
Reconstructing bimetallic carbide Mo6Ni6C for carbon interconnected MoNi alloys to boost oxygen evolution electrocatalysis†
Meng Yang
Zu
a,
Chongwu
Wang
a,
Le
Zhang
a,
Li Rong
Zheng
 b and
Hua Gui
Yang
*a
b and
Hua Gui
Yang
*a
aKey Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai, 200237, China. E-mail: hgyang@ecust.edu.cn
bBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, 100049, China
First published on 19th September 2018
Abstract
Mo6Ni6C is first utilized as a precatalyst for the OER process, reconstructing in the bulk to form small MoNi alloy clusters interconnected with atomically dispersed carbon, delivering 10 mA cm−2 at an overpotential of 190 mV for the OER and at 1.47 V for overall water splitting (mass loading of 9.8 mg cm−2).
Conceptual insightsPrevious efforts to promote the catalytic activity of metallic compounds towards the OER were mainly focused on nanostructuring or downsizing the materials to expose more active sites. Unfortunately, these strategies severely suffer from intrinsic drawbacks of poor charge transport caused by interstitial spaces between the nanoparticles. Unlike commonly-occurring surface reconstruction during the OER process, bimetallic carbide Mo6Ni6C is first utilized as a precatalyst with bulk reconstructing to form carbon interconnected MoNi alloy clusters. Furthermore, the MoNi alloys would be surface oxidized with Mo6+ modulated active NiOOH species and maintain bulk metallic MoNi alloys. As a result, nano-fragments guarantee more active sites and amorphous carbon interconnects the fragments at atomic levels, relaxing the poor charge transport caused by the interstitial spaces. Thus, this resulting catalyst delivers a benchmark current density of 10 mA cm−2 at an unprecedented low overpotential of 190 mV for more than 100 h, and at a cell voltage of 1.47 V over 200 h for overall water splitting. The high catalytic activity and long-time durability make it one of the most effective OER electrocatalysts, showing the potential for bimetallic carbides to be promising precatalysts for the OER. |
1. Introduction
The oxidation of water to molecular oxygen is the key process in solar-to-chemical conversion technologies, mainly including water splitting, carbon dioxide reduction and metal–air batteries.1,2 Unfortunately, the kinetics of the oxygen evolution reaction (OER) are sluggish, even when facilitated by high-efficiency and precious-metal-containing catalysts. Breakthroughs are urgently needed to explore high-activity, robust-stability and low-cost catalysts.3–6 To date, except for the widely studied transition metal oxide/hydroxide OER catalysts,7–12 abundant transition metal compounds such as sulfides,13 selenides,14 carbides,15 nitrides16 and phosphides,17 and even some bimetallic compounds18–21 have been explored as highly efficient OER precatalysts.5,22 In general, good performance mainly originates from the intrinsic metallicity of these compounds as the core and the excellent activity of the oxidized layer species as the shell after in situ surface reconstruction.To further promote the catalytic activity of these metallic compounds, general approaches have been focused on nanostructuring or downsizing the catalytic materials to expose more active sites.15,23,24 Nonetheless, these strategies severely suffer from intrinsic drawbacks, namely, the poor charge transport caused by interstitial spaces between the nanoparticles. Unlike commonly-occurring surface reconstruction during the OER process, some OER precatalysts featuring unique structures would be collapsed in the bulk, which might increase the number of active sites and simultaneously avoid disadvantages of stacked particles.25 Considering that the high-valence metals (such as tungsten or molybdenum) are usually subtly incorporated into OER catalysts to form homogeneously dispersed multimetal materials,10,26 and at the same time, the NiMo alloys are recorded as benchmark alkaline HER electrocatalysts because of their excellent intrinsic metallic properties,27,28 we rationally attempted to utilize bimetallic carbide Mo6Ni6C as a OER precatalyst with bulk reconstructing under OER conditions, to build up carbon interconnected MoNi alloys, which could generate high-valence Mo6+ incorporated NiOOH species. Intrinsically, Mo6Ni6C behaves as a large unit cell of a cubic crystal, which would break into nano-fragments of MoNi alloys after bulk reconstruction, guaranteeing more active sites. At the same time, the carbon element in the cubic crystal would transform into amorphous carbon to interconnect the fragments at an atomic level, relaxing the poor charge transport caused by the interstitial spaces. Therefore, we anticipate that Mo6Ni6C could act as a new category of precatalyst, to further boost OER activity.
Herein, for the first time, we rationally used bimetallic carbide Mo6Ni6C (Mo6Ni6C/NF) as a precatalyst to motivate the OER, which could be consequently reconstructed into MoNi alloys interconnected with amorphous carbon under OER conditions. Furthermore, the MoNi alloys would get oxidized on the surface to form Mo6+ incorporated NiOOH species during the pre-oxidation process. At the same time, small MoNi alloy particles, entirely separated by and intimately contacted on an in situ generated carbon substrate, would keep metallic characteristics in the bulk, which synergistically boost the charge transfer process and expose more active sites. Thus, the resulting oxidized derived catalyst (O-MoNi-C/NF, mass loading of 9.8 mg cm−2) could exhibit high OER activity, requiring a low overpotential (η10) of 190 mV to achieve a current density (j) of 10 mA cm−2, which surpasses the commercial IrO2 and other reported benchmarked OER catalysts. More importantly, the O-MoNi-C/NF catalyst exhibits outstanding stability to maintain a j of 10 mA cm−2 for at least 100 h in alkaline water. Notably, O-MoNi-C/NF is further demonstrated in a water splitting device context by pairing it with Mo6Ni6C as a hydrogen evolution reaction (HER) catalyst, which delivers a j of 10 mA cm−2 at 1.47 V for over 200 h degradation tests. These findings in our work demonstrate the possibility to utilize bimetallic carbides as precatalysts for efficient and durable OER, offering a new insight into the fabrication of high-performance OER catalysts for many electrochemical energy-conversion devices.
2. Results and discussion
2.1. Structural characterization
The Mo6Ni6C/NF sample was synthesized by a simple hydrothermal method, followed by annealing at 700 °C for 2 h under the mixture gas of H2/Ar (v/v, 15/85) in a tube furnace. To obtain O-MoNi-C/NF, a pre-oxidation step was applied to the pristine Mo6Ni6C/NF electrode by employing a potential of 1.6 V (all potentials were referenced to the reversible hydrogen electrode, and details are illustrated in the ESI†). As shown in Fig. S1 (ESI†), the corresponding i–t curve for the electrochemical oxidation at 1.6 V for 30 min mainly consists of three parts: during the first step, the current density is very high and almost keeps steady, revealing that the oxidation and the reconstruction processes are occurring in the large unit cell of Mo6Ni6C. In the second step, the current density decreases as time increases, implying that the reconstruction and the OER processes are taking place at the same time; for the third and last step, the current density reaches a stable state without change, demonstrating that a continuous OER process occurred to generate O2. The corresponding video for the pre-oxidation process is shown in Movie S1 (ESI†).Before fragmentation, the material remains as cubic Mo6Ni6C (Fig. 1a and Fig. S2, ESI†), with Mo–Mo bonds in the structure. The structure schematic illustration of the key parts of the O-MoNi-C/NF structure is shown in Fig. 1b and c. After the pre-oxidation process, the large unit cell of Mo6Ni6C collapses into the nano-fragments of MoNi3 and MoNi4 (Fig. S3, ESI†), with a carbon element homogeneously distributed throughout the whole system in the form of amorphous structures. Amorphous carbon functions as the electron bridges to interconnect the isolated nanoparticles, penetrating into the gaps to interlink MoNi3 and MoNi4, thus enhancing the charge transport. At the same time, as the conductive substrate, amorphous carbon separates MoNi3 and MoNi4 from aggregation, therefore more active sites could be exposed. Meanwhile, the MoNi alloys would get oxidized during the pre-oxidation step, forming Mo6+ activated oxide/oxyhydroxide layers on the surface to promote the rapid OER process.
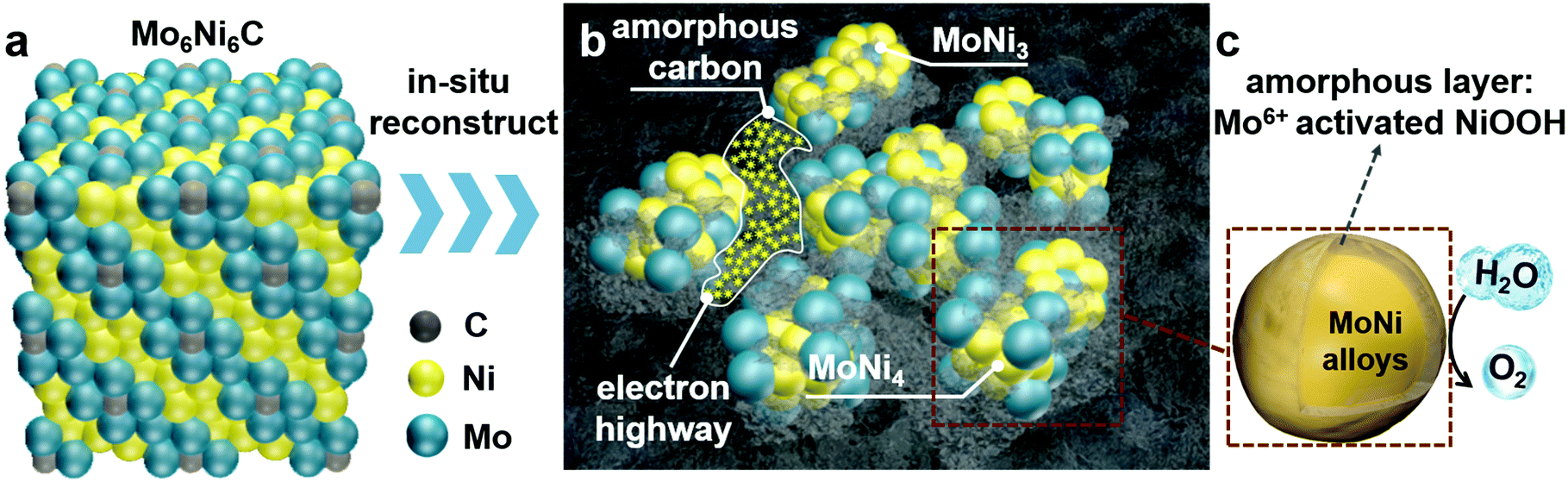 | ||
| Fig. 1 Structure analysis of Mo6Ni6C (pre-OER) and O-MoNi-C/NF (post-OER). Schematic representation of (a) cubic Mo6Ni6C, (b) the O-MoNi-C/NF system composed of in situ generated MoNi alloys and amorphous carbon, which interconnects small MoNi alloy clusters to boost the electron transfer process and (c) MoNi alloy clusters with oxidized amorphous layers. | ||
X-ray diffraction (XRD) patterns confirm the existence of MoNi3 and MoNi4 alloys in the O-MoNi-C/NF sample. As shown in Fig. 2a, before the OER process, the material exhibits the crystalline phase of Mo6Ni6C (JCPDS No. 65-4436). After pre-oxidation treatment, the material shows the characteristic peaks of MoNi3 (JCPDS No. 65-2587) and MoNi4 (JCPDS No. 65-1533). The low-intensity peaks of the Ni signals were also observed, which resulted from the surface exfoliation of the material when sonicated off from the substrate. Furthermore, the broad diffraction peak located at around 22° is assigned to the in situ generated amorphous carbon during the pre-oxidation procedure (Fig. S4, ESI†),29 which was further evidenced by Raman spectra (Fig. S5, ESI†).30 It is noted that no MoOx or NiOOH was reflected in the XRD data, mainly because of the poor crystallinity of the generated MoOx and NiOOH on the oxidized surface. Raman spectroscopy and Fourier transform infrared (FT-IR) spectroscopy were conducted to evidence the presence of MoOx and NiOOH. As shown in Fig. S6 (ESI†), characteristic peaks located at ∼470 and ∼550 cm−1 confirm the existence of nickel oxyhydroxide (NiOOH).31 Absorption bands at ∼1385, ∼824 and ∼634 cm−1 are assigned to MoOx,32 observed in the FT-IR spectra (Fig. S7, ESI†). Based on these results, after the pre-oxidation treatment, the large unit cell of Mo6Ni6C collapses into small parts of MoNi3 and MoNi4 with oxidized species on the surface, interconnected by in situ generated amorphous carbon.
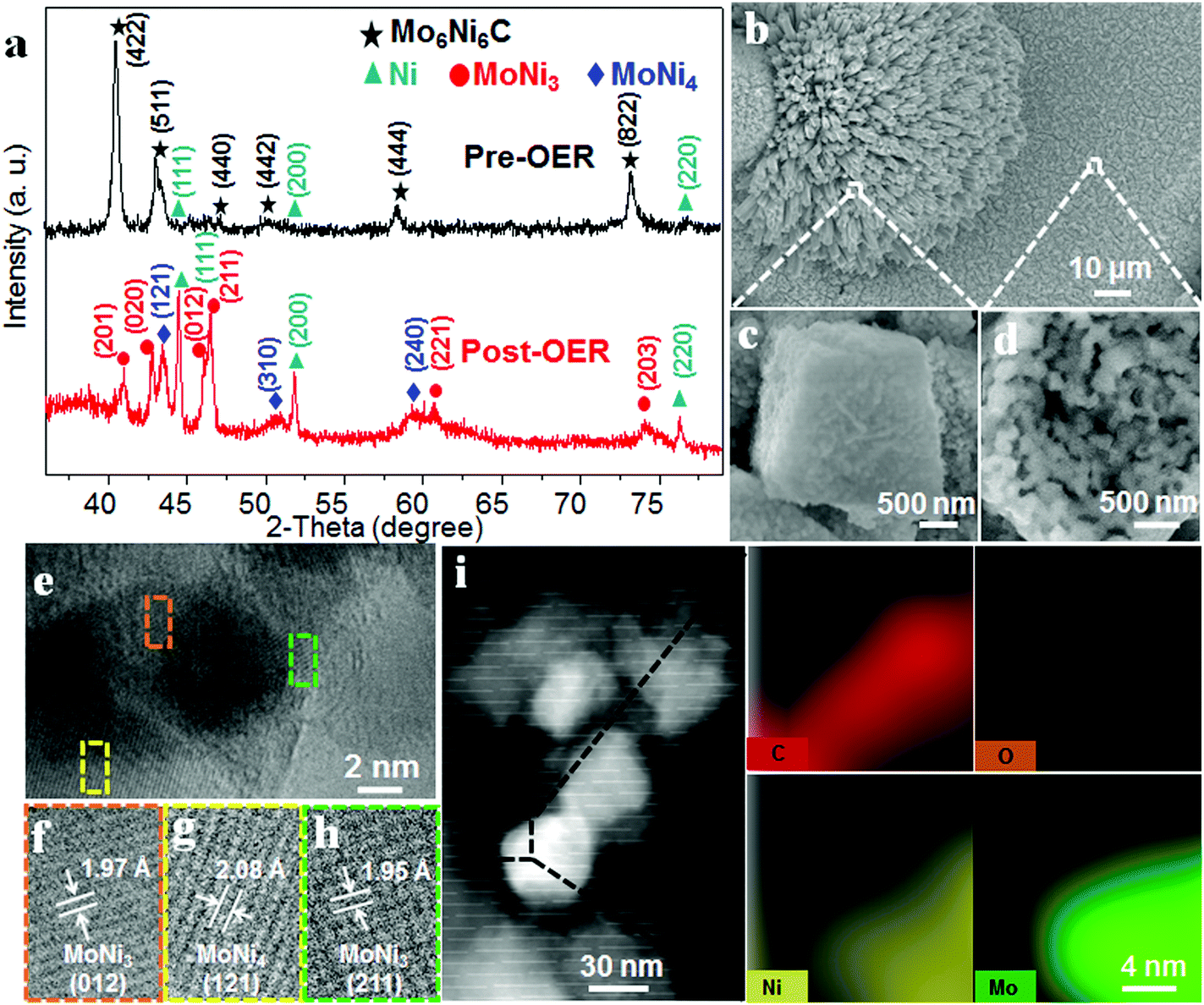 | ||
| Fig. 2 XRD, SEM, TEM and HAADF-STEM measurements of O-MoNi-C. (a) XRD patterns of Mo6Ni6C (pre-OER) and O-MoNi-C (post-OER). SEM images (b–d), and TEM and HRTEM images (e–h) of O-MoNi-C at different magnifications. (i) HAADF-STEM image and the corresponding EELS elemental mappings of C, O, Ni and Mo for O-MoNi-C. | ||
Scanning electron microscopy (SEM) images and the corresponding elemental mappings for O-MoNi-C/NF (Fig. 2b and Fig. S8, ESI†) reveal that the materials uniformly cover the surfaces of the Ni foam. Furthermore, the high-resolution SEM images clearly show that O-MoNi-C/NF is similar to the pristine Mo6Ni6C/NF (Fig. 2c, d and Fig. S9, ESI†), which consists of some clusters of rods with a mean diameter of around 1 μm. The only difference is that the surface of O-MoNi-C/NF appeared as flocculent, without clear boundaries between the nanoparticles compared to Mo6Ni6C, implying the surface rearrangements for the NiOOH species and the amorphous carbon in the system. A typical transmission electron microscopy (TEM) image of O-MoNi-C/NF is shown in Fig. S10 (ESI†), which is consistent with the SEM image of a rod with a diameter of about 1 μm. TEM and high-resolution TEM (HRTEM) images (Fig. 2e–h) further confirm that a series of lattice fringes give interplanar distances of 0.195, 0.197 and 0.208 nm, corresponding to the (211) and (012) crystallographic planes of MoNi3 and the (121) plane of MoNi4, respectively, which agrees well with the XRD results. The elemental distributions of C, Mo, Ni and O of O-MoNi-C/NF were investigated by EELS elemental mappings in the HAADF-STEM mode. Fig. 2i shows the HAADF-STEM image and the corresponding EELS elemental mappings of O-MoNi-C/NF, demonstrating the homogeneous distribution of carbon surrounding all the MoNi alloy nanoparticles.
In order to test the surface valence changes between Mo6Ni6C/NF and O-MoNi-C/NF, X-ray photoelectron spectroscopy (XPS) characterizations were conducted. The elements of Ni and Mo in O-MoNi-C/NF display higher valence states than that of the precatalyst Mo6Ni6C/NF (Fig. 3a and d). After the OER process, the peaks for metallic Ni disappear, and the oxidized Ni characteristic peak shifts to a more positive valence state.23,33 Likewise, compared to Mo6Ni6C/NF, the Mo element in O-MoNi-C/NF mainly exhibits the valence states of Mo6+, which is in the highest oxidation state. As illustrated in Fig. S11 (ESI†), benefiting from the prominent ability of drawing electrons of high-valence Mo6+, the 3d metal Ni would easily stay at its high-valence state as active sites for the OER, and thus Mo6+ serves as a modulator to boost the OER process.10,26,34–36 These results are in agreement with the XRD, Raman and FT-IR observation.
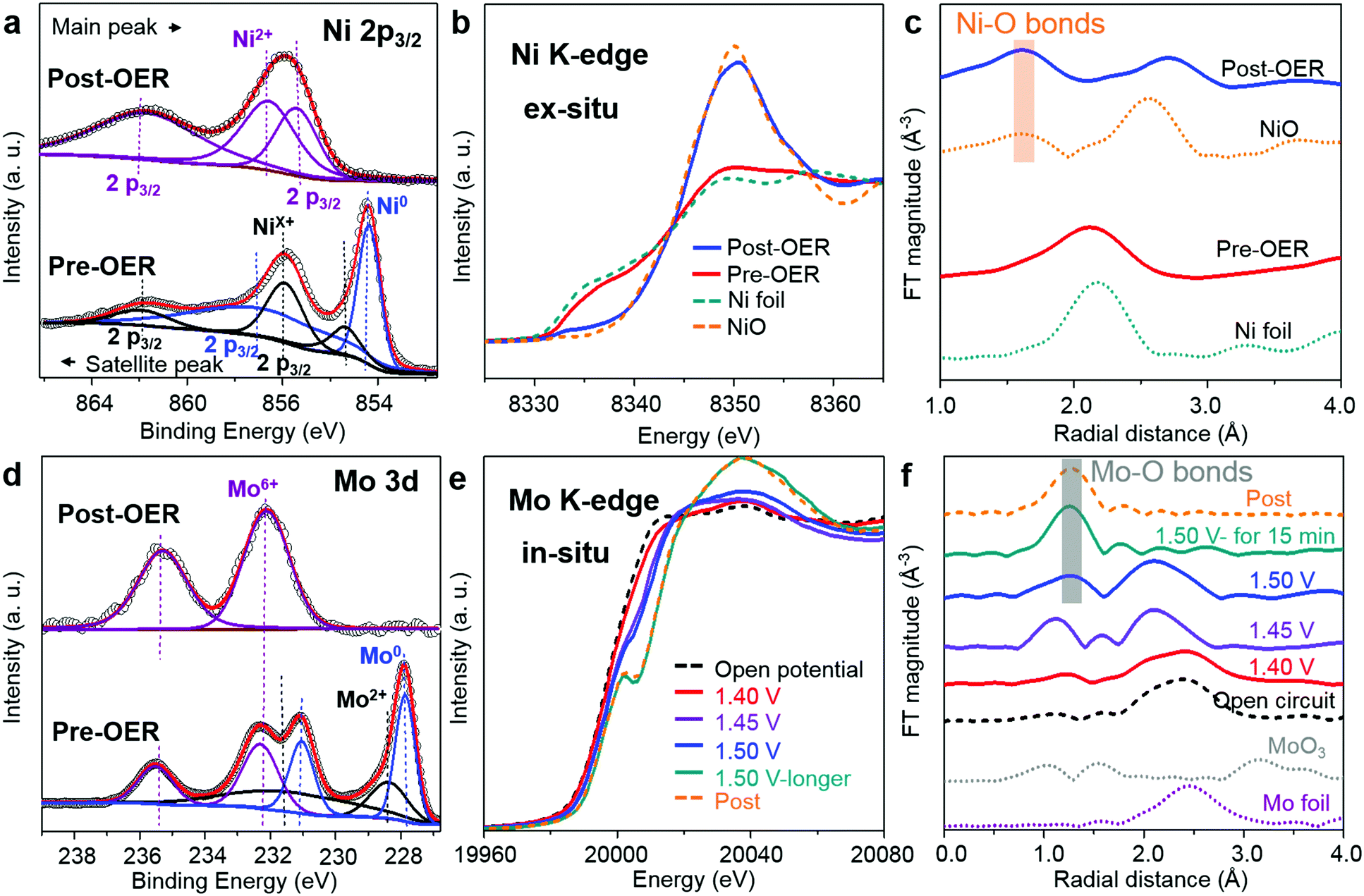 | ||
| Fig. 3 XPS and XAFS measurements of Mo6Ni6C and O-MoNi-C. (a) XPS spectra of Mo6Ni6C (pre-OER) and O-MoNi-C (post-OER) for the Ni 2p3/2 region. (b) Ni K-edge XANES spectra and (c) FT of Ni K-edge EXAFS for the samples of Mo6Ni6C (pre-OER), O-MoNi-C (post-OER), Ni foil and NiO. (d) XPS spectra of Mo6Ni6C (pre-OER) and O-MoNi-C (post-OER) for Mo 3d. (e) In situ Mo K-edge XANES spectra and (f) FT of Mo K-edge EXAFS for the samples under the working conditions of the OER process. | ||
XAFS characterization was carried out to further probe the bulk fine structures of the catalysts. In Fig. 3b, before the OER process, the Ni element in Mo6Ni6C exhibits similar valence states as Ni foil, indicating its intrinsic metallic properties. However, for the O-MoNi-C/NF sample, the valence of Ni is close to that of NiO, which is caused by oxidation during the pre-treating and OER process. As shown in Fig. 3c and Fig. S12a (ESI†), the Fourier transforms (FTs) of the XAFS spectra and the corresponding FTs of k3-weighted EXAFS oscillations collected at the Ni K-edge of the O-MoNi-C/NF sample reveal that the Ni–Ni bonds disappear and the Ni–O bonds emerge, drawing a similar conclusion. Moreover, to figure out the transformation during the OER proceedings, in situ XAFS characterization for the Mo element was implemented. The typical experimental setup for the in situ XAFS measurements in transmission and fluorescence modes and a simple schematic of the in situ cell are shown in Fig. S13 (ESI†). Fig. 3e reveals the change of the valence states during the in situ OER process by increasing the applied potential. Under open circuit conduction, Mo6Ni6C possesses the lowest valence states for the Mo element. With the potential increasing from 1.40 V to 1.50 V, the valence states for Mo increased. After maintaining the potential at 1.50 V for about 15 min, the Mo element almost reaches a stable higher valence state, similar to that of the Mo element in the post-OER sample. Correspondingly, the emergence of the Mo–O bonds and the disappearance of the Mo–Mo bonds share the same trends as the in situ XANES results (Fig. 3f and Fig. S12b, ESI†). Overall, the XAFS results unambiguously prove that during the pre-oxidation process, Mo6Ni6C was reconstructed into MoNi alloys interconnected by amorphous carbon to guarantee fast charge transport, with oxidized species on the surface to ensure distinguished intrinsic OER activity. Specifically, surface NiOOH species acted as active sites and high-valence Mo6+ further subtly modulated the oxyhydroxides to reach an optimized adsorption energy for the OER intermediates.
2.2. OER electrocatalytic performance
To evaluate the OER performances of O-MoNi-C/NF, we conducted a series of electrochemical tests in a typical three-electrode system with 1.0 M KOH aqueous solution as the electrolyte. The linear sweep voltammetry (LSV) curves show that O-MoNi-C/NF exhibits the best OER catalytic activity among all the control samples, requiring the lowest η10 of 190 mV (η10 of 290 mV for IrO2/NF, 335 mV for MoNi4/C/NF and 420 mV for blank NF, as shown in Fig. 4a and b), also standing at the forefront of the OER field (Table S1, ESI†). For comparison, the several repeated LSV curves for the initial Mo6Ni6C/NF before the oxidation process are shown in Fig. S14 (ESI†). For the first scan, the current density is very high and almost keeps steady with increasing potential, implying that the oxidation and the reconstruction processes are occurring in the large unit cell of Mo6Ni6C, and almost no OER process happens. For the second to fourth scans, the starting current density decreases on repeatedly scanning the LSV curves, revealing that the reconstruction process is gradually completing, and the OER process is taking place at the same time. For the fifth and last scans, the LSV curve reaches a stable state, and keeps almost the same as O-MoNi-C/NF, which is obtained after the electrochemical oxidation at 1.6 V for 30 min. In particular, to compare the intrinsic activity of each catalyst, the electrochemically active surface area (ECSA) is estimated from the measurements of the double-layer capacitance (see more details in the ESI,† Fig. S15–S17). The ECSAs for each sample are shown in Table S2 (ESI†), and the ECSA-corrected LSV curves are shown in Fig. S18 (ESI†). As we can see, even normalized by ECSA, the j of O-MoNi-C/NF is still larger than that of IrO2/NF and MoNi4/C/NF, further revealing that O-MoNi-C/NF is intrinsically more active than the compared samples. The Tafel slope of O-MoNi-C/NF is 65.35 mV dec−1, which is much lower than that of all the other controlled samples (Fig. 4c), suggesting fastest OER kinetics. Correspondingly, the Nyquist spectra further reveal that O-MoNi-C/NF features the smallest serial resistance and charge transfer resistance (Fig. S19, ESI†), further demonstrating that O-MoNi-C/NF possesses the fastest charge transport process among all the controlled catalysts. It is noteworthy that to evaluate the stability of O-MoNi-C/NF, galvanostatic measurements were conducted in 1.0 M KOH (Fig. 4d). A continuous OER process occurred to generate O2 at j of 10 mA cm−2. The O-MoNi-C/NF electrode could maintain a j of 10 mA cm−2 for more than 100 h without apparent deactivation. In addition, the faradaic efficiency (FE) for oxygen evolution of this catalyst was evaluated, which was determined to be nearly 100% during the 1 hour of electrolysis (see more details in the ESI,† Fig. S20 and S21). In general, such activity and stability of O-MoNi-C/NF are located in the top positions of the reported OER electrocatalysts, as compared in Table S1 (ESI†).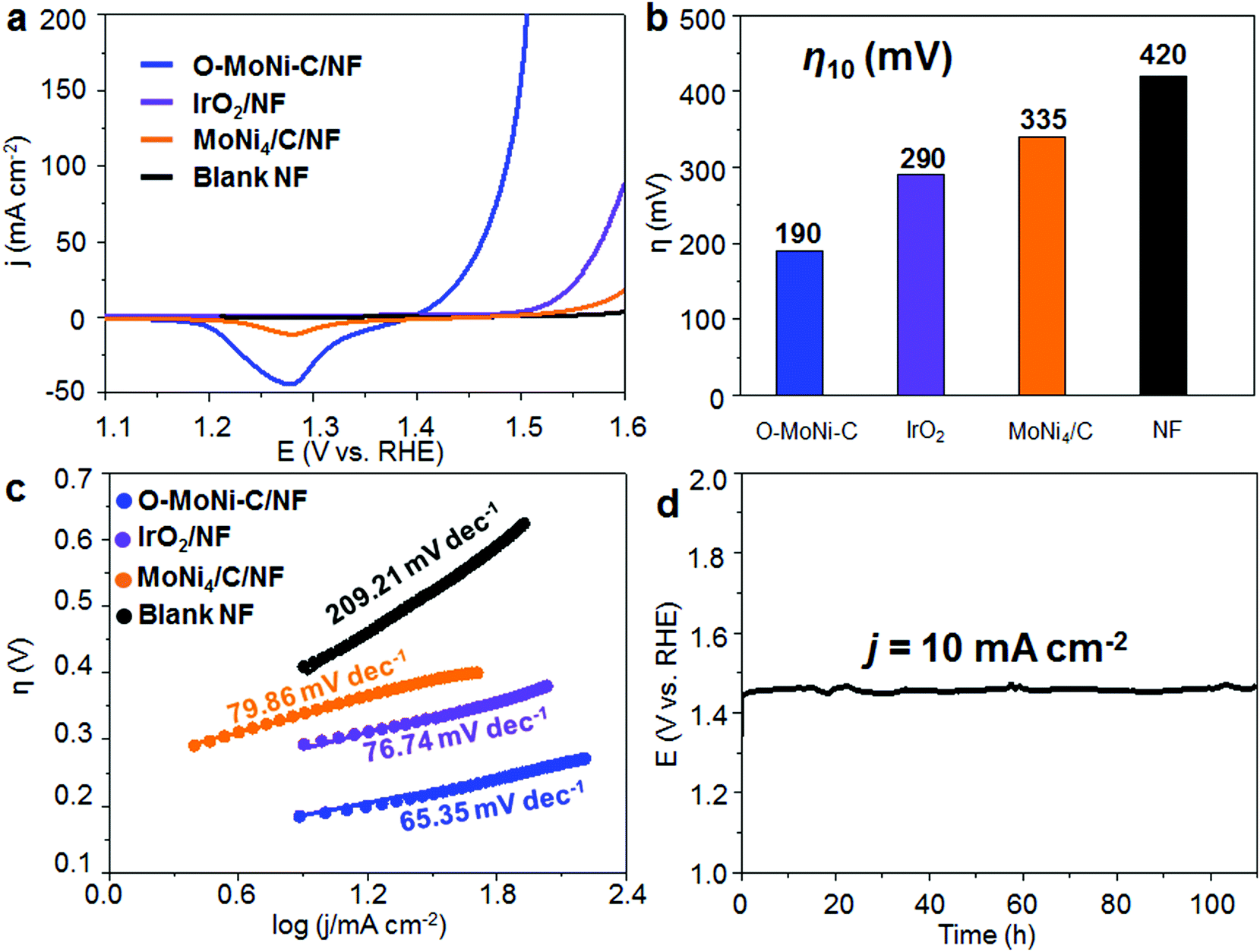 | ||
| Fig. 4 OER performance of O-MoNi-C in 1.0 M KOH. (a) LSV curves, (b) comparison of η10 and (c) Tafel plots for OER for the samples of O-MoNi-C/NF, IrO2/NF, MoNi4/C/NF and blank Ni foam. (d) Chronopotentiometric curve of O-MoNi-C/NF at a j of 10 mA cm−2 for the continuous OER process in 1.0 M KOH. | ||
2.3. Electrocatalytic overall water splitting performance
Furthermore, to test the viability of bimetallic carbide as the OER precatalyst for large-scale water electrolysis, we constructed the Mo6Ni6C/NF as both a cathode and anode at the same time to construct an alkaline electrolyzer. Mo6Ni6C/NF has been proved to be a high performance HER catalyst before in 1.0 M KOH electrolyte, requiring −34 mV overpotential to drive hydrogen production at a j of −10 mA cm−2, as shown in Fig. S22 (ESI†), which was also discussed in our previous work.37 As for the home-made single cell, after the pre-oxidation treatment at a cell voltage of around 1.6 V for 30 min, the resultant electrolyzer exhibited excellent performance, delivering a j of 10 mA cm−2 at approximately 1.47 V over continuous 200 h at room temperature (Fig. 5). To the best of our knowledge, such activity and stability for the materials to construct an electrolyzer for overall water splitting have never been reported before, as compared in Table S3 (ESI†).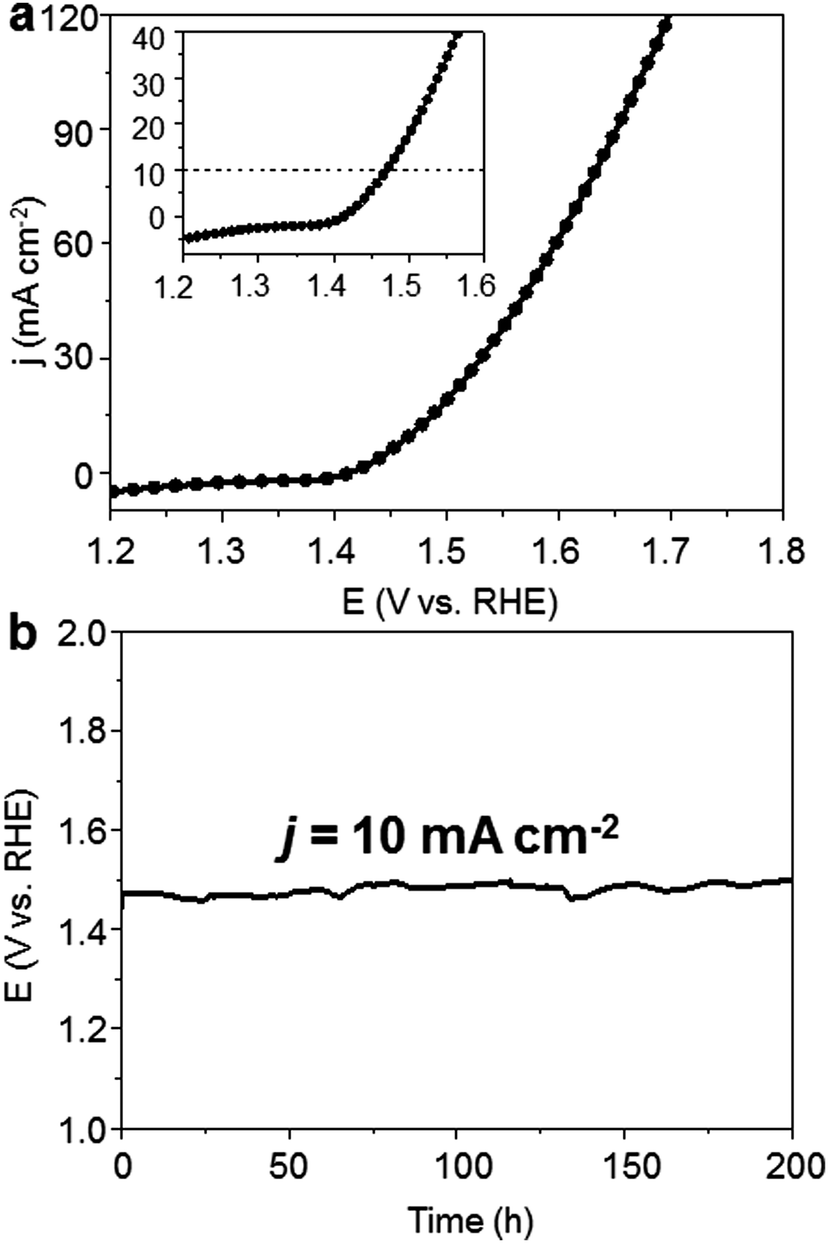 | ||
| Fig. 5 Overall water splitting performance of O-MoNi-C/NF combined with Mo6Ni6C/NF in 1.0 M KOH. (a) LSV curve for an overall water splitting electrolyzer, which uses Mo6Ni6C/NF as the cathode and O-MoNi-C/NF as the anode in 1.0 M KOH. (b) Chronopotentiometric curve of the electrolyzer at the j of 10 mA cm−2 for continuous water splitting in 1.0 M KOH. | ||
3. Conclusions
In conclusion, for the first time, bimetallic carbide Mo6Ni6C has been demonstrated as a high-performance and low-cost precatalyst for oxygen production. Impressively, it exhibits a low overpotential of 190 mV in 1.0 M KOH at a j of 10 mA cm−2 with a mass loading of 9.8 mg cm−2, and shows no evidence of degradation for continuous operation at a steady current for more than 100 h. Furthermore, an efficient water electrolyzer was demonstrated by using Mo6Ni6C/NF as both an anode and a cathode, achieving a j of 10 mA cm−2 at 1.47 V when continuously working for more than 200 h. The high catalytic activity and long-time durability of O-MoNi-C/NF make it one of the most effective OER electrocatalysts, and at the same time, showing the potential for bimetallic carbide to be a promising precatalyst for the OER. These findings represent an important advance towards expanding the scope of high-performance oxygen-evolving catalysts for the large-scale application of many electrochemical energy-conversion devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21573068), the National Natural Science Funds for Distinguished Young Scholars (51725201) and the Fundamental Research Funds for the Central Universities (222201718002). The authors also thank the crew of the 1W1B beamline of the Beijing Synchrotron Radiation Facility for the helpful assistance in the XANES measurements and data analyses.Notes and references
- S. Jin, J. M. Kevin, A. G. Hubert, B. G. John and S.-H. Yang, Science, 2011, 334, 1383–1385 CrossRef PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Science, 2010, 110, 6474–6502 CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- J. Liu, Y. Zheng, Y. Jiao, Z. Wang, Z. Lu, A. Vasileff and S. Z. Qiao, Small, 2018, 14, e1704073 CrossRef PubMed.
- J. Liu, D. Zhu, C. Guo, A. Vasileff and S.-Z. Qiao, Adv. Energy Mater., 2017, 7, 1700518 CrossRef.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed.
- D. Zhen, B. Zhao, H.-C. Shin, Y. Bu, Y. Ding, G. He and M. Liu, Adv. Mater. Interfaces, 2017, 4, 1700146 CrossRef.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. G. D. Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. D. Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- Y. Liu, C. Xiao, M. Lyu, Y. Lin, W. Cai, P. Huang, W. Tong, Y. Zou and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 11231–11235 CrossRef CAS PubMed.
- S. Zhao, R. Jin, H. Abroshan, C. Zeng, H. Zhang, S. D. House, E. Gottlieb, H. J. Kim, J. C. Yang and R. Jin, J. Am. Chem. Soc., 2017, 139, 1077–1080 CrossRef CAS PubMed.
- K. Xu, H. Ding, H. Lv, P. Chen, X. Lu, H. Cheng, T. Zhou, S. Liu, X. Wu, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 3326–3332 CrossRef CAS PubMed.
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef CAS PubMed.
- M. Ledendecker, S. K. Calderón, C. Papp, H.-P. Steinrück and M. Antonietti, and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- P. He, X.-Y. Yu and X. W. D. Lou, Angew. Chem., Int. Ed., 2017, 56, 3897–3900 CrossRef CAS PubMed.
- A. Sivanantham, P. Ganesan and S. Shanmugam, Adv. Funct. Mater., 2016, 26, 4661–4672 CrossRef CAS.
- Y.-J. Tang, C.-H. Liu, W. Huang, X.-L. Wang, L.-Z. Dong, S.-L. Li and Y.-Q. Lan, ACS Appl. Mater. Interfaces, 2017, 9, 16977–16985 CrossRef CAS PubMed.
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi, X. Kang, G. I. N. Waterhouse, L.-Z. Wu, C. H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1502585 CrossRef.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., Int. Ed., 2015, 54, 9351–9355 CrossRef CAS PubMed.
- Z. Zhang, X. Wang, G. Cui, A. Zhang, X. Zhou, H. Xua and L. Gu, Nanoscale, 2014, 6, 3540–3544 RSC.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef CAS PubMed.
- P. F. Liu, S. Yang, L. R. Zheng, B. Zhang and H. G. Yang, Chem. Sci., 2017, 8, 3484–3488 RSC.
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed.
- Y. Y. Chen, Y. Zhang, X. Zhang, T. Tang, H. Luo, S. Niu, Z. H. Dai, L. J. Wan and J. S. Hu, Adv. Mater., 2017, 29, 1703311 CrossRef PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- H. B. Wu, B. Y. Xia, L. Yu, X.-Y. Yu and X. W. D. Lou, Nat. Commun., 2015, 6, 6512 CrossRef CAS PubMed.
- W. Zhu, X. Yue, W. Zhang, S. Yu, Y. Zhang, J. Wang and J. Wang, Chem. Commun., 2016, 52, 1486–1489 RSC.
- L.-L. Feng, G. Yu, Y. Wu, G.-D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- B. You, N. Jiang, M. Sheng, M. W. Bhushan and Y. Sun, ACS Catal., 2016, 6, 714–721 CrossRef CAS.
- S. Wang, J. Wang, M. Zhu, X. Bao, B. Xiao, D. Su, H. Li and Y. Wang, J. Am. Chem. Soc., 2015, 137, 15753–15759 CrossRef CAS PubMed.
- R. Ma, Y. Zhou, Y. Chen, P. Li, Q. Liu and J. Wang, Angew. Chem., Int. Ed., 2015, 54, 14723–14727 CrossRef CAS PubMed.
- Y. Pi, Q. Shao, P. Wang, F. Lv, S. Guo, J. Guo and X. Huang, Angew. Chem., Int. Ed., 2017, 56, 4502–4506 CrossRef CAS PubMed.
- M. Y. Zu, P. F. Liu, C. Wang, Y. Wang, L. R. Zheng, B. Zhang, H. Zhao and H. G. Yang, ACS Energy Lett., 2018, 3, 78–84 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mh00664d |
| This journal is © The Royal Society of Chemistry 2019 |