
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emergence of non-monotonic deep cavity cavitand assembly with increasing portal methylation†
Alexander
Saltzman
 a,
Du
Tang
a,
Bruce C.
Gibb
b and
Henry S.
Ashbaugh
*a
a,
Du
Tang
a,
Bruce C.
Gibb
b and
Henry S.
Ashbaugh
*a
aDepartment of Chemical and Biomolecular Engineering, Tulane University, New Orleans, LA 70118, USA. E-mail: hanka@tulane.edu
bDepartment of Chemistry, Tulane University, New Orleans, LA 70118, USA
First published on 27th September 2019
Abstract
Octa-acid (OA) and tetra-endo-methyl octa-acid (TEMOA) are deep cavity cavitands that readily form multimeric complexes with hydrophobic guests, like n-alkanes, in aqueous solution. Experimentally, OA displays a monotonic progression from monomeric to dimeric complexes with n-alkanes of increasing length, while TEMOA exhibits a non-monotonic progression from monomeric, to dimeric, to monomeric, to dimeric complexes over the same range of guest sizes. Previously we have conducted simulations demonstrating this curious behavior arises from the methyl units ringing TEMOA's portal to its hydrophobic pocket barring the possibility for two alkane chains to simultaneously bridge between two hosts in a dimer. Here we expand our prior simulation study to consider the partially methylated hosts mono-endo-methyl octa-acid, 1,3-di-endo-methyl octa-acid, and tri-endo-methyl octa-acid to examine the emergence of non-monotonic assembly behavior. Our simulations demonstrate a systematic progression of non-monotonic assembly with increasing portal methylation. This behavior is traced to the progressive destabilization of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complexes (two hosts assembled with two guests) rather than stabilizing other potential host/guest complexes that could be formed.
:2 complexes (two hosts assembled with two guests) rather than stabilizing other potential host/guest complexes that could be formed.
Design, System, ApplicationThe programming of aqueous phase assembly processes constitutes a grand challenge in soft matter physics. Deep-cavity cavitands, a class of water-soluble, bowl-like supramolecular hosts, readily bind non-polar guests via hydrophobic interactions to build well-defined complexes. Here we report a molecular simulation study of the impact of methylation about the rim of cavitand host pockets on the stoichiometry of their assemblies with n-alkanes. While non-methylated hosts exhibit a monotonic assembly pattern with increasing n-alkane size from monomeric to dimeric host complexes, increasing rim methylation progressively leads to the onset of non-monotonic assembly patterns, where the monomeric complex is reemergent for intermediate guest chain lengths. The effective use of a “throttle” between dimerized hosts hints at a novel route for manipulating host/guest assembly, forcing guests to thread constrictions that stabilize/destabilize specific complex structures. |
1 Introduction
The ability to self-assemble supramolecular complexes into discrete, well-defined structures is a grand research challenge. While metal/ligand1 and hydrogen bonding2–4 offer strong, specific interactions to direct self-assembly processes, the toxicity of heavy metals and promiscuity of hydrogen bonds in polar solvents limits the ability to utilize these interactions in aqueous systems with potential biological applications. Driven by the meager solubility of non-polar moieties in water, hydrophobic interactions offer a potentially useful route toward supramolecular assembly in water. In difference to metal/ligand and hydrogen bonding, hydrophobic interactions are non-specific; making it difficult to build directed structures in solution. In this case, the complex interplay between supramolecular shape, potential host/guest packing effects, and hydrophobic interactions offers a potentially large landscape to explore for building discrete assemblies.Motivated by these challenges, Gibb has explored the relationship between the functionalization of water-soluble deep cavity cavitand hosts and the complexes formed with n-alkane guests.5–8 Specifically, his group has examined octa-acid (host 0 in Fig. 1) and tetra-methyl-endo-octa-acid (host 4 in Fig. 1), which differ only by the presence of four methyl units that ring the portal to the hydrophobic, guest binding pocket of the cavitand. The n-alkane complexes of host 0 display a straightforward progression of assembly states with increasing guest length: methane (C1, where the subscript indicates the number of carbons in the n-alkane) does not bind the host; ethane (C2) forms a monomeric 1:1 complex (denoted i:j, where i and j indicate the number of host and guest molecules in a complex. Complexes with one host are referred to as simple, monomeric host–guest complexes, while those with two hosts are referred to as dimeric capsular complexes. These host/guest complexes are illustrated in Fig. 2); propane (C3) through octane (C8) form dimeric 2:2 complexes; while larger guests form dimeric 2:1 complexes. This progression from monomeric to dimeric assemblies is a monotonic assembly pattern. Host 4, on the other hand, exhibits a decidedly non-monotonic progression of assemblies with increasing guest length: C1 and C2 form monomeric 1:1 complexes; C3 through C6 form dimeric 2:2 complexes; C7 and C8 form monomeric 1:1 complexes; while C9 and longer guests form dimeric 2:1 complexes. In difference to host 0, that at most only forms dimeric complexes, host 4 can also form tetrameric and hexameric complexes with alkanes C17 and longer.8
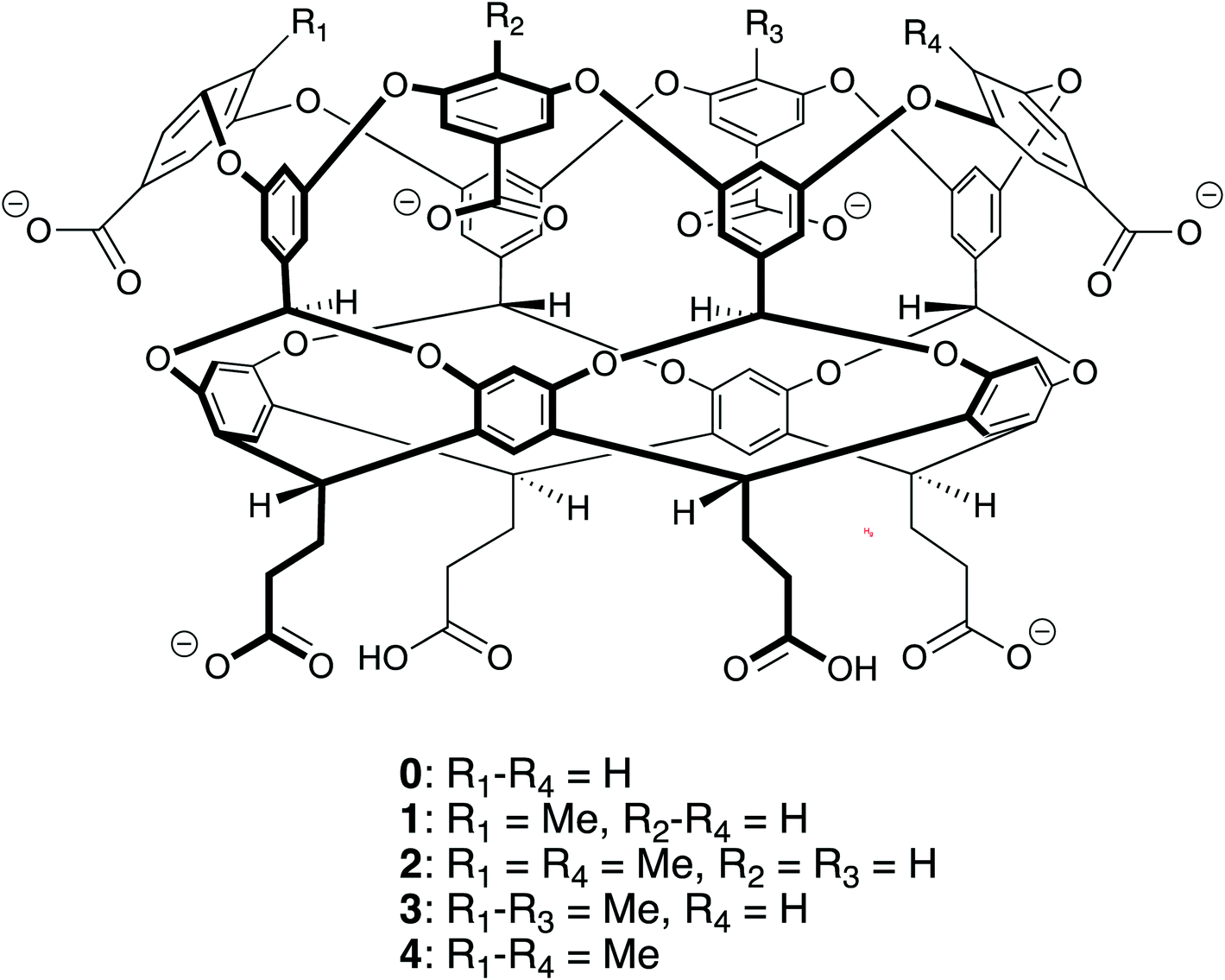 | ||
| Fig. 1 Chemical structures of the hosts octa-acid (0), mono-endo-methyl octa-acid (1), 1,3-di-endo-methyl octa-acid (2), tri-endo-methyl octa-acid (3), tetra-endo-methyl octa-acid (4) used in this study. | ||
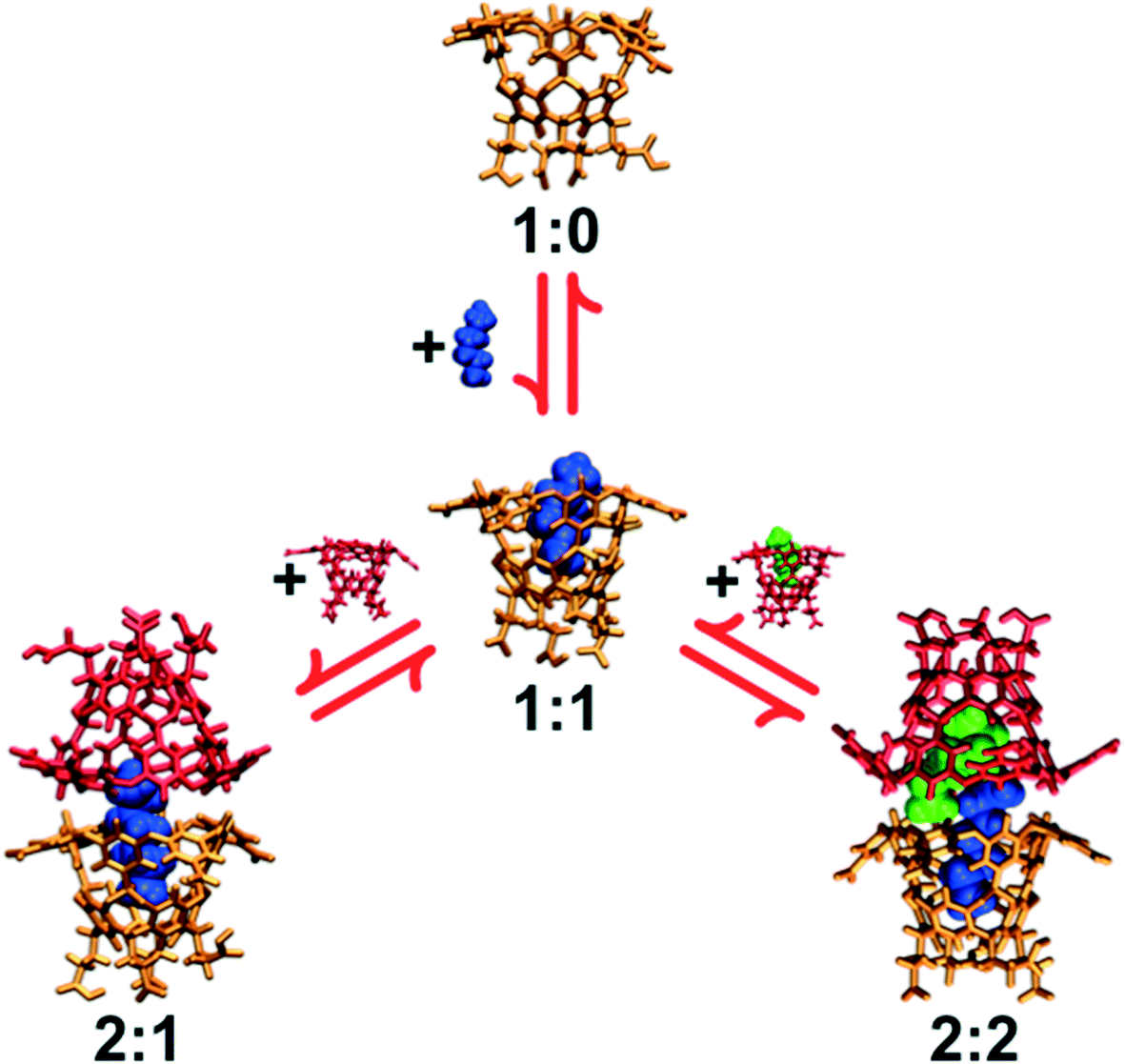 | ||
| Fig. 2 Simulation snapshots of the cavitand host (host 0) complexed with alkane guests (C7). The hosts are represented by the orange and red licorice images and the guests are represented by the green and blue van der Waals surface images. The assembly morphology (i:j) is identified below their pictures. The complexes are arranged following the reaction network embodied by eqn (1). | ||
Ashbaugh and Gibb reported molecular simulation studies of cavitand assembly with n-alkanes in water,9 breaking down the association process into elementary steps to understand the factors stabilizing distinct host/guest complexes. These simulations accurately captured the distinct monotonic versus non-monotonic assembly patterns of hosts 0 and 4. The non-monotonic assembly of 4 was found to arise from destabilization of the 2:2 complex. This destabilization was shown to result from the added methyl groups choking the portal region at the equator of the complex dimer, and limiting the ability of two alkane guests to thread between the hosts. 2:2 complex destabilization begins with guest C6, which is comparable in length to the depth of an individual host pocket. The 1:1 complex subsequently reemerges over the 2:1 complex since the 2:1 complex requires alkanes C9 and longer to bridge between two hosts. Within host 0, guests longer than ∼C15 must adopt a J-shaped conformational motif with a reverse turn in their main-chain. Interestingly however, simulations demonstrated that due to the portal narrowing within dimers of 4, alkanes cannot adopt such motifs.10,11 As a result, 2:1 complexes with guests C16 or larger are destabilized relative to the corresponding capsular complex with 0. A follow up simulation study of the transfer of alkanes into tetrameric and hexameric complexes of host 4 demonstrated that guest packing preferences can tilt the assembly equilibrium towards those larger assemblies,12 in agreement with experiment. These molecular simulations subsequently highlighted guest packing within confined host/guest complexes as a useful strategy for directing the stabilization of distinct assembly morphologies.
A question that follows from our previous studies is: what is the impact of partial methylation of the cavitand portal on their assemblies with n-alkanes? To address this question, we have performed a theoretical study of mono-endo-methyl-octa-acid (1), 1,3-di-endo-methyl-octa-acid (2), and tri-endo-methyl-octa-acid (3) (Fig. 1) complexed with n-alkanes from C1 to C14 to form 1:1, 2:1, and 2:2 complexes in aqueous solution. Molecular dynamics simulations were conducted to evaluate free energies for forming 1:1, 2:1, and 2:2 host/guest complexes along the association pathways illustrated in Fig. 2. These association free energies are subsequently utilized by a reaction network model we previously developed to predict the distribution for host/guest complexes formed as a function of the host methylation and guest length. While hosts 1 through 3 can in principle be synthesized, the cavitands would be formed in statistical yields and could not be readily purified. Here then, theory offers insight into the origin of the non-monotonic assembly in this interesting class of supramolecular complexes that cannot be met by synthetic means.
2 Methods
2.1 Molecular dynamics simulations
Molecular dynamics simulations of n-alkanes complexed with hosts 0, 1, 2, 3, and 4 (Fig. 1) were performed using GROMACS 5.13 The simulations for hosts 0 and 4 were previously reported in ref. 9. Here we extend that study to consider hosts with intermediate degrees of methylation following the same simulation procedures as the previous study. The alkanes were modeled using the L-OPLS all-atom force field, which accurately reproduces the thermodynamic and conformational properties of long alkanes.14 In difference to models that predict longer alkanes that are too rigid, we find the L-OPLS force field more freely explores the conformational landscapes of the confined guests. While we cannot guarantee that the guests have exhaustively explore their entire landscape over the course of our simulations, we obtain reproducible, experimentally consistent results using the simulation protocols described below. The series of n-alkane guests from methane (C1) to tetradecane (C14) were considered. The hosts were simulated using the generalized Amber force field (GAFF)15 with partial charges obtained from the AM1-BCC calculations.16 The net charge of each cavitand was set to −6e to match the expected protonation state at pH 7.17 This charge state was obtained by deprotonating the four benzoic acid groups around the rim of the cavitand and two of the four groups at the base of the hosts (Fig. 1). Six sodium cations per host, modeled using GAFF, were included to neutralize the host charge. Water was modeled using the TIP4P/EW potential.18 GROMACS topology files for hosts 0–4 are provided in the ESI.† Non-bonded Lennard-Jones interactions were truncated beyond a separation of 9 Å with a mean-field dispersion correction for longer-range contributions to the energy and pressure. Electrostatic interactions were evaluated using the particle mesh Ewald summation method with a real space cutoff of 9 Å.19 Simulations were conducted in the isothermal–isobaric ensemble at 25 °C and 1 bar, where the temperature and pressure were controlled using the Nosé–Hoover thermostat20,21 and Parrinello–Rahman barostat,22 respectively. Bonds involving hydrogens for the hosts and guests were constrained using the LINCS algorithm,23 while water was held rigid using the SETTLE algorithm.24 The equations of motion were integrated using a time step of 2 fs.Complex stability was characterized by evaluating potentials-of-mean force (PMF) between hosts and guests. A PMF quantifies the interaction free energy between components along a designated reaction trajectory, which here lies along the host's four-fold (C4) rotational axis of symmetry.9 We consider three distinct PMFs (Fig. 2): the interaction between a single alkane and cavitand to form a 1:1 complex; the interaction between an empty cavitand (1:0) and a 1:1 alkane/cavitand complex to form a 2:1 complex; and the interaction between two 1:1 alkane/cavitand complexes to form the corresponding 2:2 complex. In the first set of simulations, we determined the PMF between a host and guest (C1 to C14) from bulk water. In these simulations the cavitand and guest were solvated by 2600 water molecules in a cubic simulation box. Restraint potentials were applied to two dummy atoms along the C4-axis of each host to align the cavitand along the z-axis of the simulation box. The first “bottom” dummy atom was determined by the average position of the four atoms connecting the four feet of the cavitand to the bottom row of aromatic rings, while the second “top” dummy atom was determined by the average positions of the four carbon atoms on the second row of aromatic rings closest to the cavitand portal (see ESI† Fig. S1). The dummy atom at the bottom of the binding pocket was spatially restrained with a harmonic force constant of 100000 kJ mol−1 nm−2, while the vector connecting the bottom atom to the top was fixed along the z-axis using a harmonic constraint of 50000 kJ mol−1 nm−2. The PMF was determined over a series of overlapping windows spanning from bulk water into the host pocket using umbrella sampling.25 The guest center was restrained to the C4-axis of the host using a harmonic potential acting normal to the symmetry axis with a force constant of 100000 kJ mol−1 nm−2. In the case of guests with an odd number of carbon atoms, the center was taken as the middle carbon along the chain backbone (i.e., carbon number (n + 1)/2). For guests with an even number of carbons, a dummy atom was placed between the n/2 and n/2 + 1 carbons to serve as the restraint center. Sample windows were simulated from 5 Å deep-inside the cavitand pocket, measured from the center of the top plane defined by the four carbon atoms on the second row of aromatic rings closest to the cavitand mouth, to 15 Å out into bulk solvent. Forty overlapping windows were used along the z-axis with the harmonic umbrella potential minimum separated in 0.5 Å increments and a force constant of 15000 kJ (mol−1 nm−2).25 Each simulation window was equilibrated for 1 ns, followed by a 15 ns production run. We have found in our previous simulations of cavitand/alkane interactions that this simulation time is sufficient to obtain reproducible, converged results,9,10,12 suggesting the guest conformational landscape has been well explored. System configurations were saved every 0.2 ps for post-simulation analysis. The PMF for forming the 1:1 complex was reconstructed from the overlapping windows using the weighted histogram analysis method.26
In the second set of simulations, we evaluated the PMF between an empty cavitand (1:0) and a second cavitand in a 1:1 complex with a C1 to C14 guest in water. The two cavitands were oriented with their binding pockets facing one another aligned along their C4-axes to form a dimeric 2:1 host/guest assembly. Both hosts were aligned with the simulation box's z-axis, using the same restraints as in the 1:1 complexation simulations. No restraint was applied to the guest, however, which was held within its host pocket via hydrophobic interactions. Sample windows were simulated from distances ranging from the center of the two cavitand faces, which established a separation of zero, to 13 Å into the bulk water. Twenty-seven overlapping windows were simulated, with the harmonic umbrella potential minimum separated in 0.5 Å increments and a force constant of 15000 kJ (mol−1 nm−2). The same simulation procedures and PMF reconstruction methods were used here as for the 1:1 complexation study. In addition, we considered the PMF between two empty hosts devoid of guests to form a 2:0 dimer. Approximately 3000 TIP4P/EW water molecules were used to solvate these complexes.
In the third set of simulations, we evaluated the PMF between two 1:1 host/guest complexes to form a 2:2 complex (Fig. 2). As in the 2:1 complexation simulations, the cavitands were oriented with their binding pockets facing one another aligned along their C4-axes. For host 1, 2, and 3 we simulated alkane guests up to C11, C10, and C10 in length, respectively. Longer guests exhibited increasingly destabilizing repulsive interactions. As above, no restraints were placed on the guests. The same simulation procedures, PMF reconstruction methods, and numbers of hydration waters were used here as for the 2:1 complexation study.
2.2 Host/guest assembly model
We previously developed a reaction network model to predict the distribution of monomeric and dimeric host/guest assembly states as a function of the alkane guest length. Here we outline the essential elements of that model. A complete development of the model can be found in ref. 9. The host/guest assembly process is broken down into a series of four reactions (Fig. 2) that dictates the assembly equilibrium between guests (G) and the possible monomeric and dimeric host/guest complexes (1:0, 1:1, 2:0, 2:1, and 2:2 assemblies)| 1:0 + G ⇌ 1:1, | (1a) |
| 1:0 + 1:0 ⇌ 2:0, | (1b) |
| 1:1 + 1:0 ⇌ 2:1, | (1c) |
| 1:1 + 1:1 ⇌ 2:2. | (1d) |
The equilibrium constants for these reactions, K1:1, K2:0, K2:1, and K2:2, are evaluated as a Boltzmann weighting of the minima in the PMFs (ωi:j) evaluated from the corresponding simulations described above. The equilibrium constants for the monomeric and dimeric host/guest assembly reactions subsequently are
| K1:1 = αexp(−ω1:1/RT) and K2:j = βexp(−ω2:j/RT), | (2) |
The free host concentration, [1:0], is determined from the solution of a quadratic equation (see ESI† for full derivation)
| 2(K2:0 + K2:1K1:1[G] + K2:1K21:1[G]2)[1:0]2 + (1 + K1:1[G])[1:0] − [1]total = 0. | (3) |
The total host concentration, [1]total, corresponds to the amount of host added to solution distributed amongst all potential assembly states, i.e., [1]total = [1:0] + [1:1] + 2([2:0] + [2:1] + [2:2]). We assume [1]total = 3 mM, which corresponds to a typical experimental concentration. The alkane guest concentration was assumed to be saturated as described by the relationship
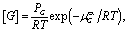 | (4) |
:1] = K1:1 [1:0][G].
3 Results & discussion
Pairwise PMFs for formation of 1:1, 2:1, and 2:2 complexes in water as a function of distance along the reaction coordinate between host 2 and C8 at 25 °C and 1 atm are reported in Fig. 3. Each of these free energy profiles exhibit minima at least 70 kJ mol−1 (28 RT) deep as a result of significant hydrophobic interactions between the hosts and guests. The strength of these interactions suggest that these complexes are long lived once assembled. The minimum of the 1:1 PMF falls at distance slightly less than zero since the center-of-mass of C8 can enter the binding pocket below the rim of the pocket's portal. The minima of the dimeric complexes sits 3 to 4 Å above the rim of the portal, roughly corresponding to the distance of closest approach between the rim carbons of opposing host “hemispheres”. The minimum of the 2:2 complex lies slightly to the right of that of the 2:1, however, due to tight packing of two guests within the capsule interior. The 1:1, 2:1, and 2:2 PMFs for all host/guest pairs examined here are qualitatively similar to those for host 2 and C8, though differ in quantitative detail.
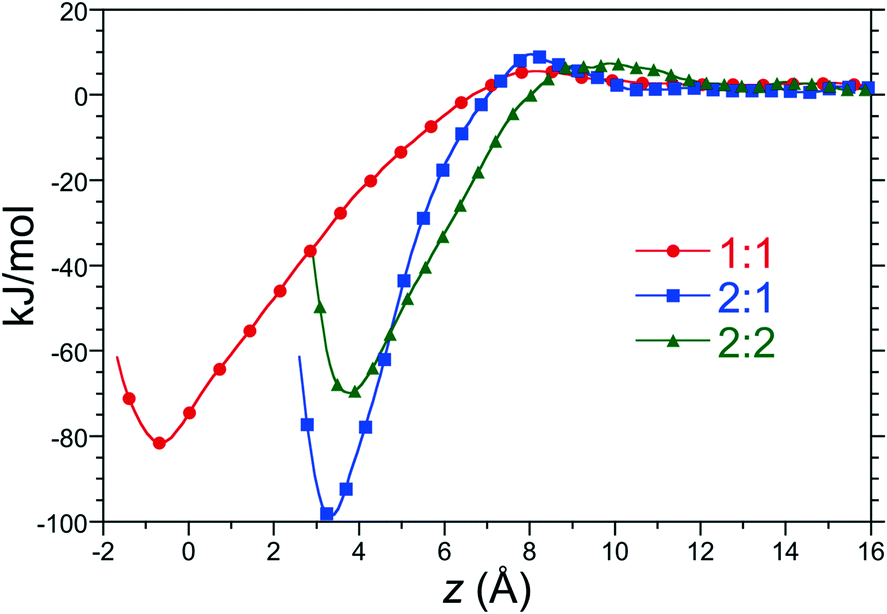 | ||
| Fig. 3 Potentials-of-mean force between host 2 and C8 to form 1:1, 2:1, and 2:2 complexes in aqueous solution. The figure symbols are defined in the legend. Error bars are comparable in size to the symbols. | ||
As previously demonstrated, the relative stability of the distinct host/guest assembly states is dominated by the free energy minima of the PMFs, denoted ω1:1, ω2:1, and ω2:2, respectively. The 1:1, 2:1, and 2:2 PMF minima for hosts 0 through 4 as a function of the alkane guest chain length are compared in Fig. 4. The PMF minima for 1:1 complex formation for all the hosts exhibit an increasing attraction with guest chain length beginning with methane that plateaus for guests approximately longer than pentane (Fig. 4a). The PMF minima for the shorter guests (∼C4 and shorter) examined are comparable for all the hosts simulated. The plateau begins roughly for guests longer than the depth of the binding pocket. Longer guests subsequently are unable to stuff more methylene units within the pocket away from water and thereby do not gain any additional benefit for forming a 1:1 complex. Interestingly, the plateau for the methyl functionalized hosts (1 through 4) is more attractive than that for host 0, but are approximately the same as one another. Previously, we attributed the greater attraction between host 4 for longer guests compared to host 0 to increased van der Waals interactions between the methyl units of host 4 and the guests.9 We might then expect the plateau for hosts 1 through 3 to systematically deepen with increasing methylation between the host 0 and 4 limits, which we do not observe. This interpretation, however, does not consider the solvent's role on directing hydrophobic host/guest interactions. Moreover, we do not observe systematic variations with host methylation for the PMF minima of the short guests despite the systematic chains in host/guest van der Waals interactions. A definitive conclusion regarding the dependence of the 1:1 complexation plateau on host methylation is thus not immediately apparent.
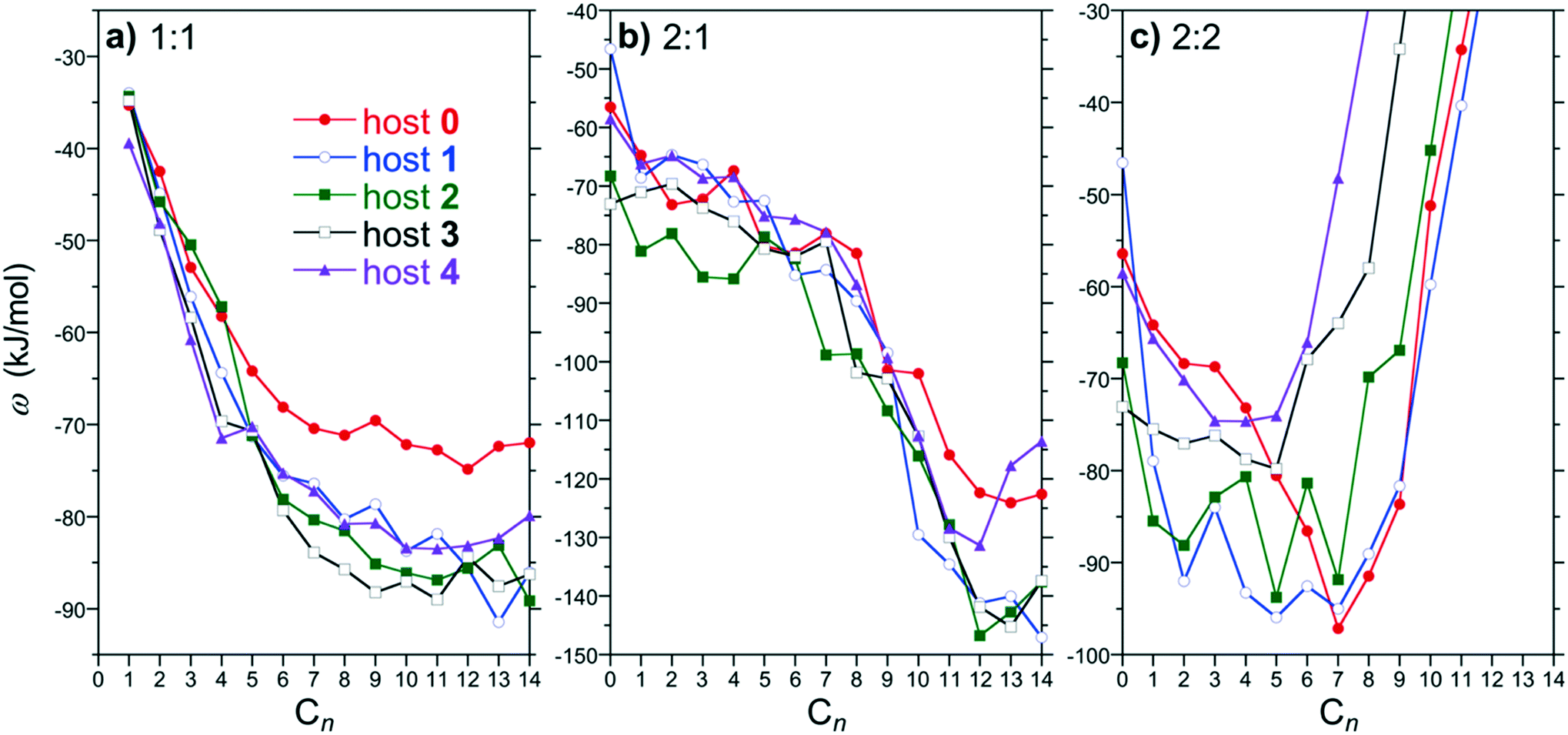 | ||
| Fig. 4 Potential-mean force minima for forming 1:1, 2:1, and 2:2 complexes as a function of the guest chain length for hosts 0 through 4. Figures a, b, and c report results for ω1:1, ω2:1, and ω2:2, respectively. The symbols identifying hosts 0, 1, 2, 3, and 4 are defined by the legend in a. Error bars, which range from 2.5 to 5 kJ mol−1 (1 to 2 RT), are neglected for clarity. Note that the free energy for forming a guest free 2:0 dimer corresponds to the potential-mean force minima for guest C0 (no guest) reported in b and c. | ||
The 2:1 PMF minima are attractive for all guest/host combinations considered (Fig. 4b). For guests up to C8, ω2:1 weakly decreases with increasing alkane chain length with little distinction between the free energies for any of the hosts within the simulation noise. Beginning with C9, ω2:1 drops precipitously with increasing chain length further stabilizing the 2:1 complexes. This guest size corresponds to the point at which a guest readily spans between the two hosts to gain additional favorable van der Waals interactions to stabilize the complex. Similar to the 1:1 free energies, ω2:1 drops to lower levels with increasing chain lengths for the methylated hosts (1 through 4) compared to that for host 0. While ω2:1 for hosts 1 through 3 are practically indistinguishable, ω2:1 for host 4 exhibits a minimum at C12 after which the complexation free energy increases with increasing guest length. In our previous work considering guests up to C16 the 2:1 complex of host 4 ultimately becomes unstable.9 Experimentally8 and from simulation12 this host transitions from a dimeric 2:1 complex to a tetrameric 4:2 complex for sufficiently long guests. Host 0, on the other hand, only forms dimeric complexes with increasing guest length. Based on these observations, we may anticipate that ω2:1 for hosts 1, 2, or 3 may exhibit a free energy minimum with increasing alkane chain length that destabilizes the dimer in favor of a tetramer. That lies beyond the scope of the present study, however.
The most significant assembly PMF changes are observed for 2:2 complexation (Fig. 4c). Generally, ω2:2 for all hosts is attractive for shorter chains and then dramatically diverges towards more positive free energies beyond a characteristic alkane chain length. This divergence was previously demonstrated to be correlated with constriction of the portal region at the dimeric complex equator by the endo-methyl rim groups. That is the endo-methyls choke the portal region between hosts blocking guests in opposing hosts threading through the portal. The divergence begins for guests C6 and longer for the host 4 dimer, which has the narrowest portal. This guest length corresponds to the depth of an individual host pocket as inferred from the ω1:1 plateau (Fig. 4a). While the divergence length for hosts 0 and 1 are similar, the divergence systematically shifts to increasingly shorter guest lengths for 2, 3, and 4. This observation agrees with the interpretation of the portal region becoming progressively constricted as more endo-methyls are added. We may anticipate then that the 2:2 complexes will become increasingly unstable with increasing methylation, tipping the balance towards other assemblies.
The PMF minima reported in Fig. 4 can be utilized within the host/guest assembly model described above to predict the distribution of complexes between 1:0, 1:1, 2:0, 2:1, and 2:2 for a given guest. The predicted population of assemblies for host 3 as a function of the alkane chain length is reported in Fig. 5. This host/guest system exhibits non-monotonic assembly characteristics with increasing guest chain length. Specifically, the dimeric 2:2 complex dominates for guests from C2 to C5, the monomeric 1:1 complex dominates for guests C6 to C8, and the dimeric 2:1 complex dominates for guests C9 and longer. Interestingly, even in the absence of any guest (C0), the dimeric 2:0 complex is predicted to exhibit a population comparable to the free 1:0 host within the simulation error. Guest free dimers are not observed experimentally. Moreover, our previous study of hosts 0 and 4 predicted a negligible population of 2:0 complexes. Based on the expectation that the guest is a necessary element of the assembly to draw the two hosts together, it is likely that that the predicted population of 2:0 complexes is erroneous. Indeed, no 2:0 complexes are predicted for guests longer than C1. We attribute this prediction to the fact that small errors on the order of RT are sufficient to shift the populations of complexes observed, especially for the shorter guests. Examining the 2:0 PMF minimum for hosts 2 and 3 we observe predicted free energies ∼10 kJ mol−1 (4 RT) more stable than those for hosts 0, 1, and 4 (Fig. 4c). Given the lack of systematic variation in the PMF minimum with increasing hosts methylation, it appears the predicted overstabilization may be diminished with increasing simulation run times. Another potential source of error may result from the simplified reaction coordinate along the principle axis of host symmetry assumed to evaluate the equilibrium constants within the host/guest assembly model (eqn (2)). To alleviate this difficulty here after we assume K2:0 = 0 within the host guest assembly model, eliminating the potential for forming 2:0 complexes. The impact of this assumption is to redistribute the population of assembly states amongst the 1:0, 1:1, 2:1, and 2:2 complexes for C0 (no guest) and C1. No complex population differences were observed for longer alkanes. Thus, this assumption has no impact on the onset of non-monotonic dimer-to-monomeric-to dimeric complex assembly patterns for longer guests.
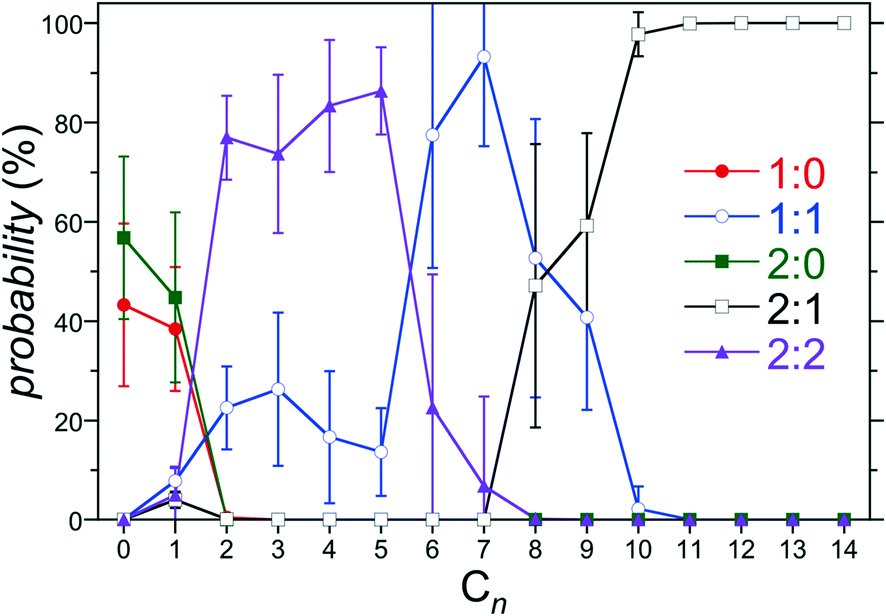 | ||
| Fig. 5 Population of 1:0, 1:1, 2:0, 2:1, and 2:2 complexes for host 3 as a function of the alkane guest chain length predicted from the host/guest assembly model using the free energies reported in Fig. 4. The symbols for each complex are defined in the legend. The error bars indicate one standard error. | ||
The predicted distributions of 1:0, 1:1, 2:1, and 2:2 complexes as a function of the alkane guest chain length are reported in Fig. 6 for hosts 0 through 4. As previously documented, the dominant (most populous) complexes observed for host 0 progresses from 1:0 for C1; to 1:1 for C2 and C3; to 2:2 for C4 through C8; to 2:1 for C9 and longer guests (Fig. 6a). Thus host 0 is predicted to exhibit monotonic assembly from monomeric to dimeric complexes with increasing alkane chain length, as observed experimentally. Similar monotonic assembly is observed for host 1, although the population of 1:1 assemblies is suppressed for the shortest guests (Fig. 6b). Interestingly, a small (∼10%) population of 1:1 complex is observed for C9 near the transition between 2:2 and 2:1 assemblies, hinting at the potential of a reemergent population of monomeric complex. This population of 1:1 complex near the 2:2 to 2:1 transition grows to ∼40% of the total for 2 (Fig. 6c), albeit with its peak maximum shifted to C8. This population of 1:1 complex finally becomes dominant for guests from C6 through C8 complexed with 3 (Fig. 6d). The host subsequently exhibits full non-monotonic assembly from monomeric, to dimeric, to monomeric, then back to dimeric complexation with increasing alkane chain length. Interestingly, the reemergence of the 1:1 population for longer guests is accompanied with growth in the 1:1 population for shorter guests as well, with a minimum between the short and long chain peaks. This trend continues for host 4, which exhibits a progression from 1:1 complexes for C1 and C2; to 2:2 complexes for C3 through C5, to 1:1 complexes for C6–C8; to 2:1 complexes for C9 and longer guests (Fig. 6e). The 1:1 population minimum between the short and long chain guests is more clearly defined for host 4 than for host 3, continuing the systematic trends observed with increasing host methylation.
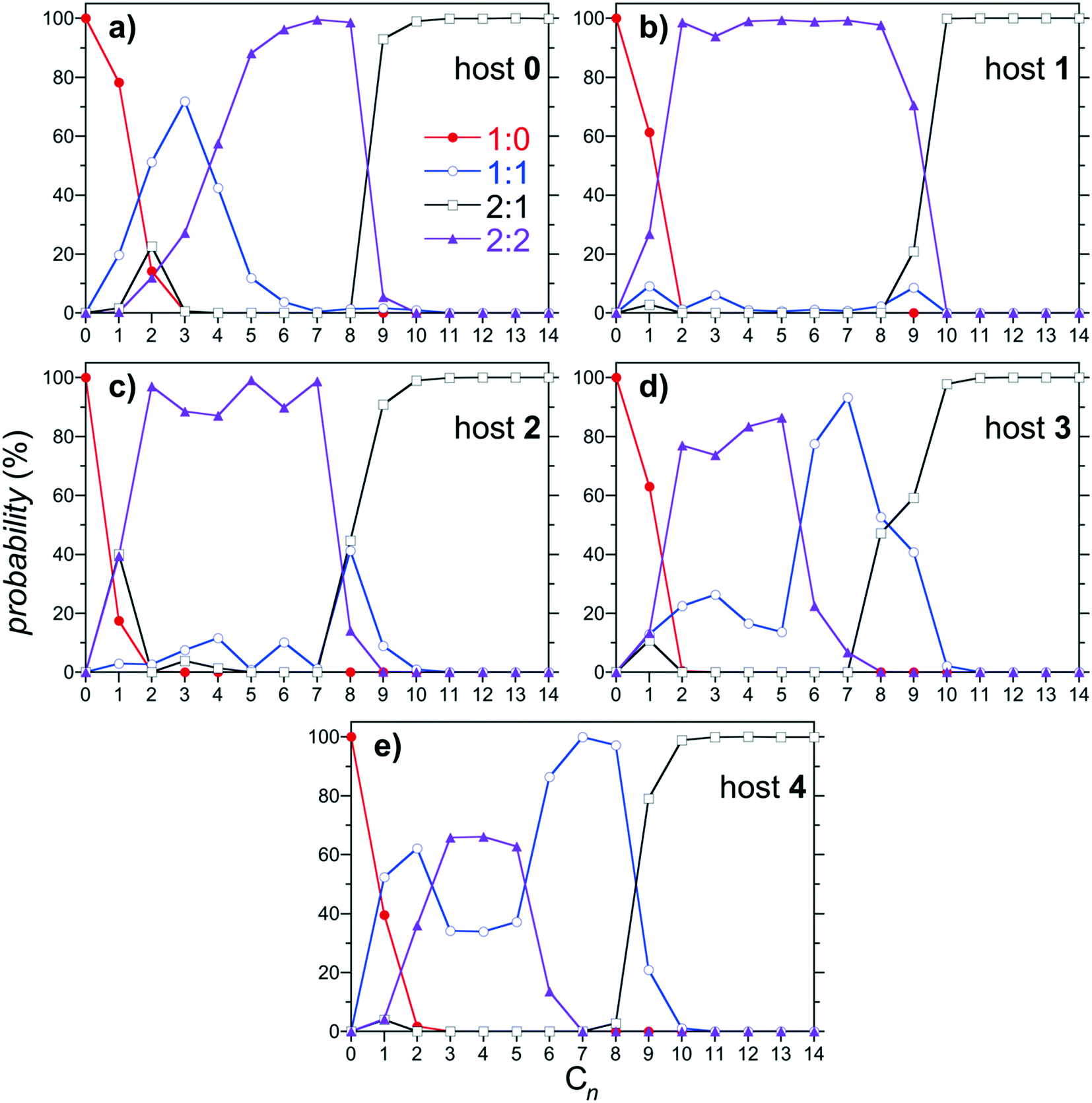 | ||
| Fig. 6 Population of 1:0, 1:1, 2:1, and 2:2 complexes as a function of the alkane guest chain length predicted from the host/guest assembly model using the free energies reported in Fig. 4. Figures a, b, c, d, and e report results for hosts 0, 1, 2, 3, and 4, respectively. The symbols for each complex are defined in the legend in a. Error bars, which are comparable to those reported in Fig. 4, are neglected for clarity. | ||
The emergence of the non-monotonic assembly pattern can be more directly visualized by evaluating the mean host aggregation number as
| 〈N〉 = p1:0 + p1:1 + 2(p2:1 + p2:2), | (5) |
:j is the probability a host is in an i:j complex (note that 2:0 complexes were neglected, but could be included within the parentheses if they contributed substantively to the dimer population). The host aggregation numbers are plotted in Fig. 7. Host 0 displays a near monotonic progression from monomers to dimers with increasing guest chain length. Beginning with host 1 a dimple in the dimer population appears at intermediate length guests, which gets deeper with increasing host methylation. By host 4 the dimer population for guest C7 is zero, for all intents and purposes, and the monomer population is fully reemergent. Interestingly, for hosts 1 through 4 the population of dimers from ∼C2 to ∼C5, associated with 2:2 complexes, becomes progressively suppressed with increasing methylation. We might surmise in this case that the 2:2 complex population could be further suppressed with increased methylation, barring the fact the host 4 has the greatest possible endo-methylation for this class of hosts.
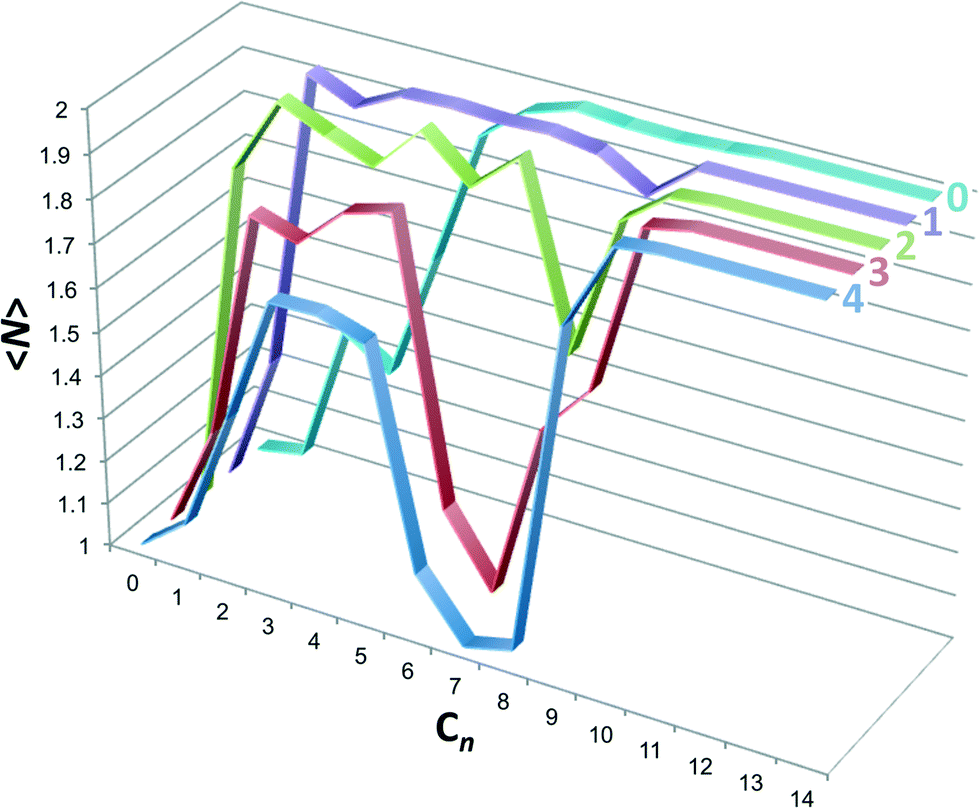 | ||
| Fig. 7 Host aggregation number, 〈N〉, as a function of the alkane guest chain length predicted from the probabilities reported in Fig. 6. Graphs for each host are identified by the text at the top right-hand side. | ||
4 Conclusions
We have presented a molecular simulation and reaction network modeling study of the complexation of deep cavity cavitand hosts with n-alkane guests to examine the onset of non-monotonic assembly patterns in biomimetic materials. Beginning with the unmethylated cavitand (host 0), which displays a monotonic transformation from monomeric to dimeric complexes with increasing alkane length, successive hosts from 1 through 4 exhibit progressively non-monotonic assembly patterns with increasing host methylation, transforming from monomeric, to dimeric, to monomeric, to dimeric complexes with increasing alkane length. The reentrance of the monomeric assembly morphology at intermediate guest lengths (C6 to C8) is associated with the systematic destabilization of the 2:2 complex association free energy rather than stabilization of the 1:1 or 2:1 complexes. We attribute 2:2 complex destabilization to increasing methylation constricting the portal region between dimerized hosts diminishing the ability for two guests to thread between opposing sides of the complex. For the maximally narrowed portal of 4, a fully reemergent monomeric 1:1 complex is observed, while only partially reemergent monomers are observed for the intermediately methylated hosts (1 through 3). These results hint at potential routes for manipulating host/guest assembly patterns that force guests to navigate narrow host portal constrictions to either stabilize or destabilize distinct complexes.
Conflicts of interest
No potential conflicts of interest were reported by the authors.Acknowledgements
We gratefully acknowledge financial support from the NSF (CBET-1403167). We also thank the Louisiana Optical Network Initiative who provided computational support.References
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Self-assembly of tetravalent Goldberg polyhedra from 144 small components, Nature, 2016, 540, 563–567 CrossRef CAS PubMed.
- L. R. MacGillivray and J. L. Atwood, A chiral spherical molecular assembly held together by 60 hydrogen bonds, Nature, 1997, 389, 469–472 CrossRef CAS.
- D. Ajami and J. Rebek, Longer guests drive the reversible assembly of hyperextended capsules, Angew. Chem., Int. Ed., 2007, 46, 9283–9286 CrossRef CAS PubMed.
- K. Tiefenbacher, D. Ajami and J. Rebek, Self-Assembled Capsules of Unprecedented Shapes, Angew. Chem., Int. Ed., 2011, 50, 12003–12007 CrossRef CAS PubMed.
- C. L. D. Gibb and B. C. Gibb, Well-defined, organic nanoenvironments in water: The hydrophobic effect drives a capsular assembly, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS PubMed.
- C. L. D. Gibb and B. C. Gibb, Straight-chain alkanes template the assembly of water-soluble nano-capsules, Chem. Commun., 2007, 1635–1637 RSC.
- H. Y. Gan, C. J. Benjamin and B. C. Gibb, Nonmonotonic assembly of a deep-cavity cavitand, J. Am. Chem. Soc., 2011, 133, 4770–4773 CrossRef CAS PubMed.
- H. Y. Gan and B. C. Gibb, Guest-mediated switching of the assembly state of a water-soluble deep-cavity cavitand, Chem. Commun., 2013, 49, 1395–1397 RSC.
- D. Tang, J. W. Barnett, B. C. Gibb and H. S. Ashbaugh, Guest controlled nonmonotonic deep cavity cavitand assembly state switching, J. Phys. Chem. B., 2017, 121, 10717–10725 CrossRef CAS.
- J. W. Barnett, B. C. Gibb and H. S. Ashbaugh, Succession of alkane conformational motifs bound within hydrophobic supramolecular capsular assemblies, J. Phys. Chem. B., 2016, 120, 10394–10402 CrossRef CAS.
- S. M. Liu, D. H. Russell, N. F. Zinnel and B. C. Gibb, Guest packing motifs within a supramolecular nanocapsule and a covalent analogue, J. Am. Chem. Soc., 2013, 135, 4314–4324 CrossRef CAS PubMed.
- J. W. Barnett, D. Tang, B. C. Gibb and H. S. Ashbaugh, Alkane guest packing drives switching between multimeric deep-cavity cavitand assembly states, Chem. Commun., 2018, 54, 2639–2642 RSC.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1-2, 19–25 CrossRef.
- S. W. I. Siu, K. Pluhackova and R. A. Bockmann, Optimization of the OPLS-AA force field for long hydrocarbons, J. Chem. Theory Comput., 2012, 8, 1459–1470 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and testing of a general amber force field, J. Comput. Phys., 2004, 25, 1157–1174 CAS.
- A. Jakalian, D. B. Jack and C. I. Bayly, Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- J. Ewell, B. C. Gibb and S. W. Rick, Water inside a hydrophobic cavitand molecule, J. Phys. Chem. B., 2008, 112, 10272–10279 CrossRef CAS PubMed.
- H. W. Horn, W. C. Swope, J. W. Pitera, J. D. Madura, T. J. Dick, G. L. Hura and T. Head-Gordon, Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew, J. Chem. Phys., 2004, 120, 9665–9678 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald - An N.log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- S. Nosé, A unified formulation of the constant temperature molecular-dynamics methods, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Canonical dynamics - Equilibrium phase-space distributions, Phys. Rev. A, 1985, 31, 1695–1697 CrossRef.
- M. Parrinello and A. Rahman, Polymorphic transitions in single-crystals - A new molecular-dynamics method, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. Fraaije, LINCS: A linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- S. Miyamoto and P. A. Kollman, SETTLE - An analytical version of the shake and rattle algorithm for rigid water models, J. Comput. Chem., 1992, 13, 952–962 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, D. Bouzida, R. H. Swendsen, P. A. Kollman and J. M. Rosenberg, The weighted histogram analysis method for free-energy calculation on biomolecules. 1. The method, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- H. B. Luo and K. Sharp, On the calculation of absolute macromolecular binding free energies, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 10399–10404 CrossRef CAS.
- H. J. Woo and B. Roux, Calculation of absolute protein-ligand binding free energy from computer simulations, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6825–6830 CrossRef CAS PubMed.
- B. Widom, Potential-distribution theory and the statistical mechanics of fluids, J Phys Chem, 1982, 86, 869–872 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full derivation of eqn (3); dummy atom placement to align cavitands and guests along their reaction coordinate; GROMACS topology files for simulating hosts 0–4. See DOI: 10.1039/c9me00076c |
| This journal is © The Royal Society of Chemistry 2020 |