minimal adverse effect towards neuronal cells. Considering the good optical properties, the excellent targeting efficiency towards the GBM margin and interior as well as the great biocompatibility with neuronal cells, FA-cRGD-TNSP NPs possess great potential for providing intraoperative fluorescence guidance and preventing recurrence by PTT treatment after tumour resection in future clinical applications.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Singapore National Research Foundation (R279-000-444-281, R279-000-483-281, R-719-000-018-281) and the National University of Singapore (R279-000-433-281) for financial support.Notes and references
- S. Roy, D. Lahiri, T. Maji and J. Biswas, South Asian J. Cancer, 2015, 4, 163 CrossRef PubMed.
- E. C. Holland, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6242 CrossRef CAS.
- R. M. Young, A. Jamshidi, G. Davis and J. H. Sherman, Ann. Transl. Med., 2015, 3, 121 Search PubMed.
- D. Krex, B. Klink, C. Hartmann, A. von Deimling, T. Pietsch, M. Simon, M. Sabel, J. P. Steinbach, O. Heese and G. Reifenberger, Brain, 2007, 130, 2596 CrossRef PubMed.
- J. Scott, N. Rewcastle, P. Brasher, D. Fulton, J. MacKinnon, M. Hamilton, J. Cairncross and P. Forsyth, Ann. Neurol., 1999, 46, 183 CrossRef CAS PubMed.
- H. I. Robins, S. Chang, N. Butowski and M. Mehta, Curr. Oncol. Rep., 2007, 9, 66 CrossRef CAS PubMed.
- D. Khosla, Ann. Transl. Med., 2016, 4, 54 CrossRef PubMed.
- P. Guo, J. Lan, J. Ge, Q. Nie, L. Guo, Y. Qiu and Q. Mao, Exp. Cell Res., 2014, 320, 200 CrossRef CAS PubMed.
- R. Stupp, M. E. Hegi, W. P. Mason, M. J. van den Bent, M. J. Taphoorn, R. C. Janzer, S. K. Ludwin, A. Allgeier, B. Fisher and K. Belanger, Lancet Oncol., 2009, 10, 459 CrossRef CAS PubMed.
- S. K. Carlsson, S. P. Brothers and C. Wahlestedt, EMBO Mol. Med., 2014, 6, 1359 CrossRef CAS PubMed.
- A. Lilja, K. Bergstr, B. Sp and Y. Olsson, J. Comput. Assist. Tomogr., 1981, 5, 625 CrossRef CAS PubMed.
- P. J. Kelly, C. Daumas-Duport, D. B. Kispert, B. A. Kall, B. W. Scheithauer and J. J. Illig, J. Neurosurg., 1987, 66, 865 CAS.
- L. Lunsford, A. Martinez and R. Latchaw, Acta Radiol., Suppl., 1986, 369, 154 CAS.
- H. Ishihara, H. Kubota, R. L. Lindberg, D. Leppert, S. M. Gloor, M. Errede, D. Virgintino, A. Fontana, Y. Yonekawa and K. Frei, J. Neuropathol. Exp. Neurol., 2008, 67, 435 CrossRef CAS PubMed.
- Y. Gu, C. M. Dee and J. Shen, Front. Biosci., 2011, 3, 1216 CrossRef.
- E. Karathanasis and K. B. Ghaghada, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 678 CAS.
- L. G. Dubois, L. Campanati, C. Righy, I. D’Andrea-Meira, T. C. L. D. S. E. Spohr, I. Porto-Carreiro, C. M. Pereira, J. Balça-Silva, S. A. Kahn, M. F. Dos Santos, M. D. A. R. Oliveira, A. Ximenes-da-Silva, M. C. Lopes, E. Faveret, E. L. Gasparetto and V. Moura-Neto, Front. Cell. Neurosci., 2014, 8, 418 Search PubMed.
- O. van Tellingen, B. Yetkin-Arik, M. C. de Gooijer, P. Wesseling, T. Wurdinger and H. E. de Vries, Drug Resist. Updat., 2015, 19, 1 CrossRef CAS PubMed.
- S. Song, G. Mao, J. Du and X. Zhu, Drug Deliv., 2016, 23, 1404 CrossRef CAS PubMed.
- A. Mangraviti, S. Y. Tzeng, K. L. Kozielski, Y. Wang, Y. Jin, D. Gullotti, M. Pedone, N. Buaron, A. Liu and D. R. Wilson, ACS Nano, 2015, 9, 1236 CrossRef CAS PubMed.
- J. L. Munoz, S. A. Bliss, S. J. Greco, S. H. Ramkissoon, K. L. Ligon and P. Rameshwar, Mol. Ther. Nucleic Acids, 2013, 2, e126 CrossRef PubMed.
- A. Rodriguez, S. B. Tatter and W. Debinski, Pharmaceutics, 2015, 7, 175 CrossRef CAS PubMed.
- R. Gabathuler, Neurobiol. Dis., 2010, 37, 48 CrossRef CAS PubMed.
- L. Zou, H. Wang, B. He, L. Zeng, T. Tan, H. Cao, X. He, Z. Zhang, S. Guo and Y. Li, Theranostics, 2016, 6, 762 CrossRef CAS PubMed.
- N. S. Abadeer and C. J. Murphy, J. Phys. Chem. C, 2016, 120, 4691 CrossRef CAS.
- X. Cai, A. Bandla, D. Mao, G. Feng, W. Qin, L. D. Liao, N. Thakor, B. Z. Tang and B. Liu, Adv. Mater., 2016, 28, 8760 CrossRef CAS PubMed.
- W. Lu, M. P. Melancon, C. Xiong, Q. Huang, A. Elliott, S. Song, R. Zhang, L. G. Flores, J. G. Gelovani and L. V. Wang, Cancer Res., 2011, 71, 6116 CrossRef CAS PubMed.
- S.-K. Baek, A. R. Makkouk, T. Krasieva, C.-H. Sun, S. J. Madsen and H. Hirschberg, J. Neurooncol., 2011, 104, 439 CrossRef CAS PubMed.
- K. Yang, S. Zhang, G. Zhang, X. Sun, S. T. Lee and Z. Liu, Nano Lett., 2010, 10, 3318 CrossRef CAS PubMed.
- Y. Wang, K. Wang, J. Zhao, X. Liu, J. Bu, X. Yan and R. Huang, J. Am. Chem. Soc., 2013, 135, 4799 CrossRef CAS PubMed.
- Z. Xu, J. Jin, L. K. Siu, H. Yao, J. Sze, H. Sun, H. F. Kung, W. S. Poon, S. S. Ng and M. C. Lin, Int. J. Pharm., 2012, 426, 182 CrossRef CAS PubMed.
- A. Srivatsan, M. Ethirajan, S. K. Pandey, S. Dubey, X. Zheng, T.-H. Liu, M. Shibata, J. Missert, J. Morgan and R. K. Pandey, Mol. Pharm., 2011, 8, 1186 CrossRef CAS PubMed.
- B. Oller-Salvia, M. Sanchez-Navarro, E. Giralt and M. Teixido, Chem. Soc. Rev., 2016, 45, 4690 RSC.
- P. A. Lapchak, P. D. Boitano, P. V. Butte, D. J. Fisher, T. Hölscher, E. J. Ley, M. Nuño, A. H. Voie and P. S. Rajput, PLoS One, 2015, 10, e0127580 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mh00946e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |