
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards efficient initiators for two-photon induced polymerization: fine tuning of the donor/acceptor properties†
Brigitte
Holzer
 *a,
Markus
Lunzer
ab,
Arnulf
Rosspeintner
c,
Giuseppe
Licari
c,
Maximilian
Tromayer
ab,
Sergej
Naumov
d,
Daniel
Lumpi
a,
Ernst
Horkel
a,
Christian
Hametner
a,
Aleksandr
Ovsianikov
b,
Robert
Liska
a,
Eric
Vauthey
c and
Johannes
Fröhlich
a
*a,
Markus
Lunzer
ab,
Arnulf
Rosspeintner
c,
Giuseppe
Licari
c,
Maximilian
Tromayer
ab,
Sergej
Naumov
d,
Daniel
Lumpi
a,
Ernst
Horkel
a,
Christian
Hametner
a,
Aleksandr
Ovsianikov
b,
Robert
Liska
a,
Eric
Vauthey
c and
Johannes
Fröhlich
a
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, A-1060 Vienna, Austria. E-mail: brigitte.holzer@tuwien.ac.at
bDepartment of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, CH-1211 Geneva, Switzerland
cInstitute of Materials Science and Technology, TU Wien, Getreidemarkt 9/308, A-1040 Vienna, Austria
dLeibniz Institute of Surface Engineering (IOM), Permoserstrasse 15, D-04318 Leipzig, Germany
First published on 12th February 2019
Abstract
In this work we present the design, synthesis and systematic investigation of the optical properties of symmetric triphenylamine (TPA)-substituted thiophenes. The use of electron-donating (–OMe, –tBu, –Me, –TMS), -neutral (–H) or -withdrawing (–F, –CN, –SO2Me) substituents gives rise to D–A–D based two-photon absorption (2PA) chromophores. The photophysical properties of these compounds, including one-photon absorption and 2PA using two-photon-excited fluorescence, were investigated in different organic solvents with varying polarity. The maximum 2PA cross sections prove to be strongly dependent on the nature of the TPA substituent and range between ∼173 GM (Goeppert-Mayer units) and 379 GM. Although most of the investigated substances also exhibit high fluorescence quantum yields, two-photon absorption screening tests of an acrylate monomer formulation revealed the efficiency of these materials as 2PA photoinitiators. These results are supported by quantum chemical calculations of the spin density distribution indicating that the mechanism of polymerization initiation using acrylate monomer is favored by strong localization of the unpaired electrons in the triplet state on the C2 carbon of the thiophene moiety.
Design, System, ApplicationPrinting cubic centimetre volumes with sub-micrometre feature sizes at fast writing speeds by means of two-photon induced polymerization still constitutes a challenge particularly in regard to commercial applications. In this context, this work covers the design, synthesis and systematic investigation of the optical properties of symmetric triphenylamine-substituted thiophenes as a new substance class of two-photon absorption photoinitiators. The introduction of a variety of electron-donating, -neutral and -withdrawing substituents on the triphenylamine moiety gives rise to D–A–D two-photon absorption chromophores and allows for tailoring their photophysical properties. Two-photon absorption screening tests of an acrylate monomer formulation reveal the efficiency of these materials as two-photon absorption photoinitiators over a broad processing window. An in-depth analysis based on quantum chemical calculations provides mechanistic information on the polymerization initiation process using acrylate monomers. The straightforward synthetic route combined with high two-photon absorption efficiency renders this compound class an attractive platform for two-photon-based applications also beyond two-photon induced polymerization. Hence, the structural versatility and capability of our molecular design may contribute to further developments in other areas of functional organic materials. |
Introduction
Materials exhibiting high two-photon absorption (2PA) are increasingly used in various fields of materials and biomedical sciences as they offer unique properties;1,2 applications include three-dimensional optical data storage,3–5 optical power limiting,6 two-photon fluorescence imaging,7 photodynamic therapy8,9 and multiphoton lithography.10–14 The key feature of these two-photon induced processes is based on the nonlinearity of the underlying absorption. In the past decade, multiphoton lithography has gained scientific attention due to its high potential for industrial applications as well as the benefits of this technique.15–17 Two-photon induced photopolymerization (2PP) enables true 3D printing within a monomer formulation featuring excellent spatial control and takes advantage of well-defined penetration depth by the use of a long wavelength excitation source (typically ∼800 nm).18,19 While the size of the activated volume (voxel) can be below the diffraction limit, positions outside of this volume are not affected. Consequently, this polymerization technique offers high resolution while minimizing unwanted thermal or photochemical initiation and degradation processes usually associated with excitation of UV-photoinitiators.20 Nevertheless, the development of powerful two-photon absorption photoinitiators (2PA PIs) featuring high two-photon cross sections (σ2PA) constitutes a significant challenge in ensuring high efficiency of the 2PP process especially with regard to scanning speeds of several cm s−1.15,20Generally, the nature of the excited state in organic molecules crucially determines their optical properties. Therefore, rational design strategies for tailored 2PA PIs may allow for efficient photopolymerization. Powerful 2PA PIs have been developed by combining (i) strong donor (D) and/or acceptor (A) chromophore groups as well as (ii) building blocks with good molecular coplanarity.2,21,22 Triphenylamine (TPA)-based donors/acceptors proved to be promising subunits to compose molecules with large σ2PA.23–25 Furthermore incorporation of aromatic heterocycles in dipolar D–π–A or quadrupolar D–π–D, A–π–A, D–π–A–π–D or A–π–D–π–A type molecules gave rise to extended π-conjugated networks with enhanced optical properties, increasing both linear extinction coefficient and σ2PA.19,21,26 Particularly the application of thiophene as planar π-linker yielded materials with high σ2PA.2,27,28
Recently, we published on TPA-substituted oligothiophenes constituting a new substance class suitable for tailoring emission properties (Scheme 1A, BRA-1T, R denoting different substituents).29–31 First photophysical results also revealed the strong two-photon absorption of these compounds32 suitable for 2PP (Scheme 1B). In this article, we investigate the photophysical properties including the excited state dynamics of these novel 2PA PIs and correlate these findings to the electronic nature of the substituent R on the TPA moiety (varying from electron-donating (–OMe, –tBu, –Me, –TMS), electron-neutral (–H) to electron-withdrawing (–F, –CN, –SO2Me)).
 | ||
| Scheme 1 A Synthetic pathways towards triphenylamine-substituted thiophene motifs BMOA-1T, BtBuA-1T, BMA-1T, BTMSA-1T, BHA-1T, BFA-1T, BCNA-1T and BSO2MA-1T bearing different substituents ranging from electron-donating (–OMe > –tBu > –Me > –TMS) to -neutral (–H) to -withdrawing residues (–F > –CN > –SO2Me). Reaction yields are included for each target compound. i: KOtBu, allyl[1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene]chloropalladium(II), (IPr)Pd(allyl)Cl, isopropanol/water, reflux; ii: KOtBu, DMSO, 120 °C. B Darkfield ultraviolet microscopy image of defined woodpile structure obtained from speed power screening tests of BHA-1T. | ||
Measurements of optical properties (e.g. linear absorption spectra, fluorescence quantum yields and two-photon absorption spectra) of the obtained materials are correlated to the substituent's nature and are supported by density functional theory (DFT) calculations. The obtained materials are characterized with respect to their applicability in 2PP structuring tests.
Results and discussion
Design and synthesis
In general, TPA building blocks constitute promising core structures with large σ2PA related to their great number of π-electrons and strong electron-donating ability.23 In this study, the electronic properties of the TPA subunit were systematically altered by the incorporation of different substituents ranging from electron-donating to electron-withdrawing residues (–OMe > –tBu > –Me > –TMS > –H > –F > –CN > –SO2Me). The synthetic assembly of these potential D–A–D 2PA PIs was realized for compounds BMOA-1T, BtBuA-1T, BMA-1T, BTMSA-1T, BHA-1T and BFA-1Tvia Suzuki cross-coupling reactions and for compounds BCNA-1T and BSO2MA-1T by nucleophilic aromatic substitution (Scheme 1A). While the syntheses of BMOA-1T, BMA-1T, BHA-1T and BFA-1T have been reported earlier,29,32 this work presents the synthetic approach towards all target compounds: a general Suzuki cross-coupling procedure developed in our group tolerating a broad spectrum of functionalized TPA boronic acid esters31 was applied for the synthesis of BMOA-1T, BtBuA-1T, BMA-1T, BTMSA-1T, BHA-1T and BFA-1T from 1a–f and 2,5-dibromothiophene 2. Since the synthesis of triphenylamineboronic acid pinacolates bearing electron-withdrawing substituents proved to be troublesome in earlier studies,31 nucleophilic aromatic substitution was chosen as an alternative synthetic approach towards BCNA-1T and BSO2MA-1T starting from amine 3 and fluorobenzenes 4a,b using KOtBu as base in DMSO at 120 °C (see ESI†).Linear optical properties
Electronic absorption spectra of all target compounds were measured in n-hexane (n-Hex), acetonitrile (ACN) and tetrahydrofuran (THF). The absorption spectra of all compounds are virtually independent of solvent polarity in terms of band shape, band position (see Fig. 1) and intensity over the entire observed wavelength range. In particular, a strong absorption band around 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 (400 nm) is observed for BMOA-1T, BtBuA-1T, BMA-1T, BHA-1T and BFA-1T. This band is increasingly blue-shifted for BTMSA-1T, BCNA-1T and BSO2MA-1T. Fig. 1 shows that the lowest energy absorption band exhibits a distinct red-shift when going from electron-withdrawing to donating TPA substituents. In total, little more than a 1100 cm−1 red-shift is observed when going from BSO2MA-1T to BMOA-1T (see Fig. 3b). However, the transition dipole moment of this lowest energy absorption transition is unaltered by the nature of the substituent.
000 cm−1 (400 nm) is observed for BMOA-1T, BtBuA-1T, BMA-1T, BHA-1T and BFA-1T. This band is increasingly blue-shifted for BTMSA-1T, BCNA-1T and BSO2MA-1T. Fig. 1 shows that the lowest energy absorption band exhibits a distinct red-shift when going from electron-withdrawing to donating TPA substituents. In total, little more than a 1100 cm−1 red-shift is observed when going from BSO2MA-1T to BMOA-1T (see Fig. 3b). However, the transition dipole moment of this lowest energy absorption transition is unaltered by the nature of the substituent.
 | ||
| Fig. 1 Absorption and emission spectra of all samples in n-Hex (blue), tetrahydrofuran (red) and acetonitrile (green). The low solubility of BSO2MA-1T did not allow recording spectra in n-Hex. | ||
Comparison of the absorption spectra of BMA-1T with those of its building blocks (TPA33 and TPA-substituted thiophene TPA-T,34,35Fig. 2) allows for a tentative assignment of the observed bands. First, the short-wavelength band at approximately 33000 cm−1 (300 nm) can be ascribed to the local transition of the substituted TPA moiety (Fig. 2d). Depending on the electronic nature of the substituents on the TPA moiety this transition can be red-shifted to 29000 cm−1 (350 nm) for the cyano and sulfone group33 leading to a larger extinction coefficient of the corresponding absorption band in the case of the cyano substituted compound.
 | ||
| Fig. 2 a) Excitation anisotropy of BMA-1T in n-Hex. b) Spectral decomposition of the absorption spectrum of BMA-1T using line shape functions based on the displaced harmonic oscillator model and corresponding residuals (vertically offset grey line). c) Absorption spectrum of 4-[5-(3-bromophenyl)-2-thienyl]-N,N-bis(4-methylphenyl)benzenamine (TPA-T, in n-Hex),35 its spectral decomposition and the corresponding residuals (vertically offset). d) Absorption spectrum of triphenylamine (in THF). | ||
In fact, as can be observed in Fig. 2 the extinction coefficient of this band roughly doubles that of the unmodified TPA (ε = 20000 M−1 cm−1) owing to the presence of two TPA moieties in all symmetric BRA-1T molecules.36 Secondly, the broad and structureless lowest energy transition centered around 25000 cm−1 (400 nm) seems to be composed of two distinct bands with considerably different transition dipole moments (Fig. 2). Comparing a spectral decomposition of said absorption band for e.g.BMA-1T with that of the single branch D–π–A counterpart TPA-T reveals, that for BMA-1T indeed a weak high-energy shoulder can be identified, which is not observed for TPA-T (see the ESI† for spectral decompositions of all samples). A distinct change in the fluorescence excitation anisotropy of BMA-1T at 28600 cm−1 (350 nm) provides additional evidence for the above observation.
A qualitative explanation of these spectral changes can be given within the framework of the Frenkel exciton model.37 Herein, the excitonic interaction between the two TPA branches in BRA-1T is expected to split the lowest energy transition into two bands: a strongly allowed one-photon (1P) transition at lower energy than that of the single branch, and a one-photon forbidden, but two-photon (2P) allowed transition (see below) at higher energy. The sum of the squared transition dipole moments for these two transitions is expected to be twice that of the single branch.37 A straightforward spectral decomposition could be performed for all spectra with sufficient separation of the two “excitonic” bands from the local TPA transition. Indeed, for these compounds the so obtained transition dipole moments obey this prediction (see Table 1).
| ν 1PA | λ 1PA | ε | ϕ F | τ | ν 2PA | λ 2PA | σ 2PA | ν 1 | μ 10 , | ν 2 | μ 20 , | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [103 cm−1] | [nm] | [M−1 cm−1] | [ns] | [103 cm−1] | [nm] | [GM] | [103 cm−1] | [D] | [103 cm−1] | [D] | ||
|
a Wavenumber of the 1PA maximum.
b Wavelength of the 1PA maximum.
c Extinction coefficient (the molar extinction coefficients have been obtained using an all-optical method suggested in ref. 41 and outlined in the ESI).
d Fluorescence quantum yield.
e Fluorescence lifetime.
f Wavenumber at which the maximum 2PA value is obtained (due to the limited measurement range this does not necessarily correspond to a band maximum).
g Wavelength at which the maximum 2PA value is obtained (due to the limited measurement range this does not necessarily correspond to a band maximum).
h 2PA cross section at the 2PA maximum, values in parenthesis correspond to 12500 cm−1 (800 nm).
i Wavenumber of the first vibronic transitions of electronic transition x.
j Transition dipole moment for the x ← 0 absorption transition.
k Measured in n-Hex and taken from ref. 35.
l Calculations are given in the ESI.
|
||||||||||||
| BMOA-1T | 25.0 | 400 | 66200 |
0.57 | 1.29 | 28.9 | 346 | 379 (28) | 23.43 | 9.8 | 28.53 | 4.2 |
| BtBuA-1T | 25.3 | 396 | 65100 |
0.44 | 0.85 | 28.9 | 346 | 358 (28) | 23.67 | 9.8 | 29.18 | 3.5 |
| BMA-1T | 25.3 | 396 | 65400 |
0.40 | 0.79 | 28.9 | 346 | 331 (30) | 23.71 | 9.8 | 29.19 | 3.9 |
| BTMSA-1T | 25.4 | 394 | 66100 |
0.40 | 0.75 | 28.8 | 347 | 345 (24) | 23.82 | 9.8 | 30.92 | 3.7 |
| BHA-1T | 25.6 | 390 | 66500 |
0.34 | 0.65 | 29.4 | 340 | 301 (18) | 24.00 | 9.7 | 29.78 | 4.7 |
| BFA-1T | 25.8 | 387 | 70700 |
0.33 | 0.64 | 29.4 | 340 | 301 (19) | 24.12 | 9.7 | 29.14 | 5.1 |
| BCNA-1T | 28.4 | 352 | 97000 |
0.39 | 0.65 | 29.4 | 340 | 213 (13) | 24.54 | 9.8 | ||
| BSO 2 MA-1T | 29.0 | 345 | 68100 |
0.36 | 0.64 | 29.4 | 340 | 173 (11) | 24.57 | 9.5 | ||
| TPA-T | 26.2 | 381 | 35000 |
n.m. | n.m. | n.m. | n.m. | n.m. | n.d. | 7.3 | ||
With the exception of a constant energy offset38 (4400 cm−1 with respect to the low energy band maximum) the quantum mechanical calculations reproduce the experimental spectra in terms of relative band positions and intensities exceptionally well, as can be seen in Fig. 3a and b and 4.
 | ||
| Fig. 3 Substituent dependence of a) experimental (full, in THF) and calculated (open, gas phase) absorption transition dipole moment, μ10, b) the relative position of the first absorption band with respect to substituent R = H in THF (calculated in gas phase – open; experimental – full), c) energy difference of the highest energy vibronic band of the first absorption band between acetonitrile (ACN) and n-hexane (n-Hex), and d) energy difference of the lowest energy vibronic band of the emission band between acetonitrile and n-Hex. The substituents have been arranged from left to right according to decreasing electron-donating ability. Due to the limited solubility no absorption data for BCNA-1T and BSO2MA-1T and no emission data for BSO2MA-1T in n-Hex are available. | ||
 | ||
| Fig. 4 One- (blue) and two-photon (red) absorption spectra. The calculated stick spectra – representing the energy of the corresponding electronic states – have been red-shifted by 4400 cm−1 and a single common scaling factor for 1PA and 2PA (4.4) has been applied (see ESI† for further details) to show these data in the same graph. | ||
Notably, all compounds show quantum yields in the range from 0.33 to 0.57 and fluorescence lifetimes in the range from 0.64 to 1.29 ns. Absorption and excitation spectra are congruent to below 33000 cm−1 (300 nm), thus justifying the assumption of validity of Kasha's rule39 for the samples under investigation. The emission spectra in apolar n-Hex show significant vibronic structure, thus contrasting with the rather structureless absorption spectra. As a consequence, absorption and emission spectra do not show mirror symmetry, pointing towards significant changes in the ground and excited state geometries.32 However, except for the methoxy-substituted BMOA-1T, the emission transition dipole moments do not depend on the solvent polarity, which is in line with the findings by Beckwith et al.32
Fig. 3 clearly shows, that contrary to the quite negligible absorption solvatochromism (Fig. 3c) the samples exhibit a distinct fluorescence solvatochromism (Fig. 3d), which becomes more pronounced with increasing electron-donating ability of the TPA substituent. These observations have recently been assigned as manifestations of the absence (e-withdrawing) or presence (e-donating) of excited state symmetry breaking, which had been independently probed via the dynamics of the emission transition dipole moment.32
In essence, stronger electron-donating substituents on the TPA unit (i) shift the optical bandgap to lower energies, (ii) do not significantly alter the transition dipole moment of this lowest energy absorption transition, but (iii) lead to an increased probability for excited state symmetry breaking and thus pronounced fluorescence solvatochromism.
Two-photon absorption
Two-photon absorption spectra of the compounds BMOA-1T, BtBuA-1T, BMA-1T, BTMSA-1T, BHA-1T, BFA-1T, BCNA-1T and BSO2MA-1T were measured in THF using wavelengths from 680 to 960 nm, corresponding to transition wavenumbers from 29400 to 20800 cm−1 applying the two-photon excited fluorescence technique reported by Makarov et al.40Fig. 4 shows that the 2PA and 1PA spectra are complementary in the wavenumber range of the two lowest energy transitions, i.e. the 2PA is weak where the 1PA is strong and vice versa. This feature can be especially well observed for those samples where the local TPA transition is located at sufficiently high energies to not overlap.
However, for BTMSA-1T, BCNA-1T and BSO2MA-1T, just as in the 1PA spectrum, the local TPA transition is expected to show up as well and distort the spectra. Fig. 4 also shows, that – at least within the observed wavenumber range – two trends can be observed as a function of the TPA substituent group: firstly, the less electron-donating the substituent group is, the more the position of the 2PA maximum is shifted to the blue as similarly observed for the 1PA spectra. Judging from the onset of the strong 2PA band, this blue-shift is significantly more pronounced than for the strong 1PA band (2000 instead of 1000 cm−1). Secondly, quite in contrast to the 1PA observations, it seems that the 2PA cross section also diminishes with less and less electron-donating substituents (from 380 GM at 29000 cm−1 for BMOA-1T to 170 GM at 30600 cm−1 for BSO2MA-1T). The observed wavenumber range does not allow for the unambiguous observation of the 2PA maxima of all samples. However, as all of the above observations are almost quantitatively reproduced by the results of quantum mechanical calculations (Fig. 3, 4 and ESI† Fig. S17, Table S1) these can be used to gain further insight. It is clear that additional weaker (approx. 50 GM) 2PA bands show up at slightly higher energy than the strong 2PA transition for BTMSA-1T and at slightly lower energy for BCNA-1T and BSO2MA-1T and that the intensity of the 2P-allowed transition considerably decreases upon increasing the electron withdrawing character of the substituent.
Exited state dynamic
Rational design of 2PA PIs may allow for modulation of singlet/triplet excited states of chromophores. In order to investigate the effect of the substituents of the TPA moiety on the fluorescence decay dynamics, transient absorption measurements were carried out for compounds BMOA-1T, BMA-1T and BFA-1T. To gain insight into the fate of the first electronically excited state, the rate constants of all relevant deactivation processes were determined, i.e. fluorescence (F), internal conversion (IC) and intersystem crossing (ISC), for the three representative compounds (Table 2) using:with ϕF,ISC and τ being the fluorescence or intersystem crossing quantum yield and the fluorescence lifetime. Fig. 5 shows that the rate constant for fluorescence (radiative decay) kF for all compounds exhibits only a relatively small variation, while the associated errors on the per se small internal conversion rates kIC do not allow for observing any clear trend with the S0–S1 energy gap, νϕ0.

| R | Solvent | ν f0 | τ F | ϕ F | ϕ ISC |
|---|---|---|---|---|---|
| [103 cm−1] | [ns] | ||||
| a Transition energy for the highest energy vibronic transition of fluorescence. b Fluorescence lifetime. c Quantum yield for fluorescence. d Quantum yield for intersystem crossing. | |||||
| OMe | n-Hex | 22.36 | 0.52 | 0.28 | 0.61 |
| THF | 21.47 | 1.29 | 0.57 | 0.36 | |
| ACN | 21.09 | 2.57 | 0.69 | 0.17 | |
| Me | n-Hex | 22.71 | 0.49 | 0.28 | 0.60 |
| THF | 22.04 | 0.85 | 0.40 | 0.48 | |
| ACN | 21.64 | 1.27 | 0.50 | 0.35 | |
| F | n-Hex | 23.19 | 0.50 | 0.32 | 0.61 |
| THF | 22.57 | 0.65 | 0.33 | 0.60 | |
| ACN | 22.26 | 0.89 | 0.39 | 0.51 | |
 | ||
| Fig. 5 Dependence of the rate constants, kF, kIC and kISC, on the S0–S1 energy gap for BMOA-1T, BMA-1T and BFA-1T in ACN, THF and n-Hex. The grey circles denote the data points for samples not tabulated in Table 2, using only fluorescence data. Note, that the corresponding points in the ISC-panel denote the sum of IC and ISC. | ||
However, a dramatic decrease in the intersystem crossing rates kISC upon decreasing the energy gap by changing the solvent polarity could be observed. Rather than being attributable to the S0–S1 energy gap this decrease is most likely associated to a change in the S0–Tn gap. Thus, the intersystem crossing yield strongly depends on both, the nature of the TPA substituent as well as the polarity of the solvent. The higher the donor strength of the substituent (OMe > Me > F) and the more polar the solvent (ACN > THF > n-Hex), the smaller is the intersystem crossing quantum yield. Assuming that the triplet state constitutes the reactive species for the polymerization initiation, the two-photon triplet yield, i.e. ϕT = ϕISCσ2PA can be defined as a figure of merit.
While the two-photon absorption cross sections can be expected to depend only marginally on solvent polarity,42 the intersystem crossing rate constants, on the other hand, do depend strongly. It can thus be concluded, that the efficiency of potential 2PA PIs strongly depends on the rate of the intersystem crossing and thus on the polarity of the monomer formulation. As has been shown, given the quadrupolar nature of the electronic ground state of compounds BMOA-1T, BMA-1T and BFA-1T the position of the absorption (one- and two-photon) does not change upon changing the solvent polarity. However, electron-donating groups enhance the dipolar character of the TPA–thiophene part in the excited state, increasing the 2PA cross section (Table 1), but decreasing the ISC yield (Table 2) especially in polar solvents due to symmetry breaking and solvent relaxation.
These findings are further supported by DFT calculations for the model structures BHA-1T, BMOA-1T and BSO2MA-1T bearing electron-donating, neutral and withdrawing substituents. The optimized structures of ground and triplet state, electron distributions of the occupied and unoccupied molecular orbitals (MOs) involved in the population of the lowest excited states, calculated UV/vis spectra as well as spin density distribution of the optimized triplet states of the new 2PA PIs are given in the ESI† (Fig. S20–S26). The calculated energy scheme for the population of the reactive excited triplet state of the studied 2PA PIs is given in Fig. 6. After excitation into the S1 exited state, the triplet state T2 is populated via intersystem crossing. Internal conversion to the T1 state, which then undergoes further relaxation through adjustment of the molecular structure to the changes of electron distributions through the excitation, results in the relaxed T1 state. This optimized triplet state is an actually photochemically reactive state potentially responsible for the initiating of the polymerization process. The possible mechanism of initiation will be discussed further (see Fig. 8). The calculations show that the first excited state S1 is populated mostly by HOMO–LUMO π-electron excitation for all studied 2PA PIs. The MOs involved in the population of the first excited S1 (π, π*) and T1 (π, π*) of all studied 2PA PIs are given in the ESI† (Fig. S21, S23 and S25).
 | ||
| Fig. 6 Jablonski energy diagram of the population of the reactive triplet by two-photon excitation of studied 2PA PIs (calculated in THF at the TDM06-2X/6-311+G(d,P)/PBF level); f-oscillator strength. | ||
Two-photon induced polymerization testing
Printing cubic centimeter volumes with (sub-)micrometer feature sizes at high writing speeds still remains a challenge particularly in regard to commercial applications. As the laser power required for two-photon polymerization increases proportionally to the square root of the writing speed, for high throughput mass production, specifically designed highly sensitive 2PA PIs are required.15 An efficient method to evaluate the activity of 2PA PIs comprises the fabrication of defined 3D test structures applying various laser powers and writing speeds.In order to assess the ideal processing window of the investigated compounds BMOA-1T, BtBuA-1T, BMA-1T, BTMSA-1T, BHA-1T, BFA-1T, BCNA-1T and BSO2MA-1T; defined woodpiles (lateral dimension: 50 μm, 5 μm hatch distance, 0.7 μm layer distance, 20 layers) were fabricated using a fs-pulsed NIR laser (∼800 nm, 72 fs pulse duration) in an acrylate-based test resin formulation by means of 2PP. In earlier studies, B3FL (2,7-bis[[4-(dibutylamino)phenyl]ethynyl]-9H-fluoren-9-one) was shown to outperform commercially available 1PA initiators such as Irgacure 369 already in low concentrations in two-photon-induced photopolymerization processes.43,44 For this reason, B3FL was used as reference 2PA PI (the two-photon absorption spectrum of B3FL is given in the ESI,† in Fig. S18). The laser powers and the writing speeds were varied in a range of 10–100 mW (measured after passing the 20× microscope objective) and 0.316–316 mm s−1, respectively. The obtained 3D woodpile structures were analyzed by scanning electron microscopy (SEM).
The quality of the micro-fabricated woodpiles was evaluated using a four colour classification reported earlier45 (Fig. 7): class A (green) defines excellent structures with fine hatch lines and class B (yellow) good structures with thicker hatch lines or slightly contorted shapes. Objects rated as class C (red) have identifiable shapes but with small errors (e.g. holes and burst regions caused by overexposure), whereas woodpiles rated as class D (blue) are not identifiable as such anymore (close to polymerization threshold). All investigated 2PA PIs BMOA-1T, BtBuA-1T, BMA-1T, BTMSA-1T, BHA-1T, BFA-1T, BCNA-1T and BSO2MA-1T produce defined woodpile structures rendering this substance class an ideal platform for potent tailored 2PA PIs.
 | ||
| Fig. 7 Speed power screening of all target 2PA PI compounds and our previously published efficient 2PA PI B3FL. Woodpile structures were fabricated using a fs-pulsed NIR laser (∼800 nm, 72 fs pulse duration). In these SEM pictures (originals and magnification of woodpiles of a test series using BMA-1T are given in Fig. S27–29†) the abscissa describes the writing speed in semi-logarithmic steps from 0.316 to 316 mm s−1 (0.316, 1, 3.16, 10, 31.6, 100 and 316 mm s−1) and the ordinate the laser powers ranging from 10–100 mW (10, 20, 30, 40, 50, 60, 70, 80, 90 and 100 mW). A mixture (1:1 w/w) of triacrylates ETA and TTA was used as test resin. | ||
Whereas compounds BMOA-1T, BtBuA-1T, BMA-1T and BTMSA-1T bearing electron-donating substituents on the TPA generally showed a broader processing window, compounds BFA-1T, BCNA-1T and BSO2MA-1T with electron-withdrawing substituents only gave perfect woodpile structures at comparably lower laser powers and moderate writing speeds making the latter compounds less qualified for 2PP at the parameters used. Notably, BHA-1T bearing an electronically neutral substituent exhibits excellent performance at both low and high writing speeds making it the most applicable 2PA PI of the investigated compounds. Compared to one of our previously published highly efficient 2PA PIs, B3FL,43BHA-1T shows an even broader processing window, especially at higher writing speeds rendering it the most promising dye for 2PP.
Mechanistic considerations
Although, similar photosensitizers have been reported in the literature21,25 in general there are scarce reports on the actual two-photon absorption mechanism.46,47 In order to overcome this shortage of any experimental hints on the nature and origin of the photopolymerization process, quantum chemical calculations were performed for the model 2PA PIs BRA-1T to understand the possible initiation mechanism via the photochemically reactive relaxed triplet state T1 in more detail. As calculations show, all reactions resulting in monomolecular bond scissions or intramolecular rearrangements are strongly endergonic and therefore do not lead to formation of reactive radicals (Fig. 8 for the case of BHA-1T).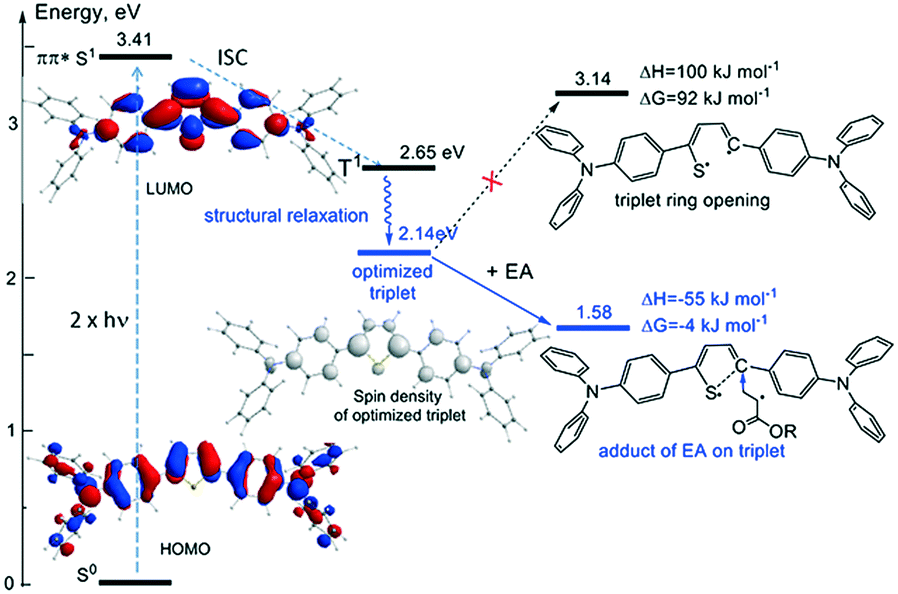 | ||
| Fig. 8 Calculated energy scheme for the possible two-photon initiation of BHA-1T as 2PA PI, which can lead to formation of the propagating radical through addition of a monomer acrylate (ethyl acrylate (EA) as model structure) followed by C–S bond scission. | ||
Yet, the possible thiophene ring opening in triplet state was calculated to be strongly endergonic, with Gibbs free energies for the bond cleavage up to −92 kJ mol−1, therefore rendering 2PP unlikely. Nevertheless, the spin density distribution of un paired electrons in the triplet state shows strong localization on the C2 position of the thiophene ring indicating this to be the most reactive position for a possible addition reaction (see Fig. 8 for the case of BHA-1T). As calculated for the compounds BHA-1T, BMOA-1T and BSO2MA-1T, the addition reaction of monomer acrylate (ethyl acrylate (EA) as model structure) to the C2 position of the thiophene ring followed by ring opening is energetically favourable and leads to the exergonic formation of a propagating radical, thus, rendering photopolymerization feasible.
Conclusion
In this study a series of thiophenes bearing p-substituted triarylamines with various electronically different substituents capable of initiating polymerization upon two-photon excitation has been synthesized. From a systematic evaluation of structure–property relationships via electronic spectroscopy it can be concluded that strong electron-donating substituents on the TPA unit (i) shift the optical bandgap to lower energies, (ii) do not significantly alter the transition dipole moment of this lowest energy absorption transition, but (iii) lead to an increased probability for excited state symmetry breaking and thus pronounced fluorescence solvatochromism. Especially in polar solvents a decrease in ISC yield could be determined originating from symmetry breaking and solvent relaxation. Therefore, it can be concluded, that the efficiency of potential 2PA PIs strongly depends on the rate of the intersystem crossing and thus on the polarity of the used monomer formulation. Furthermore, when altering the electronic properties of the TPA substituent from donating to accepting, a hypsochromic shift of the experimental 2PA maxima can be observed, which is in strong agreement with the performed DFT calculations. This also suggests a decrease in 2PA strength in the same order of electronic properties.Although most target compounds also exhibit high quantum yields of fluorescence, two-photon induced polymerization tests revealed the efficiency of these materials as 2PA photoinitiators. All compounds showed relatively broad processing windows constituting a prerequisite for printing cubic volumes with (sub)micrometer feature sizes at fast writing speeds. When comparing the performance of these 2PA photoinitiators with previously characterized state-of-the-art materials, such as B3FL, the obtained results suggest also the applicability of this new substance class for printing sub-micrometer structures. Calculations of the spin density distribution point towards a polymerization initiation mechanism resulting from strong localization of the unpaired electrons in the triplet state on the C2 carbon of the thiophene moiety, which may allow the exergonic addition of acrylate monomers and thus the propagation of radicals.
The straightforward synthetic route combined with high 2PA efficiency renders this compound class an attractive platform for two-photon-based applications also beyond two-photon induced polymerization. Hence, the structural versatility and capability of our molecular design may contribute to further developments in other areas of functional organic materials.
Experimental
Synthesis
Experimental procedures and materials characterization of all 2PA PI target compounds are given in the ESI.†Computational details
Density functional theory (DFT) calculations were carried out for BHA-1T, BMOA-1T and BSO2MA-1T using M06-2X functional48 which is parameterized for nonmetals, main-group thermochemistry, kinetics, noncovalent interactions and electronic excitation energies as implemented in Jaguar 9.1 program49 package. The molecular geometries and frequency analyses of all studied molecules were calculated in solvent using Jaguar's Poisson–Boltzmann solver (PBF)50 at M06-2X/6-31G(d,p)/PBF level of theory. The reaction enthalpies (ΔH) and the changes of the Gibbs free energy (ΔG) of the reactions were calculated as the difference of the calculated total enthalpies H and the Gibbs free energy G of the reactions. The electronic transition spectra were calculated in solvent with the time-dependent (TD)51 TDDFT M06-2X/6-311+G(d,p)/PBF method. The energies of the lowest relaxed triplet states (T1) were calculated as the difference of zero point (ZPE) corrected electronic energies between the most stable structures of the singlet (S0) and optimized triplet states.For the calculation of 1PA and 2PA properties in Fig. 2B and 3 and Fig. S17 and Table S1,† DFT calculations based on long-range corrected hybrid CAM-B3LYP52 functional and the cc-pVDZ basis set were performed in the gas phase. All molecules exhibited C2 point group symmetry. Geometrical optimizations were performed using the Gaussian09 software (Rev.D)53 with default convergence criteria and an ultra-fine numerical integration grid. The D3 version of Grimme's dispersion54 was included during the geometry optimizations. The compounds were optimized in the gas phase. The excitation energies and 2PA strengths were obtained as vertical excitations in Dalton 2015.1 code.55 These calculations were performed in the gas phase using the respective optimized structures from the Gaussian optimizations. For the stick spectra, used for comparison with the experimental spectra in Fig. 4 integrated one-photon and two-photon extinction coefficients were used (see ESI† for their calculations).
Steady-state measurements
Absorption spectra were measured on a Cary 50 spectrometer, whereas fluorescence emission and excitation (anisotropy) spectra were recorded on a FluoroMax-4 (Horiba Scientific) with a R2658 PMT. All emission spectra were corrected for the wavelength dependent sensitivity of the detection using a set of secondary emissive standards.565 μL of a concentrated stock solution of the samples in THF were pipetted directly into a 1 cm quartz cuvette containing approximately 2 mL of solvent. After mixing, the longest-wavelength peak of the resulting absorbance spectrum remained between 0.3 and 0.7 in optical density for all solvents and samples. For the emission measurements, solutions were prepared in pure solvents and (when lifetimes exceeded 2 ns) were bubbled with nitrogen for 10 min in order to purge the samples of oxygen. Emission quantum yields were determined using Coumarin 152 in deaerated n-hexane as reference (ϕ = 1.0).57
Time-resolved fluorescence
Fluorescence dynamics on the nanosecond time-scale was measured using time-correlated single photon counting (TCSPC). Excitation was performed at 395 nm using 60 ps pulses at 40 MHz produced by a laser diode (Picoquant, LDH-P-C-400B, driven by PDL 800-B). The fluorescence was collected at 90°, its polarization set by a foil polarizer to magic angle with respect to the vertically polarized excitation and filtered through bandpass filters located in front of the photomultiplier tube (Hamamatsu, H5783-P-01). The detector output was connected to the input of a TCSPC computer board module (Becker and Hickl, SPC-130-EM). The full width at half maximum (fwhm) of the instrument response function (IRF), as determined by a dilute solution of ludox, was around 200 ps.Transient absorption (TA) spectroscopy
TA measurements were performed with two pump-probe setups. The fs–ps TA setup used to record spectra up to 1.5 ns with a wavelength dependent IRF of ca. 100–200 fs (fwhm) has been described in detail elsewhere.32 Excitation was performed using 400 nm pulses generated by frequency doubling part of the output of a standard 1 kHz Ti:Sapphire amplified system (Spectra Physics, Spitfire). The ns–μs TA setup, used to record spectra up to 1 μs with an IRF of 350 ps (fwhm) has been described previously.58 Excitation was performed at 355 nm using a passively Q-switched, frequency doubled Nd:YAG laser (Teem Photonics, Powerchip NanoUV) producing pulses with a repetition rate of 500 Hz repetition rate and a pulse duration of 300 ps. In both setups, probing was achieved using white light pulses generated by focusing 800 nm pulses in a CaF2 plate and polarized at magic angle relative to the pump pulses. The sample solutions were located in a quartz cell (Starna) with 1 mm optical path length and were continuously stirred by nitrogen bubbling.Triplet quantum yields were determined monitoring the amount of ground state bleach (GSB) at 385 nm immediately after the laser pulse (10–20 ps) and between 10–20 ns. The cross-contamination by overlapping excited state absorption features was considered by determining the necessary amount of GSB to obtain artifact-free excited state absorption spectra.
Two-photon excited fluorescence
Two-photon cross sections were determined measuring two-photon excitation spectra using a setup similar to the one described by Makarov et al.40 In detail, the output of a TOPAS-Prime in combination with a NirUVis frequency mixer (both from Light Conversion, seeded by the output of the above mentioned amplified laser system, Spectra Physics) was used as excitation source. The excitation intensity was adjusted using a combination of a broadband zero-order halfwave plate and a Glan–Taylor polarizer, and the polarization set to vertical. The beam was slightly focused by a (f = 20 cm) lens, which was placed 10 cm before the sample. The pump power was monitored using a powermeter (Thorlabs PM100A) equipped with a thermal sensor (Thorlabs S302C) behind the sample. The fluorescence was focused onto the entrance slit of a monochromator (0.25 m Cornerstone, Oriel, grating 74166 Newport) equipped with a multi-pixel photon-counter avalanche photodiode detector (Hamamatsu S-10362-11-050U) using a spherical mirror (Ø = 75 mm, f = 150 mm). The output signal was preamplified (SR240, Stanford Research Systems), processed with a gated boxcar-integrator and averager module (SR250, SRS), digitized (SR245, SRS) and recorded on a computer. The two-photon cross section at a given wavelength, σ2PA,x(λ), was calculated according to Makarov et al.40
Here Ix(λ,λobs) is the (two-photon induced) fluorescence intensity at excitation wavelength, λ, and observation wavelength, λobs, for either sample or reference and cx and ϕx are the concentration and differential fluorescence quantum yield (at the observation wavelength) of sample and reference. Coumarin 153 in dimethyl sulfoxide and Rhodamine 6G in methanol were used as reference spectra.59 The sample concentrations in THF were below 5 × 10−6 M.
Multiphoton processing
For two-photon induced polymerization for the fabrication of 3D objects, a Ti:sapphire laser from High Q Lasers providing NIR pulses at 797.5 nm with a pulse duration of 72 fs was used. The system was operated at a repetition rate of 73 MHz and a maximum output power of 427 mW. Multiphoton processing with this system was carried out at laser powers from 10–100 mW (measured after passing the microscope objective). The laser was focused by a 20× microscope objective (NA = 0.8), and the sample was mounted on a high-precision piezoelectric XYZ scanning stage with 200 nm positioning accuracy. For all samples the same fabrication process was implemented: the optical material was drop-cast from a liquid acrylate-based test resin formulation (5 μmol g−1 of the 2PA PI in a 1:1 (w/w) mixture of trimethylolpropane triacrylate (TTA, Genomer 1330) and ethoxylated-(20/3)-trimethylolpropane triacrylate (ETA, Sartomer 415)) onto a methacrylate functionalized glass substrate (3-(trimethoxysilyl)propyl methacrylate, Sigma-Aldrich). As BTMSA-1T proved to be less soluble, a concentrated formulation of this 2PA PI in the resin was applied. Subsequently, the samples were exposed to the laser beam, and the focus was moved across the two-photon sensitive material, which led to embedded 3D structures (lateral dimension: 50 × 50 μm, 5 μm hatch distance, 0.7 μm z-layer distance, 20 layers) inside the liquid formulation. After multiphoton processing, the unexposed monomer was removed by rinsing of the structures with ethanol. The resulting microstructures, particularly their shape and dimensions, were studied by means of scanning electron microscopy (SEM).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr. K. Föttinger for supporting the photo-physical characterization, A. Gaubitzer, M. Mathuber, Dr. B. Pokorny and T. Schwartz for contributing to the synthetic ex-periments. Dr. F. Plasser is acknowledged for fruitful discussions. The authors thank Dr. M. Marchetti-Deschmann for MALDI-HRMS and E. Eitenberger for SEM measurements. M. Lunzer is grateful for partial funding of this work by research scholarships (“Forschungsstipendium”) of the TU Wien. This work was supported by the University of Geneva and the Swiss National Science Foundation (Project No. 200020-165890).References
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed
.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed
- E. Walker and P. M. Rentzepis, Nat. Photonics, 2008, 2, 406–408 CrossRef CAS
- J. Lott, C. Ryan, B. Valle, J. R. Johnson, D. A. Schiraldi, J. Shan, K. D. Singer and C. Weder, Adv. Mater., 2011, 23, 2425–2429 CrossRef CAS PubMed
- L. Li, P. Wang, Y. Hu, G. Lin, Y. Wu, W. Huang and Q. Zhao, Spectrochim. Acta, Part A, 2015, 139, 243–252 CrossRef CAS PubMed
- T.-C. Lin, Y.-F. Chen, C.-L. Hu and C.-S. Hsu, J. Mater. Chem., 2009, 19, 7075–7080 RSC
- W. Denk, J. Strickler and W. Webb, Science, 1990, 248, 73–76 CrossRef CAS PubMed
- H. A. Collins, M. Khurana, E. H. Moriyama, A. Mariampillai, E. Dahlstedt, M. Balaz, M. K. Kuimova, M. Drobizhev, V. X. D. Yang, D. Phillips, A. Rebane, B. C. Wilson and H. L. Anderson, Nat. Photonics, 2008, 2, 420–424 CrossRef CAS
- J. R. Starkey, A. K. Rebane, M. A. Drobizhev, F. Meng, A. Gong, A. Elliott, K. McInnerney and C. W. Spangler, Clin. Cancer Res., 2008, 14, 6564–6573 CrossRef CAS PubMed
- A. Ovsianikov, Z. Li, J. Torgersen, J. Stampfl and R. Liska, Adv. Funct. Mater., 2012, 22, 3527 CrossRef CAS
- J.-P. Malval, S. Achelle, L. Bodiou, A. Spangenberg, L. C. Gomez, O. Soppera and F. R. Guen, J. Mater. Chem. C, 2014, 2, 7869–7880 RSC
- M. Tromayer, P. Gruber, M. Markovic, A. Rosspeintner, E. Vauthey, H. Redl, A. Ovsianikov and R. Liska, Polym. Chem., 2017, 8, 451–460 RSC
-
Multiphoton Lithography: Techniques, Materials and Applications, ed. J. Stampfl, R. Liska and A. Ovsianikov, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016 Search PubMed
- S. Theis, A. Iturmendi, C. Gorsche, M. Orthofer, M. Lunzer, S. Baudis, A. Ovsianikov, R. Liska, U. Monkowius and I. Teasdale, Angew. Chem., Int. Ed., 2017, 56, 15857–15860 CrossRef CAS PubMed
- C. Barner-Kowollik, M. Bastmeyer, E. Blasco, G. Delaittre, P. Müller, B. Richter and M. Wegener, Angew. Chem., Int. Ed., 2017, 56, 15828–15845 CrossRef CAS PubMed
- R. J. Narayan, A. Doraiswamy, D. B. Chrisey and B. N. Chichkov, Mater. Today, 2010, 13, 42–48 CrossRef CAS
- J. Torgersen, X.-H. Qin, Z. Li, A. Ovsianikov, R. Liska and J. Stampfl, Adv. Funct. Mater., 2013, 23, 4542–4554 CrossRef CAS
- B. H. Cumpston, A. P. Sundaravel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, S. I.-Y. Lee, D. McCord-Maughon, J. Qin, H. Röckel, M. Rumi, X.-L. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51–54 CrossRef CAS
- D. Warther, S. Gug, A. Specht, F. Bolze, J.-F. Nicoud, A. Mourot and M. Goeldner, Bioorg. Med. Chem., 2010, 18, 7753–7758 CrossRef CAS PubMed
- Z. Li, N. Pucher, K. Cicha, J. Torgersen, S. C. Ligon, A. Ajami, W. Husinsky, A. Rosspeintner, E. Vauthey, S. Naumov, T. Scherzer, J. Stampfl and R. Liska, Macromolecules, 2013, 46, 352–361 CrossRef CAS
- S. Zheng, L. Beverina, S. Barlow, E. Zojer, J. Fu, L. A. Padilha, C. Fink, O. Kwon, Y. Yi, Z. Shuai, E. W. V. Stryland, D. J. Hagan, J.-L. Bredas and S. R. Marder, Chem. Commun., 2007, 1372–1374 RSC
- R. Nazir, P. Danilevicius, A. I. Ciuciu, M. Chatzinikolaidou, D. Gray, L. Flamigni, M. Farsari and D. T. Gryko, Chem. Mater., 2014, 26, 3175–3184 CrossRef CAS
- Y. Zhang, M. Jiang, G.-C. Han, K. Zhao, B. Z. Tang and K. S. Wong, J. Phys. Chem. C, 2015, 119, 27630–27638 CrossRef CAS
- M. G. Vivas, D. L. Silva, J. Malinge, M. Boujtita, R. Zaleśny, W. Bartkowiak, H. Ågren, S. Canuto, L. De Boni, E. Ishow and C. R. Mendonca, Sci. Rep., 2014, 4, 4447 CrossRef PubMed
- M. Albota, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed
- O.-K. Kim, K.-S. Lee, H. Y. Woo, K.-S. Kim, G. S. He, J. Swiatkiewicz and P. N. Prasad, Chem. Mater., 2000, 12, 284–286 CrossRef CAS
- C.-F. Chow, RSC Adv., 2013, 3, 18835–18843 RSC
- H. Zhou, F. Zhou, S. Tang, P. Wu, Y. Chen, Y. Tu, J. Wu and Y. Tian, Dyes Pigm., 2012, 92, 633–641 CrossRef CAS
- D. Lumpi, E. Horkel, F. Plasser, H. Lischka and J. Fröhlich, ChemPhysChem, 2013, 14, 1016–1024 CrossRef CAS PubMed
- B. Holzer, J. Bintinger, D. Lumpi, C. Choi, Y. Kim, B. Stöger, C. Hametner, M. Marchetti-Deschmann, F. Plasser, E. Horkel, I. Kymissis and J. Fröhlich, ChemPhysChem, 2017, 18, 549–563 CrossRef CAS PubMed
- D. Lumpi, B. Holzer, J. Bintinger, E. Horkel, S. Waid, H. D. Wanzenböck, M. Marchetti-Deschmann, C. Hametner, E. Bertagnolli, I. Kymissis and J. Fröhlich, New J. Chem., 2015, 39, 1840–1851 RSC
- J. S. Beckwith, A. Rosspeintner, G. Licari, M. Lunzer, B. Holzer, J. Fröhlich and E. Vauthey, J. Phys. Chem. Lett., 2017, 8, 5878–5883 CAS
- S. Easwaramoorthi, P. Thamaraiselvi, K. Duraimurugan, A. J. Beneto, A. Siva and B. U. Nair, Chem. Commun., 2014, 50, 6902–6905 RSC
- Y. J. Chang and T. J. Chow, Tetrahedron, 2009, 65, 9626–9632 CrossRef CAS
- J. Moreno, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, S. Hecht and S. A. Kovalenko, J. Chem. Phys., 2015, 143, 024311 CrossRef CAS PubMed
-
K. Hirayama, Handbook of ultraviolet and visible absorption spectra of organic compounds, Plenum Press Data Vision, New York, 1971 Search PubMed
- C. Katan, F. Terenziani, O. Mongin, M. H. V. Werts, L. Porrès, T. Pons, J. Mertz, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. A, 2005, 109, 3024–3037 CrossRef CAS PubMed
- F. J. Avila Ferrer, J. Cerezo, E. Stendardo, R. Improta and F. Santoro, J. Chem. Theory Comput., 2013, 9, 2072–2082 CrossRef CAS PubMed
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef CAS PubMed
- M. Drobizhev, S. Tillo, N. S. Makarov, T. E. Hughes and A. Rebane, J. Phys. Chem. B, 2009, 113, 855–859 CrossRef CAS PubMed
- G. Wicks, A. Rebane and M. Drobizhev, Proc. SPIE, ed. C. E. Tabor, F. Kajzar, T. Kaino and Y. Koike, 2014, p. 89830R Search PubMed.
- Z. Li, M. Siklos, N. Pucher, K. Cicha, A. Ajami, W. Husinsky, A. Rosspeintner, E. Vauthey, G. Gescheidt, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3688–3699 CrossRef CAS
- N. Pucher, A. Rosspeintner, V. Satzinger, V. Schmidt, G. Gescheidt, J. Stampfl and R. Liska, Macromolecules, 2009, 42, 6519–6528 CrossRef CAS
- Z. Li, N. Pucher, K. Cicha, J. Torgersen, S. C. Ligon, A. Ajami, W. Husinsky, A. Rosspeintner, E. Vauthey, S. Naumov, T. Scherzer, J. Stampfl and R. Liska, Macromolecules, 2013, 46, 352–361 CrossRef CAS
- Y. Lu, F. Hasegawa, Y. Kawazu, K. Totani, T. Yamashita and W. Toshiyuki, FIBER, 2004, 60, 165–172 CrossRef CAS
- L. H. Nguyen, M. Straub and M. Gu, Adv. Funct. Mater., 2005, 15, 209–216 CrossRef CAS
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
-
Jaguar; version 9.1, Schrodinger Inc., New York, 2016 Search PubMed
- D. J. Tannor, B. Marten, R. Murphy, R. A. Friesner, D. Sitkoff, A. Nicholls, B. Honig, M. Ringnalda and W. A. Goddard, J. Am. Chem. Soc., 1994, 116, 11875–11882 CrossRef CAS
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenaes, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjaergaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnaes, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CAS
- J. A. Gardecki and M. Maroncelli, Appl. Spectrosc., 1998, 52, 1179–1189 CrossRef CAS
- K. Rechthaler and G. Köhler, Chem. Phys., 1994, 189, 99–116 CrossRef CAS
- B. Lang, S. Mosquera-Vázquez, D. Lovy, P. Sherin, V. Markovic and E. Vauthey, Rev. Sci. Instrum., 2013, 84, 073107 CrossRef PubMed
- S. de Reguardati, J. Pahapill, A. Mikhailov, Y. Stepanenko and A. Rebane, Opt. Express, 2016, 24, 9053–9066 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8me00101d |
| This journal is © The Royal Society of Chemistry 2019 |