
Design of solvent-free functional fluids via molecular nanoarchitectonics approaches
Akira
Shinohara
 ab,
Chengjun
Pan
a,
Lei
Wang
a and
Takashi
Nakanishi
*ac
ab,
Chengjun
Pan
a,
Lei
Wang
a and
Takashi
Nakanishi
*ac
aShenzhen Key Laboratory of Polymer Science and Technology, College of Materials Science and Engineering, Shenzhen University, Shenzhen 518060, China
bKey Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
cInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba 305-0044, Japan. E-mail: nakanishi.takashi@nims.go.jp
First published on 9th January 2019
Abstract
Solvent-free functional fluidic materials have recently attracted considerable attention owing to their high deformability and facile processability. The counterintuitive softness of functional organic molecules/materials that are intrinsically rigid, such as polycyclic hydrocarbons, aromatic macrocycles, fullerenes, and conjugated polymers, is a significant advantage towards various softness-oriented applications. For example, the development of optoelectronically active fluids has shifted the attention from hard matter-based devices that barely bend and stretch under actions required in flexible/stretchable electronics. This minireview presents a summary of the latest reports and current challenges of functional molecular/polymeric fluids from their molecular engineering approaches or the nanoarchitectonics of functional fluids to the state-of-the-art functions derived from the fluids.
 Akira Shinohara Akira Shinohara | Akira Shinohara (right) obtained his PhD in 2017 from the University of Yamanashi (Japan) under the supervision of Prof. Hideyuki Shinmori. His doctoral research focused on the design and investigation of photodynamic functions of porphyrin–gold nanoparticle composites. At present, he is a postdoctoral researcher in the research team of Prof. Lei Wang at Shenzhen University, China, where he has been working since 2017. His current research involves investigating and understanding solvent-free functional polymer fluids. |
| Chengjun Pan Chengjun Pan (centre) received his PhD degree in 2014 from the University of Tsukuba (Japan) under the supervision of Prof. Masayuki Takeuchi and Prof. Kazunori Sugiyasu. In the same year, he joined the Organic Materials Group, National Institute for Materials Science (NIMS), as a postdoctoral researcher. Since 2015, he has been working at Shenzhen University as an assistant professor, and his current research interest focuses on the design and creation of functional conjugated polymers with unique optical, electronic, and mechanical properties. |
| Lei Wang Lei Wang (left) received his PhD degree in 2006 from the Guangzhou Institute of Chemistry, Chinese Academy of Sciences (China). In the same year he moved to Shenzhen University as an assistant professor, and he was promoted as a full professor in 2011. His current research interest focuses on organic thermoelectric materials and proton exchange membranes. |
 Takashi Nakanishi | Takashi Nakanishi received his Ph.D. degree in a shorter period from Nagasaki University in 2000. Thereafter, he did JSPS postdoctoral research at Houston University, USA, and at Oxford University, UK, and joined NIMS, Japan, in 2004. Since 2016, he has held the position of a group leader at MANA, NIMS. In 2007–2010, he was also a group leader at the Max Planck Institute of Colloids and Interfaces, Germany, and in 2007–2011, he was a researcher of PRESTO, Japan Science and Technology Agency. He also has several visiting professor experiences at Warsaw University of Technology in Poland, Institute for Molecular Science in Japan, Hokkaido University in Japan, and Shenzhen University in China. His research interest focuses on functional organic soft materials with precisely controlled self-assembly, in particular non-assembled functional molecular liquids, and their optoelectronic functions. |
Design, System, ApplicationThe molecular design strategy described in this minireview is mainly based on alkylated-π systems, especially bulky and flexible branched alkyl chains attached on optoelectronically-active π-conjugated chromophores; thus the resulting state of the compounds is a solvent-free, room temperature liquid. Since a chromophore unit is wrapped with bulky substituents, the intrinsic optoelectronic function can be transferred directly to the bulk liquid state. The developed soft, deformable fluids are now considered as attractive components in flexible, bendable, and stretchable devices. |
1. Introduction
The recent development of functional organic molecular/polymeric materials significantly has been focused on softening towards softness-derived applications, such as for use in robots,1 actuators,2 and sensors.3 Conversely, conventional inorganic equivalents are designed with significantly hard and rigid materials. Hence, they exhibit limited fabricability and processability. Simultaneously, organic molecular/polymeric materials present significant advantages such as tailorability, resource-saving production, low weight, and facile- and low-cost processing like conjugated polymer engineering.4 While softening highly functional organic materials, it is necessary to maintain the units providing the desired function (e.g. optoelectronic functions,5 spin-activity,6 continuous molecular networks,7 and cages or porous frameworks possessing guest inclusion ability8) under various utilisation conditions such that they are independent of the shape and geometry of substrates/devices. Although practically all organic materials are recognised as softer and more flexible when compared to traditional inorganic counterparts, they are mostly insufficiently flexible owing to their inherent rigidity and/or highly ordered structures that lead to poor resistance under large deformations such as stretching and bending. In this course, an attractive prospect involves altering the rigid mechanical properties albeit maintaining their excellent functions in organic molecular/polymeric materials through molecular design/engineering approaches. The increased potential in solvent-free functional ‘fluidic’ materials presents a significant advantage in this field and aids in shifting the attention from hard matter-based devices that barely stretch and bend under actions required in flexible/stretchable softness-derived applications.It is difficult to determine when solvent-free functional fluidic materials were first discovered. However, there have been a few examples of the ‘counterintuitive’ fluidity of functional materials over the past few decades. Research areas exploring solvent-free functional organic fluids are in the development stages. Early-stage studies, especially those on small molecule-based functional liquid materials, are summarised in recent review articles.5 Nevertheless, in this minireview, we focus on the ‘molecular/polymeric design approaches’ of optoelectronically active small π-conjugated molecules and also polymeric fluid substances which exhibit enhanced optoelectronic and mechanical functions. The highlighted studies describe ongoing and cutting-edge research at their current stage. It should be noted that this is the first review article describing the latest developments in functional polymer fluids.
2. Molecular design
2.1. Engineering of side chains
The interruption and hindering of intermolecular interactions is an essential methodology to soften intrinsically rigid functional molecules and polymers (e.g. π-conjugated molecules and polymers), which are often well-organised into ordered structures. The attachment of large and sterically hindered bulky substituents, such as a dendron unit9 on the rigid π-conjugated moiety, hampers intermolecular π–π interactions but does not soften the materials very efficiently. The large entropy in the materials produces disordered, low-melting point, and low-viscosity matter. In order to reduce cohesive forces between neighbouring molecules, the incorporation of low-melting point, ‘soft’ substituents is the most reliable approach. Substituents used for this purpose are briefly classified into two categories, namely ion-pair and non-ionic groups. In the former, weak ionic interactions are introduced to produce ionic liquids with relatively low viscosity.10 The latter involves covalent substitutions by flexible and bulky side chains to prevent aggregation among neighbouring molecules and results in solvent-free condensed fluids that exhibit relatively high viscosities and low melting points (Tm). Long-/branched-alkyl, oligo(ethylene glycol) (OEG)11–13 and poly(dimethylsiloxane) (PDMS)14,15 substituents are widely used to fabricate low-melting point functional materials. These types of non-ionic and low-melting point substituents are advantageous in terms of their chemical and electrochemical inertness when compared to ionic equivalents.Linear long-alkyl chains are routinely used to soften and improve the solubility of π-conjugated molecules and polymers. The alkylation of π-conjugated molecules is a common approach for fabricating thermotropic liquid crystals that can form self-assembled and long-range ordered structures. Conversely, branched long-alkyl chains with the chemical formula –CH2–CH((CH2)n−1CH3)–(CH2)n+1CH3, which are abbreviated as –CnCn+4 and known as Guerbet alcohol-based branched alkyl chains, were recently used as more flexible, bulky, and low-melting point substituents.16 The branching at the β-position and resulting low symmetry (generally used as a racemic mixture) inhibited close and regular molecular arrangements among neighbouring molecules and increased amorphousness in the solvent-free condensed state. Solvent-free and room-temperature (25 °C) liquid fulleropyrrolidine derivatives were serendipitously discovered by our group by attaching a 2,4,6-tris(n-alkyloxy)phenyl group (Fig. 1A and B).17a The structural requirements for this series of room-temperature liquid C60 and C70 derivatives were also investigated.17b With increasing length of the alkyl chains, the Tm of some C60 derivatives significantly decreased from 147 °C to −36.5 °C for 1 to 3 (Fig. 1C). However, a further increase in the length of alkyl chains increased the Tm from −36.5 °C to 21.5 °C for 3 to 6. For C70 derivatives with the same substituent motif as the C60 derivative, a similar phase behaviour was observed to that described above. The Tm gradually increased with increasing length of the alkyl chains from −10.5 °C to 20.6 °C for 10 to 12. It was noted that unlike C60 systems, C70 derivatives are obtained as isomeric mixtures due to the elliptic geometry of C70. This is why the Tm values are similar to those of the corresponding C60 derivatives despite the extended π-system of the C70 unit and their larger molecular size. Our group also synthesised liquid C60 derivatives 7–9 attached by Guerbet alcohol-based branched alkyl chains (7, 8) or hyperbranched iso-octadecyl chains (9).18 When compared with the three linear alkyl chain-substituted derivatives 2–6, both 7 and 8 generated room-temperature liquids with only two substituting branched chains. More significantly, 7 and 8 exhibited lower Tm values (≤120 °C) and also lower viscosity than those of 2–6. Interestingly, the viscosity of 7 (260 Pa s) was significantly lower than that of 8 (1500 Pa s) although 7 consists of shorter branched alkyl chains. This finding emphasises the importance of bulky and flexible alkyl chain-substitution at the ortho-position on the phenyl unit to produce less viscous C60 liquids. However, a further increase in the branching extent (9) increased both the Tm (12 °C) and viscosity (128![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa s). The viscosity obtained in 9 even exceeded that of linear chain-substituted C602–6. This is because the hyperbranched-structure induced high intra- and inter-molecular friction during deformation (flowing). Therefore, both the Tm and the viscosity of alkylated C60 derivatives were effectively reduced by suitable branching and choosing the proper substitution position of the alkyl side chains.
000 Pa s). The viscosity obtained in 9 even exceeded that of linear chain-substituted C602–6. This is because the hyperbranched-structure induced high intra- and inter-molecular friction during deformation (flowing). Therefore, both the Tm and the viscosity of alkylated C60 derivatives were effectively reduced by suitable branching and choosing the proper substitution position of the alkyl side chains.
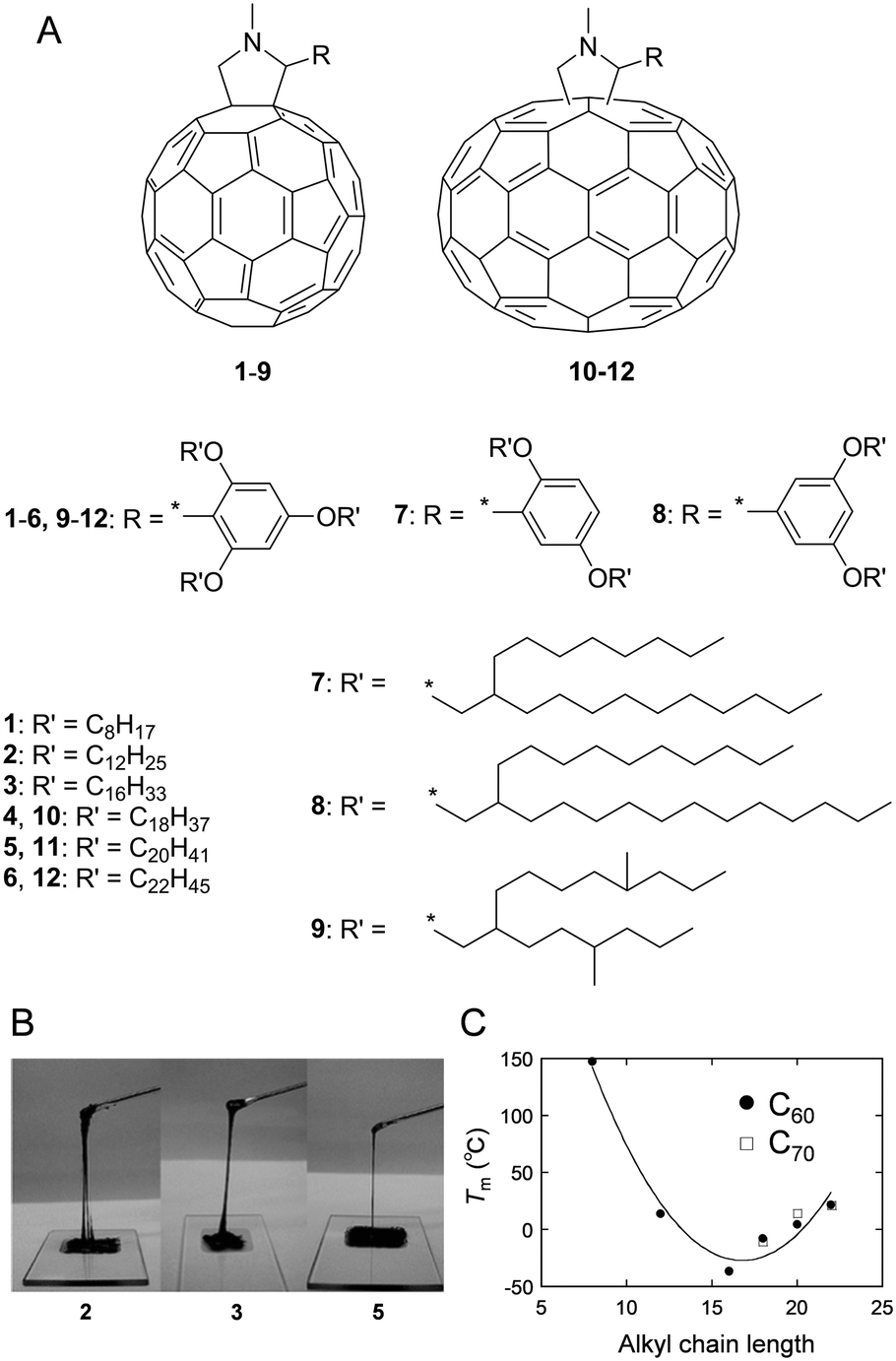 | ||
| Fig. 1 (A) Structure of C60 (1–9) and (b) C70 (10–12) derivatives. (B) Photographs of room-temperature liquid fullerenes 2, 3 and 5. Adapted with permission from ref. 17a © 2006 American Chemical Society. (C) Relationship between the melting points (Tm) and alkyl chain lengths of the fullerene. Adapted with permission from ref. 17b © 2013 American Chemical Society. | ||
OEGs refer to oligomers of ethylene oxide composed of –CH2CH2O– repeating units known as low-melting point compounds. In contrast to linear alkyl chains, replacement of carbon atoms with oxygen atoms significantly decreases the Tm due to the following possibilities that inhibit the crystallisation of the OEG chains:19 i) the electron lone pairs on the oxygen atoms which provide an intermolecular repulsive force, ii) difference in rotational freedom, and iii) lower symmetry in the molecular structure. Octa(ethylene glycol) (HO(CH2CH2O)8H) behaves as a liquid at room temperature (Tm = 22 °C) while its n-alkanediol analogue 1,24-tetracosanediol (HO(CH2)24OH) exhibits a significantly higher Tm (108 °C). Given the low-melting point property of OEGs, the Tm of intrinsically solid π-conjugated molecules effectively decreased. Carbazole (Tm = 246 °C) and pyrene (Tm = 145–148 °C) were liquefied by attaching a tri(ethylene glycol) monomethyl ether chain (Fig. 2, 13 (ref. 11) and 14 (ref. 12)). In order to liquefy more cohesive and crystalline molecules that exhibit expanded π-conjugated systems, it was expected that longer or multiple OEG substituents are required. Phthalocyanine (Pc) is a macrocyclic molecule with a highly planar expanded π-plane and does not exhibit a Tm up to the decomposition temperature (≳300 °C) under ambient pressure. Snow et al. reported room-temperature liquid Pcs 15–17 periphery-substituted by OEG (mean number of repeating units = 8.8) chains at their α- and β-positions on the Pc ring.1315–17 show Tm values close to 10 °C, indicating that the OEG substituent dominates this property.
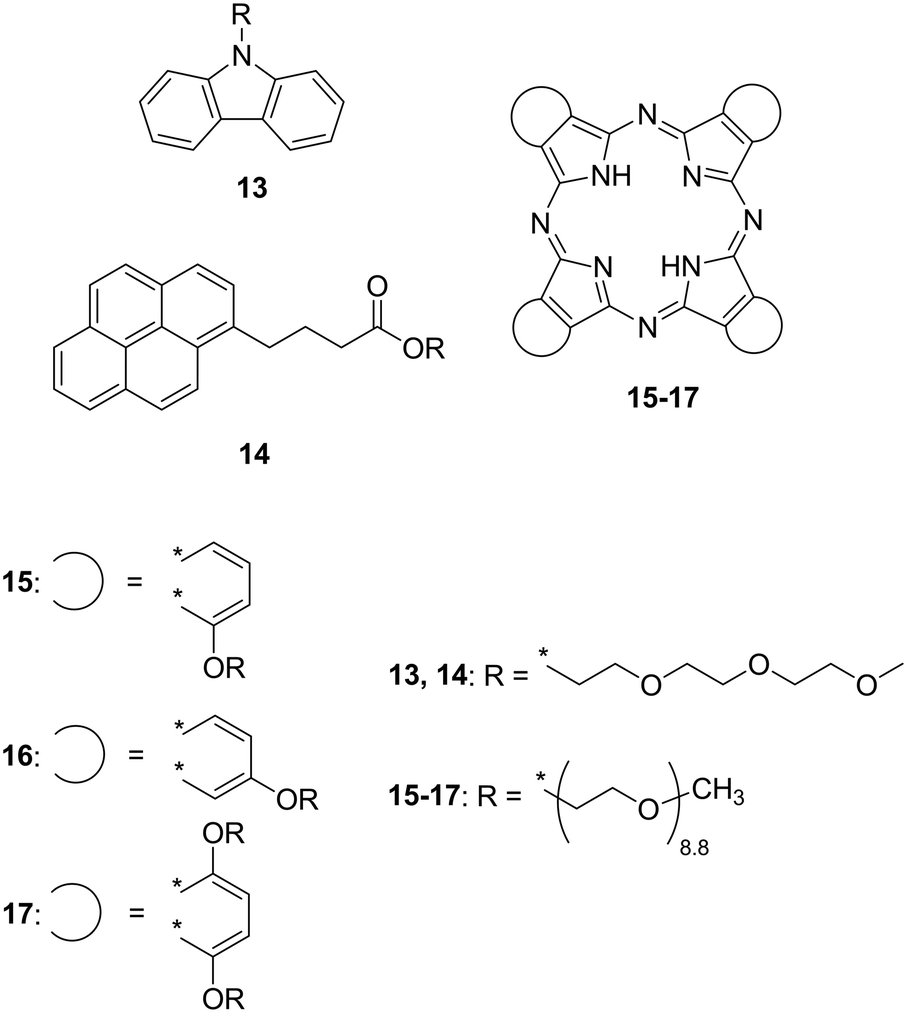 | ||
| Fig. 2 Chemical structures of oligo(ethylene glycol) (OEG)-substituted carbazole (13), pyrene (14), and phthalocyanines (15–17) that exhibit fluidic behaviour at room temperature. | ||
PDMS and its derivatives are also effective side chains to produce room-temperature functional fluids due to their low-melting point feature and also their high chemical and thermal stability. The binding energy of the Si–O (466 kJ mol−1) bond significantly exceeds those of C–C (346 kJ mol−1) and C–O (358 kJ mol−1). Furthermore, PDMS is the most widely used silicon-based organic polymer and is especially known for its unusual fluidic properties. PDMS behaves as a watery fluid at room temperature with a viscosity of 0.1 Pa s (comparable to olive oil), and its molecular weight reaches 10000 g mol−1 (over 100 –Si–O– repeating units). When compared to the bond length for C–C (1.54 Å) or for C–O (1.43 Å), the Si–O (1.64 Å) bond is slightly longer, and hence, siloxanes exhibit low barriers for rotation about the Si–O bonds due to low steric hindrance. By attaching PDMS chains, rigid and solid π-conjugated molecules are effectively liquefied. Maya et al. reported a series of PDMS-substituted Pcs and their lead(II) complexes 18–21 (Fig. 3A).14 All silicone lead Pcs are viscous green liquids at room temperature. Metal-free Pc 21 was obtained as a blue coloured liquid by post-synthetic treatment of 18 with trifluoroacetic acid. Furthermore, PDMS facilitates liquefying extremely aggregated carbon allotropes. Giannelis et al. reported PDMS-substituted multi-walled carbon nanotubes (MWCNTs) that exhibit fluidic behaviour in the absence of a diluent or solvent. The solvent-free CNT fluids are synthesised via the oxidation of CNTs by nitric acid following attaching a corona of flexible PDMS (molecular weight = 5000 g mol−1) chains onto the CNT surface.15 The CNT functionalised with PDMS (22) is a viscous tar-like liquid at room temperature (Fig. 3B) and is dispersible at high concentrations (10 mg mL−1) in toluene, thereby providing homogeneous black organosols. Despite its high content of CNTs (∼85 wt% in the solvent-free state), the material maintains a liquid state at room temperature due to low-melting point PDMS chains.
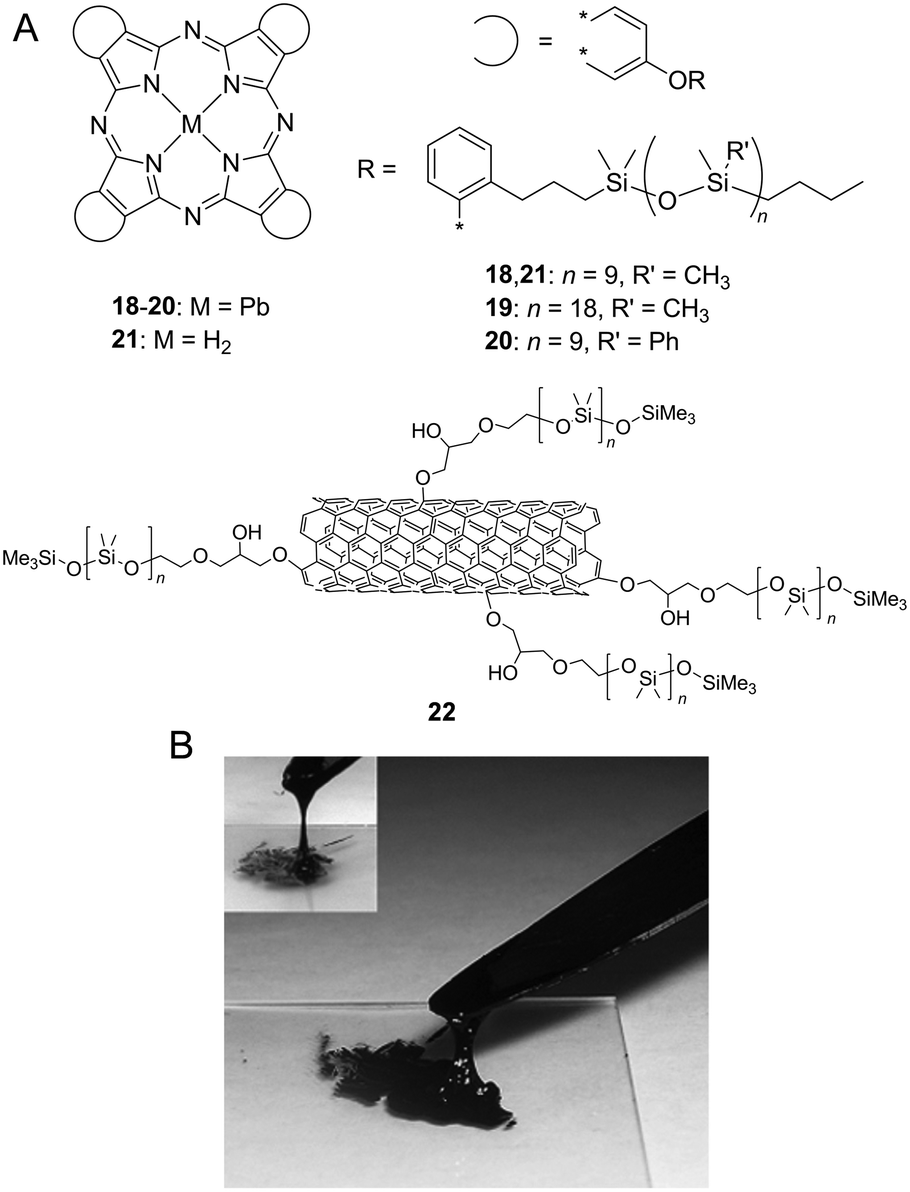 | ||
| Fig. 3 (A) Chemical structure of poly(dimethylsiloxane) (PDMS)-substituted liquid phthalocyanines (18–21) and multi-walled carbon nanotubes (MWCNTs) (22). (B) Photograph of 22 at room temperature. Reproduced from ref. 15 with permission from the © 2007 Elsevier B.V. | ||
2.2. Scale-up synthesis of alkyl–π liquids
A large quantity (for e.g., exceeding 1 g) of fluid material is required for the full characterisation of fluid matter. Therefore, the large-scale production of desired fluids is a necessary procedure. Although the introduction of branched alkyl chains is a simple bimolecular nucleophilic substitution (SN2) reaction (e.g., C-, N-, and O-alkylation), the SN2 reaction is inherently accompanied by a bimolecular elimination (E2) reaction of the starting alkyl halide (Scheme 1). This leads to difficulties in the purification of the crude material and especially in the large-scale synthesis of alkyl–π liquids because both products and impurities (mainly composed of the starting alkyl halide and olefin as an eliminative product) are liquid; consequently, recrystallisation cannot be employed. In addition, the polarities of the desired compound and the impurities are generally close to each other because of aliphatic (low-polarity) alkyl chains.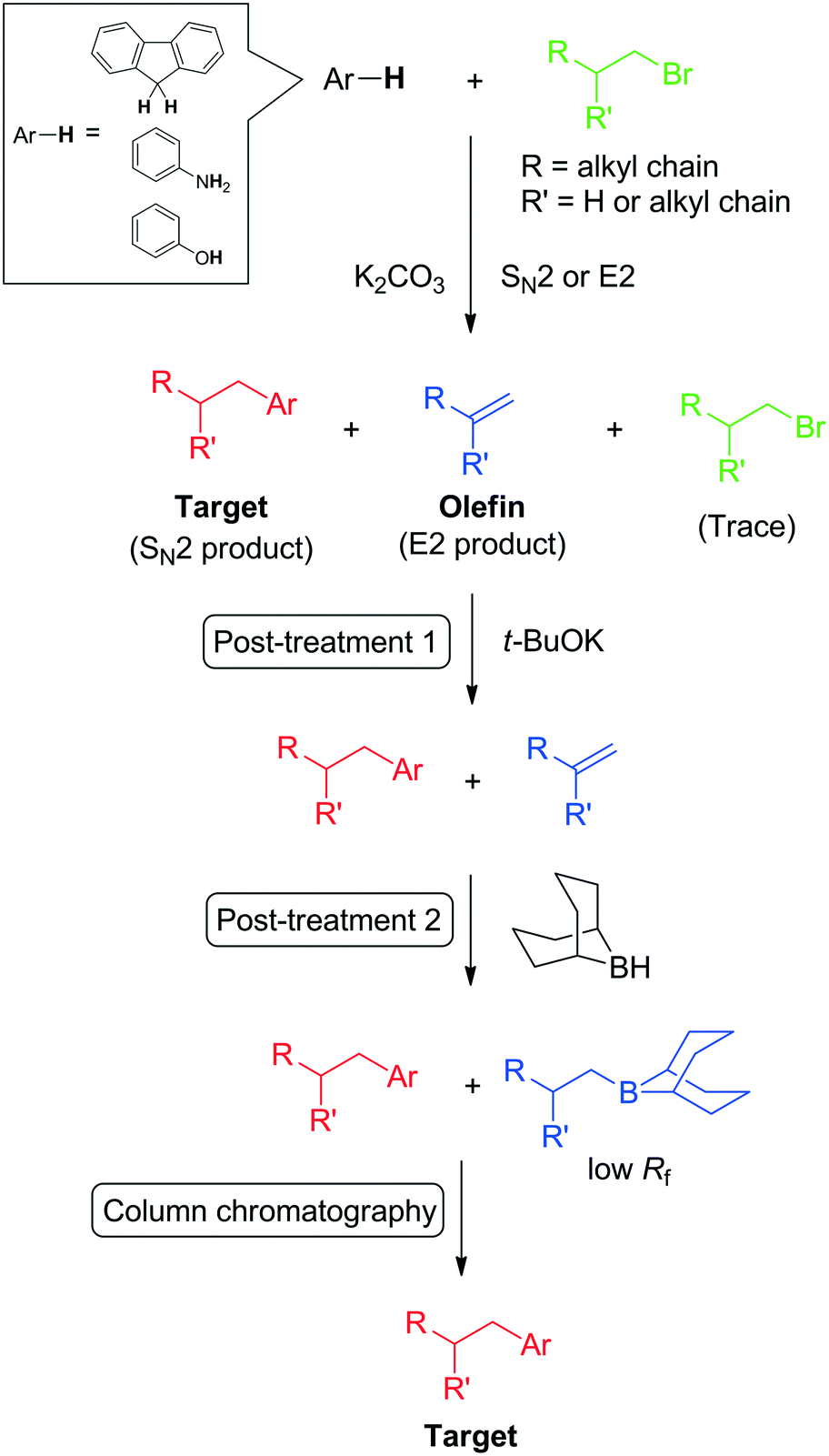 | ||
| Scheme 1 Post-treatment technique for SN2 alkylation of aromatics with alkyl halides. Here the Ar moiety contains C, N, and O elements at the position of alkyl chain substitution. | ||
We recently reported post-treatments for the purification of alkyl–π compounds using common chemicals to remove unreacted alkyl halide and by-products (olefins) from the reaction mixtures.20 The post-treatment procedure involves the following: 1) complete conversion of trace alkyl halide to olefin via a reaction with potassium tert-butoxide; and 2) hydroboration of the olefins with a 0.5 M solution of 9-borabicyclo[3.3.1]nonane (9-BBN) in tetrahydrofuran (THF). The resulting alkyl-boron compound exhibited a significantly lower retention factor (Rf) than the desired alkyl–π compound and was readily removed by column chromatography on silica gel. Thus, the target alkylated π-compounds can be collected as the first fraction. Notably, products can be purified in one short column after these post-treatments even for gram-scale synthesis.
2.3. Characterisation techniques of fluidic materials
In contrast to crystalline materials which exhibit distinct solid–liquid transition (i.e. Tm), materials that exhibit high amorphousness (e.g. non-crystalline polymers) typically do not exhibit Tm. As opposed to Tm, amorphous materials exhibit a discontinuous jump in heat capacity at a specific temperature, which is known as the glass transition temperature (Tg). Below the Tm or Tg, materials exhibit nearly ideal elasticity (≳109 Pa in modulus).21 During heating, the aforementioned substances lose their stiffness at Tg, and this loss is characterised by a significant decrease in the storage (elastic) modulus, thereby resulting in a liquid or viscoelastic state. In order to investigate their fluidic characteristics, sinusoidal oscillatory rheology is the first choice. If the shear loss (viscous) modulus (G′′) exceeds the shear storage (elastic) modulus (G′), then the substance is in a liquid state. However, additional structural characterisations, such as polarised optical microscopy (POM), small-angle X-ray scattering (SAXS), and X-ray diffraction (XRD), are typically required to distinguish the isotropic phase and anisotropic mesophase.As a typical liquid alkyl–π sample, we introduced alkylated-pyrene liquids at room temperature and characterisation methodologies to evaluate their fluid state. Our group synthesised 1-monophenyl-substituted (23, 24) and 1,3,6,8-tetraphenyl-substituted (25–27) pyrene derivatives (Fig. 4A).22 They exhibited viscous liquid states, with glassy transitions ranging from −63.5 to −43.0 °C (determined by differential scanning calorimetry, DSC). POM images of all fluids 23–27 at room temperature did not provide any birefringent texture, thereby indicating a lack of long-range ordered and assembled domains. The absence of a long-range order of molecules was further supported by small and wide-angle X-ray scattering (SWAXS), which exhibited only broad halos. The random orientation of pyrene cores was further evidenced by flash-photolysis time-resolved microwave conductivity (FP-TRMC). Based on FP-TRMC profiles, 23–27 exhibited pseudo-photoconductivity maxima on the order of 10−5–10−4 cm2 V−1 s−1 and a prompt deactivation faster than 0.3 μs. The aforementioned values were significantly lower than those reported for other crystalline and liquid crystalline compounds.23 Furthermore, DSC, POM, SWAXS, and FP-TRMC analyses indicated that 23–27 are amorphous fluids with random molecular orientations at room temperature although they exhibited different alkyl chain substituent motifs. We also performed a rheological study to confirm the fluidic behaviour of 23–27 (Fig. 4B and C). In dynamic frequency sweeps, G′′ always exceeded G′ in the measured angular frequency (ω) range, and the complex viscosity (|η*|) of liquid pyrenes was constant, indicating Newtonian liquid behaviour. This set of analytical techniques is the standard procedure and is highly recommended to researchers who are developing new fluidic materials.
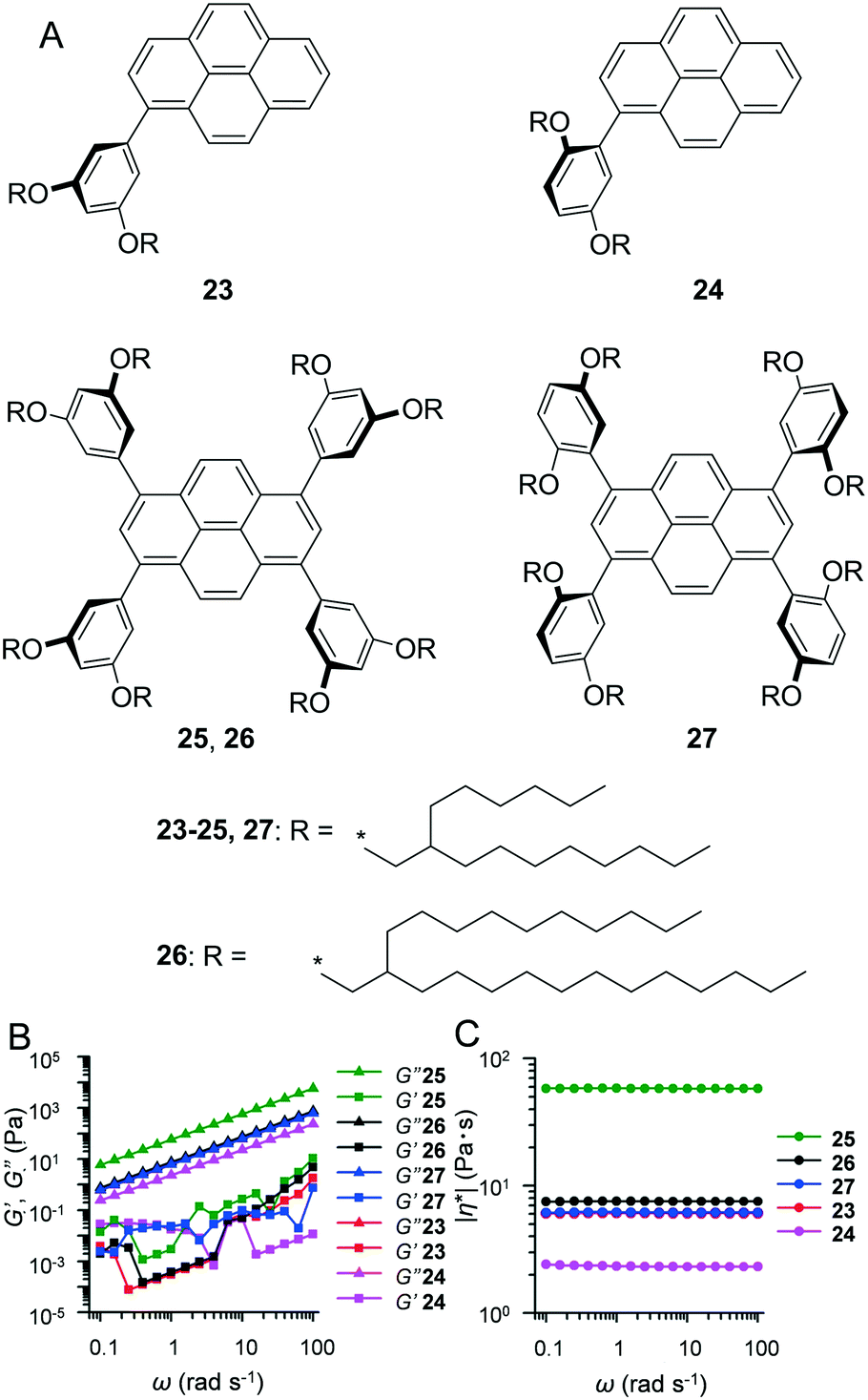 | ||
| Fig. 4 (A) Chemical structures of liquid pyrene derivatives 23–27. (B) Variation of storage elastic modulus (G′, square), viscous loss modulus (G′′, triangle) and (C) complex viscosity (|η*|, circle) (right) as a function of angular frequency (ω) for neat samples of 23 (red), 24 (pink), 25 (green), 26 (black) and 27 (blue) with a constant strain amplitude (γ0) = 0.25 at 25 °C. Reproduced from ref. 22 with permission from the © 2017 under a Creative Commons Attribution 4.0 Unported (CCBY) license. | ||
2.4. Preventing the formation of supercooled liquids
Metastable states in stimuli-responsive materials are extensively used in several applications.24 However, materials in metastable states are detrimental to applications that require consistent performance under various conditions. Supercooled liquids (SCLs) constitute a typical example of a kinetically trapped isotropic liquid even below its Tm. In order to guarantee the reliable performance of functional fluid-based devices, techniques to reveal an indistinguishable supercooled state and molecular design strategies to produce kinetically stable functional fluids are necessary.9,10-Diphenylanthracene (DPA) derivatives substituted with Guerbet alcohol-based branched alkyl chains of different lengths (CnCn+4) were successfully liquefied with n = 6 (30), 8 (31), and 10 (32) due to the effective reduction of intermolecular π–π interactions (Fig. 5A).25 However, 31, which was previously reported as a room-temperature liquid,26 unexpectedly solidified after ageing under ambient conditions for over several months (Fig. 5B). Moreover, 32 was obtained as a liquid but solidified within 4 weeks. Based on the phase behaviours of 28 to 30 in DSC, the lengthening alkyl chains reduced the Tm. Conversely, from 30 to 32, lengthening the alkyl chains enhanced the crystallisation tendency of the melts, which can be attributed to increased van der Waals interactions among longer alkyl chains.27 Furthermore, 30 did not exhibit solidification during storage at ambient temperature throughout the experimental period (exceeding 1 year). The viscosity of the liquid was influenced by the alkyl–DPA ratio. As confirmed by the order of |η*| of the melts at 30 °C of 32 (3.0 Pa s) < 31 (4.4 Pa s) < 30 (7.1 Pa s) (Fig. 5D), higher alkyl chain contents increased molecular softening and thus increased fluidity. Thus, 32 and 31 can experience faster molecular diffusion, which facilitates crystallisation. When the length of alkyl chains increased from 30 to 32, the low-q halo in the SWAXS profile became narrower (Fig. 5C, the full width at half maximum was calculated to be 0.26 Å−1 for 30, 0.22 Å−1 for 31, and 0.19 Å−1 for 32), thereby suggesting improved regularity of intermolecular distances. This type of improvement was potentially caused by the more uniform molecular shape, such as more globular-like, with longer alkyl side chains, which can also contribute to the enhanced crystallisation tendency. The brominated equivalent of 31 and 33 did not exhibit crystallisation under the investigated conditions including long-term ageing (>1 year, at ambient temperature). The suppressed crystallisation of 33 is partially attributed to its decreased π-skeleton symmetry (31: D2h, 33: C2h) and higher complex viscosity (9.0 Pa s) compared to those of 31. The importance of thermal and SWAXS studies is highlighted in this study and the techniques are useful in quickly distinguishing the real thermodynamically stable state of fluidic materials. In fact, the supercooled state is occasionally indistinguishable by its spectroscopic features.
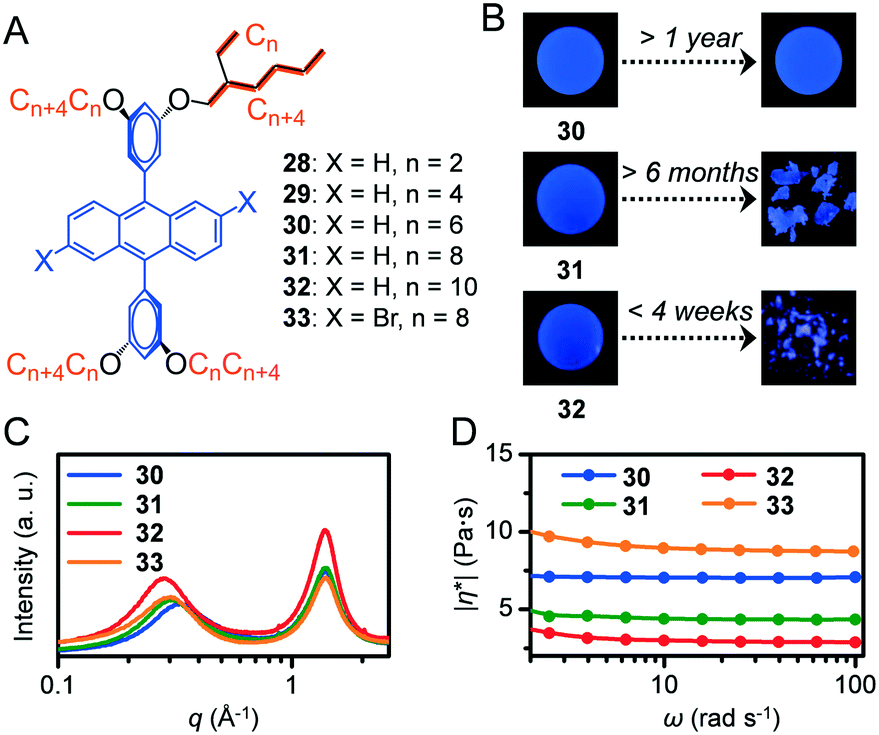 | ||
| Fig. 5 (A) Chemical structures of 9,10-diphenylanthracene derivatives 28–33. (B) Photographs of 30, 31, and 32 as fresh (left) and after long term ageing at ambient temperature (right) obtained under irradiation of a UV lamp (365 nm). (C) SWAXS profiles and (D) complex viscosity of 30 (blue), 31 (green), 32 (red), and 33 (orange) at 30 °C. Reproduced from ref. 25 with permission from the © 2018 under a Creative Commons Attribution 3.0 Unported (CCBY) license. | ||
2.5. Effect of regioisomerism
We reported 1- and 2-alkoxy-substituted naphthalenes 34–39 (Fig. 6A) with a Guerbet alcohol-based branched alkyl chain of different lengths (CnCn+4, n = 4, 6 and 8).28G′′ exceeded G′ throughout the range of applied angular frequencies (ω), thereby indicating liquid behaviour. The |η*| values were in the range of 38.4 (34) to 67.0 mPa s (39) at 25 °C (ω = 10 rad s−1). The aforementioned values were significantly lower than those in the previously reported alkylated π-conjugated molecules from our laboratory such as alkylated oligo(p-phenylenevinylenes) (0.64–34.6 Pa s),29 fullerenes (260–128000 Pa s),17,18 pyrene (2.3–58.0 Pa s),22 or anthracenes (0.28–84.0 Pa s).30 The aforementioned lower |η*| values can be explained by the smaller size of the π moiety.31
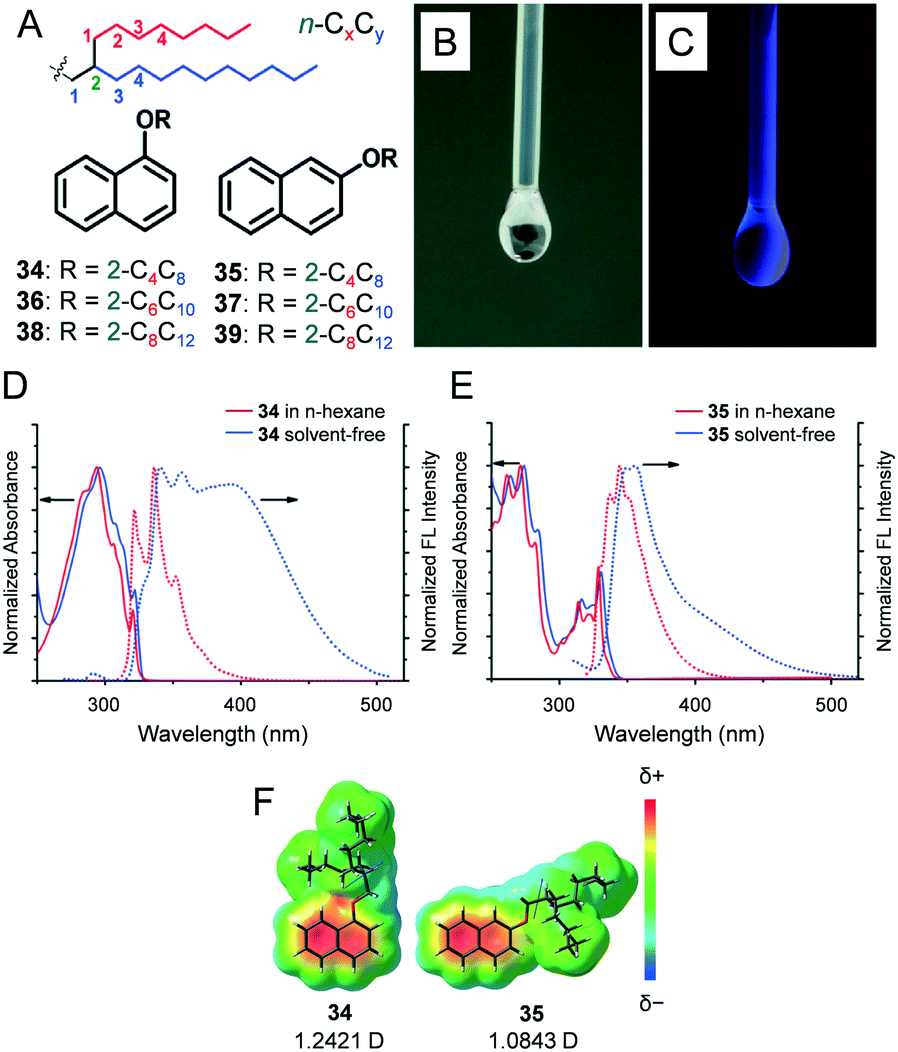 | ||
| Fig. 6 (A) Chemical structures of regioisomeric liquid naphthalenes (34–39). The odd series represents the 1-regioisomers and the even series is the 2-regioisomers. The chain lengths chosen for the study are C4C8, C6C10, and C8C12. Photographs of 34 dropping down through a PTFE needle under (B) ambient light and (C) UV (254 nm) light. Normalised UV-visible absorption (solid lines) and fluorescence (dotted lines) spectra of (D) 34 and (E) 35 in n-hexane solution (0.1 mM, red lines) and in the solvent-free liquid state (blue lines), respectively. (F) Dipole moments (blue arrows) and electrostatic potential maps of liquid naphthalenes 34 and 35 calculated at the MP2/6-31G(d)//B3LYP/ 6-311G(d,p) level. Reproduced from ref. 28 with permission from the Royal Society of Chemistry. | ||
A dilute solution (0.1 mM) of 34 in n-hexane exhibited structureless absorption features with an absorption maximum at 294 nm (Fig. 6D) while 35 exhibited more vibronic features (Fig. 6E). Spectroscopic investigations in the solvent-free liquid state permitted an examination of the inherent effect of regioisomerism on the bulk optical properties of liquid naphthalenes. Solvent-free samples of both 34 and 35 exhibited absorption spectra redshifted by only a few nanometres when compared with those samples in solutions. The vibronic structures were similar, thereby indicating very weak intermolecular interactions in the ground state. In contrast, the fluorescence spectrum of 34 exhibited features corresponding to the monomer mixed with broad excimer-like emissions, while monomer-enriched emissions with significantly smaller excimer features were evident in 35. Theoretical calculations suggested that the dipole moment of 34 (1.2421 D) slightly exceeded that of 35 (1.0843 D) (Fig. 6F). Diamagnetic organic molecules tend to cancel their dipole moments, and hence, we speculate that 34 exhibits enhanced π–π stacking interactions when compared with 35.
3. Advanced functions in molecular liquids
3.1. Spin-activity and electrochromism
As an attractive example of solvent-free functional fluid possessing advanced functions, a room-temperature liquid double-decker lutetium(III)–phthalocyanine complex (Pc2Lu) that exhibits spin-active nature and electrochromic properties was reported recently.6 Pc2Lu is a neutral organic radical species composed of a Lu3+ centre sandwiched with two Pc2− aromatic macrocycles. 40 shows non-Newtonian liquid behaviour due to subtle intermolecular π–π interactions among neighbouring Pc units. The electron spin resonance (ESR) spectrum of 40 in a dichloromethane (10−4 M) solution exhibited an ESR signal at g = 2.0027 ± 0.0002 and wide peaks of width approximately 0.9 ± 0.1 mT (Fig. 7B). Similarly, its neat sample at room temperature exhibited an almost identical feature. The aforementioned ESR results indicated that the bulky branched alkyl chains contribute to sufficient weakening of the intermolecular Pc2Lu–Pc2Lu interactions. During chemical (Fig. 7C) or electrochemical reduction in a thin film of 40, two new absorption peaks gradually appeared at 745 and 670 nm for 40. The aforementioned redox processes were significantly faster when compared to those in the solution due to the limited electrochemically active diffusion layer in the film.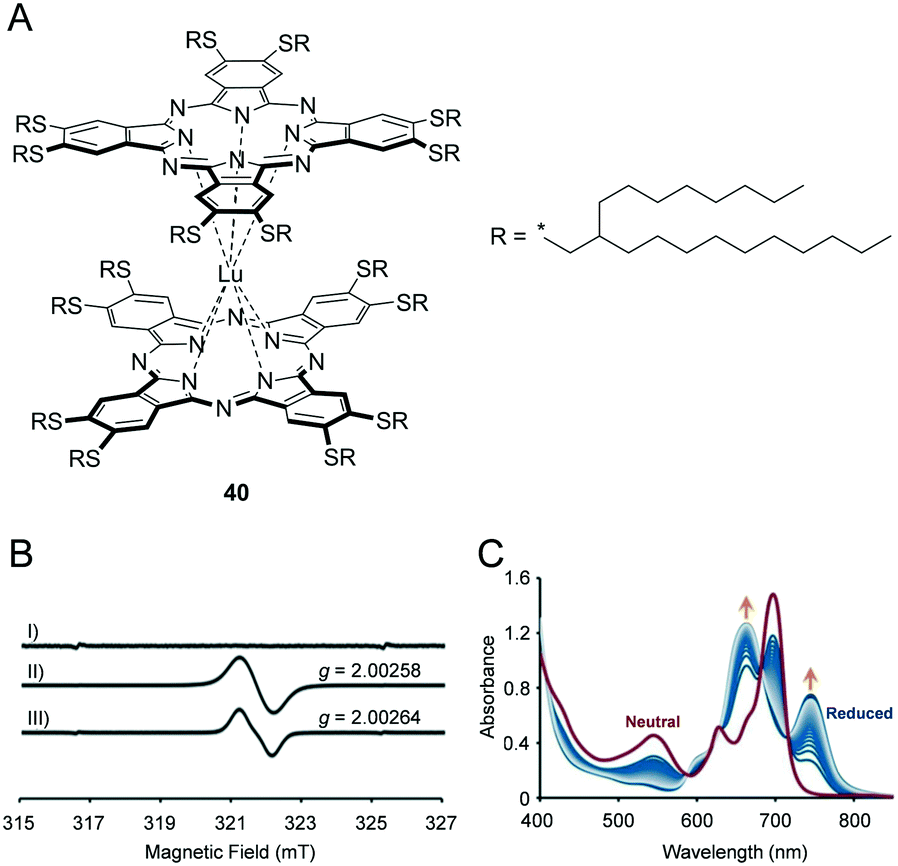 | ||
| Fig. 7 (A) Chemical structure of 40. (B) ESR spectra of I) a thin film of one-electron reduced species of 40, II) a thin film of neutral 40, III) 40 in dichloromethane (10−4 M). (C) UV-vis spectral changes of a thin film of 40 during chemical reduction by Na2S2O4 (from red line to blue lines). Reproduced from ref. 6 © 2018 with permission of John Wiley and Sons. | ||
3.2. Photoinduced isothermal liquefaction
Thermally reversible solid–liquid phase transition (i.e. thermal melting) is a common occurrence for most chemical compounds. However, photoinduced liquefaction under isothermal conditions is significantly limited. Azobenzenes are a class of the most common photochromic molecules that exhibit reversible trans-/cis-isomerisation driven by light. Photoinduced isomerisation from the thermally stable trans-isomer to the metastable (kinetically trapped) cis-isomer significantly changes their mechanical and chemical properties as well as photochemical properties. The aforementioned compounds were employed for optically functional systems such as microscopic phase transitions from an ordered solid or liquid crystalline state to an isotropic state transition.Noritake et al. reported photochemically reversible liquefaction–solidification cycles at room temperature.32 Alkylated dimeric (41) and trimeric (42) azobenzenes exhibited crystal-to-liquid phase transitions by photoirradiation, while the monomer equivalent did not exhibit the aforementioned behaviour (Fig. 8). A bright synchrotron X-ray study elucidated structural features governing the photoinduced crystal–liquid phase transition.33 Akiyama et al. also reported similar photoinduced liquefaction–solidification cycles using a series of azobenzene-appended sugar alcohols.34 The trans-isomers of 43–47 were solid at room temperature although they melted to their cis-form by exposure to UV light under isothermal conditions. The obtained fluids gradually returned to the solid trans-form (about two days). The reversible liquid–solid transition property was successfully applied as a photoresponsive adhesive.35
 | ||
| Fig. 8 Chemical structures of azobenzene derivatives showing isothermal solid–liquid phase transition induced by photoisomerisation (41–47), possessing hydrophobic amphiphilicity (48) and heat storage nature (49). | ||
A room-temperature liquid azobenzene via substitution by a 2-octyldedecyl (C8C12) group (48) was also examined, and its hydrophobic–amphiphilicity feature in solution was reported.36 In addition, Kimizuka et al. synthesised a similar liquid azobenzene attached with a 2-ethylhexyl (C2C6) group (49).37 The trans-isomer of 49 is a yellowish liquid with a low Tg of −63 °C and stable up to 220 °C without thermal decomposition. The enthalpy of isomerisation (ΔH) and energy density of cis-49 are calculated to be 52 kJ mol−1 and 47 W h kg−1, respectively, and this suggests potential applications as solar thermal fuel under solvent-free conditions.
3.3. Permanent porosity in liquid
In addition to the above discussed π-conjugated system-based optoelectronic functions in solvent-free fluids, geometrical functions including porosity are an alternative interest in the emerging liquid science. James et al. reported unique non-volatile ‘porous liquids’ that were composed of a rigid organic cage architecture and had permanent porosity.8 This provided a clear contrast with common liquids that exhibit limited porosity due to their poorly defined and transient intermolecular cavities. The cage molecule 50 comprises a rigid cage architecture with 15-crown-5 (15C5) moieties at the periphery, which defines a molecular pore space, and is dissolved at a high concentration in 15C5 (∼44 wt%, 50:15C5 = 1:12 in molar ratio) as a non-volatile solvent (Fig. 9). The cage molecule exhibits a cavity with a diameter of approximately 5 Å at its centre that is accessible through four access windows with a diameter of approximately 4 Å. The viscosity of the mixture liquid ranged from 20 mPa s to >140 mPa s in the range of 25 °C to 50 °C. Molecular dynamics (MD) simulation and positron (e+) annihilation lifetime spectroscopy (PALS) experiments suggested that the cage in the porous liquid is empty because the 15C5 molecule is significantly larger than the cavity. The solubility of methane in the porous liquid (52 μmol g−1 at 30 °C) significantly exceeds that in pure 15-crown-5 (6.7 μmol g−1), thereby indicating future applications in gas storage/separation technologies.
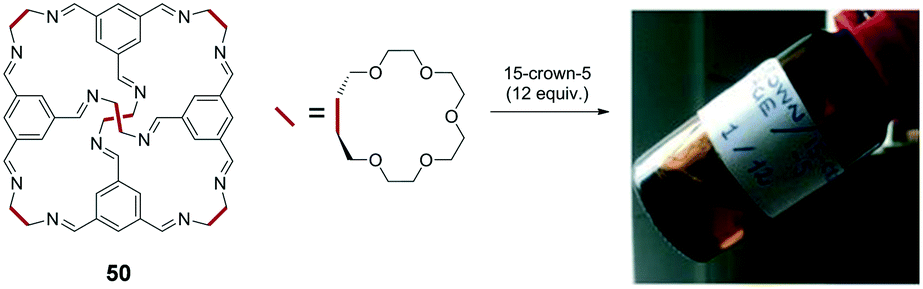 | ||
| Fig. 9 Structure of cage molecule 50 and its solution ‘porous liquid’ in 15C5 (50:15C5 = 1:12). Reproduced from ref. 8 with permission from the © 2015 Springer Nature. | ||
3.4. Rotaxanes
In a complexation process in host–guest (H–G) supramolecular systems, the high stability and efficiency of the H–G complex (i.e. significantly higher associative rate constant when compared with the dissociative rate constant) are essential in their practical application. However, the solvation of H and/or G molecules acts as a barrier for the complexation. Ogoshi et al. proposed solvent-free and non-volatile liquid host molecules, termed as cyclic host liquids (CHLs). The CHLs based on pillararene (51, 52) and cyclodextrins (CDs, 53–55) were obtained by attaching hydrophilic triethylene glycol chains (Fig. 10).38 Given the absence of a solvent, liquid rotaxane 56 was obtained by the threading of aliphatic chains following cationation interlocking under solvent-free conditions with excellent yield (91%, isolated).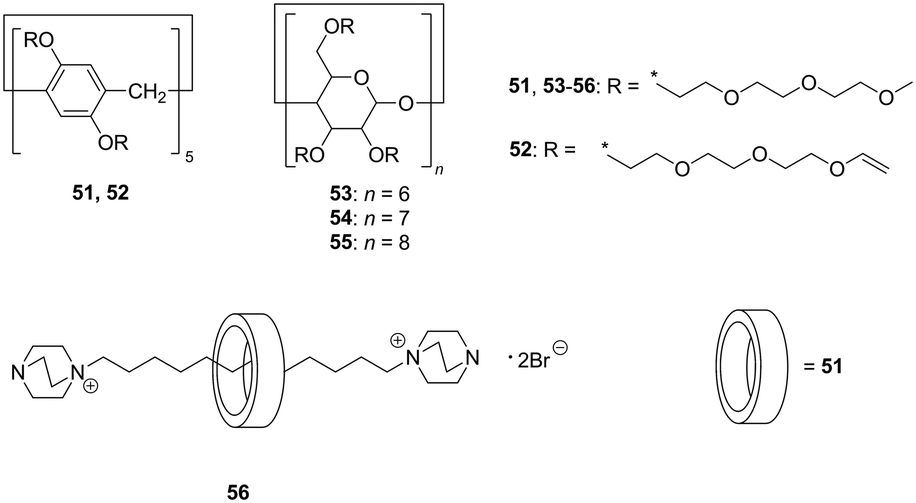 | ||
| Fig. 10 Cyclic host liquids (CHLs) based on pillar[5]arene (51, 52), α-, β-, and γ-cyclodextrins (53–55) and liquid[2]rotaxane (56). | ||
4. Current challenges
Despite the aforementioned significant achievements in functional molecular fluids, functional polymeric materials still face more difficult challenges towards reliable and predictable molecular design principles. Polymeric materials exhibit enhanced optoelectronic and mechanical functionality (e.g. electron/hole mobility, viscoelasticity) that are in clear contrast with their monomeric counterpart. However, polymeric materials unsurprisingly exhibit significantly high elastic modulus due to their rigid framework and strong intra- and/or inter-chain interactions. Therefore, the functionality–fluidity (softness) trade-off is not yet settled in functional polymeric materials. In this section, we discuss the challenges in detail and provide an outlook for future studies.4.1. Conjugated polymers
Conjugated polymers, such as poly(thiophene) (PT), poly(fluorene) (PF), poly(phenylene) (PP), poly(phenylenevinylene) (PPV), and poly(phenylene ethynylene) (PPE), have attracted significant interest in organic optoelectronic applications owing to their characteristic electronic conductivity along the defect-free π-conjugated main chain. However, the softening of conjugated polymers is significantly difficult when compared to their monomeric/oligomeric counterparts because increases in the degree of polymerisation significantly stiffen the bulk material due to the planar geometry (long persistence length) and strong inter-chain interactions (e.g. cofacial π–π interaction). Indeed, most conjugated polymers exist as glassy solids at room temperature even while using low-melting point monomers.39 It should be noted that the softening of conjugated polymers was previously achieved by exploiting copolymerisation with soft polymeric chains,40 blending with the matrix,41 insertion of a flexible spacer unit,42 and the use of solvents to form lyogels.43 We highlight a few challenges related to the fabrication of solvent-free and soft conjugated polymers that exhibit an ‘intact’ conjugated backbone.Kwak et al. reported conjugated polymers based on poly(diphenylacetylene) (PDPA) derivatives (57, 58 in Fig. 11A) that exhibit a highly twisted backbone due to steric repulsion between the adjacent bulky phenyl groups.44a The same group also developed soft conjugated polymers comprising sulfonated PDPA 59 as anionic polyelectrolytes and cationic surfactants (octadecyl)x(methyl)y ammonium bromides (OxMyABs).44b The Tm after complexation was 12.5 °C, 14.3 °C, and 6.1 °C for 59-O1M3AB, 59-O2M2AB, and 59-O3M1AB, respectively.
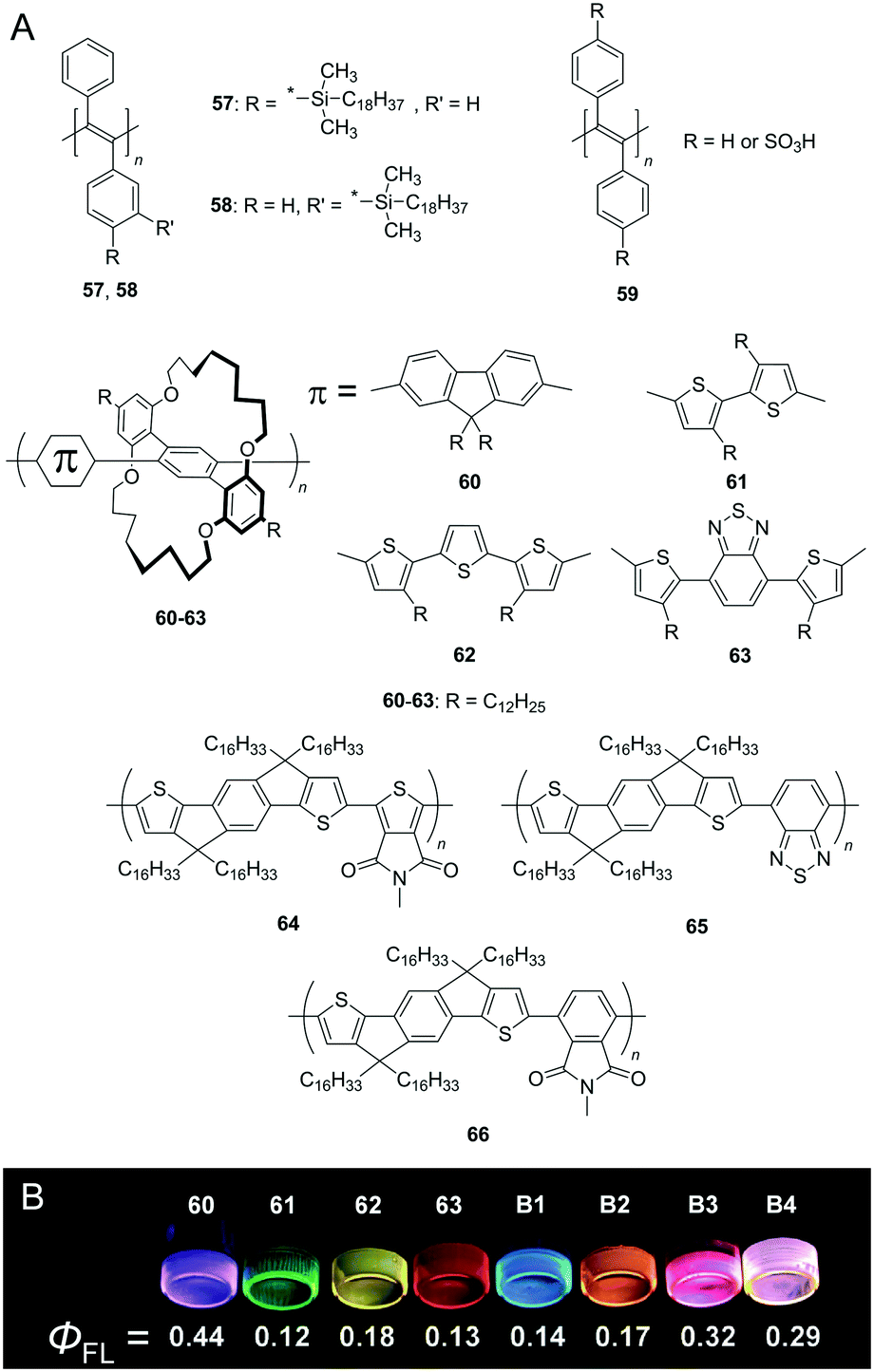 | ||
| Fig. 11 (A) Chemical structures of conjugated polymers (57–63). (B) Photographs of the films of 60–63 and blend films B1–B4 taken under UV (365 nm) illumination. The blending ratios 60/61/62/63 of B1, B2, B3, and B4 are 100/10/0/0, 0/0/100/5, 100/0/0/5, and 200/0/3/1, respectively. Reproduced with permission from ref. 45 © 2013 John Wiley and Sons. | ||
Sugiyasu et al. reported thiophene-based unique insulated molecular wires (IMWs).4560–63 are emissive in the complete visible region and exhibit high fluorescence quantum yield (ΦFL) of up to 44% even in the film state, while IMWs in the other studies exhibit generally approximately 10–20% ΦFL (Fig. 11B). A fluorescent flexible self-standing film was obtained after the thermal annealing of a drop-cast film of 62 at 80 °C. A small piece of the film can sustain weight exceeding 200 g (>5 MPa tensile stress), is stretchable (>300% of tensile strain), and is even foldable without the formation of any cracks.
In the fabrication of conjugated polymer-based soft devices, active components should maintain their functions during deformation such as bending and stretching. Flexibility (bendability) is easily achievable by processing a material into a thin film. However, it is typically difficult to achieve stretchability because tensile deformation mainly results in permanent alterations to the microscopic structure. Recently, Luscombe et al. systematically investigated the molecular design and charge mobility of low-elastic-modulus (<450 MPa) alkylated-indacenodithiophene (alkyl-IDT)-containing donor–acceptor conjugated polymers 64–66.46 The acceptor portion was varied to control the dihedral angle between the donor and acceptor units (φDA). With increases in the φDA, 66 (φDA = 35.9°) exhibited excellent stretchability with crack-onset strain of >100%. However, the charge mobility of 66 (μ = 0.06 ± 0.03 cm2 V−1 s−1) was significantly lower than that of 64 (μ = 0.20 ± 0.10 cm2 V−1 s−1) and 65 (μ = 0.17 ± 0.09 cm2 V−1 s−1), which exhibited high charge mobility but poor stretchability. The formation of highly ductile conjugated polymers typically involves a trade-off between flexibility/deformability and conducting/semiconducting functions because the softening moieties generally act as an electrical insulator.
4.2. Coordination polymers
The melting of substances is a process during which cohesive forces working in solids loosen at a specific temperature (Tm) to form an isotropic liquid. In the case of melting small molecules, polymers, metals, and salts, cohesive forces including van der Waals forces, hydrogen bonds, metallic bonds, and ionic bonds loosen to turn into the molten state. The melting of the solids that exhibit covalent bonds as a cohesive force, namely, covalent crystals, is significantly limited. Examples include diamond, silicon, silicon oxide, and silicon carbide. Coordinating polymers are an important class of crystalline organic–inorganic hybrid materials for gas storage/separation, heterogeneous catalysis, and other applications. Kitagawa et al. reported the melting of crystalline coordinating polymers7a although the melting of coordinating polymers is not typically observed because most coordinating polymers decompose irreversibly when heated with few exceptions.47 A proton conductive coordinating polymer 67 exhibited a reversible solid–liquid phase transition with a sharp endothermic peak (onset = 154 °C, DSC). Rapid cooling of the melt (67′ in Fig. 12) resulted in a supercooled glassy state (67′′ in Fig. 12). The supercooled state was metastable at room temperature while recrystallisation was promoted at 100 °C. The authors synthesised coordinating polymers with different organic moieties and metal centres to extract key factors involved in melting. The diversity observed for the aforementioned coordinating polymers 68–72 despite their structural similarity indicates that thermal behaviour is sensitive to slight alteration in their chemical composition (Table 1). The absence of melting for 71 or 72 indicates that the melting of phosphate-containing inorganic–organic hybrid materials is not necessarily common. In the aforementioned coordinating polymers that were composed of zinc ions, phosphate, and azoles, well-balanced composition, ionicity, and bond strength play critical roles in the melting of coordinating polymers. There is a paucity of studies on the excellent compatible electrical properties and deformability in conjugated polymers, and thus further studies are necessary to overcome this softness–conductivity trade-off.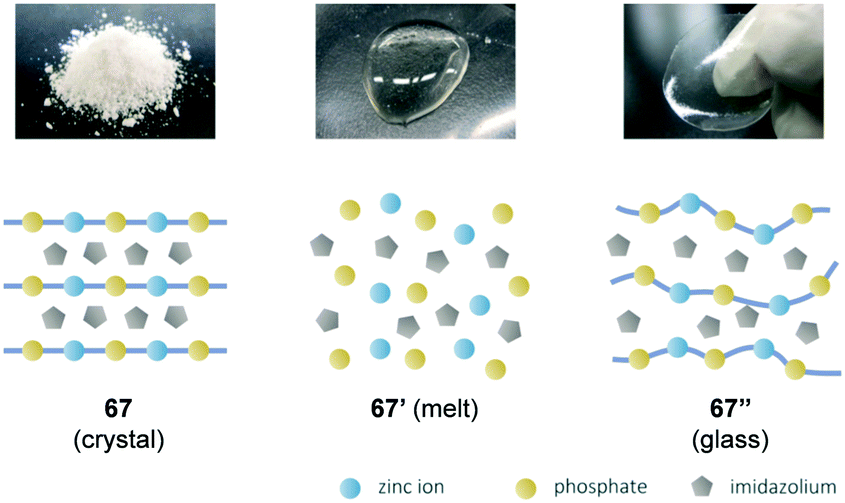 | ||
| Fig. 12 Schematic illustration of the structures of 67 (crystal), 67′ (molten state) and 67′′ (glassy state). Adapted with permission from ref. 7a © 2015 American Chemical Society. | ||
| Compound | Behaviour when heated | T m (°C) | Structural dimension | Array around the metal ion |
|---|---|---|---|---|
| H2Im: imidazolate, HTr: 1,2,4-triazolate, HBim: benzimidazolate, H(2-MeBim): 2-methylbenzimidazolate, H(2-ClBim): 2-chlorobenzimidazolate. | ||||
| [Zn(HPO4)(H2PO4)2]·2H2Im (67) | Melt | 154 | 1D | T d |
| [Zn(H2PO4)2(HTr)2] (68) | Melt | 184 | 2D | O h |
| [Zn3(H2PO4)6(H2O)3]·HBim (69) | Melt | 164 | 1D | O h |
| [Zn3(H2PO4)6(H2O)3]·H(2-MeBim) (70) | Melt | 97 | 1D | O h |
| [Zn3(H2PO4)6(H2O)3]·H(2-ClBim) (71) | Solidify | n/a | 1D | O h |
| [Co3(H2PO4)6(H2O)3]·HBim (72) | Decompose | n/a | 1D | O h |
4.3. Supersoft and superelastic ‘drygels’
Elastomers are an indispensable class of soft materials in our daily life. However, the lower limit of elastic modulus in existing elastomers corresponds to ca. 105–106 Pa (ref. 48) although theoretical expectations suggest that soft elastomers can be readily prepared by increases in the molar mass of a network strand via reductions in the crosslinking density. The aforementioned approaches are primarily hampered by two reasons. First, the crosslinking process leads to permanent trapping of chain entanglements that then behave as topological crosslinks. Second, although employing bottlebrush architecture is a reliable approach to increase the linear density, conventional solid side chains stiffen when the diameter increases and increase the persistent length and entanglement densities. Sheiko et al. recently reported viscoelastic polymers 73–77 that are composed of a rigid dense bottlebrush framework (Fig. 13A).49 Increases in the linear density by employing a dense poly(butyl acrylate) (PBA) bottlebrush architecture generally results in rigidifying the polymer backbone and high Tg. However, supersoft and superelastic ‘dry gels’ are generated via flexible side chains. Although AFM studies are not equivalent to conformations in bulk melts, they qualitatively corroborate the increase in diameter and rigidity of molecular bottlebrushes with increases in the side chain length (nsc) (Fig. 13C).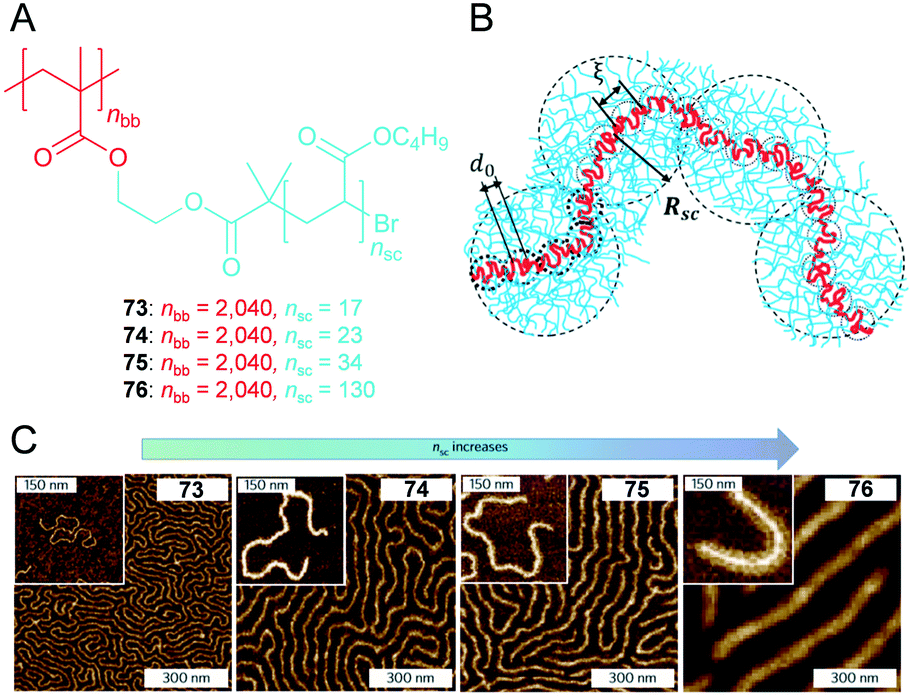 | ||
| Fig. 13 (A) Chemical structure of poly(butyl acrylate) (PBA) bottlebrushes 73–76 with the same backbone length (nBB) but different side chain lengths (nSC). (B) Simulated conformation of a bottlebrush molecule in a melt in a loosely grafted bottlebrush regime. (C) AFM height images of the PBA bottlebrushes on a mica substrate. Large images: Langmuir–Blodgett monolayers. Insets: Single molecules prepared by spin-casting. Reproduced from ref. 49 with permission from the © 2015 Springer Nature. | ||
The architectural reduction of entanglement density within bottlebrush melts provides access to a new range of properties for the design of elastomers with unprecedentedly low elastic modulus and high deformability. In order to expand the supersoft concept from melts into solid materials and to demonstrate the universality of this method as it relates to differing chemical structures, the authors synthesised a series of PDMS bottlebrushes with systematically controlled crosslinking densities by using atom-transfer radical polymerisation of mono- and di-functional macromonomers (Fig. 14A). Crosslinked PDMS bottlebrush 77 (8 mol% crosslinked) exhibited significantly low elastic modulus (103 Pa in magnitude) and significantly higher compressibility when compared to the poly(acrylamide) hydrogel (Fig. 14B and C). Although neither PBA nor PDMS exhibits optoelectronic functions, a unique combination of high flexibility and decreased entanglement density of bottlebrushes exhibited the potential for the creation of a new class of single component (additive-free) polymeric materials including intrinsically rigid functional polymers with low modulus and high deformability.
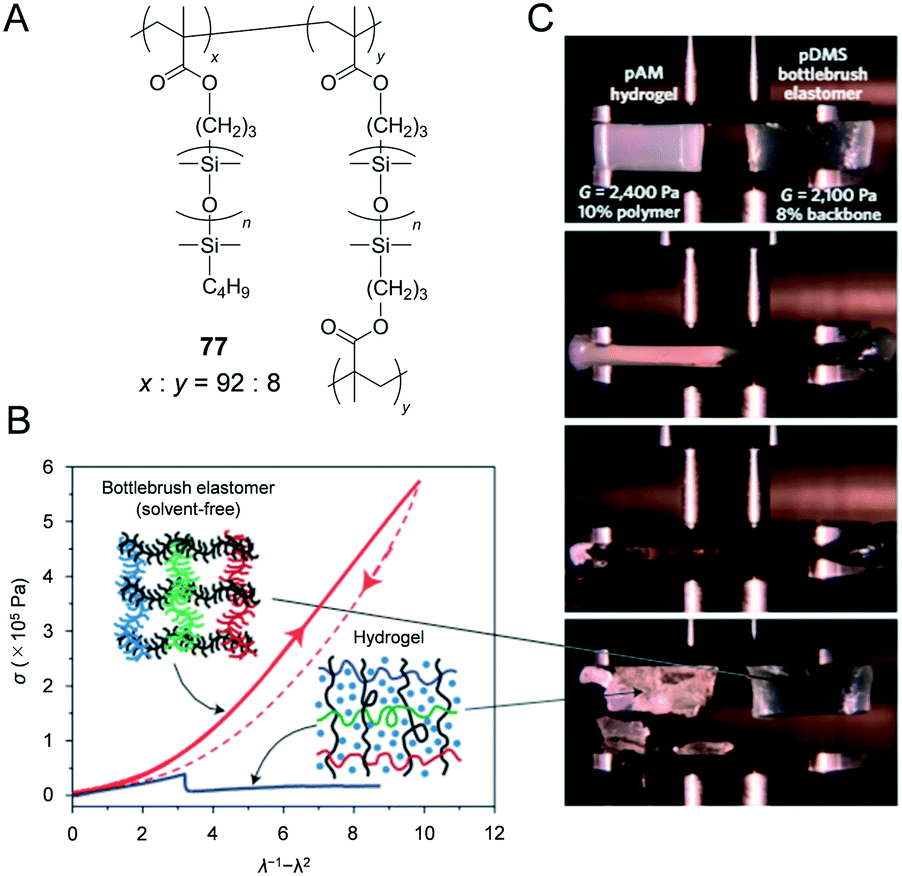 | ||
| Fig. 14 (A) Structure of poly(dimethylsiloxane) (PDMS) crosslinked bottlebrush (77). (B) Stress–strain diagram during compression of 77 and poly(acrylamide) (PAM) hydrogel as a comparison. 77 exhibits significantly higher fracture energy than the hydrogel. (C) A compression behaviour of the poly(acrylamide) (PAM) hydrogel (shear modulus = 2400 Pa) and 77 (shear modulus = 2100 Pa). Reproduced from ref. 49 with permission from the © 2015 Springer Nature. | ||
Conclusions
In this minireview, recent progress in the design, synthesis, and engineering of various functional molecular/polymeric fluids was discussed. In the last decade, molecularly engineered functional fluids have rapidly spread due to their unprecedented fluidic and optoelectronic functions as well as reliability and excellent durability. Various molecular nanoarchitectonics strategies to ‘soften’ highly functional compounds are performed by employing linear-/branched-alkyl chains, OEG or PDMS chains on an intrinsically stiff and rigid functional unit. This is commonly utilised to improve the lack of processability and homogeneous fabrication of organic materials. Despite the aforementioned fascinating properties, only a few studies focus on the development of functional fluid applications when compared with the tremendous progress in conventional optoelectronically active π-conjugated molecules and their ordered assemblies. The history of the aforementioned functional fluids and fabrication into flexible/stretchable devices has just commenced and further exploration, improvements, and novel development are highly desirable. Thus, it is reasonably expected that more sophisticated device systems will be constructed with excellent deformability and bending/stretching functions that originate from the fluidic characteristics as well as specific functions including luminescence, semiconductance, electrochromicity, adhesiveness, and thermal energy- and gas-storage properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (JSPS KAKENHI Grant Number JP18H03922) from the MEXT, Japan, National Science Foundation of China (No. 51803126), Shenzhen Science and Technology Research Grant (No. JCYJ20170302150144008), Start-up Foundation of Shenzhen University (No. 2016003), and Science Foundation of Guangdong Province (No. 2017A030310397).References
- (a) M. Wehner, R. L. Truby, D. J. Fitzgerald, B. Mosadegh, G. M. Whitesides, J. A. Lewis and R. J. Wood, Nature, 2016, 536, 451 CrossRef CAS PubMed; (b) S. I. Rich, R. J. Wood and C. Majidi, Nat Electron, 2018, 1, 102 CrossRef; (c) E. J. Markvicka, M. D. Bartlett, X. Huang and C. Majidi, Nat. Mater., 2018, 17, 618 CrossRef CAS PubMed.
- (a) K. Kumar, C. Knie, D. Bléger, M. A. Peletier, H. Friedrich, S. Hecht, D. J. Broer, M. G. Debije and A. P. H. J. Schenning, Nat. Commun., 2016, 7, 11975 CrossRef CAS PubMed; (b) L. T. de Haan, J. M. N. Verjans, D. J. Broer, C. W. M. Bastiaansen and A. P. H. J. Schenning, J. Am. Chem. Soc., 2014, 136, 10585 CrossRef CAS PubMed; (c) E. Smela, O. Inganäs and I. Lundström, Science, 1995, 268, 1735 CrossRef CAS PubMed.
- (a) S. Bauer, S. Bauer-Gogonea, I. Graz, M. Kaltenbrunner, C. Keplinger and R. Schwödiauer, Adv. Mater., 2014, 26, 149 CrossRef CAS PubMed; (b) T. Yamada, Y. Hayamizu, Y. Yamamoto, Y. Yomogida, A. Izadi-Najafabadi, D. N. Futaba and K. Hata, Nat. Nanotechnol., 2011, 6, 296 CrossRef CAS PubMed; (c) J. T. Muth, D. M. Vogt, R. L. Truby, Y. Mengüç, D. B. Kolesky, R. J. Wood and J. A. Lewis, Adv. Mater., 2014, 26, 6307 CrossRef CAS PubMed.
- (a) G. Wang, N. Persson, P. Chu, N. Kleinhenz, B. Fu, M. Chang, N. Deb, Y. Mao, H. Wang, M. A. Grover and E. Reichmanis, ACS Nano, 2015, 9, 8220 CrossRef CAS PubMed; (b) G. Wang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Angew. Chem., Int. Ed., 2018 DOI:10.1002/ange.201808976.
- (a) S. S. Babu and T. Nakanishi, Chem. Commun., 2013, 49, 9373 RSC; (b) A. Ghosh and T. Nakanishi, Chem. Commun., 2017, 53, 10344 RSC.
- A. Zielinska, A. Takai, H. Sakurai, A. Saeki, M. Leonowicz and T. Nakanishi, Chem. – Asian J., 2018, 13, 770 CrossRef CAS PubMed.
- (a) D. Umeyama, S. Horike, M. Inukai, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2015, 137, 864 CrossRef CAS PubMed; (b) R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F.-X. Coudert, Nat. Mater., 2017, 16, 1149 CrossRef CAS PubMed.
- N. Giri, M. G. Del Pópolo, G. Melaugh, R. L. Greenaway, K. Rätzke, T. Koschine, L. Pison, M. F. Costa Gomes, A. I. Cooper and S. L. James, Nature, 2015, 527, 216 CrossRef CAS PubMed.
- (a) S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Müllen, F. Meghdadi, E. J. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946 CrossRef CAS PubMed; (b) N. Cumpstey, R. N. Bera, P. L. Burn and I. D. W. Samuel, Macromolecules, 2005, 38, 9564 CrossRef CAS; (c) J. Qu, J. Zhang, A. C. Grimsdale, K. Müllen, F. Jaiser, X. Yang and D. Neher, Macromolecules, 2004, 37, 8297 CrossRef CAS.
- H. Ohno and K. Fukumoto, Acc. Chem. Res., 2007, 40, 1122 CrossRef CAS PubMed.
- (a) K. Kubota, S. Hirata, Y. Shibano, O. Hirata, M. Yahiro and C. Adachi, Chem. Lett., 2012, 41, 934 CrossRef CAS; (b) S. Hirata, H. Heo, Y. Shibano, O. Hirata, M. Yahiro and C. Adachi, Jpn. J. Appl. Phys., 2012, 51, 041604 Search PubMed.
- C. Shim, S. Hirata, J. Oshima, T. Edura, R. Hattor and C. Adachi, Appl. Phys. Lett., 2012, 101, 113302 CrossRef.
- A. W. Snow, J. S. Shirk and R. G. S. Pong, J. Porphyrins Phthalocyanines, 2000, 4, 518 CrossRef CAS.
- (a) E. M. Maya, J. S. Shirk, A. W. Snow and G. L. Roberts, Chem. Commun., 2001, 615 RSC; (b) E. M. Maya, A. W. Snow, J. S. Shirk, R. G. S. Pong, S. R. Flom and G. L. Roberts, J. Mater. Chem., 2003, 13, 1603 RSC.
- A. B. Bourlinos, V. Georgakilas, N. Boukos, P. Dallas, C. Trapalis and E. P. Giannelis, Carbon, 2007, 45, 1583 CrossRef CAS.
- (a) A. J. O'Lenick Jr., J. Surfactants Deterg., 2001, 4, 311 CrossRef; (b) M. J. Hollamby and T. Nakanishi, J. Mater. Chem. C, 2013, 1, 6178 RSC.
- (a) T. Michinobu, T. Nakanishi, J. P. Hill, M. Funahashi and K. Ariga, J. Am. Chem. Soc., 2006, 128, 10384 CrossRef CAS PubMed; (b) T. Michinobu, K. Okoshi, Y. Murakami, K. Shigehara, K. Ariga and T. Nakanishi, Langmuir, 2013, 29, 5337 CrossRef CAS PubMed.
- H. Li, S. S. Babu, S. T. Turner, D. Neher, M. J. Hollamby, T. Seki, S. Yagai, Y. Deguchi, H. Möhwald and T. Nakanishi, J. Mater. Chem. C, 2013, 1, 1943 RSC.
- H. G. Elias, Macromolecules – I: Structure and Properties, Plenum, N. Y., 1984 Search PubMed.
- K. Okamoto, F. Lu and T. Nakanishi, Bull. Chem. Soc. Jpn., 2018, 91, 1258 CrossRef CAS.
- W. Hu, Polymer Physics: A Molecular Approach, Springer-Verlag, Wien, 2013 Search PubMed.
- F. Lu, T. Takaya, K. Iwata, I. Kawamura, A. Saeki, M. Ishii, K. Nagura and T. Nakanishi, Sci. Rep., 2017, 7, 3416 CrossRef PubMed.
- (a) A. Saeki, S. Seki, T. Takenobu, Y. Iwasa and S. Tagawa, Adv. Mater., 2008, 20, 920 CrossRef CAS; (b) Q. Xiao, T. Sakurai, T. Fukino, K. Akaike, Y. Honsho, A. Saeki, S. Seki, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2013, 135, 18268 CrossRef CAS PubMed.
- (a) H. Wang, H. K. Bisoyi, L. Wang, A. M. Urbas, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2018, 57, 1627 CrossRef CAS PubMed; (b) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CrossRef CAS PubMed.
- F. Lu, K. Jang, I. Osica, K. Hagiwara, M. Yoshizawa, M. Ishii, Y. Chino, K. Ohta, K. Ludwichowska, K. J. Kurzydłowski, S. Ishihara and T. Nakanishi, Chem. Sci., 2018, 9, 6774 RSC.
- P. Duan, N. Yanai and N. Kimizuka, J. Am. Chem. Soc., 2013, 135, 19056 CrossRef CAS PubMed.
- K. Chung, M. S. Kwon, B. M. Leung, A. G. Wong-Foy, M. S. Kim, J. Kim, S. Takayama, J. Gierschner, A. J. Matzger and J. Kim, ACS Cent. Sci., 2015, 1, 94 CrossRef CAS PubMed.
- B. Narayan, K. Nagura, T. Takaya, K. Iwata, A. Shinohara, H. Shinmori, H. Wang, Q. Li, X. Sun, H. Li, S. Ishihara and T. Nakanishi, Phys. Chem. Chem. Phys., 2018, 20, 2970 RSC.
- S. S. Babu, J. Aimi, H. Ozawa, N. Shirahata, A. Saeki, S. Seki, A. Ajayaghosh, H. Möhwald and T. Nakanishi, Angew. Chem., Int. Ed., 2012, 51, 3391 CrossRef PubMed.
- S. S. Babu, M. J. Hollamby, J. Aimi, H. Ozawa, A. Saeki, S. Seki, K. Kobayashi, K. Hagiwara, M. Yoshizawa, H. Möhwald and T. Nakanishi, Nat. Commun., 2013, 4, 1969 CrossRef PubMed.
- C. Allain, J. Piard, A. Brosseau, M. Han, J. Paquier, T. Marchandier, M. Lequeux, C. Boissieére and P. Audebert, ACS Appl. Mater. Interfaces, 2016, 8, 19843 CrossRef CAS PubMed.
- (a) Y. Norikane, Y. Hirai and M. Yoshida, Chem. Commun., 2011, 47, 1770 RSC; (b) E. Uchida, K. Sasaki, Y. Nakamura, R. Azumi, Y. Hirai, H. Akiyama, M. Yoshida and Y. Noritake, Chem. – Eur. J., 2013, 19, 17391 CrossRef CAS PubMed.
- M. Hoshino, E. Uchida, Y. Norikane, R. Azumi, S. Nozawa, A. Tomita, T. Sato, S. Adachi and S. Koshihara, J. Am. Chem. Soc., 2014, 136, 9158 CrossRef CAS PubMed.
- H. Akiyama and M. Yoshida, Adv. Mater., 2012, 24, 2353–2356 CrossRef CAS PubMed.
- H. Akiyama, S. Kanazawa, Y. Okuyama, M. Yoshida, H. Kihara, H. Nagai, Y. Norikane and R. Azumi, ACS Appl. Mater. Interfaces, 2014, 6, 7933 CrossRef CAS PubMed.
- M. J. Hollamby, M. Karny, P. H. H. Bomans, N. A. J. M. Sommerdjik, A. Saeki, S. Seki, H. Minamikawa, I. Grillo, B. R. Pauw, P. Brown, J. Eastoe, H. Möhwald and T. Nakanishi, Nat. Chem., 2014, 6, 690 CrossRef CAS PubMed.
- K. Masutani, M. Morikawa and N. Kimizuka, Chem. Commun., 2014, 50, 15803 RSC.
- (a) T. Ogoshi, T. Aoki, R. Shiga, R. Iizuka, S. Ueda, K. Demachi, D. Yamafuji, H. Kayama and T. Yamagishi, J. Am. Chem. Soc., 2012, 134, 20322 CrossRef CAS PubMed; (b) T. Ogoshi, T. Aoki, S. Ueda, Y. Tamura and T. Yamagishi, Chem. Commun., 2014, 50, 6607 RSC.
- (a) B. Tanto, S. Guha, C. M. Martin, U. Scherf and M. J. Winokur, Macromolecules, 2004, 37, 9438 CrossRef CAS; (b) J. Jo, C. Chi, S. Höger, G. Wegner and D. Y. Yoon, Chem. – Eur. J., 2004, 10, 2681 CrossRef CAS PubMed.
- (a) J. Oh, S. Rondeau-Gagné, Y. Chiu, A. Chortos, F. Lissel, G. N. Wang, B. C. Schroeder, T. Kurosawa, J. Lopez, T. Katsumata, J. Xu, C. Zhu, X. Gu, W. Bae, Y. Kim, L. Jin, J. Chung, J. B.-H. Tok and Z. Bao, Nature, 2016, 539, 411 CrossRef CAS PubMed; (b) P. Baek, N. Aydemir, Y. An, E. W. C. Chan, A. Sokolova, A. Nelson, J. P. Mata, D. McGillivray, D. Barker and J. Travas-Sejdic, Chem. Mater., 2017, 29, 8850 CrossRef CAS.
- (a) Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu, N. I. Rabiah, Z. Chen, J. W. Chung, C. Linder, M. F. Toney, B. Murmann and Z. Bao, Sci. Adv., 2017, 3, e1602076 CrossRef PubMed; (b) J. Xu, S. Wang, G. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B.-H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59 CrossRef CAS PubMed.
- (a) J. Y. Oh, S. Rondeau-Gagné, Y. Chiu, A. Chortos, F. Lissel, G. N. Wang, B. C. Schroeder, T. Kurosawa, J. Lopez, T. Katsumata, J. Xu, C. Zhu, X. Gu, W. Bae, Y. Kim, L. Jin, J. W. Chung, J. B.-H. Tok and Z. Bao, Nature, 2016, 539, 411 CrossRef CAS PubMed; (b) S. Savagatrup, X. Zhao, E. Chan, J. Mei and D. J. Lipomi, Macromol. Rapid Commun., 2016, 37, 1623 CrossRef CAS PubMed.
- (a) N. Adachi, Y. Takewaki, H. Shirai and M. Kimura, Polym. J., 2009, 41, 132 CrossRef CAS; (b) R. C. Huber, A. S. Ferreira, J. C. Aguirre, D. Kilbride, D. B. Toso, K. Mayoral, Z. H. Zhou, N. Kopidakis, Y. Rubin, B. J. Schwartz, T. G. Mason and S. H. Tolbert, J. Phys. Chem. B, 2016, 120, 6215 CrossRef CAS PubMed.
- (a) Y. Jin, J. Bae, K. Cho, W. Lee, D. Hwang and G. Kwak, Adv. Funct. Mater., 2014, 24, 1928 CrossRef CAS; (b) Y. Jin, J. Yoon, T. Sakaguchi, C. Lee and G. Kwak, Adv. Funct. Mater., 2016, 26, 4501 CrossRef CAS.
- C. Pan, K. Sugiyasu, Y. Wakayama, A. Sato and M. Takeuchi, Angew. Chem., Int. Ed., 2013, 52, 10775 CrossRef CAS PubMed.
- Y. Li, W. K. Tatum, J. W. Onorato, Y. Zhang and C. K. Luscombe, Macromolecules, 2018, 51, 6352 CrossRef CAS.
- S. Horike, D. Umeyama, M. Inukai, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 7612 CrossRef CAS PubMed.
- (a) S. K. Patel, S. Malone, C. Cohen, J. R. Gillmor and R. H. Colby, Macromolecules, 1992, 25, 5241 CrossRef CAS; (b) R. Everaers, New J. Phys., 1999, 1, 12 CrossRef.
- W. F. M. Daniel, J. Burdyńska, M. Vatankhah-Varnoosfaderani, K. Matyjaszewski, J. Paturej, M. Rubinstein, A. V. Dobrynin and S. S. Sheiko, Nat. Mater., 2016, 15, 183 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |