
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoupling segmental relaxation and ionic conductivity for lithium-ion polymer electrolytes
Dominic
Bresser
 *ab,
Sandrine
Lyonnard
c,
Cristina
Iojoiu
d,
Lionel
Picard
e and
Stefano
Passerini
*ab
*ab,
Sandrine
Lyonnard
c,
Cristina
Iojoiu
d,
Lionel
Picard
e and
Stefano
Passerini
*ab
aKarlsruhe Institute of Technology (KIT), 76021 Karlsruhe, Germany. E-mail: dominic.bresser@kit.edu; stefano.passerini@kit.edu
bHelmholtz Institute Ulm (HIU), 89081 Ulm, Germany
cCEA, CNRS, SyMMES, University Grenoble Alpes, 38000 Grenoble, France. E-mail: sandrine.lyonnard@cea.fr
dCNRS, Grenoble INP, LEPMI, University Grenoble Alpes, 38000 Grenoble, France. E-mail: cristina.iojoiu@lepmi.grenoble-inp.fr
eCEA, LITEN, DEHT, STB, University Grenoble Alpes, 38054 Grenoble, France. E-mail: lionel.picard@cea.fr
First published on 29th April 2019
Abstract
The use of polymer electrolytes instead of liquid organic systems is considered key for enhancing the safety of lithium batteries and may, in addition, enable the transition to high-energy lithium metal anodes. An intrinsic limitation, however, is their rather low ionic conductivity at ambient temperature. Nonetheless, it has been suggested that this might be overcome by decoupling the ion transport and the segmental relaxation of the coordinating polymer. Here, we provide an overview of the different approaches to achieve such decoupling, including a brief recapitulation of the segmental-relaxation dependent ion conduction mechanism, exemplarily focusing on the archetype of polymer electrolytes – polyethylene oxide (PEO). In fact, while the understanding of the underlying mechanisms has greatly improved within recent years, it remains rather challenging to outperform PEO-based electrolyte systems. Nonetheless, it is not impossible, as highlighted by several examples mentioned herein, especially in consideration of the extremely rich polymer chemistry and with respect to the substantial progress already achieved in designing tailored molecules with well-defined nanostructures.
 Dominic Bresser | Dominic Bresser is serving as group leader at the Helmholtz Institute Ulm (HIU), affiliated with the Karlsruhe Institute of Technology (KIT), Germany, focusing especially on the development of alternative anode materials, polymer electrolytes, and aqueous cathode processing technologies for lithium batteries. Prior to this, he held a postdoctoral position at CEA in Grenoble, France, where he was studying new macromolecular electrolytes, after he completed his PhD at Muenster University, Germany, during which he studied nanostructured lithium-ion active materials. He is Co-author of >60 peer-reviewed publications (h-index: 26; Scopus), 4 book chapters, and 18 patents and patent applications. |
 Sandrine Lyonnard | Dr. Sandrine Lyonnard received her Ph.D. in Solid State Physics from the University of Orsay, France. She was appointed at the Commissariat à l'Energie Atomique et aux Energies Alternatives (CEA) to develop neutron and X-rays scattering/imaging techniques for the characterization of the structure and (ion/solvent) dynamics in soft matter materials, e.g. ionic polymers, self-assembled and nanostructured materials. In recent years, her focus is on operando studies of proton exchange fuel cells and lithium ion batteries at Large Scale Facilities. She is serving as group leader at the Fundamental Research Department of CEA in Grenoble. She (co)authored >60 peer-reviewed publications and 2 book chapters. |
 Cristina Iojoiu | Cristina Iojoiu is Senior Research Scientist at National Center for Scientific Research (CNRS) in the Laboratory of Electrochemistry and Physical-chemistry of Materials and Interfaces (LEPMI), Grenoble, France. Her activity is focused on the design and physicochemical and electrochemical characterization of ionomers, salts, ionic liquids and polymer electrolytes dedicated to energy applications such as: batteries (lithium-polymer, lithium-ion, Mg, Ca, Na, redox flow) and fuel cells (protonic and alkaline conducting membranes). She is co-author of >75 peer-reviewed papers and 5 book chapters and co-inventor of 15 patents. |
 Lionel Picard | Lionel Picard is a CEA researcher since 2010, after receiving his Ph.D. in Material Sciences (polymers and composites specialty) from Univ. Lyon 1. His expertise combines the development of Li-ion batteries up to the pre-commercial scale with a deep knowledge of polymer chemistry including synthesis, physicochemical and electrochemical characterization, and processing. He has gained several years of experience already in leading a group of 20 people active in the development of (gel) polymer electrolytes and innovative post-Li-ion batteries (Li-S & Li-Air). Recently, he has also actively fostered the research and development of organic active materials and nanostructured single-ion conducting macromolecular electrolytes at CEA and lead several industry and publicly funded research projects in this field. He is a CEA expert since 2013 in polymers for energy storage and generations. He has published 7 peer-reviewed papers and has a h-index of 6. He is co-inventor of 25 patents, dealing with his different research subjects, most of which are extended. He supervises 6 PhD student (3 defended and 3 ongoing) and 18 MSc students. |
 Stefano Passerini | Stefano Passerini is Professor at the Karlsruhe Institute of Technology (KIT), Helmholtz Institute Ulm (HIU, Germany) since January 1, 2014. Formerly Professor at the University of Muenster (Germany), he cofounded the MEET battery research centre (Muenster, Germany). His research activities are focused on electrochemical energy storage in batteries and supercapacitors. He is co-author of more than 500 scientific papers (h-index: 76; Scopus), a few book chapters and several international patents. In 2012, he has been awarded the Research Award of the Electrochemical Society Battery Division. In 2019, he has been admitted to the Leopoldina Akademie. |
Design, System, ApplicationPolymer electrolytes are considered key for enabling the transition to high-energy lithium metal batteries. However, decoupling the Li+-ion transport from the segmental relaxation of the coordinating polymer is a must to achieve high conductivities at room and sub-ambient temperatures. Here, we provide an overview of the different approaches to achieve such a decoupling taking advantage of the extremely rich polymer chemistry and the substantial progress already achieved in designing tailored molecules with well-defined nanostructures. |
1. Introduction
Electrochemical energy storage in batteries and, in particular, lithium-ion batteries is considered key for satisfying the mobility needs of our society, thus, contributing to the successful transition to renewable energy sources only.1–5 To fully satisfy the corresponding requirements, however, further improvement concerning the energy and power density as well as safety is required. In this regard, polymer-based electrolytes may help to address at least two of these aspects, i.e., energy density and safety, allowing for the use of metallic lithium as anode and replacing easily flammable and chemically unstable liquid organic electrolytes, respectively.6–10 Polyethylene oxide (PEO) is the most investigated and commercially employed polymer electrolyte due to its high stability towards reduction, including the contact with metallic lithium, low cost, and relatively facile handling and processing.11–13 Nevertheless, it is affected by a rather low electrochemical stability towards oxidation limiting its application to LiFePO4-type cathodes.14,15 Additionally, its ionic conductivity at ambient temperature is rather limited. In fact, for PEO-based systems, it commonly remains well below 10−4 S cm−1 and, thus, significantly below the minimum of 10−3 S cm−1 required for practical applications.8,9,13 This is essentially related to its dependency on the segmental relaxation of the cation-coordinating polymer chains, i.e., the lack of substantial segmental motion at temperatures substantially lower than the melting point of the polymer.12,13,16–19 Many efforts have focused on lowering the glass transition temperature (Tg), however, resulting in poorer mechanical stability.20–26 Recently, it has been proposed that this intrinsic conductivity limitation might be overcome by decoupling charge transport and segmental relaxation, for instance, by designing well-ordered ion conduction channels within the polymer substrate.Herein, we review the progress in this field, starting from a brief recapitulation of the “PEO-type” ion conduction mechanism, followed by an overview of the main approaches to realize (partially) decoupled ionic conductivities within the past years. The manuscript is concluded by a short outlook on the approaches and fundamental parameters to be considered for realizing further progress to finally achieve suitable ionic conductivities at (near-)room temperature.
2. Dependency of segmental relaxation and ionic conductivity in PEO-based electrolytes
It has been commonly agreed that the ion conduction in “dry” polymer electrolytes, in particular polyethers like PEO, occurs essentially in the amorphous state via the segmental motion of the Li+ coordinating polymer chains,13,17,18,27 (see Fig. 1). On the contrary, the crystalline phase was considered to be only poorly conducting due to the high energy barrier for the required cooperative motion of Li+ between the preferred sites.28 As a result, the temperature-dependent ionic conductivity follows the non-linear Vogel–Fulcher–Tammann (VFT) equation,17,27,29i.e., a modified Arrhenius equation proposed for describing the temperature-dependence of viscosity in amorphous glasses.30–32 Additionally, the conducting lithium salt generally slows down the segmental dynamics leading to an increase of the Tg.27,33 This renders the achievement of suitable ionic conductivities at ambient temperature (i.e., ≥10−3 S cm−1) even further challenging since the segmental motion is sufficiently pronounced only for temperatures significantly exceeding the Tg of the polymer electrolyte (see Fig. 1, right panel).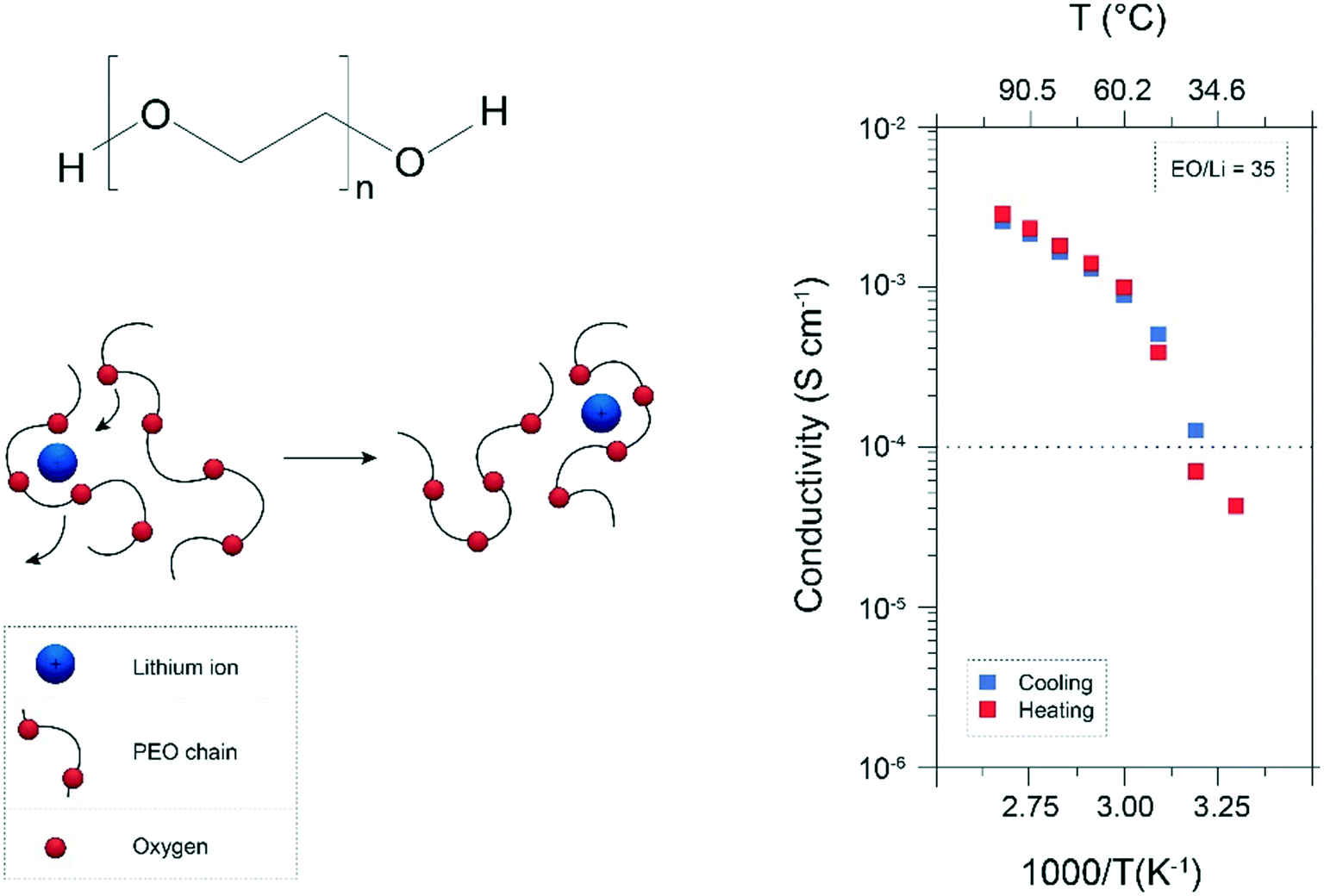 | ||
| Fig. 1 Chemical formula of PEO and schematic illustration of the Li+ conduction via segmental motion of the cation coordinating PEO chains (left panel). Also shown is the ionic conductivity as a function of temperature, exemplarily for PEO35:LiCF3SO3 (ref. 34) (reproduced by permission of The Royal Society of Chemistry). | ||
The underlying mechanism can be illustrated by the free volume model, depicted in Fig. 2.35,36 Above the Tg, the polymer chains are in a state of local segmental motion, resulting in the availability of free volume in direct vicinity of the moving chain segment. This free volume provides the opportunity for an intermolecular coordination of lithium cations, eventually resulting in Li+ ions “hopping” from one coordination site to another in presence of an electric field, i.e., the transfer of the cation from one polymer chain to another due to the sufficiently reduced energy barrier (see left panel of Fig. 2).37
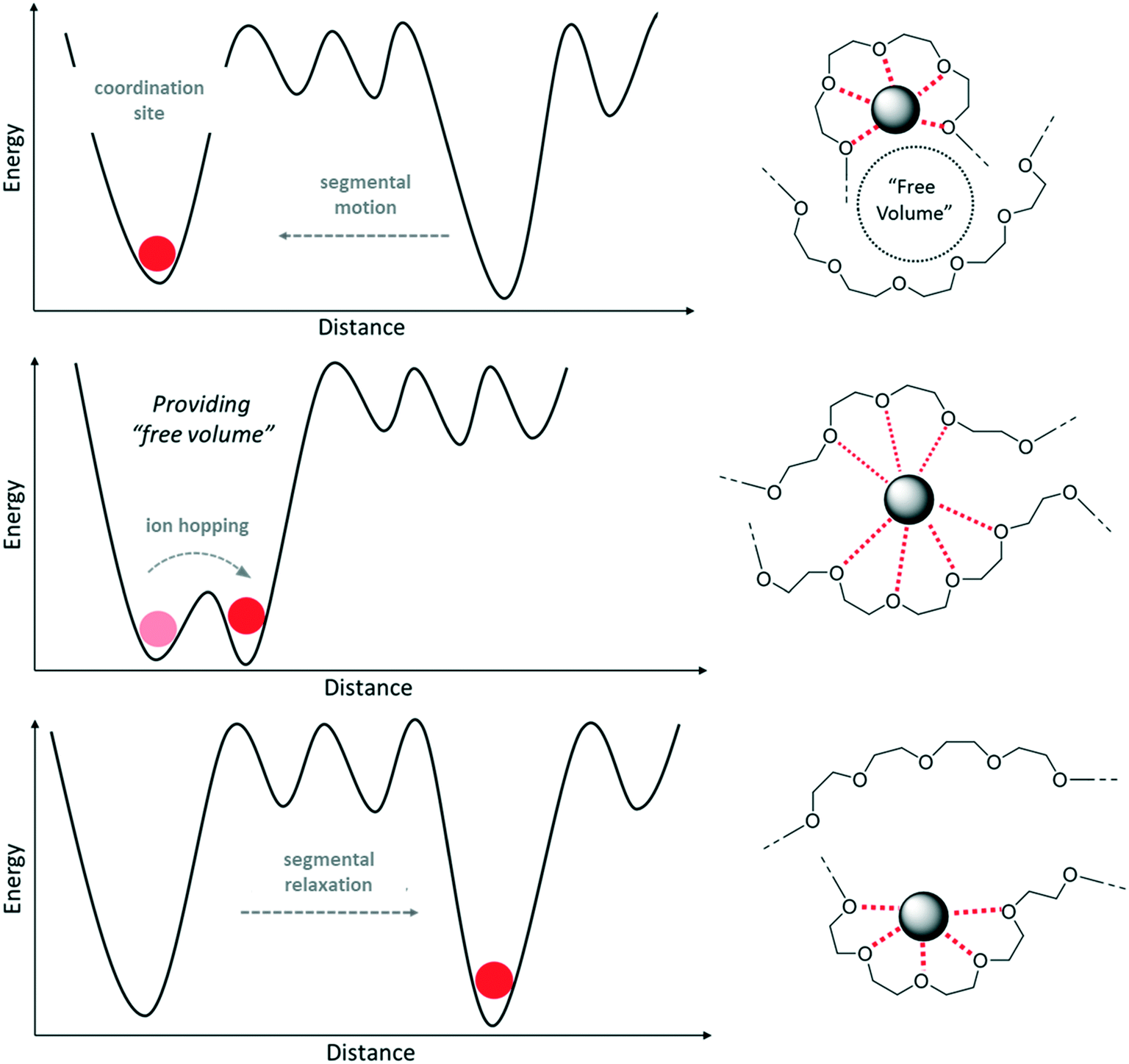 | ||
| Fig. 2 Energy profile and schematic illustration of the ion transport mechanism within (PEO-type) polymer electrolytes based on the free volume model. | ||
In an attempt to overcome this limitation, numerous studies have investigated the conduction mechanism in more detail, targeting the in-depth understanding of the effective parameters and their interplay. Insightful findings have been obtained by means of molecular dynamics simulations. Brooks et al.,38 for instance, studied electrolyte systems based on LiTFSI (lithium bis(trifluoromethanesulfonyl)imide) and PEO. Using atomistic simulations, they focused the investigations on the interplay between the Li+ cation and TFSI− anion motion along the PEO chain and the hopping between different chains, the polymer motion, as well the intra- and inter-chain diffusion as a function of the LiTFSI concentration and the PEO molecular weight. Based on this thorough investigation they found that the inter-chain hopping is, in fact, the most effective Li+ transport mechanism, while the oscillation and/or shift of less-mobile ions between several chains has a potentially negative impact on the segmental polymer dynamics. Moreover, the authors highlighted that very flexible PEO chains might coordinate the lithium cations “too strongly”, thus, hindering the effective Li+ ion conduction. They suggested that the incorporation of more rigid backbones (in the polymer itself or as blend) might allow for a faster diffusion relative to the polymer matrix. Complementary studies were conducted by Wheatle et al.39,40 regarding the influence of the polymer's dielectric constant and polarity. They used all-atom39 and coarse-grained40 molecular dynamics simulations to understand the competition between ion aggregation and polymer segmental dynamics in LiTFSI-doped polyethers. According to their findings, the ionic conductivity in such systems is very sensitive towards the polymer polarity, as the ions tend to cluster into large aggregates in case of low dipole moments, leading to highly correlated ionic motion and decreased ionic mobility. Similarly, a too high dipole moment slows down the segmental dynamics of the polymer – also resulting in a reduced ionic conductivity. Accordingly, the polymer is ideally characterized by a sufficiently high dipole moment to avoid ion aggregation, but sufficiently low to allow for fast segmental motion. Interestingly, they found that PEO is one of the most suitable polymers out of those studied in this regard, further supporting its advantageous properties. Hence, it is not surprising that most studies focused on improving the ionic conductivity of PEO-based electrolyte systems by decreasing the Tg and preventing the crystallization of the electrolyte. Approaches to enhance the ionic conductivity of PEO at lower temperatures include the addition of liquid or solid plasticizers41–50 or the modification of the polymer matrix via copolymerization or crosslinking.51–56
Bruce and co-workers,57–60 however, challenged this general trend and showed that lithium ions can move in short-chain crystalline PEO/lithium salt complexes along well-defined nanochannels. These are constituted by the PEO helices coordinating the Li+ cations, in which the latter can move by hopping akin to the Arrhenius-type ion conduction mechanism in ceramic electrolytes.17,61 Precisely, the ionic motion is coupled to the local modes of the PEO molecule, serving as gatekeeper for the cation transport within and the anion transport between these helices (see Fig. 3).58
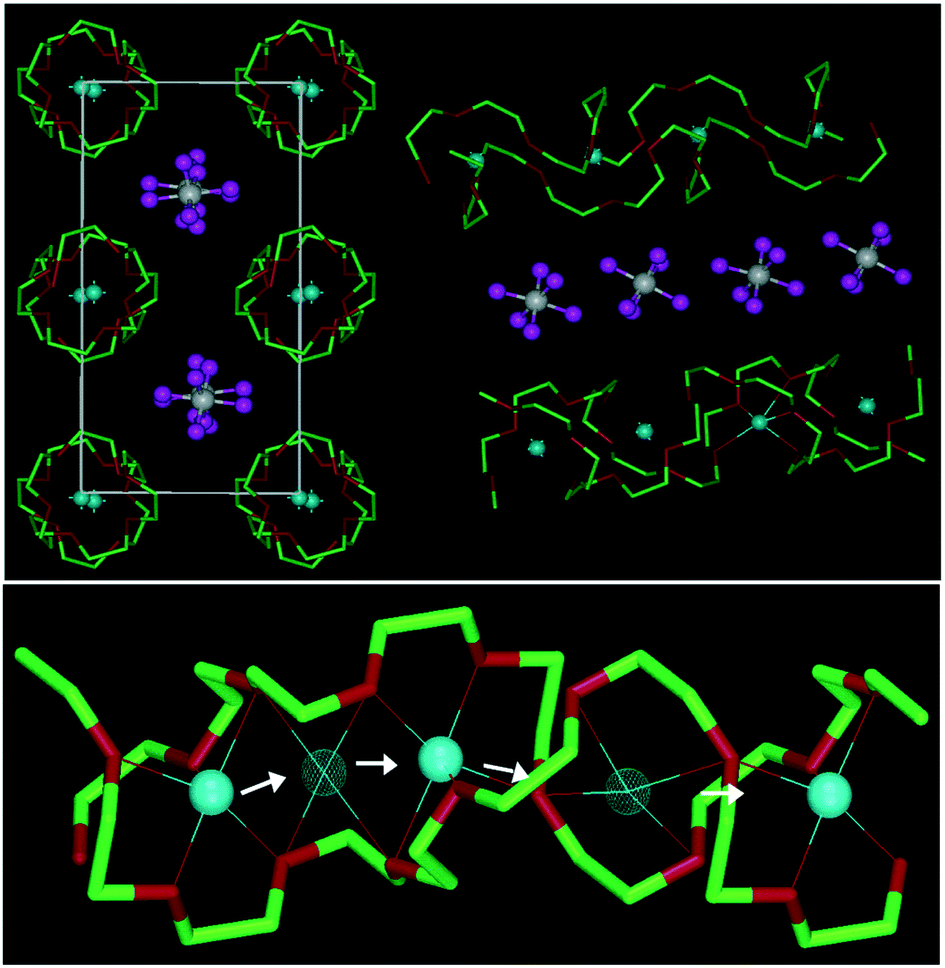 | ||
| Fig. 3 Upper panel: Illustration of the structure of PEO6:LiAsF6 (left) along the rows of lithium cations coordinated by the PEO helices with the anions in-between and (right) in the side view. Lower panel: Schematic presentation of the diffusion of lithium cations within the PEO helices from one coordination site to another (light blue spheres: Li+; white spheres: As5+; magenta spheres: F−; green: carbon: red: oxygen) (reprinted with permission from Stoeva et al.60 Copyright 2003 American Chemical Society). | ||
These findings were supported theoretically by using single-point ab initio electronic structure calculations62 and experimentally by combined stretching/conductivity experiments63–65 as well as an investigation of the anisotropy of ion conduction in single-crystalline PEO.66 The latter two studies revealed that the ion conduction mechanism in crystalline PEO is highly dependent on the molecular alignment of the PEO chains – at least below the melting point. Similarly, an in-depth investigation of Fullerton-Shirey and Maranas67 employing different complementary scattering techniques (incl. quasi-elastic neutron scattering, QENS) on semi-crystalline PEO comprising different lithium salts (e.g., LiClO4 or LiTFSI) evidenced two types of dynamics. The fast PEO segmental mobility and slow restricted rotations of protons around the Li+ cations indicated that one or two PEO chains are embedding the lithium ions in self-assembling helical or cylindrical objects. Moreover, they found that the maximum conductivity recorded could not be attributed to the fast PEO segmental mobility only. Instead, they proposed that also the directed lithium transport within the crystalline domains contributes to the overall ionic conductivity, supporting the partial decoupling of conductivity and segmental dynamics. Nevertheless, the corresponding anion appears also to play a decisive role for the charge transport dynamics, as unveiled by further QENS studies by Saboungi and co-workers,68 indicating the dual-ion transport in PEO/lithium salt complexes as essential. Although the mechanism is rather different from ceramic electrolytes, in which only one ionic species is mobile, the conductivity of the polymer electrolyte could be improved by following similar approaches. The isovalent doping, i.e., the introduction of small quantities of LiTFSI into the LiAsF6/PEO complex, resembling the effect of defects in crystalline ceramic ion conductors, was found to enhance the ionic conductivity.58
3. Decoupling segmental relaxation and charge transport
a) The ‘polymer-in-salt’ approach
Angell and co-workers have been the first to propose an alternative to the ‘salt-in-polymer’ approach by reversing the ratio of the conducting salt and the dissolving polymer, i.e., the so-called ‘polymer-in-salt’ systems.25 Accordingly, they dissolved large amounts of the conducting salt in relatively small amounts of PEO or polypropylene oxide (PPO). In particular, around 0.1–0.2 units of the polyether per mole of salt were used to ensure sufficient elastomeric properties. Such composites revealed ionic conductivities of up to 10−4 S cm−1 at ambient temperature. These are accompanied by high transport numbers (around 0.9) as such systems allow the rather small lithium cations to move independently of their surroundings, i.e., be decoupled from the segmental motion of the polymer. Similarly, Wei and Shriver22 reported electrolyte systems based on rigid polymers (i.e., poly(1,3-dioxolan-2-one-4,5-diyl oxalate)) with a high concentration of lithium salt (LiCF3SO3; 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio). Forsyth, MacFarlane and co-workers further extended this approach to other rigid polymers, for instance, poly(acrylonitrile) (comprising more than 60 wt% of LiCF3SO3).69,70 They observed substantially increased conductivities with respect to the “diluted” systems – especially for temperatures approaching the Tg. Accordingly, it has been generally proposed that the rather irregular polymer structure of such rigid polymers results in a frustrated close packing of the polymer chains. This induces a relatively large free volume within the crystalline electrolyte systems, providing a high density of polar groups to enable a reduced activation energy for the cation hopping from one polar group to another. It may be noted that later studies on high concentrations of the conducting salt dissolved in ionic liquids also revealed a decoupled lithium transport (in this case from viscosity),71 indicating that this approach is not limited to polymer-based systems only.
:1 molar ratio). Forsyth, MacFarlane and co-workers further extended this approach to other rigid polymers, for instance, poly(acrylonitrile) (comprising more than 60 wt% of LiCF3SO3).69,70 They observed substantially increased conductivities with respect to the “diluted” systems – especially for temperatures approaching the Tg. Accordingly, it has been generally proposed that the rather irregular polymer structure of such rigid polymers results in a frustrated close packing of the polymer chains. This induces a relatively large free volume within the crystalline electrolyte systems, providing a high density of polar groups to enable a reduced activation energy for the cation hopping from one polar group to another. It may be noted that later studies on high concentrations of the conducting salt dissolved in ionic liquids also revealed a decoupled lithium transport (in this case from viscosity),71 indicating that this approach is not limited to polymer-based systems only.
b) Frustrated polymers and polymerized ionic liquids
The great importance of frustrated polymer packing for decoupling segmental relaxation and ion conductivity has been investigated also for ‘salt-in-polymer’ systems. Among others, Sokolov and co-workers highlighted that the decoupling correlates with the fragility of the polymer system.72–74 Precisely, their studies demonstrated that fragile polymer systems – i.e., rather rigid polymer structures like poly(vinyl chloride), polycarbonate, polystyrene, or poly(methyl methacrylate) and their derivatives – reveal a (partial) decoupling of diffusion-controlled motions, including ion conduction, from segmental dynamics. Such a decoupling increases for an increasing fragility of the polymer and temperatures approaching the Tg – which is analogous to non-polymeric materials.72–75 For such systems, ionic motion occurs in the relatively large free volume of the rather loose polymer structure, resulting from the frustrated, thus inefficient, chain packing.73,74In subsequent studies, Sokolov and co-workers correlated this decoupling to the superionic behavior using a modified Walden plot analysis, suggesting that strongly coupled ion conduction, as observed for amorphous PEO, faces intrinsic limitations for achieving suitable ionic conductivities at ambient temperatures.76,77 The classic Walden rule is based on the assumption that the ionic conductivity is directly related to the viscosity of the electrolyte – i.e., the structural relaxation in case of polymers. Accordingly, PEO-type electrolytes show, as expected, the ideal Walden-type behavior. However, the Walden plot analysis also reveals that the targeted molar conductivity can be achieved only for very short relaxation times of less than 10−8 s, i.e., at around 80 °C (see Fig. 4a).76 Differently, rather rigid polymers, like polystyrene derivatives, reveal superionic behavior at low temperatures (see Fig. 4b). If, moreover, the conductivity is “normalized” by referring to the concentration of free ions only (i.e., excluding ion pairs; indicated as “true” molar conductivity in Fig. 4b), these rigid polymer electrolytes show a superionic behavior across the whole temperature range comparable to superionic inorganic ion conductors like (AgI)0.5(AgPO3)0.5. However, it should be noted that the concentration of free ions is rather low (about 0.01%) and the actual ionic conductivity still remains inferior to PEO-based systems.76,77 Accordingly, an increased number of free ions, e.g., by increasing the solvating properties of such rigid polymers accompanied by a rather low Tg, is required to achieve suitable ionic conductivities at ambient temperature.77,78
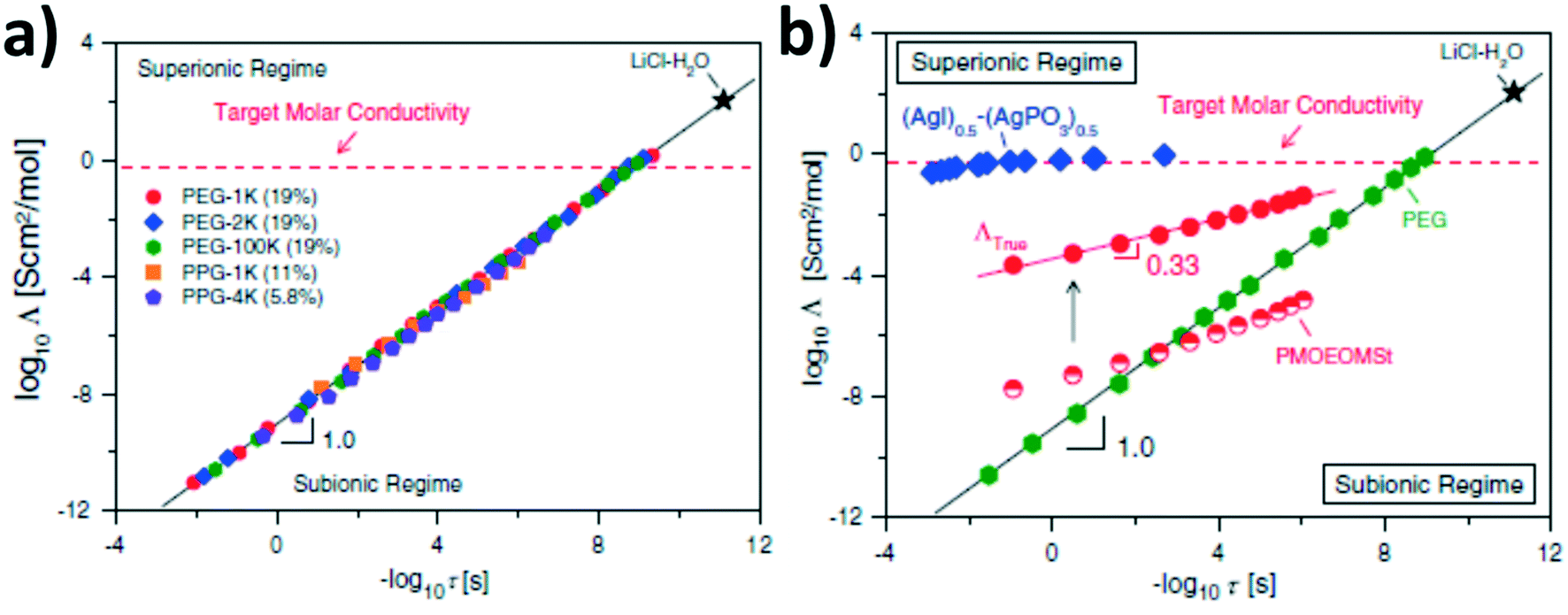 | ||
| Fig. 4 Modified Walden plots, correlating the molar conductivity (Λ) to the rate of structural (segmental) relaxation (1/τ) for (a) polyethylene glycol (PEG) based electrolytes with varying molecular weight and comprising LiClO4 as conducting salt in the given weight percentage and (b) a comparison of poly[4-(2-methoxyethoxy)methyl styrene] (PMOEOMSt) comprising 0.3 wt% LiClO4 (semi-filled symbols in red) and (AgI)0.5(AgPO3)0.5 (in blue) as well as PEG (100000 g mol−1) containing 19.4 wt% of LiClO4 (in green) as reference system for a fully decoupled (inorganic) and fully coupled lithium-ion conductor, respectively. The filled red circles represent the true molar conductivity (ΛTrue) for PMOEOMSt taking into consideration the free-ion concentration. The ideal Walden line represents a diluted aqueous solution of LiCl and the dashed horizontal line indicates the target molar conductivity needed for achieving an ionic conductivity of 10−3 S cm−1 (reprinted from Wang et al.,76 Copyright 2014, with permission from Elsevier). | ||
One approach to reach a higher charge concentration relies on the polymerization of ionic liquids (poly(IL)s),79–82 resulting in an intrinsically higher number of (free) ions contributing to the charge transfer. Most of the studies in this field focus on polymerized cations, i.e., the anion is the only mobile species if no lithium salt is added, the following considerations may provide some fundamental guidelines for lithium battery electrolytes. Among the first to realize that the temperature-dependent ion conduction in poly(IL)s is not exactly following the trend for the Tg have been Colby and co-workers83,84 as well as Winey and co-workers.85 These latter systematically studied the effect of a varying alkyl chain length (n = 2, 4, 8; see Fig. 5) on the polymer structure and ionic conductivity. A closer investigation of the correlation between the backbone-to-backbone distance and the ionic conductivity in these systems revealed that the Tg is, indeed, not the only factor determining the ionic conductivity, though playing a dominant role.86 In addition, the intermolecular anion hopping was found to significantly contribute to the overall conductivity, especially at temperatures approaching the Tg, in agreement with the earlier reported findings of Sokolov and co-workers. For such a reason, the intermolecular distance between the cationic groups as “hopping sites” plays a decisive role, i.e., the larger the distance, the higher the energy barrier for the hopping process and the lower the contribution to the overall charge transport.
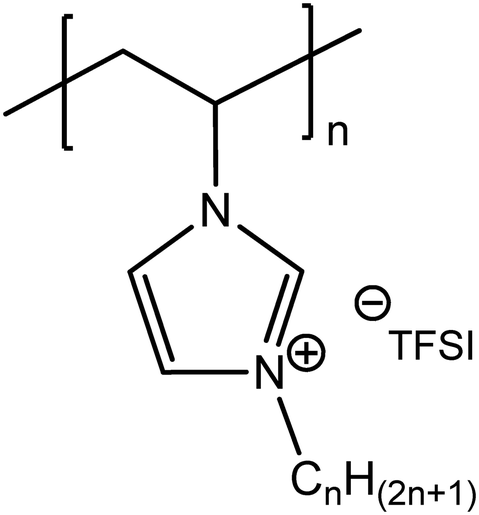 | ||
| Fig. 5 Chemical structure of 1-alkyl-3-vinylimidazolium bis-(trifluoromethanesulfonyl)imide-derived (CnVIm-TFSI) homopolymers. | ||
While Winey and co-workers86 suggested that this energy barrier increases with an increasing alkyl chain length, Delhorbe et al.87 investigated an extended set of CnVImTFSI poly(IL)s with n = 2, 4, 6, 8, and 10, showing that such “linear” conclusions have to be taken with care. According to their results, the ion conduction in such systems is governed by a rather complex interplay of physicochemical properties (including the Tg), the charge carrier concentration, and the eventual formation of nanostructures. As a matter of fact, the modified Walden plot analysis proposed by Sokolov and co-workers76,77 reveals that the greatest degree of decoupling is achieved for n = 2 and 8, although all systems investigated are characterized by superionic behavior across the whole temperature range (Fig. 6). Following the earlier work on rigid polymers73–77 as well as rather recent studies,88,89 Delhorbe et al.87 assigned this finding to the frustration in polymer packing related to the bulky imidazolium cation being is closely bonded to the polymer backbone, which result in sufficient free volume for the anions to move. Moreover, they reported additional studies for such poly(IL)s including LiTFSI, showing that the addition of the lithium salt may not lead to substantial changes in the ion conduction mechanism.
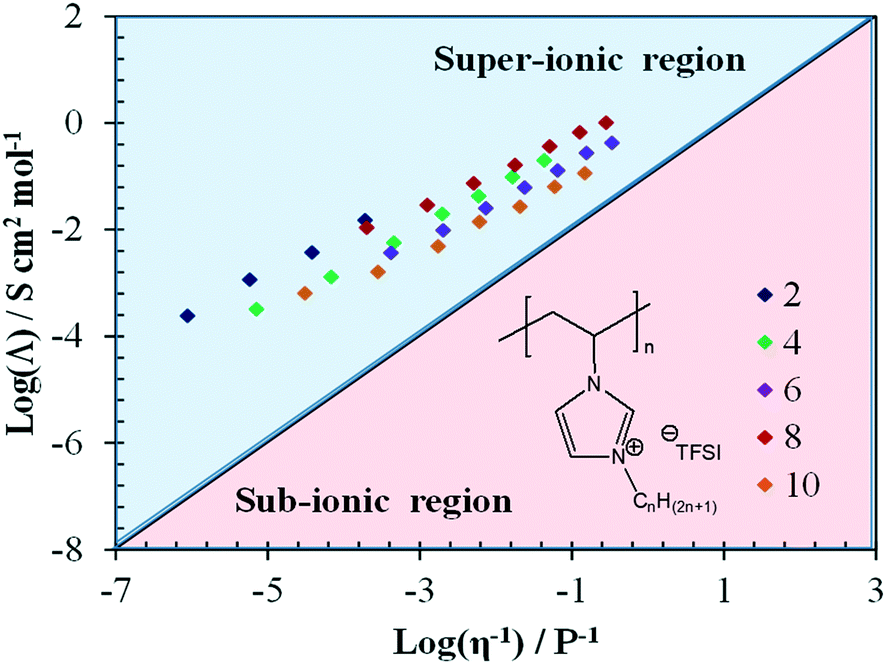 | ||
| Fig. 6 Walden plot (molar conductivity vs. fluidity) for different CnVIm-TFSI-derived poly(IL)s with n = 2, 4, 6, 8, and 10; the “ideal” Walden line with a slope of 1, obtained for 10 mM aqueous solution of LiCl serves as reference (reprinted with permission from Delhorbe et al.87 Copyright 2017 American Chemical Society). | ||
Comparable results have been obtained by Sokolov and co-workers for ammonium-based poly(IL)s, also revealing superionic behavior independent of the pendant group, particularly when approaching the Tg and for increasing molecular weights.90,91 While the actual ionic conductivity decreased for the latter, a normalization with the Tg revealed that the segmental-relaxation independent conductivity increased up to a polymerization degree of ten before stabilizing.91 Correlating the degree of decoupling with the flexibility (i.e., the fragility) of the poly(IL) electrolyte by comparing the relatively rigid vinyl-based poly(IL)s and the rather flexible siloxane-type poly(IL)s, confirmed that there is a direct relationship between the chain packing frustration (due to the polymer fragility) and the decoupled ionic motion within the resulting free volume.92 As a matter of fact, the authors observed a decreased decoupling under elevated pressure, especially for rather flexible poly(IL)s. For rigid poly(IL) electrolyte, however, this effect was less pronounced, highlighting the importance of the free volume.
In conclusion, the governing factor for such decoupling is essentially the same for poly(IL)s as for the formerly studied neutral polymers. Mogurampelly et al.93 confirmed this very recently by means of atomistic molecular dynamics simulations employing quantum-mechanically parametrized force fields. In addition to the experimental findings, these authors stressed the importance of the average lifetime of ion-association as the underlying timescale. Studying exemplarily imidazolium-based poly(IL)s, they reported that the decoupled anion transport occurs via intra- and intermolecular ion hopping, involving the formation and breaking of ion-association with four cationic monomeric units of two different polymer chains. If such ion-ion correlations are too strong, however, the decoupled ion conduction remains limited.94 In fact, despite the great progress in understanding the relationship between polymer fragility and charge transfer, the ionic conductivity of these electrolytes remains significantly lower than that of PEO-based electrolytes.
c) Realization of anisotropic conduction pathways
The realization of static and anisotropic pathways for the efficient ionic motion17via supramolecular ordering, similar to highly efficient organic electron conductors,95 has been earlier suggested as the key to fully decouple ion conduction and polymer relaxation. Significant contribution to this field was provided by Wright and co-workers, who have been also the first to propose the use of PEO as ion conductor.96 The focus of their studies has been on polyether-type polymers, comprising aromatic mesogenic moieties to introduce liquid-crystalline behavior in order to realize the targeted structural ordering (Fig. 7a, left panel).97–102 These well-organized electrolytes showed enhanced ionic (cationic and anionic) conductivities along the 2D ionic layers, which could be further improved by mechanical shearing as a result of the increased ordering.101,102 Further improvement of the conductivity was realized by introducing a second polymer (Fig. 7b, right panel), which essentially led to an increase of the internal space of the ionophilic polyether layer by expanding the ionophobic layer.103–105 Remarkably, the authors reported an ionic conductivity of 10−4 S cm−1 at 40 °C within the plane, which appeared to have an Arrhenius-type temperature dependency, and only 10−12 to 10−13 S cm−1 perpendicular to the plane. This confirms the highly anisotropic, potentially decoupled charge transport.103,105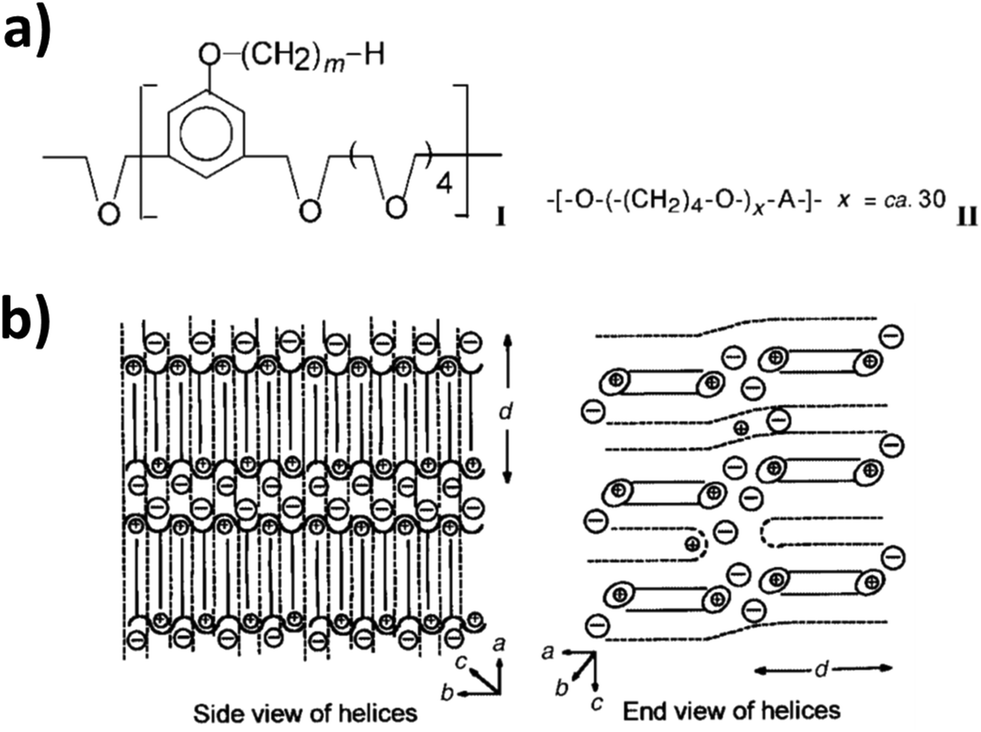 | ||
| Fig. 7 (a) General formula of the (I) amphiphilic liquid-crystalline polymers investigated (poly[2,5,8,11,14-pentaoxapentadecamethylene(5-alkyloxy-1,3-phenylene)]); with m commonly being 16 and (II) the blending polymer (poly(tetramethylene oxide)); with A being either –CH2– or –CH2C(NCH2)CH2–. (b) A schematic illustration of 2D ordering. The solid lines refer to the alkyl chain covalently bonded to the liquid-crystalline polymer I, while the dashed lines refer to additionally introduced polymer II. The alkyl side chains of I interdigitate in a hexagonal lattice layer between the helices formed by the polyethers, encapsulating the lithium cations. The anions are located in the interhelical space (reproduced from ref. 104 with permission from The Royal Society of Chemistry). | ||
Following the same approach, Imrie, Ingram, and their co-workers targeted the decoupling of ion conduction from segmental relaxation by realizing supramolecular ordering in liquid-crystalline complexes. In their studies, they focused on lithium salt complexed molecules comprising a biphenyl-type mesogenic moiety attached via a flexible alkyl-based spacer to a PEO backbone (see Fig. 8a).106–108
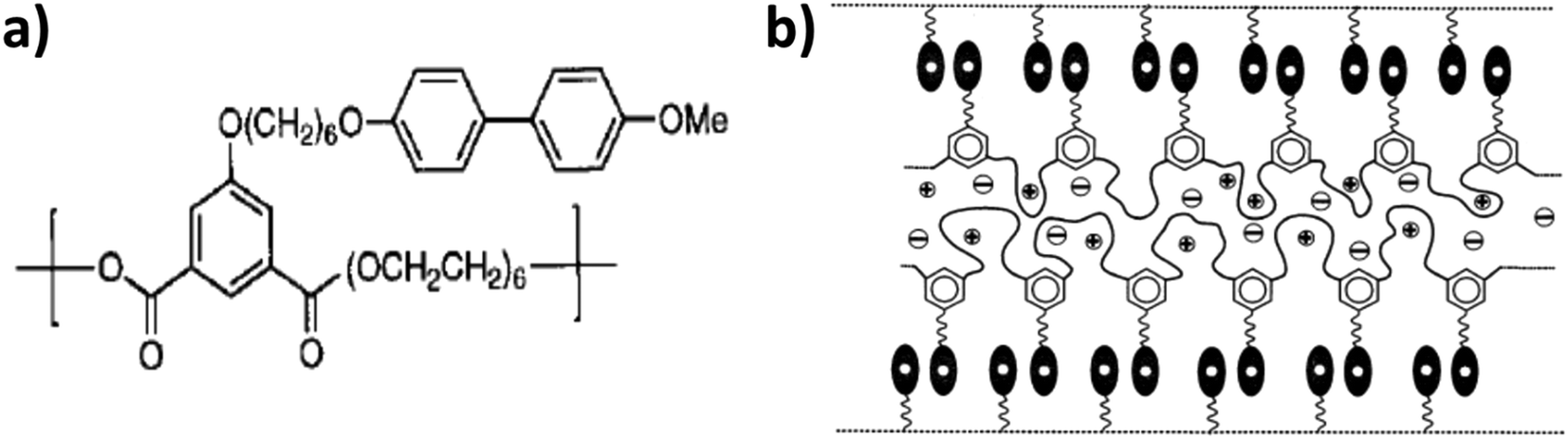 | ||
| Fig. 8 (a) Molecular structure of the liquid-crystalline polymer investigated (from Imrie108 Copyright © 1999 by John Wiley Sons, Inc. reprinted by permission of John Wiley & Sons, Inc.). (b) Proposed smectic bilayer nanostructure for the lithium salt complexed liquid-crystalline molecule described in panel (a) (reprinted from Imrie and Ingram,109 Copyright 2001, with permission from Elsevier). | ||
The comparison with amorphous polymers revealed a great degree of decoupled charge transfer, if not full decoupling, in the liquid-crystalline material. As a matter of the fact, this electrolyte did not show a substantial decrease in conductivity when approaching the Tg. Additionally, the temperature dependency of the ionic conductivity essentially followed the Arrhenius equation,108,109 which is characteristic for segmental-relaxation independent ionic motion.17,61 Interestingly, they observed a change in slope for the conductivity as a function of temperature for the first-order phase transition rather than for the Tg. Accordingly, they proposed that the molecules self-organize in a smectic phase, bilayer structure (Fig. 8b) supported by several complementary techniques like polarized optical microscopy and X-ray scattering. The pendant liquid-crystalline side groups jacket the polyether side chains, preventing their collapse upon cooling. This eventually enables the ionic mobility also in the solid state (i.e., below the Tg).108,109 These findings and the conceived interpretation are remarkable. They indicate that lithium cations might be conducted in polyethers independent of the segmental relaxation also above the Tg. This is a rather interesting subject for further studies to clarify the precise lithium transport mechanism in such systems.
Another group that has largely contributed to the development of liquid-crystalline lithium-ion conductors providing anisotropic conduction pathways, is the group of Kato, Ohno, and co-workers.110,111 For instance, they reported the synthesis of biphenyl-based liquid-crystalline complexes of a lithium salt and twin oligomers or mesogenic dimeric molecules comprising oxyethylene spacers, both providing smectic mesophases in a certain temperature range.112,113 Comparable to the “common” approach for block copolymer electrolytes,114,115 they introduced a non-conducting phase to ensure a certain mechanical stability and an ion-conducting phase based on polyether moieties to dissolve the lithium salt (herein, LiCF3SO3).112,113 These electrolytes achieved rather high ionic conductivities following this approach (e.g., >10−4 S cm−1 at 50 °C (ref. 113)). The realization of free-standing membranes, to facilitate handling and processing, was successfully obtained by employing liquid-crystalline, in situ polymerizable materials as well PEO-containing moieties for the lithium-ion conduction.116 The general strategy involved three essential steps: (i) the complexation of the monomer with the lithium salt, (ii) the macroscopic alignment (e.g., by applying an electric field117 or mechanical shearing118), and (iii) the in situ polymerization (e.g., by means of UV irradiation).116,118–120 However, the ionic conductivity decreased as a result of the polymerization, like for poly(IL)s.116,120 To overcome this limitation, new molecules were designed based on tris(alkoxy)phenyl groups and imidazolium salts with acrylate groups at the periphery (see Fig. 9a).118 These resulted resulting in 1D ion-conductive polymer films with ion nanochannels perpendicular (or, for comparison, parallel) to the film surface. While the direct comparison with the horizontally aligned sample revealed a clearly enhanced ionic conductivity, confirming the highly anisotropic charge transport, suitable ionic conductivities were essentially achieved for rather high temperatures (>10−3 S cm−1 at 150 °C). Nevertheless, using instead polymerizable ammonium moieties complexed with LiBF4 in a molar ratio of 4:1 (monomer:LiBF4) led to the realization of 3D bicontinuous, cubic liquid-crystalline networks (see Fig. 9b). Thanks to this extended network of ion conduction pathways, such electrolyte systems provided higher ionic conductivities (3.1 × 10−4 S cm−1 at 90 °C) than the hexagonal columnar phase investigated for comparison purposes.119
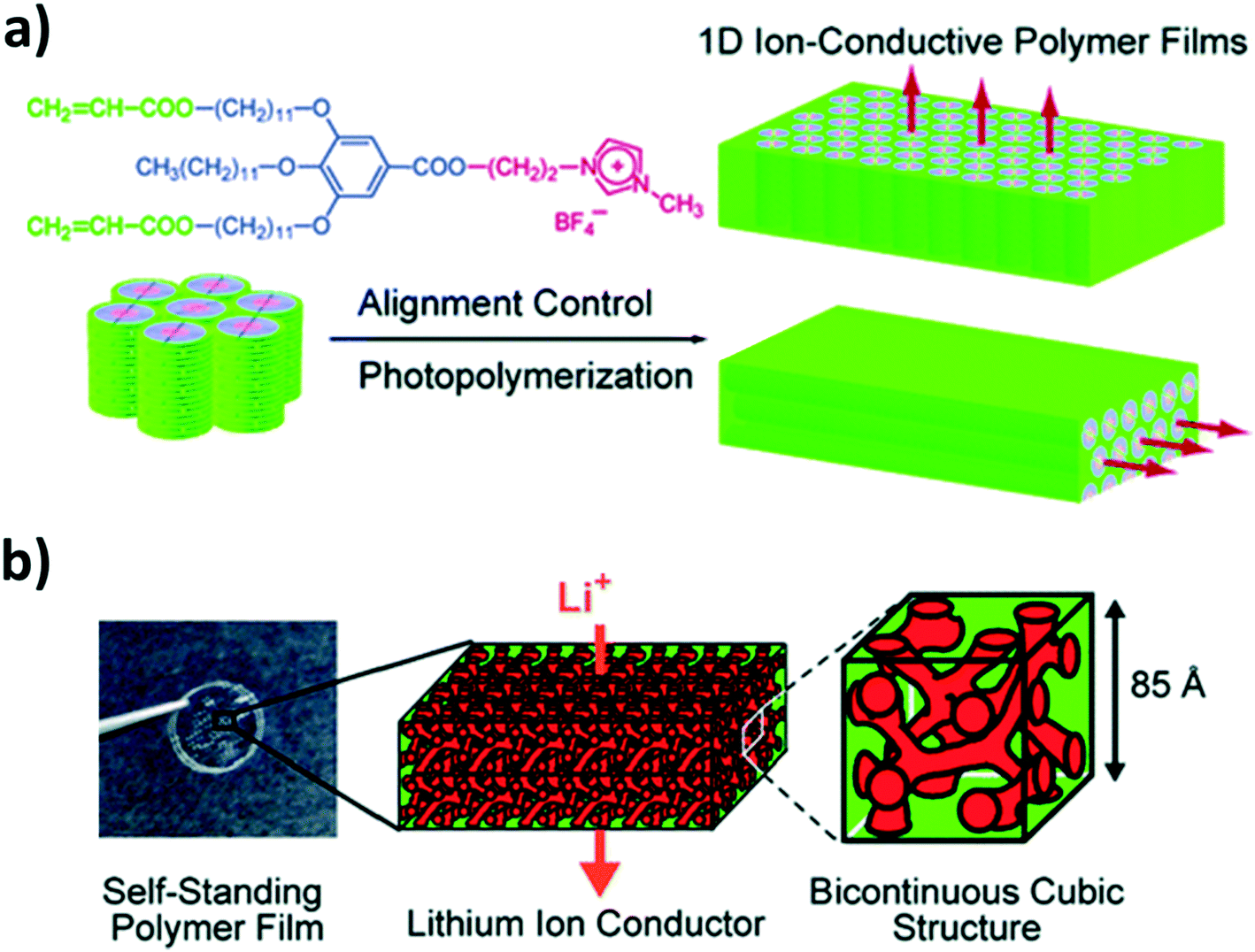 | ||
| Fig. 9 (a) Chemical formula of the liquid-crystalline molecule providing 1D ion conduction pathways and the general strategy of fixing the macroscopic alignment by photopolymerization to realize 1D nanochannels either perpendicularly or horizontally oriented to the film substrate (reprinted with permission from Yoshio et al.118 Copyright 2006 American Chemical Society). (b) Photograph of the self-standing polymer electrolyte membranes and schematic illustration of the 3D bicontinuous cubic liquid-crystalline structure at different magnifications (reprinted with permission from Ichikawa et al.119 Copyright 2011 American Chemical Society). | ||
Despite the outstanding achievements in designing advanced liquid-crystalline electrolyte systems, one aspect remained commonly overlooked in recent years. This was the potential decoupling of ionic motion and segmental relaxation as well as the contribution of the commonly more mobile anion.101,102 Only a few studies have continued focusing on this aspect for liquid-crystalline electrolyte systems. Among these are Stoeva et al.,121 who investigated discotic liquid crystal triblock copolymers based on a central main chain triphenylene-based liquid-crystalline block capped at both ends by PEO blocks (MW = 2000 g mol−1) and doped with LiClO4 in the 6:1 EO:Li ratio. Such material provides a phase-separated morphology consisting of a columnar hexagonal liquid-crystalline phase and PEO-rich domains. Interestingly, these self-standing polymer films revealed an ion conduction mechanism, which appeared to be closely related to that of conventional PEO-type electrolytes. Similarly, a VFT-type conduction mechanism was demonstrated by Wang et al.122 for flexible, discotic liquid crystal-based crosslinked polymer electrolytes. Polymer electrolytes with different compositions were obtained by one pot photopolymerization of triphenylene-based discotic liquid crystals grafted with vinyl functions to polyethylene glycol diacrylate with a molecular weight varying from 200 to 1000 g mol−1 and LiTFSI. More efficient ion transport was reported in polymer electrolytes with ordered structures obtained by annealing and macroscopic aligned self-assembled columns as well as higher flexibility, i.e., longer polyethylene glycol chains. Differently, Majewski et al.123 observed a rather Arrhenius-type temperature dependence of the ionic conductivity for hexagonally-packed, cylindrical liquid-crystalline polymers based on the mesogenic biphenyl moiety and (cylindrical) PEO chains dissolving LiClO4. While the ionic motion was confined to the interior of the PEO domains, according to the authors a remarkable improvement in conductivity was obtained by macroscopically aligning the liquid-crystalline orientation up to the millimeter range. Such an improvement stood up to the clearing point, i.e., the transition to the isotropic phase.
Some insights into this rather different behavior, despite the common ion conduction in the polyether domains, might be gained from a rather different system – PEO-based single-ion conductors with the anionic function covalently tethered to the polymer backbone. Molecular dynamics simulations showed that relatively large ion clusters may form chain-like structures, serving as charge conduction pathways, in single-ion conductors with an anionic sulfonate function covalently bonded to the PEO backbone by isophthalate groups.121 In such a case, the conduction process might be decoupled from the polymer motion, occurring via structural diffusion, including the transfer of charges without cation moving – comparable to the proton transfer in poly(perfluorosulfonic acid) or phosphoric acid. Accordingly, the realization of highly ordered ion-aggregate networks might improve the total conductivity by enhancing correlated ion transport, as suggested by coarse-grained molecular dynamics simulations.124 Furthermore, LaFemina et al.125 showed for essentially the same polymer that the lithium dynamics are dominated by either VFT or Arrhenius behavior. This depends on the length of the polyether spacer, the total ion content, and the degree of ionic aggregation. Beside the lithium-ion/polymer interaction in general, the potential ion pairing and aggregation impact the number of lithium ions available for diffusive motion. The formation of ionic aggregates initially slows down the lithium diffusion until, eventually, these aggregates start to overlap. This enables the lithium diffusion between these clusters, as reflected by an increase of the diffusion coefficient. As a consequence, the rate-determining step for low and high ion contents is the separation of ion pairs and the transfer of lithium cations between the aggregates, respectively.
d) The utilization of solvent molecules as ‘single-ion transporters’
Following another approach to (partially) decouple the ion transport from the segmental relaxation of the polymer, a few groups have recently started to develop ionic polymers (also referred to as ‘ionomers’) with the anionic function covalently tethered to the (block co-)polymer backbone. In this systems the lithium cations are essentially “transported” by the incorporated highly dielectric solvent molecules (e.g., ethylene carbonate (EC) or propylene carbonate (PC) – potentially in a mixture with linear organic carbonates like dimethyl carbonate (DMC)). One of the great advantages of this concept is the (near) single-ion conductivity avoiding detrimental charge polarization thus potentially leading to homogeneous lithium deposition, i.e., preventing dendritic metal deposition.126–130 Following this approach Rohan et al. studied EC/PC-swollen ionic polymers based on polystyrene131 or polysiloxane,132 providing conductivities around 10−3 S cm−1 at ambient temperature. Similarly, Qin et al.133 synthesized and investigated polyborate-based electrolytes offering high ionic conductivity (1.8 × 10−4 S cm−1 at 30 °C) upon incorporation of a mixture of EC and DMC. Comparable results were obtained by Li et al.134 for the electrospun poly(diaminodiphenylsulfone)-type electrolyte. Noteworthy, however, all these systems provide Li+ transference numbers (t+) below 0.9 and require the incorporation of additional polymers like highly dielectric poly(vinylidene difluoride) (PVdF) or its hexafluoropropylene-copolymer to realize suitable mechanical properties. Differently, Oh et al.135 recently reported a porous poly(arylene ether)-based single-ion electrolyte drenched with a mixture of diethyl carbonate (DEC), EC, and PC (1:1:1 by volume). The polymer swollen with 92 wt% of the organic carbonates mixture, does not require the presence of PVdF and offers an excellent ionic conductivity (more than 10−3 S cm−1 at 25 °C) with t+ approaching unity (0.98). Nonetheless, one severe constraint remained: the electrochemical (in-)stability towards oxidation, limiting the application of such polymer electrolytes to LiFePO4 as the cathode active material.131–135
This limitation has been overcome only very recently by Nguyen et al.,136 who synthesized multi-block co-poly(arylene ether sulfone) single-ion conductors based on alternating ionophobic and ionophilic blocks. The resulting films are self-standing and nanophase-separated. The incorporated EC selectively swells the ionophilic domains, allowing the effective Li+ coordination (see Fig. 10). In addition to high ionic conductivities (>10−3 S cm−1 at 30 °C, depending on the EC content) and t+ = 1, these membranes allowed for the stable cycling of Li[Ni1/3Co1/3Mn1/3]O2 for more than 200 cycles. Interestingly, the authors observed a correlation between the polymer nanostructure and the overpotential upon lithium deposition, which, however, is a rather unexplored field of research so far.
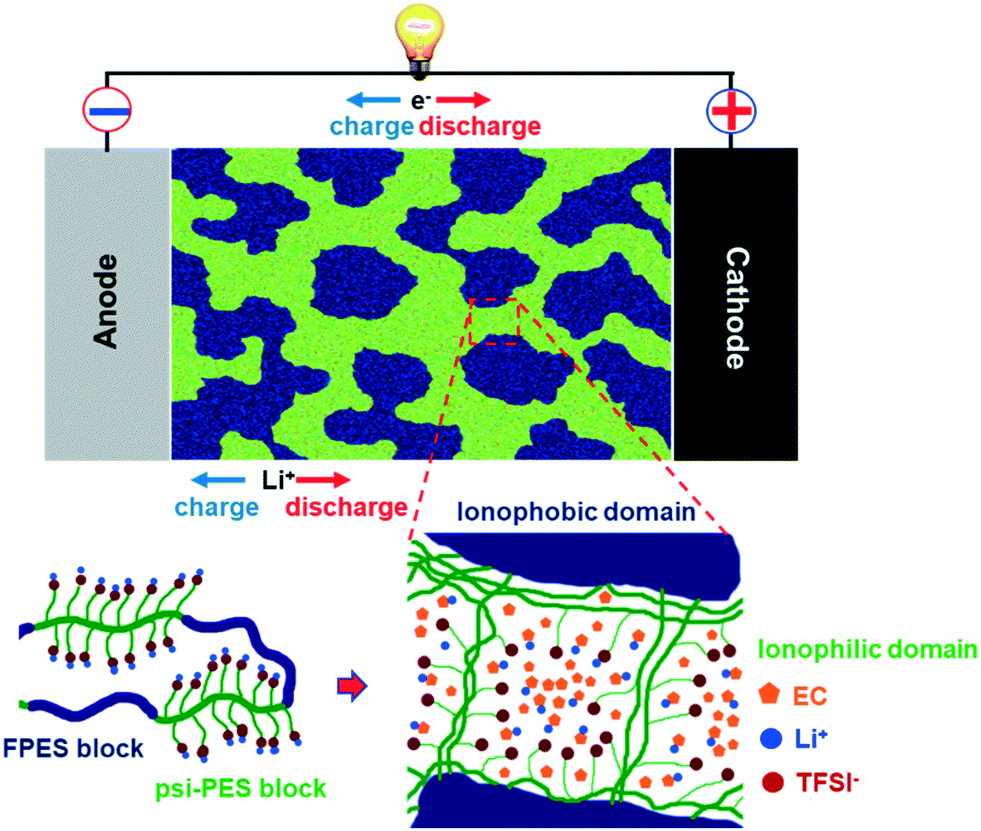 | ||
| Fig. 10 Schematic illustration of a nanophase-separated ionomer-based electrolyte according to Nguyen et al.,136 for which the comprised EC molecules selectively swell the ionophilic domains, thus effectively coordinating the Li+ cations (reproduced by permission of The Royal Society of Chemistry). | ||
4. Remaining challenges and perspective
As reviewed herein, there are different promising approaches to decouple ion conduction in polymer-based electrolyte systems – each of them with its own advantages and remaining challenges. Nevertheless, the realization of defined conduction pathways and/or the use of very mobile small molecules acting as “transporter” appear presently as the most promising approaches. In spite of “solvent-in-salt” electrolytes, these systems operate with lithium concentrations similar to those used in the state-of-the-art lithium-ion batteries. Thus, no great additional costs are expected, however, two great challenges are still to be addressed. Overcoming the conductivity limitations originating from the charge transfer across grain boundaries between rather randomly oriented domains characterized by anisotropic conduction pathways is certainly the first. A potential solution could be the achievement of high, ideally full, degree of alignment,112i.e., “single-crystalline” electrolytes with perfectly anisotropic ion conduction pathways (as achieved already for liquid-crystalline systems). Alternatively, very small coherent domains with an isotropic orientation and high interconnecting defect densities may address the challenge.137 Especially for the latter, the realization of 3D (e.g., bicontinuous cubic or gyroid) conduction pathways rather than (smectic) layered 2D or columnar 1D ion transport channels appear advantageous.The second challenge regards designing new “transporter” molecules with advanced electrochemical and thermal properties compared to the standard organic carbonates. Here, one may consider the design of small molecules mimicking the important polymer characteristics – comparable to the example of ionic liquids and poly(IL)s. Also, the incorporation of small molecules tailored in a highly complementary fashion, could “complete” the set of requirements for practical electrolyte applications. In any case, it is anticipated that the richness of organic and polymer chemistry will allow for great and rapid progress in this highly attractive and rapidly evolving research field. A key aspect, however, that certainly deserves further investigation is the development of an in-depth understanding of the interplay between the polymer nanostructure and the ion conduction mechanism. This is especially true for the (liquid-)crystalline electrolyte systems, including the contribution of the anion in case of lithium salt complexes. In an attempt to overcome any potentially detrimental impact of the anion, we have recently developed a new (proof-of-concept) electrolyte system, mimicking the single-ion conductivity in ceramic electrolytes, thus, allowing for a fully decoupled charge transport in organic electrolyte systems.138 In such liquid-crystalline electrolytes, the Li+ transport occurs solely via hopping from one anionic function to the other as confirmed by the Arrhenius-type temperature dependence of the ionic conductivity up to the transition to the isotropic phase.
Another very promising alternative consists in using oxidized conjugated polymers like polyphenylene sulfide. The introduction of several delocalized carbocations on the polymer backbone by in situ oxidation results in anchoring the anions of the lithium salt on the polymer, thus “freeing” the Li+ cations. Additionally, the rigidity of the conjugated polymer limits its coil conformation, helping creating direct pathways for the Li+ cations into the polymer. Accordingly, these electrolytes can achieve ionic conductivities exceeding 10−4 S cm−1 at 40 °C, i.e., below the Tg of these polymers.139
In fact, it appears that the smart design of suitably engineered polymer-based electrolyte systems with advanced electrochemical properties has been rapidly developing recently – certainly driven also by the great promise of all-solid-state batteries – indicating that the “golden age” of polymer electrolytes might be yet to come.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. B. and S. P. would like to acknowledge financial support from the German Federal Ministry of Education and Research (BMBF) within the FestBatt project (03XP0175B) and from the Helmholtz Association for the basic funding. S. L. and L. P. would like to thank CEA for the financial support. C. I. would like to acknowledge the Centre of Excellence of Multifunctional Architectured Materials “CEMAM” no. AN-10-LABX-44−01. In addition, the authors would like to thank Mr. Dominik Steinle for providing Fig. 2 presented herein.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- D. Bresser, K. Hosoi, D. Howell, H. Li, H. Zeisel, K. Amine and S. Passerini, J. Power Sources, 2018, 382, 176–178 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- A. Varzi, R. Raccichini, S. Passerini and B. Scrosati, J. Mater. Chem. A, 2016, 4, 17251–17259 RSC.
- D. T. Hallinan and N. P. Balsara, Annu. Rev. Mater. Res., 2013, 43, 503–525 CrossRef CAS.
- J. Kalhoff, G. G. Eshetu, D. Bresser and S. Passerini, ChemSusChem, 2015, 8, 2154–2175 CrossRef CAS PubMed.
- A. Hammami, N. Raymond and M. Armand, Nature, 2003, 424, 635–636 CrossRef CAS PubMed.
- B. Scrosati and C. A. Vincent, MRS Bull., 2000, 25, 28–30 CrossRef CAS.
- M. Armand, Solid State Ionics, 1983, 9–10(Part 2), 745–754 CrossRef CAS.
- M. Armand, Adv. Mater., 1990, 2, 278–286 CrossRef CAS.
- M. Wetjen, G.-T. Kim, M. Joost, G. B. Appetecchi, M. Winter and S. Passerini, J. Power Sources, 2014, 246, 846–857 CrossRef CAS.
- Y. Lin, Y. Cheng, J. Li, J. D. Miller, J. Liu and X. Wang, RSC Adv., 2017, 7, 24856–24863 RSC.
- M. A. Ratner and D. F. Shriver, Chem. Rev., 1988, 88, 109–124 CrossRef CAS.
- M. A. Ratner, P. Johansson and D. F. Shriver, MRS Bull., 2000, 25, 31–37 CrossRef CAS.
- W. A. Henderson and S. Passerini, Electrochem. Commun., 2003, 5, 575–578 CrossRef CAS.
- S. D. Druger, M. A. Ratner and A. Nitzan, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 3939–3947 CrossRef CAS.
- S. Béranger, M.-H. Fortier, D. Baril and M. B. Armand, Solid State Ionics, 2002, 148, 437–441 CrossRef.
- S. Zhang, Z. Chang, K. Xu and C. A. Angell, Electrochim. Acta, 2000, 45, 1229–1236 CrossRef CAS.
- X. Wei and D. F. Shriver, Chem. Mater., 1998, 10, 2307–2308 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- D. Golodnitsky, E. Strauss, E. Peled and S. Greenbaum, J. Electrochem. Soc., 2015, 162, A2551–A2566 CrossRef CAS.
- C. A. Angell, C. Liu and E. Sanchez, Nature, 1993, 362, 137–139 CrossRef CAS.
- F. L. Souza, E. Longo and E. R. Leite, ChemPhysChem, 2007, 8, 1778–1781 CrossRef CAS PubMed.
- K. I. S. Mongcopa, M. Tyagi, J. P. Mailoa, G. Samsonidze, B. Kozinsky, S. A. Mullin, D. A. Gribble, H. Watanabe and N. P. Balsara, ACS Macro Lett., 2018, 7, 504–508 CrossRef CAS.
- P. Johansson, J. Tegenfeldt and J. Lindgren, Polymer, 2001, 42, 6573–6577 CrossRef CAS.
- S. B. Aziz, T. J. Woo, M. F. Z. Kadir and H. M. Ahmed, Journal of Science: Advanced Materials and Devices, 2018, 3, 1–17 Search PubMed.
- H. Vogel, Z. Phys. Chem., 1921, 22, 645–646 CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- G. Tammann and W. Hesse, Z. Anorg. Allg. Chem., 1926, 156, 245–257 CrossRef CAS.
- G. Mao, M.-L. Saboungi, D. L. Price, M. Armand, F. Mezei and S. Pouget, Macromolecules, 2002, 35, 415–419 CrossRef CAS.
- D. Bresser, S. Passerini and B. Scrosati, Chem. Commun., 2013, 49, 10545–10562 RSC.
- W. H. Meyer, Adv. Mater., 1998, 10, 439–448 CrossRef CAS PubMed.
- J. Owen, in Comprehensive Polymer Science and Supplements, Elsevier, 1989, vol. 1, pp. 669–686 Search PubMed.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- D. J. Brooks, B. V. Merinov, W. A. Goddard, B. Kozinsky and J. Mailoa, Macromolecules, 2018, 51, 8987–8995 CrossRef CAS.
- B. K. Wheatle, J. R. Keith, S. Mogurampelly, N. A. Lynd and V. Ganesan, ACS Macro Lett., 2017, 6, 1362–1367 CrossRef CAS.
- B. K. Wheatle, N. A. Lynd and V. Ganesan, ACS Macro Lett., 2018, 7, 1149–1154 CrossRef CAS.
- W. H. Meyer, Adv. Mater., 1998, 10, 439–448 CrossRef CAS PubMed.
- J.-H. Shin, W. A. Henderson and S. Passerini, Electrochem. Commun., 2003, 5, 1016–1020 CrossRef CAS.
- J. W. Fergus, J. Power Sources, 2010, 195, 4554–4569 CrossRef CAS.
- E. Quartarone, P. Mustarelli and A. Magistris, Solid State Ionics, 1998, 110, 1–14 CrossRef CAS.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- D. Diddens and A. Heuer, J. Phys. Chem. B, 2014, 118, 1113–1125 CrossRef CAS PubMed.
- M. Joost, M. Kunze, S. Jeong, M. Schönhoff, M. Winter and S. Passerini, Electrochim. Acta, 2012, 86, 330–338 CrossRef CAS.
- D. Lin, W. Liu, Y. Liu, H. R. Lee, P.-C. Hsu, K. Liu and Y. Cui, Nano Lett., 2016, 16, 459–465 CrossRef CAS PubMed.
- R. J. Sengwa, P. Dhatarwal and S. Choudhary, Electrochim. Acta, 2014, 142, 359–370 CrossRef CAS.
- S. Murakami, K. Ueda, T. Kitade, Y. Ikeda and S. Kohjiya, Solid State Ionics, 2002, 154–155, 399–406 CrossRef CAS.
- J.-S. Chung and H.-J. Sohn, J. Power Sources, 2002, 112, 671–675 CrossRef CAS.
- Y. Ikeda, Y. Wada, Y. Matoba, S. Murakami and S. Kohjiya, Electrochim. Acta, 2000, 45, 1167–1174 CrossRef CAS.
- A. Nishimoto, M. Watanabe, Y. Ikeda and S. Kohjiya, Electrochim. Acta, 1998, 43, 1177–1184 CrossRef CAS.
- A. Thiam, C. Iojoiu, J.-C. Leprêtre and J.-Y. Sanchez, J. Power Sources, 2017, 364, 138–147 CrossRef CAS.
- Z. Gadjourova, Y. G. Andreev, D. P. Tunstall and P. G. Bruce, Nature, 2001, 412, 520–523 CrossRef CAS PubMed.
- A. M. Christie, S. J. Lilley, E. Staunton, Y. G. Andreev and P. G. Bruce, Nature, 2005, 433, 50–53 CrossRef CAS PubMed.
- C. Zhang, S. Gamble, D. Ainsworth, A. M. Z. Slawin, Y. G. Andreev and P. G. Bruce, Nat. Mater., 2009, 8, 580 CrossRef CAS PubMed.
- Z. Stoeva, I. Martin-Litas, E. Staunton, Y. G. Andreev and P. G. Bruce, J. Am. Chem. Soc., 2003, 125, 4619–4626 CrossRef CAS PubMed.
- P. Heitjans and S. Indris, J. Phys.: Condens. Matter, 2003, 15, R1257–R1289 CrossRef CAS.
- P. Johansson and P. Jacobsson, Electrochim. Acta, 2003, 48, 2279–2281 CrossRef CAS.
- S. H. Chung, Y. Wang, S. G. Greenbaum, D. Golodnitsky and E. Peled, Electrochem. Solid-State Lett., 1999, 2, 553–555 CrossRef CAS.
- D. Golodnitsky, E. Livshits and E. Peled, Macromol. Symp., 2003, 203, 27–46 CrossRef CAS.
- D. Golodnitsky and E. Peled, Electrochim. Acta, 2000, 45, 1431–1436 CrossRef CAS.
- S. Cheng, D. M. Smith and C. Y. Li, Macromolecules, 2014, 47, 3978–3986 CrossRef CAS.
- S. K. Fullerton-Shirey and J. K. Maranas, Macromolecules, 2009, 42, 2142–2156 CrossRef CAS.
- G. Mao, R. F. Perea, W. S. Howells, D. L. Price and M.-L. Saboungi, Nature, 2000, 405, 163 CrossRef CAS PubMed.
- A. Ferry, L. Edman, M. Forsyth, D. R. MacFarlane and J. Sun, J. Appl. Phys., 1999, 86, 2346–2348 CrossRef CAS.
- M. Forsyth, S. Jiazeng and D. MacFarlane, Electrochim. Acta, 2000, 45, 1249–1254 CrossRef CAS.
- G. A. Giffin, A. Moretti, S. Jeong and S. Passerini, J. Power Sources, 2017, 342, 335–341 CrossRef CAS.
- A. P. Sokolov and K. S. Schweizer, Phys. Rev. Lett., 2009, 102, 248301 CrossRef PubMed.
- A. L. Agapov and A. P. Sokolov, Macromolecules, 2011, 44, 4410–4414 CrossRef CAS.
- Y. Wang, A. L. Agapov, F. Fan, K. Hong, X. Yu, J. Mays and A. P. Sokolov, Phys. Rev. Lett., 2012, 108, 088303 CrossRef PubMed.
- K. Kunal, C. G. Robertson, S. Pawlus, S. F. Hahn and A. P. Sokolov, Macromolecules, 2008, 41, 7232–7238 CrossRef CAS.
- Y. Wang, F. Fan, A. L. Agapov, X. Yu, K. Hong, J. Mays and A. P. Sokolov, Solid State Ionics, 2014, 262, 782–784 CrossRef CAS.
- Y. Wang, F. Fan, A. L. Agapov, T. Saito, J. Yang, X. Yu, K. Hong, J. Mays and A. P. Sokolov, Polymer, 2014, 55, 4067–4076 CrossRef CAS.
- M. Adams, V. Richmond, D. Smith, Y. Wang, F. Fan, A. P. Sokolov and D. A. Waldow, Polymer, 2017, 116, 218–225 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- I. Osada, H. de Vries, B. Scrosati and S. Passerini, Angew. Chem., Int. Ed., 2016, 55, 500–513 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. Zheng, S. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- M. Lee, U. H. Choi, R. H. Colby and H. W. Gibson, Chem. Mater., 2010, 22, 5814–5822 CrossRef CAS.
- U. H. Choi, M. Lee, S. Wang, W. Liu, K. I. Winey, H. W. Gibson and R. H. Colby, Macromolecules, 2012, 45, 3974–3985 CrossRef CAS.
- M. D. Green, D. Salas-de la Cruz, Y. Ye, J. M. Layman, Y. A. Elabd, K. I. Winey and T. E. Long, Macromol. Chem. Phys., 2011, 212, 2522–2528 CrossRef CAS.
- D. Salas-de la Cruz, M. D. Green, Y. Ye, Y. A. Elabd, T. E. Long and K. I. Winey, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 338–346 CrossRef CAS.
- V. Delhorbe, D. Bresser, H. Mendil-Jakani, P. Rannou, L. Bernard, T. Gutel, S. Lyonnard and L. Picard, Macromolecules, 2017, 50, 4309–4321 CrossRef CAS.
- J. R. Sangoro, C. Iacob, A. L. Agapov, Y. Wang, S. Berdzinski, H. Rexhausen, V. Strehmel, C. Friedrich, A. P. Sokolov and F. Kremer, Soft Matter, 2014, 10, 3536–3540 RSC.
- Z. Wojnarowska, J. Knapik, J. Jacquemin, S. Berdzinski, V. Strehmel, J. R. Sangoro and M. Paluch, Macromolecules, 2015, 48, 8660–8666 CrossRef CAS.
- F. Fan, Y. Wang, T. Hong, M. F. Heres, T. Saito and A. P. Sokolov, Macromolecules, 2015, 48, 4461–4470 CrossRef CAS.
- F. Fan, W. Wang, A. P. Holt, H. Feng, D. Uhrig, X. Lu, T. Hong, Y. Wang, N.-G. Kang, J. Mays and A. P. Sokolov, Macromolecules, 2016, 49, 4557–4570 CrossRef CAS.
- Z. Wojnarowska, H. Feng, Y. Fu, S. Cheng, B. Carroll, R. Kumar, V. N. Novikov, A. M. Kisliuk, T. Saito, N.-G. Kang, J. W. Mays, A. P. Sokolov and V. Bocharova, Macromolecules, 2017, 50, 6710–6721 CrossRef CAS.
- S. Mogurampelly, J. R. Keith and V. Ganesan, J. Am. Chem. Soc., 2017, 139, 9511–9514 CrossRef CAS PubMed.
- E. W. Stacy, C. P. Gainaru, M. Gobet, Z. Wojnarowska, V. Bocharova, S. G. Greenbaum and A. P. Sokolov, Macromolecules, 2018, 51, 8637–8645 CrossRef CAS.
- D. Adam, P. Schuhmacher, J. Simmerer, L. Häussling, K. Siemensmeyer, K. H. Etzbachi, H. Ringsdorf and D. Haarer, Nature, 1994, 371, 141 CrossRef CAS.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- J. A. Siddiqui and P. V. Wright, Faraday Discuss. Chem. Soc., 1989, 88, 113–122 RSC.
- J. P. Patel, P. V. Wright, D. A. Dunmur and A. R. Tajbakhsh, Adv. Mater. Opt. Electron., 1994, 4, 265–271 CrossRef CAS.
- F. B. Dias, J. P. Voss, S. V. Batty, P. V. Wright and G. Ungar, Macromol. Rapid Commun., 1994, 15, 961–969 CrossRef CAS.
- F. B. Dias, S. V. Batty, J. P. Voss, G. Ungar and P. V. Wright, Solid State Ionics, 1996, 85, 43–49 CrossRef CAS.
- F. B. Dias, S. V. Batty, G. Ungar, J. P. Voss and P. V. Wright, J. Chem. Soc., Faraday Trans., 1996, 92, 2599–2606 RSC.
- F. B. Dias, S. V. Batty, A. Gupta, G. Ungar, J. P. Voss and P. V. Wright, Electrochim. Acta, 1998, 43, 1217–1224 CrossRef CAS.
- Y. Zheng, F. Chia, G. Ungar, T. H. Richardson and P. V. Wright, Electrochim. Acta, 2001, 46, 1397–1405 CrossRef CAS.
- Y. Zheng, F. Chia, G. Ungar and P. V. Wright, Chem. Commun., 2000, 1459–1460 RSC.
- Y. Zheng, P. V. Wright and G. Ungar, Electrochim. Acta, 2000, 45, 1161–1165 CrossRef CAS.
- G. S. McHattie, C. T. Imrie and M. D. Ingram, Electrochim. Acta, 1998, 43, 1151–1154 CrossRef CAS.
- C. T. Imrie, M. D. Ingram and G. S. McHattie, J. Phys. Chem. B, 1999, 103, 4132–4138 CrossRef CAS.
- C. T. Imrie, M. D. Ingram and G. S. McHattie, Adv. Mater., 1999, 11, 832–834 CrossRef CAS.
- C. T. Imrie and M. D. Ingram, Electrochim. Acta, 2001, 46, 1413–1417 CrossRef CAS.
- T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., 2006, 118, 44–74 CrossRef.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- T. Ohtake, K. Ito, N. Nishina, H. Kihara, H. Ohno and T. Kato, Polym. J., 1999, 31, 1155 CrossRef CAS.
- T. Ohtake, Y. Takamitsu, K. Ito-Akita, K. Kanie, M. Yoshizawa, T. Mukai, H. Ohno and T. Kato, Macromolecules, 2000, 33, 8109–8111 CrossRef CAS.
- N. S. Wanakule, A. Panday, S. A. Mullin, E. Gann, A. Hexemer and N. P. Balsara, Macromolecules, 2009, 42, 5642–5651 CrossRef CAS.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- K. Kishimoto, M. Yoshio, T. Mukai, M. Yoshizawa, H. Ohno and T. Kato, J. Am. Chem. Soc., 2003, 125, 3196–3197 CrossRef CAS PubMed.
- H. Shimura, M. Yoshio, A. Hamasaki, T. Mukai, H. Ohno and T. Kato, Adv. Mater., 2009, 21, 1591–1594 CrossRef CAS.
- M. Yoshio, T. Kagata, K. Hoshino, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2006, 128, 5570–5577 CrossRef CAS PubMed.
- T. Ichikawa, M. Yoshio, A. Hamasaki, J. Kagimoto, H. Ohno and T. Kato, J. Am. Chem. Soc., 2011, 133, 2163–2169 CrossRef CAS PubMed.
- T. Ichikawa, M. Yoshio, A. Hamasaki, S. Taguchi, F. Liu, X. Zeng, G. Ungar, H. Ohno and T. Kato, J. Am. Chem. Soc., 2012, 134, 2634–2643 CrossRef CAS PubMed.
- Z. Stoeva, Z. Lu, M. D. Ingram and C. T. Imrie, Electrochim. Acta, 2013, 93, 279–286 CrossRef CAS.
- S. Wang, X. Liu, A. Wang, Z. Wang, J. Chen, Q. Zeng, X. Jiang, H. Zhou and L. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 25273–25284 CrossRef CAS PubMed.
- P. W. Majewski, M. Gopinadhan, W.-S. Jang, J. L. Lutkenhaus and C. O. Osuji, J. Am. Chem. Soc., 2010, 132, 17516–17522 CrossRef CAS PubMed.
- K. Lu, J. K. Maranas and S. T. Milner, Soft Matter, 2016, 12, 3943–3954 RSC.
- N. H. LaFemina, Q. Chen, R. H. Colby and K. T. Mueller, J. Chem. Phys., 2016, 145, 114903 CrossRef.
- C. Brissot, M. Rosso, J.-N. Chazalviel, P. Baudry and S. Lascaud, Electrochim. Acta, 1998, 43, 1569–1574 CrossRef CAS.
- C. Brissot, M. Rosso, J.-N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81–82, 925–929 CrossRef CAS.
- M. Rosso, T. Gobron, C. Brissot, J.-N. Chazalviel and S. Lascaud, J. Power Sources, 2001, 97–98, 804–806 CrossRef CAS.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- M. D. Tikekar, S. Choudhury, Z. Tu and L. A. Archer, Nat. Energy, 2016, 1, 16114 CrossRef CAS.
- R. Rohan, Y. Sun, W. Cai, Y. Zhang, K. Pareek, G. Xu and H. Cheng, Solid State Ionics, 2014, 268, 294–299 CrossRef CAS.
- R. Rohan, K. Pareek, Z. Chen, W. Cai, Y. Zhang, G. Xu, Z. Gao and H. Cheng, J. Mater. Chem. A, 2015, 3, 20267–20276 RSC.
- B. Qin, Z. Liu, J. Zheng, P. Hu, G. Ding, C. Zhang, J. Zhao, D. Kong and G. Cui, J. Mater. Chem. A, 2015, 3, 7773–7779 RSC.
- C. Li, B. Qin, Y. Zhang, A. Varzi, S. Passerini, J. Wang, J. Dong, D. Zeng, Z. Liu and H. Cheng, Adv. Energy Mater., 2018, 1803422 Search PubMed.
- H. Oh, K. Xu, H. D. Yoo, D. S. Kim, C. Chanthad, G. Yang, J. Jin, I. A. Ayhan, S. M. Oh and Q. Wang, Chem. Mater., 2016, 28, 188–196 CrossRef CAS.
- H.-D. Nguyen, G.-T. Kim, J. Shi, E. Paillard, P. Judeinstein, S. Lyonnard, D. Bresser and C. Iojoiu, Energy Environ. Sci., 2018, 11, 3298–3309 RSC.
- K. M. Diederichsen, R. R. Brow and M. P. Stoykovich, ACS Nano, 2015, 9, 2465–2476 CrossRef CAS PubMed.
- D. Bresser, M. Leclere, L. Bernard, P. Rannou, H. Mendil-Jakani, S. Lyonnard and L. Picard, presented in part at the International Symposium on Polymer Electrolytes (ISPE) XV, Uppsala, Sweden, August, 2016 Search PubMed.
- M. Zimmerman, U.S. Pat., US9819053B1, Ionic Materials, Inc., 2013 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |