
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimization of proximity-dependent initiation of hybridization chain reaction for improved performance
Mattias
Leino
 *a,
Johan
Heldin
a,
Marie Rubin
Sander
a,
Despoina
Kermpatsou
a,
Doroteya
Raykova
a,
Björn
Koos
b and
Ola
Söderberg
*a
*a,
Johan
Heldin
a,
Marie Rubin
Sander
a,
Despoina
Kermpatsou
a,
Doroteya
Raykova
a,
Björn
Koos
b and
Ola
Söderberg
*a
aDepartment of Pharmaceutical Biosciences, Pharmaceutical Cell Biology, Uppsala University, Uppsala, Sweden. E-mail: Mattias.Leino@farmbio.uu.se; ola.soderberg@farmbio.uu.se
bKlinik für Anästhesiologie, Intensivmedizin und Schmerztherapie, Universitätsklinikum Knappschaftskrankenhaus Bochum-Langendreer, Ruhr-Universität Bochum, Bochum, Germany
First published on 11th September 2019
Abstract
Proximity based detection methods are invaluable tools in the field of molecular biology, increasing selectivity and allowing for analysis of protein interactions. ProxHCR utilizes pairs of antibodies labelled with oligonucleotides to probe for proximal binding and to initiate a hybridization chain reaction (HCR) to generate an amplified detection signal. As HCR is based upon hybridization of DNA hairpins, the performance is dependent on salt concentrations and temperature. Herein we have redesigned the proxHCR system to increase the performance and to reduce dependency on temperature and salt concentrations. The new oligonucleotides provide an increased signal when performed at physiological salt concentrations and in room temperature.
Design, System, ApplicationProximity-dependent initiation of hybridization chain reaction (proxHCR) is a method that uses antibodies conjugated with oligonucleotides to detect targets in proximity of each other. The method relies on continuous hybridizations and strand-displacements of DNA. The previous design of this system is easily influenced by changes in temperature and salt concentrations making it harder to achieve consistent data. Here, we redesign the oligonucleotide sequence to alter the activation strategy while drastically reducing the size of all involved oligo sequences. This optimization lessens the constraints of salt concentrations and temperature while consistently producing strong signal. This method can be used to investigate protein–protein interactions and post-translational modifications. |
Introduction
Immunocytochemistry (ICC)1 is an invaluable method for routine diagnostics and research. Through the use of straightforward staining protocols specific endogenous proteins can be targeted using antibodies. These antibodies, often tagged with enzymes or fluorophores, can then be visualized with a microscope allowing the user to detect and localize the targeted protein with single cell resolution.However, detection of mere expression levels might not provide information on functional states, often the protein activity is dependent on both post-translational modifications (PTMs) and protein–protein interactions (PPIs). PTM-specific antibodies may be used to detect the modified protein, but potential cross-reactivity when using a single antibody needs to be considered.2,3 Similarly, PPIs can be inferred from the proximity of two fluorescent signals, however, due to the diffraction limit this is usually impossible for conventional microscopes.4–6
To enable visualization of endogenous PPIs we developed the in situ proximity ligation assay (in situ PLA) that requires dual recognition events of two antibodies to produce a signal.7,8 Here two antibodies are each labelled with a unique DNA strand. If bound in proximity to each other, these so called proximity probes act as hybridization templates for two subsequently added circularization oligonucleotides. These oligonucleotides are then ligated into a circular single-stranded DNA molecule. Only these circular ligation products can be amplified through rolling circle amplification (RCA), generating single-stranded RCA product that can be visualized by hybridization of sequence specific fluorophore-labelled detection oligonucleotides. The individual RCA products can be enumerated using conventional epifluorescence microscopes. While in situ PLA allows for the detection of both protein-complexes and PTMs,9 it is dependent on enzymes for ligation and amplification.
We recently developed the proximity-dependent initiation of hybridization chain reaction (proxHCR) method10 that, as in situ PLA, provides the ability to visualize PPIs and PTMs. Instead of enzymatic amplification, this method utilizes the hybridization chain reaction (HCR)11,12 to generate a repetitive nicked double-stranded DNA molecule consisting of two species of fluorophore-labelled DNA oligonucleotides. The fluorophore-labelled DNA oligonucleotides in the HCR method are designed as metastable hairpins, i.e. they will remain as monomers unless an initiator is added to the system. Upon the addition of an initiator, a sequence complementary to the foothold and stem of one of the hairpins, the hairpin will open up through a strand displacement of the stem. The now exposed loop and stem can in turn act as a new initiator and bind to the second species of hairpins that will be opened up to reveal the first initiator sequence. Hence, once an initiator is introduced a chain reaction of hybridization events will commence.
In the proxHCR method, the proximity probes consist of antibodies that are tagged with two species of DNA hairpins. One of the hairpins can be opened by adding a complementary activator oligonucleotide. By opening the first hairpin, a stretch of DNA, that previously was hidden in the stem, will be revealed so it can invade a proximally bound second hairpin. This invasion opens the second hairpin, revealing the initiator sequence that had been hidden within the stem region. This sequence will act as the trigger for the HCR, yielding an amplified fluorescent signal. Thus, proximity of any two antigens can be detected. While proxHCR could completely remove the reliance on enzymes for its amplification, it still requires stringent reaction conditions to yield strong signal.10
It should also be noted that much effort has been made in order to enhance immunocytochemistry through DNA displacement based strategies. For example; ImmunoHCR has been used as an approach to enhance signal strength in normal immuno assays.13 It has also been utilized as a multiplexing strategy with fluorescent signals removable through strand displacement.14 Dual recognition has also been published, unlike our proxHCR where the initiator is exposed through sequential opening of hairpins, the initiator has been split and conjugated to two antibodies. When these two antibodies are in proximity they function as a full initiator and bind a fluorescently tagged reporter sequence while displacing a quenching sequence.15
The aim of this work was to optimize reaction conditions as well as sequence design to yield a more efficient and versatile proxHCR protocol.
Materials and methods
Design of oligonucleotide system
The oligonucleotide systems (Tables 1 and 2) layout was constructed by hand with sequences constructed in silico using the NUPACK design tool (http://www.nupack.org).16 The systems were designed in a way that proximity arm1 and arm2 would not interact without the presence of the activator. The system was further investigated in silico to ensure that arm1, arm2, activator and detection hairpins would be sufficient to start an HCR when mixed together. Proximity arm1 and arm2 for both systems were ordered with 5′ formylbenzoate modifications for antibody conjugation. Detection hairpin1 and detection hairpin2 for both systems were bought conjugated with Texas Red – X (absorption max 583 nm, emission max 603 nm) at the 5′ or 3′ respectively. All oligonucleotides were purchased from Biomers.| Oligonucleotide name | Sequence (5′-3′) |
|---|---|
| Activator | GACTCGCATTCACTGAATACAGCGGGCCTTCATGCCACAGACGA |
| Proximity arm1 | Formylbenzoate – AAAAATCGTCTGTGGCATGAAGGCCCGCTGTATTCAGTGAATGCGAGTCAGACGAATACAGCGGGCCTTCATGCCACAGACGA |
| Proximity arm2 | Formylbenzoate – AAAAAGTGGGAGTCGTCTGTAACATGAAGGCCCGCTGTATTCGTCTTACTTCATGTTACAGACGACTCCCAC |
| Detection hairpin1 | Texas Red – X-ACAGACGACTCCCACATTCTCCAGGTGGGAGTCGTCTGTAACATGAAGTA |
| Detection hairpin2 | CTGGAGAATGTGGGAGTCGTCTGTTACTTCATGTTACAGACGACTCCCAC-Texas Red – X |
| Oligonucleotide name | Sequence (5′-3′) |
|---|---|
| Activator | AGTTCCCGTTTCAGTTTCATCCC |
| Proximity arm1 | Formylbenzoate – AAAAAGGGATGAAACTGAAACGGGAACTAAGATTCGGCTTAGTTCCCG |
| Proximity arm2 | Formylbenzoate – AAAAAGAACTAAGCCGAATCCCAAAGTGGATTCGGC |
| Detection hairpin1 | Texas Red – X-TTAACCGCCGAATCCCAAAGTGGATTCGGC |
| Detection hairpin2 | GGATTCGGCGGTTAAGCCGAATCCACTTTG-Texas Red – X |
| Initiator sequence | CAAAGTGGATTCGGC |
Conjugation of antibodies
Conjugation of antibodies was performed as described before.9 In essence, anti-mouse and anti-rabbit antibodies (Jackson ImmunoResearch Laboratories) were concentrated with Amicon Ultra 10K centrifugal filter units (Merck Millipore) to a concentration >3 mg ml−1. Antibodies were then incubated for 2 h with a 25-molar excess of succinimidyl 6-hydrazinonicotinate acetone hydrazine (SANH) (Solulink) at RT. After the activation the buffer was exchanged to 150 mM NaCl and 100 mM NaH2PO4 pH 6.0 using a Zeba Spin Desalting Columns 7K MWCO (ThermoFisher Scientific). To the anti-mouse antibody proximity arm1 was added and to the anti-rabbit antibody proximity arm2 was added, both at a oligonucleotide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :antibody molar ratio of 3:1. To the oligonucleotide–antibody mixture aniline (Sigma-Aldrich) was added to a final concentration of 10 mM and incubated for 2.5 h RT. Directly following the incubation the buffer was exchanged to pH 7.0 TBS and stored at 4 °C until further use.
:antibody molar ratio of 3:1. To the oligonucleotide–antibody mixture aniline (Sigma-Aldrich) was added to a final concentration of 10 mM and incubated for 2.5 h RT. Directly following the incubation the buffer was exchanged to pH 7.0 TBS and stored at 4 °C until further use.
Cell culture
All cells were maintained in a humidified incubator at 37 °C, 5% CO2 atmosphere. The cells were grown using high glucose DMEM supplemented with Glutamax, sodium pyruvate (Cat#31966047, ThermoFisher Scientific) and 10% fetal bovine serum (FBS) (ThermoFisher Scientific) with routine passaging when confluent using 0.25% Trypsine-EDTA (ThermoFisher Scientific). HaCaT cells used for E-cadherin–β-catenin as well as calnexin–ribosomal protein S3 proximity stains were trypsinized and seeded to 8-well Lab-Tek II Chamber Slides (Sigma-Aldrich) and grown until 90–100% confluence. BJ-hTERT cells, used for platelet-derived growth factor receptor β (PDGFR-β) – pan pY and Akt–pAkt proximity assays, were seeded with 25000 cells per cm2 to chamber slides, attached overnight and starved in medium supplemented with 0.2% FBS for 24 h. For the PDGFR-β – pan pY stain, cells were pre-incubated on ice for 10 min followed by stimulation with 50 ng ml−1 PDGF-BB for 1 h. For the Akt–pAkt stain, cells were instead stimulated for 30 min at 37 °C, following starvation. Following growth/treatment, all cells were fixed with 3.7% formaldehyde (Sigma-Aldrich) on ice for 15 min, washed in PBS, dried and stored in −20 °C until further use.
In situ proxHCR stain of fixed cells
This section contains the standard protocol for proxHCR; temperatures and NaCl concentrations vary as specified by the figures. All slides were initially permeabilized with 0.2% Triton-X100 (Sigma-Aldrich) for 10 min followed by a brief wash in TBS (50 mM tris(hydroxymethyl)aminomethane, 150 mM NaCl, pH 7.5). Slides were then blocked with Odyssey blocking buffer in TBS (LI-COR) for 1 h RT. After blocking, the slides were incubated with primary antibodies diluted in Odyssey blocking buffer overnight at 4 °C. Primary antibodies used in this paper were: mouse anti-E-cadherin (1:100, BD Transduction Laboratories, #610182), rabbit anti-β-catenin (1:100, Cell signaling, #8480), mouse anti-calnexin (1:100, abcam, ab31290), rabbit anti-ribosomal protein S3 (1:50, Cell signaling, #9538), mouse anti-pan pY (1:200, cell signaling, #9411), rabbit anti-PDGFRβ (1:100, Cell signaling, #3169), mouse anti-Akt (1:100, Cell signaling, #2920), rabbit anti-Akt(pS473) (1:50, Cell signaling, #4060). Following primary incubation, the slides were washed three times in TBS. Next, secondary antibodies, conjugated with either proximity arm1 or arm2, were added at a concentration of 5 μg ml−1 mixed in TBS with 0.25× Odyssey blocking buffer and 10% dextran sulfate (DS) (Merck Millipore, MW > 500000) and incubated at RT, followed by three washes with TBS with added 0.05% Tween-20 (Sigma-Aldrich). To activate proximity arm1 10 nM of activator oligonucleotide was added in DSTBS-T (TBS with 10% DS and 0.05% tween-20) and incubated for 30 min at RT. Following a brief wash in TBS, 100 nM of each detection hairpin mixed in DSTBS-T was added and incubated for 60 min at RT. Slides were then washed twice in TBS, followed by a 10 min incubation with Hoechst-33342 (ThermoFisher Scientific) before a final wash in TBS followed by sealing the slide with Slowfade Gold antifade reagent (ThermoFisher scientific). All tests were repeated at least three times.
Imaging and image analysis
For each experimental condition, at least six images were acquired. All images were acquired using a Zeiss imager M2 microscope controlled by Zen 2 software (blue version). The camera used was a Hamatsu C11440 and the objective was a 40×/1.4 oil apochromat (Zeiss). The light source used for excitation was a HXP 120 V (Zeiss). Exposure times were kept the same and set after the brightest stain within experiments. Images here have been enhanced for visual purposes, all images within experiments are handled the same way.Gel electrophoresis
For in solution analysis of the new system all oligonucleotides were first snap cooled by heating them to 95 °C for 3 min followed by cooling in room temperature for 30 min. Next all reactions were mixed in TBS to a final concentration of 0.5 μM and allowed to react for 1 h RT. Following the incubation the mixtures were immediately mixed with DNA gel loading dye (ThermoFisher Scientific) and separated using a 4–20% TBE polyacrylamide gel (ThermoFisher Scientific) with a TBE (Fisher Scientific) running buffer at 120 V for 70 min. Subsequently, the gel was stained using SYBR gold nucleic acid stain (ThermoFisher Scientific) for 20 min. The gel was scanned using an Odyssey Fc imaging system.Results and discussion
Testing versatility of the proxHCR system
The proxHCR method consists of three main steps, binding of the proximity probes, activation of proximity arm1 that leads to the opening of arm1 and the subsequent invasion of a proximal arm2, and signal amplification via HCR. Initially we investigated the performance of the proxHCR system under different salt concentrations and temperatures, using the interaction between E-cadherin and β-catenin as a model system for PPIs (Fig. 1A). Simultaneously, the addition of dextran sulfate (DS) was also evaluated (Fig. 1A). DS is commonly used in methods utilizing HCR as signal amplification and has been shown to increase signal while also decreasing non-specific binding of the amplification hairpins.17,18 We observed that lowering salt concentration, temperature or both, greatly reduces signal strength, and that the addition of DS gives a clear increase in signal regardless of temperature or salt concentration. | ||
| Fig. 1 Previous proxHCR version performance under different conditions. Proximity between E-cadherin and β-catenin detected with the previous version of the proxHCR method, proxHCR signal in red, Hoechst-33342 in blue. (A) The complete reaction was performed at low (150 mM) or high (1 M) NaCl concentrations, low (20 °C) or high (37 °C) temperature with or without 10% dextran sulfate (DS). (B) Temperature and salt concentrations for the activation step were altered. Amplification step was performed at 1 M NaCl, 37 °C and 10% DS. (C) Temperature and salt concentrations for the amplification step were altered. Activation was performed at 1 M NaCl, 37 °C and 10% DS. Scale bars are 50 μm. | ||
To elucidate which step of the method was impacted by the change in temperature and salt concentration, the same stain was repeated. This time the entire protocol was performed at 37 °C and with 1 M NaCl with the exception of just the activation step, or just the amplification step (Fig. 1B and C). It was apparent that there are impacts of both NaCl concentration and temperature on these steps. Most notably the reduction in NaCl concentration in the activation step seems to result in the greatest loss of signal. This would indicate that the strand displacement of arm1 and arm2 during the activation was the limiting step.
Redesigning the proxHCR system
The proxHCR system starts with the binding of the activator to the loop region of proximity arm1 (Fig. 2A). This binding is followed by a strand displacement into the stem of arm1, followed by opening of the hairpin structure of arm1. The now unhybridized 3′ stem region is free to interact with arm2. Here, similar to the activator-arm1 interaction, the released strand from arm1 can hybridize to the loop region of arm2 and subsequently invade into the stem – unfolding the hairpin structure. The now liberated strand contains the initiating sequence for the HCR. All of these hybridization steps are mediated through a foothold-dependent strand displacement. A DNA strand can be displaced through a random walk process, as long as the invading strand has long enough foothold to bind to and the sequence is reverse complementary.19,20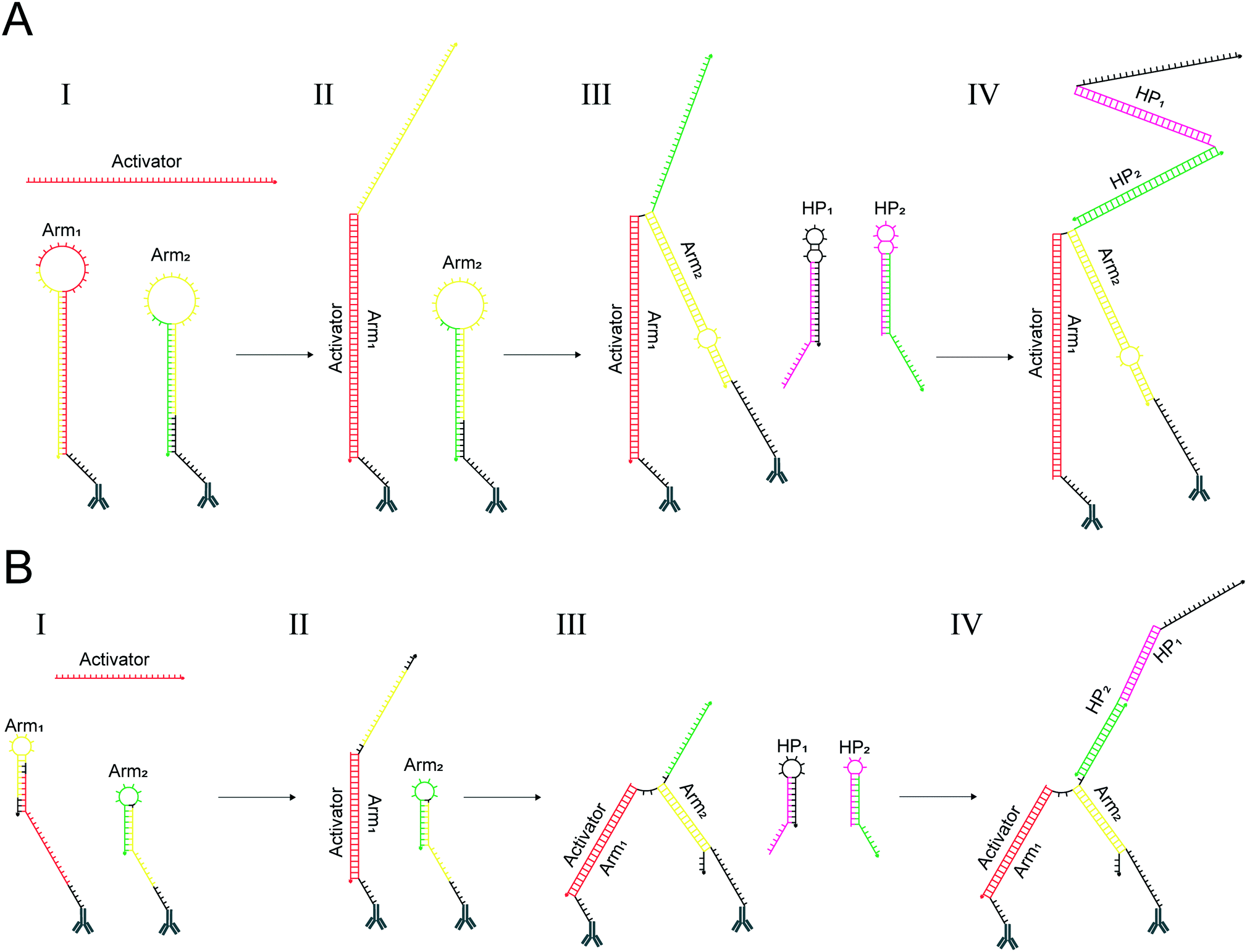 | ||
| Fig. 2 Mechanism of interaction for the previous design and the redesign of proxHCR. (A) Activation and opening of the previous proxHCR design. (I) The activator (red) binds to the loop region and invades the stem of arm1. (II) The released sequence of arm1 (yellow) can now bind the loop region of arm2, followed by a strand displacement of the stem of arm2. (III) The displacement of the arm2 stem releases an initiator sequence (green), which can bind and open hairpin2 (Hp2). (IV) Binding and opening of Hp2 triggers HCR, leading to the rapid polymerization of Hp1 and Hp2. (B) Activation and opening of the new proxHCR design. (I) The activator (red) binds to an extended external foothold of arm1, which results in a displacement of the arm1 stem. (II) The displaced stem (yellow) can now bind to the external foothold and invade the stem of arm2. (III) The now displaced strand contains the initiating (green) sequence for HCR which binds to Hp2. (IV) Binding and opening of Hp2 triggers HCR, leading to the rapid polymerization of Hp1 and Hp2. Reverse complementary sequences are displayed in the same color. | ||
Although, the kinetics of strand displacement depend on the foothold length of the accepting strand,19,21 data indicate that the kinetics plateau at a foothold length of 6–7 nucleotides.19,20 In this aspect our design already fulfils the conditions. However, strand displacement kinetics are affected by foothold placement as well, and the opening of hairpins is vastly slower when the strand displacement is initiated from the hairpin loop instead of a foothold situated outside the hairpin structure.22 Furthermore, it has been shown that the time required for strand displacement greatly increases with stem length.19 Similar results can be seen for reaction kinetics for hairpins of different stem length in HCR.12 Based on these data, we hypothesized that the two consecutive invasions of loop-based footholds, along with displacement of two longer stems could affect the kinetics of the system and thereby the time required for the assay.
Hence, we redesigned the proxHCR system to provide a more efficient and robust system (Fig. 2B). Although it is functionally similar to the previous system, i.e. two oligonucleotides (arm1 and arm2) conjugated to antibodies, an activation oligo with two amplification hairpins. The redesign comprises of three main changes; (1) moving of the footholds in arm1 and arm2 from the loop to an external position, (2) reducing stem length by moving parts of the initiator sequence to the loop of arm2 and (3) reducing the size of amplifying hairpins which also reduces the size of both arm1 and arm2.
To verify the functionality of the new system, an in solution test analysed with gel electrophoresis was performed. As seen in Fig. 3, without the addition an initiator oligonucleotide sequence the new hairpins remain metastable (lanes 7 & 8). Furthermore, arm1, arm2, the activator or both arms mixed together does not trigger HCR of hairpins (lanes 9–12). Finally, the mix of arm1, arm2 and the activator triggers the hairpin chain reaction (lane 13). The performance of the new design was compared with the one we previously published,10 using the E-cadherin and β-catenin interaction as a model system (Fig. 4). Interestingly, the new design performed better with reduced salt concentration and temperature displaying a clear stain of the E-cadherin and β-catenin interaction at the cell–cell junctions under all tested experimental conditions. Although the stain at 37 °C and 1 M NaCl was quite similar for both systems, it is a great advantage to have a method that is less dependent on changes in temperature and salt concentration. Especially when using the technique for diagnostic purposes as a point of care device it would be highly desirable to perform the analysis at physiological salt concentration and room temperature.
 | ||
| Fig. 3 In solution test of new design. Gel electrophoresis analysis of the new system separated either as single oligonucleotide (lanes 1–6) or mixtures of two or more oligonucleotides (lanes 7–13). All oligonucleotides were mixed to a final concentration of 0.5 μM and incubated for 1 h RT. | ||
 | ||
| Fig. 4 Comparison between the two designs. HaCat cells were assayed for proximity between E-cadherin and β-catenin using the previous and the redesigned proxHCR method, proxHCR signal in red, Hoechst-33342 in blue. Both activation and amplification were performed at various temperatures and NaCl concentrations as shown. Scale bars are 50 μm. | ||
In situ analysis of background signal and dual target recognition
HCR-based methods are dependent on the metastability of the participating oligonucleotide hairpins, so that the hybridization and invasion do not occur unless an initiator for the chain reaction is introduced. Given that for the proxHCR system the initiator is present, albeit hidden in the stem structure of the arm2, it is important to make sure that this sequence remains unavailable to start an amplification unless arm2 is opened by the specific hybridization of arm1. Of similar importance is the fact that arm1 must not contain a sequence that can initiate HCR on its own. These factors will determine the specificity of the assay.To determine the performance of the new proxHCR design we opted to validate the method on two well-known PPIs. Proximity between E-cadherin and β-catenin as well as calnexin and ribosomal protein S3 (RPS3) was assayed. In order to investigate if the probes caused any non-specific background, due to leakage, we also performed technical controls withholding one of the two primary antibodies (Fig. 5A and B) from the mixture. Neither of the antibodies caused any notable signal on its own, confirming that there is no detectable leakage from the proximity probes. However, we could observe strong signal for both PPIs if both primary antibodies were present (Fig. 5A and B).
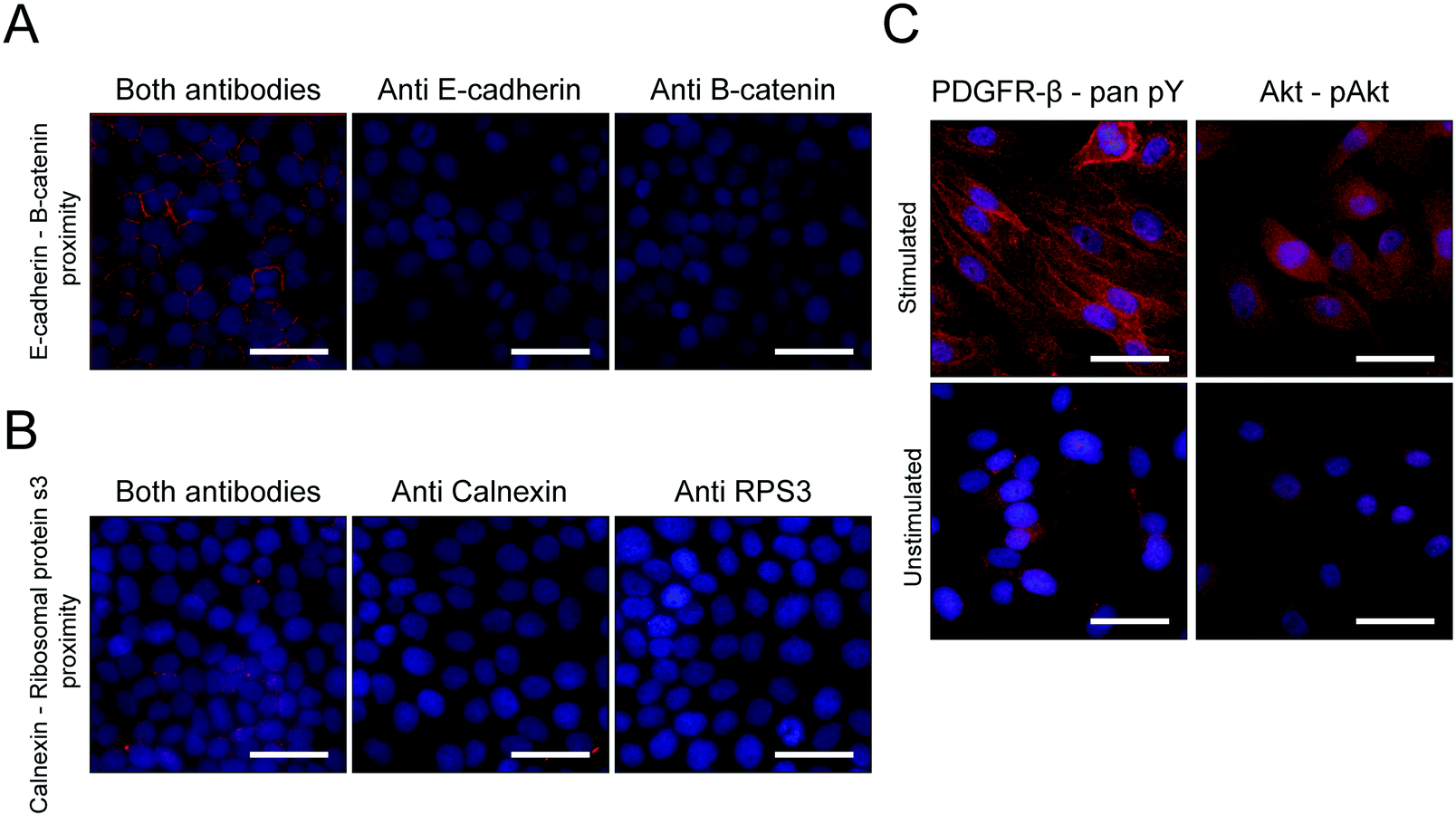 | ||
| Fig. 5 Testing the detection of various targets with the redesigned proxHCR. ProxHCR signal in red, Hoechst-33342 in blue. (A) Proximity between E-cadherin and β-catenin. Stain was performed either with both primaries or with one primary omitted. (B) Proximity between calnexin and RPS3. Stain was performed either with both primaries or with one primary omitted. (C) Proximity between Pan-phosphotyrosine and PDGFR-β, or between Akt and phosphorylated Akt in PDGF-stimulated and unstimulated BJ hTERT cells. Scale bars are 50 μm. | ||
To verify the usability of the new proxHCR design the stain was validated using well-known inducible PTMs (Fig. 5C). A clear induction of phosphorylation of PDGFR-β as well as phosphorylation of Akt upon stimulation with PDGF-BB could be visualized with the new design.
Conclusions
We have optimized our previously published proxHCR method by applying recent data regarding HCR and DNA strand displacement. The alterations in the sequences allow for staining over a larger span of temperatures and salt concentrations, including room temperature and physiological salt concentrations, without large changes in signal strength and stain quality. The sequences are also far shorter, making it more cost effective without displaying any background signal.Conflicts of interest
O. S. and B. K. are inventors on the patent covering proxHCR.Acknowledgements
This study has been funded by grants from the Swedish Foundation for Strategic Research and the Swedish Research Council.Notes and references
- A. H. Coons, H. J. Creech, J. R. Norman and E. Berliner, J. Immunol., 1942, 45, 159–170 CAS.
- K. J. Leuchowius, I. Weibrecht, U. Landegren, L. Gedda and O. Söderberg, Cytometry, Part A, 2009, 75, 833–839 CrossRef.
- S. Andersson, M. Sundberg, N. Pristovsek, A. Ibrahim, P. Jonsson, B. Katona, C.-M. Clausson, A. Zieba, M. Ramström, O. Söderberg, C. Williams and A. Asplund, Nat. Commun., 2017, 8, 15840 CrossRef CAS.
- A. Egner, S. Jakobs and S. W. Hell, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3370–3375 CrossRef CAS.
- F. Chen, P. W. Tillberg and E. S. Boyden, Science, 2015, 347, 543–548 CrossRef CAS PubMed.
- D. Raykova, B. Koos, A. Asplund, M. Gelléri, Y. Ivarsson, U. Danielson and O. Söderberg, Proteomes, 2016, 4, 36 CrossRef.
- O. Söderberg, M. Gullberg, M. Jarvius, K. Ridderstråle, K. J. Leuchowius, J. Jarvius, K. Wester, P. Hydbring, F. Bahram, L. G. Larsson and U. Landegren, Nat. Methods, 2006, 3, 995–1000 CrossRef.
- M. Jarvius, J. Paulsson, I. Weibrecht, K.-J. Leuchowius, A.-C. Andersson, C. Wählby, M. Gullberg, J. Botling, T. Sjöblom, B. Markova, A. Östman, U. Landegren and O. Söderberg, Mol. Cell. Proteomics, 2007, 6, 1500–1509 CrossRef CAS.
- A. Klaesson, K. Grannas, T. Ebai, J. Heldin, B. Koos, M. Leino, D. Raykova, J. Oelrich, L. Arngården, O. Söderberg and U. Landegren, Sci. Rep., 2018, 8, 5400 CrossRef.
- B. Koos, G. Cane, K. Grannas, L. Löf, L. Arngården, J. Heldin, C.-M. Clausson, A. Klaesson, M. K. Hirvonen, F. M. S. de Oliveira, V. O. Talibov, N. T. Pham, M. Auer, U. H. Danielson, J. Haybaeck, M. Kamali-Moghaddam and O. Söderberg, Nat. Commun., 2015, 6, 7294 CrossRef CAS.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS.
- Y. S. Ang and L. Y. L. Yung, Chem. Commun., 2016, 52, 4219–4222 RSC.
- P. Li, Z. Liu, Z. Wang, N. Tang, P. Zhou, R. Wang, F. Shao, R. Lin, X. Qi, M. Luo and Q. Feng, Nat. Methods, 2018, 15, 275–278 CrossRef.
- R. M. Schweller, J. Zimak, D. Y. Duose, A. A. Qutub, W. N. Hittelman and M. R. Diehl, Angew. Chem., Int. Ed., 2012, 51, 9292–9296 CrossRef CAS.
- Y. Tang, Z. Wang, X. Yang, J. Chen, L. Liu, W. Zhao, X. C. Le and F. Li, Chem. Sci., 2015, 6, 5729–5733 RSC.
- J. N. Zadeh, C. D. Steenberg, J. S. Bois, B. R. Wolfe, M. B. Pierce, A. R. Khan, R. M. Dirks and N. A. Pierce, J. Comput. Chem., 2011, 32, 170–173 CrossRef CAS.
- T. Yamaguchi, B. M. Fuchs, R. Amann, S. Kawakami, K. Kubota, M. Hatamoto and T. Yamaguchi, Syst. Appl. Microbiol., 2015, 38, 400–405 CrossRef CAS.
- H. M. T. Choi, V. A. Beck and N. A. Pierce, ACS Nano, 2014, 8, 4284–4294 CrossRef CAS.
- D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303–17314 CrossRef CAS.
- A. P. Mills, F. C. Simmel, A. J. Turberfield, J. L. Neumann and B. Yurke, Nature, 2002, 406, 605–608 Search PubMed.
- N. Srinivas, T. E. Ouldridge, P. Šulc, J. M. Schaeffer, B. Yurke, A. A. Louis, J. P. K. Doye and E. Winfree, Nucleic Acids Res., 2013, 41, 10641–10658 CrossRef CAS.
- S. J. Green, D. Lubrich and A. J. Turberfield, Biophys. J., 2006, 91, 2966–2975 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |