
Nanoarchitectonics through supramolecular gelation: formation and switching of diverse nanostructures
Yutao
Sang
 ac and
Minghua
Liu
*abc
ac and
Minghua
Liu
*abc
aBeijing National Laboratory for Molecular Science, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: liumh@iccas.ac.cn
bCollaborative Innovation Centre of Chemical Science and Engineering, Tianjin 300072, China
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 25th October 2018
Abstract
Supramolecular gels are currently an important class of soft materials that are attracting great interest. The gelation process has been proven to be a powerful approach for the construction of diverse and uniform architectures. Generally, these architectures are on the nanoscale and hierarchically self-assembled from small molecules. Nanoarchitectonics tries to bridge the gap between molecules and nanostructures, while supramolecular gelation provides a good platform to realize such a goal. While the design of the gelator molecules and the function of the gel are important topics, here, we focus on how small molecules can be fabricated into nanostructures through gelation, which is in fact the connection between the microstructures to the macroscopic functions. We showed meritorious examples of typical architectures constructed through the supramolecular gelation process. Then some examples showing the transformation or switching between different nanostructures are presented, which reflected the unique responsiveness of the gel system. Through these two key parts, we hope to provide useful insights into the design and control of functional nanoarchitectures via supramolecular gelation.
 Yutao Sang | Yutao Sang obtained his B.Sc. and M.Sc. degrees in Chemistry from Qingdao University, China. In 2016, he enrolled in a Ph.D course at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), under the supervision of Prof. Minghua Liu, where he is currently in the third year. His ongoing research includes supramolecular chirality, chiroptical materials and symmetric breaking of achiral molecules in gels and related systems. |
 Minghua Liu | Minghua Liu graduated from Nanjing University in 1986. He received his Ph.D. from Saitama University, Japan in 1994. After doing postdoctoral research at the Institute of Physical and Chemical Research (RIKEN) and at the Tokyo University of Agriculture and Industry, he joined the Institute of Photographic Chemistry in 1998 and then the Institute of Chemistry, CAS in 1999 and has been working as a professor to date. His research interests cover supramolecular chemistry, colloid and interface science, self-assembly and gel-based soft nanomaterials. He is particularly interested in the chirality issues in supramolecular and self-assembly systems. |
Design, System, ApplicationNanoarchitectonics refers to a unified concept combining nanotechnology and methodologies in related research fields, such as supramolecular chemistry, self-assembly, and self-organization, to satisfy the major features of nanosciences. With such a concept, supramolecular gelation provides a methodology for the precise fabrication of nanoarchitectures that bridge the molecular to macroscopic structure and function via self-assembly. Although fibrous structures occupied many of the gel systems, diverse nanostructures are in fact observed in many supramolecular gels. Thus, the gelation process is considered as one of the most effective ways to obtain diverse nanoarchitectures starting from molecules. Here, we focus on how small molecules could be fabricated into various nanostructures and try to bridge the gap between molecular designs and nanoarchitectures. Moreover, due to the inherent reversibility and flexibility that are unique to supramolecular gel systems, the transformation or switching between different nanostructures is also discussed. The formation and transformation in gel systems provide supramolecular gelation many possibilities to obtain diverse nanoarchitectures, which are supposed to be useful in a number of applications. |
1. Introduction
Nanoarchitectonics, which has been defined as the precise fabrication of nanoarchitectures, has become an interesting emerging multidisciplinary field that bridges the gap between molecular materials and nanotechnology.1–5 While nanoarchitectonics can be realized in many ways from atomic or molecular manipulation, field-induced material fabrication, hierarchical assembly, and controlled self-assembly and organization,2,6–11 supramolecular gelation provides an efficient way of obtaining nanostructures starting from molecules. Supramolecular gels, which can be considered to comprise a solid matrix that immobilizes a liquid component by surface-tension effects, have been attracting a huge amount of research attention in the past few decades.12–16Macroscopically, the most intuitionistic phenomenon for gelation is the loss of fluidity of a transparent solution, which makes the gel appear homogeneous. However, microscopically, gels generally contain two phases and are heterogeneous systems. With the help of microscopy techniques,17 including scanning electron microscopy (SEM), transmission electron microscopy (TEM) and atomic force microscopy (AFM), those solid phase or nano/micro-structured materials with wide-ranging shapes and sizes are unveiled. These nanostructures are generally self-assembled from small organic molecules, in which the molecular design and the non-covalent bond between the molecules play important roles. Through supramolecular gelation, diverse and uniform nanostructures can be obtained in a large quantity, thus, it can serve as a general way of bridging molecular engineering and nanotechnology, which is exactly the task of nanoarchitectonics.
With respect to supramolecular gels, there are essentially three important aspects. One is the design of gelator molecules. While the early stage of gel research largely depended on the serendipitous discovery of gelator molecules, now it becomes common to design gelator molecules by using a method based on gelation.18,19 The second aspect is the nanostructures formed by small molecules. Although fibrous structures occupied many of the gel systems, diverse nanostructures are in fact observed in many gel systems, which are largely dependent on the design of the gelator molecules and significantly affect the gel properties. The third is the properties and applications of the gels. As one important class of soft materials, supramolecular gels can be formed from a broader range of building blocks and have achieved applications in nearly every field from cosmetics, food, pharmaceuticals, petroleum, electronics, energy, environment, biological and many more.20–26 There are many reviews on gelator design and application of gels.18,19,21–24,27,28 However, the role of the nanostructure in the gels is not fully discussed.
Different from permanently covalently cross-linked polymer gels, the main driving force during the supramolecular gelation process is non-covalent interactions such as hydrogen bonding, solvophobic effects, metal–ligand interactions, ion pair coupling, etc.13,28–30 Within this context, the principal advantage of supramolecular gels over other materials is their inherent reversibility and flexibility in structure design. For example, the double exchange between the phase state and microscopic morphology can be easily realized, one kind of gelator molecule can sometimes form various morphologies, and switching among preformed architectures is possible.31 All of these closely depend on the local conditions such as ageing time, solvent, pH value, temperature, sonication and photo-irradiation, which provide gelation many possibilities to obtain diverse nanoarchitectures.24,32–34
In this review, with the concept of nanoarchitectonics, we will start with focusing on how small molecules could be fabricated into various nanoarchitectures and try to bridge the gap between molecular structures and nanoarchitectures. Then we focus on the transformation or switching between different nanostructures, as illustrated in Fig. 1. This is unique to gels due to their responsiveness to various stimuli. We anticipate that the examples discussed in this review article will give some hints or inspiration to the design of various nanostructured materials based on molecular engineering.
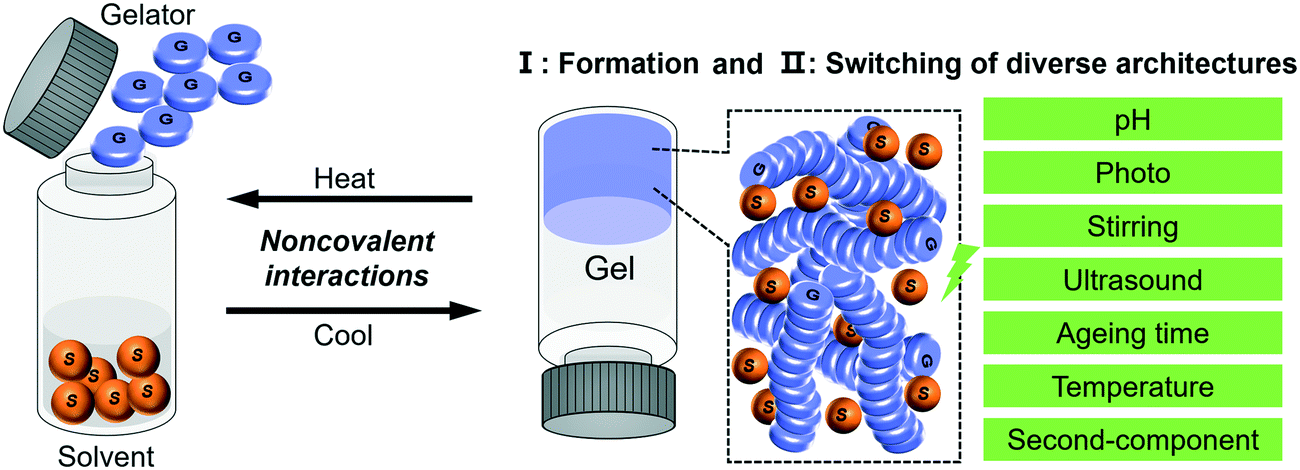 | ||
| Fig. 1 Schematic illustration of the typical gelation process and the two related aspects of focus in this review. | ||
2. Diverse nanoarchitectures via supramolecular gelation
Fig. 2 illustrates some typical architectures formed through gel formation, which can be broadly classified into several hierarchical levels and several dimensions from zero-dimension (0D) to three-dimension (3D). One-dimensional (1D) morphologies, including nanofibers, helixes, tubes and their analogue, are the most common structures observed in gels. 0D structures mainly consist of hollow or solid spheres, which are not very common in gels. Two-dimensional (2D) nano/micro-sheets, plate-like morphologies, membranous- and porous membrane-like structures can be formed as well during the gelation process. More hierarchical level nano/micro-structures such as flower-like structures usually composed of dozens of tubes, nanospheres or petals. Besides the above conventional structures, some unique structures such as trumpet-shaped tubes, dendritic nanotwists, nanotrumpets, and toruloid-like nanostructures could be also obtained through gel formation. In this section, several basic nanoarchitectures and their molecular packing are listed in order to explain the relationship between the molecular design and the nanoarchitectures. | ||
| Fig. 2 Schematic illustration of various nanoarchitectures obtained through supramolecular gelation. Based on the structural features, these basic nanoarchitectures can be classified into different hierarchical orders from zero-dimensional (0D) to three-dimensional (3D). In addition, some special architectonics (right panel) can also be formed via the gelation process. | ||
2.1 Fiber-based nanoarchitectures
Nanofibers and fiber bundles of diverse diameters are the most commonly observed nanoarchitectures during supramolecular gelation. Furthermore, a nanofiber is also the basic structure in 1D architectures, and it can be obtained from a large number of organic low-molecular-weight molecules.35–37 For example, as shown in Fig. 3A (left), well developed cylindrical fibers with a diameter of about 200 nm were formed from a C3-symmetric molecule.38 When the saturated alkyl ends were replaced by double bonds, a significant change in the morphology of the assembly was observed (Fig. 3A right). Both right- and left-handed helical fibers with a diameter of about 3 mm were generated. It should be noted that the size of the helical fibers is 15 times thicker than that of the cylindrical fibers, which suggested that the terminal double bonds enhanced the further growth of the initial cylindrical assemblies via entwinement of the side chains. Density functional theory (DFT) calculation revealed that the helical columnar assemblies were formed due to the intra- and intermolecular local dipole–dipole interactions (Fig. 3C).38 The stacked dimer exhibited a S6 symmetric structure, in which the six local dipoles of the isoxazole rings adopt a circular array. The terminal double bonds in the alkyl chain were proved to be important, which further enhanced the growth of the initial cylindrical assemblies via entwinement of the side chains.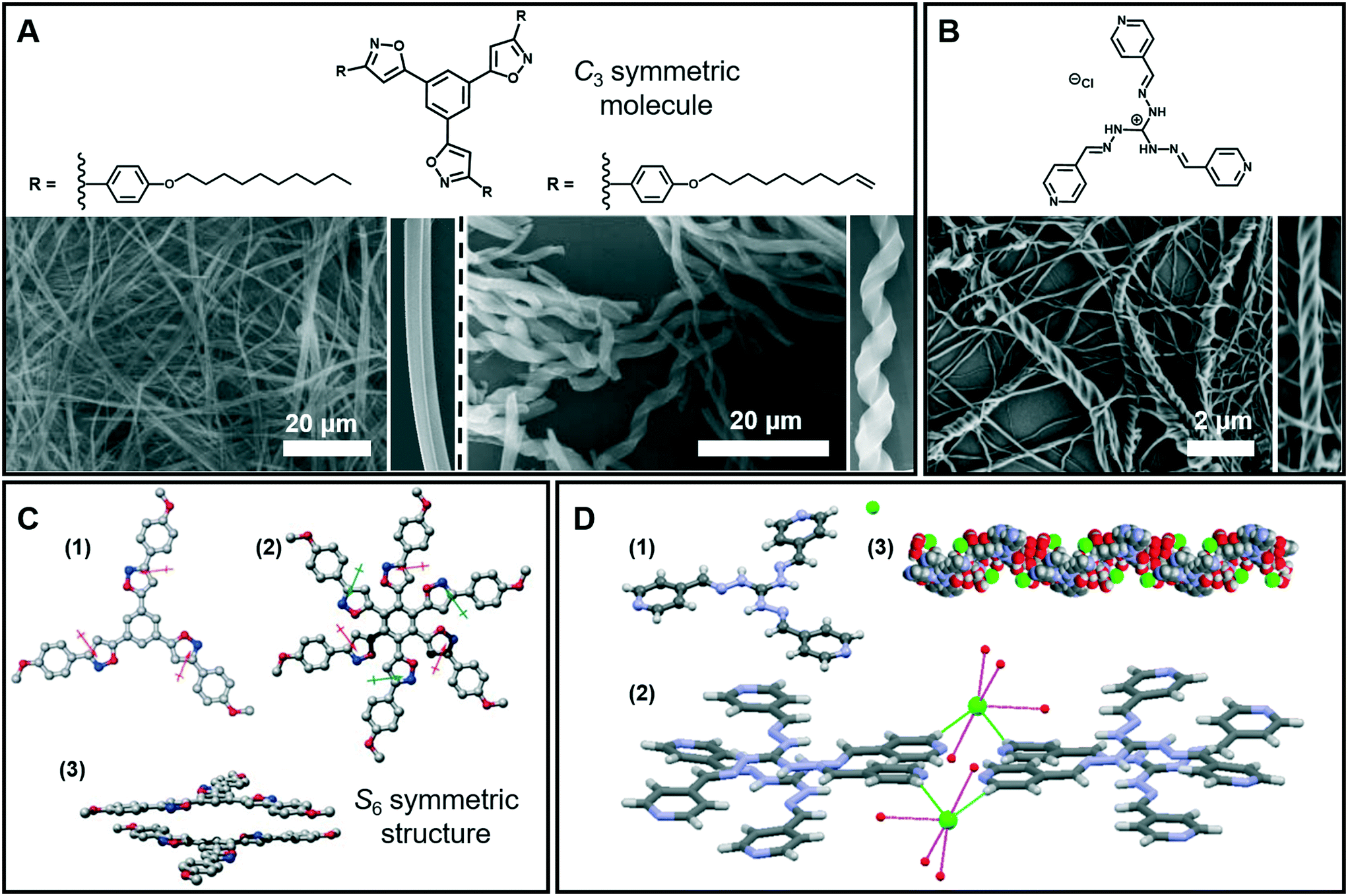 | ||
| Fig. 3 (A) Nanofibers, helical fibers and (B) super-helical fibers architectures obtained in supramolecular gels. (C) Calculated structures and stacked dimer of the molecules in (A): (1) the arrows denote the local dipole moment of each isoxazole ring; (2 and 3) top and side views of the stacked dimer. (D) Crystal structure (1), packing model (2) and 1D left-handed helical arrangements (3) of the molecule in (B). (A and C) Reprinted with permission from ref. 38. Copyright 2008, Royal Society of Chemistry. (B and D) Reprinted with permission from ref. 40. Copyright 2016, American Chemical Society. | ||
The 1D nanofibers can be bundled together and form super-helical fiber nanostructures.39–42Fig. 3B shows that another C3-symmetric gelator formed super-helical structures obviously twisted from a bunch of nanofibers with preferential orientations.40 As illustrated from the crystal structures (Fig. 3D), each Cl− ion forms four H-bonds with four-water molecules and the hydrated Cl− ion connects the two cationic units by constructing two H-bonds with terminal pyridyl hydrogen atoms. Because of the intrinsic positive charge of guanidinium units, two consecutive molecules stack in a slipped way which results in a double propeller-type arrangement, leading to a left-handed helix-like one-dimensional (1D) arrangement. Since the central guanidinium unit is cationic, the handedness of the super-helical fibers was successfully controlled by introducing an optically active counteranion.
2.2 Nanobelt and nanosheet architectures
Generally, nanobelt architectures can be regarded as the parallel packing of nanofibers,43 and when the nanobelts continuously grow, nanosheets can be obtained. Therefore, these layer structures have two clear features: one is that the planar dimensions are at least a few orders of magnitude greater than their thicknesses, and the other is the high degree of anisotropy because the molecules within a layer are integrated by stronger intermolecular interactions than binding forces between layers.44Fig. 4A shows the nanobelt structures formed from tetraphenylethylene (TPE)-capped dipeptides.45 From the TEM image of the supramolecular gels, it clearly indicates that the gel is comprised of nanobelts with an average width of about 295 nm. The optimized tetramer structure shown in Fig. 4B suggested that the π–π interactions in the TPE packing were significantly important. Moreover, the visualization of intermolecular interactions of a dimer indicated a continuous wave function overlap with the electronic attraction effect, van der Waals effect and some weak repellent effect (Fig. 4B). It was found that the intermolecular electrostatic interactions between the negatively charged carboxylates and partially positively charged hydrogen atoms on tetraphenylethylene are necessary in the formation of the nanobelts.45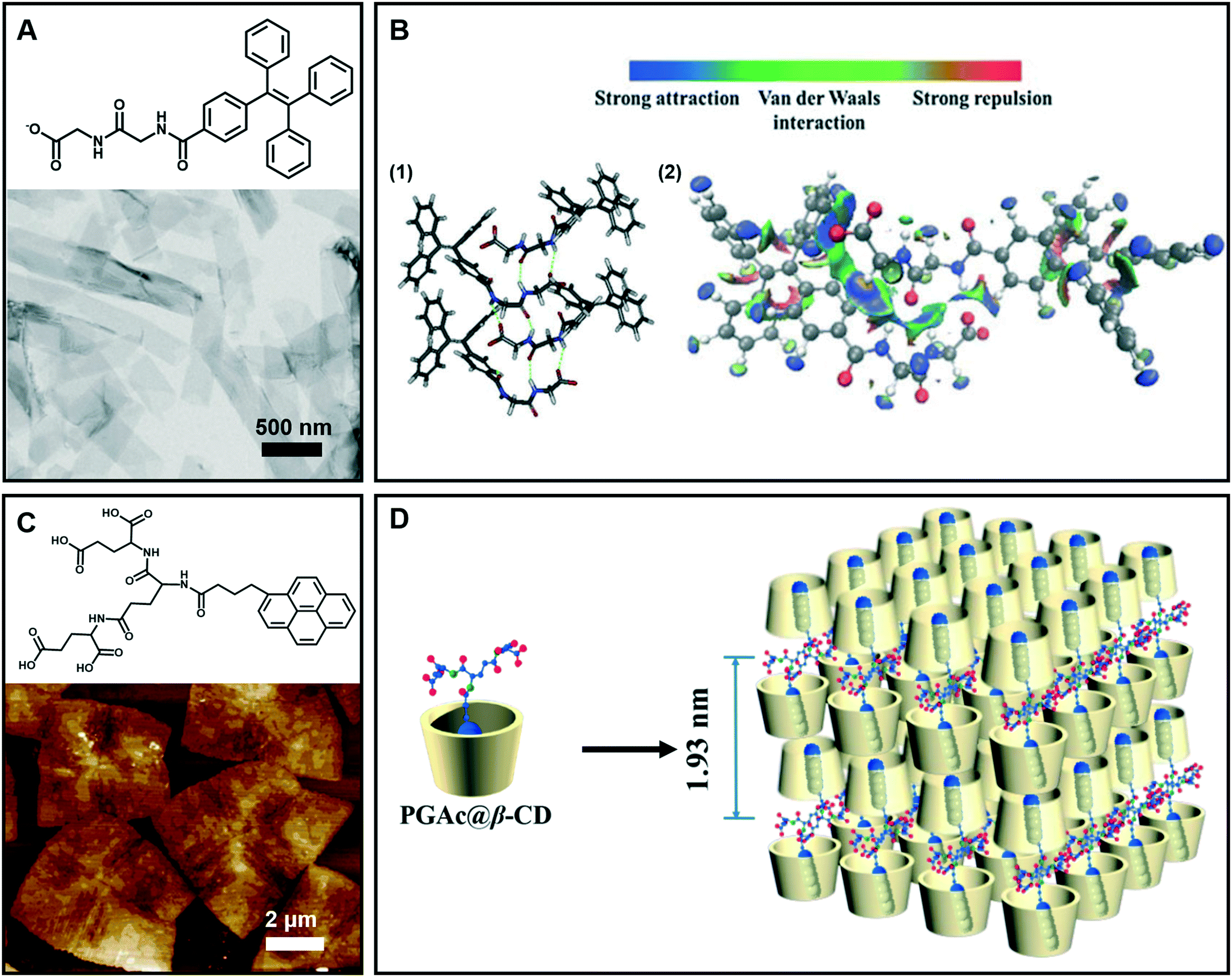 | ||
| Fig. 4 (A) Nanobelt and (C) nanosheet architectures obtained in supramolecular gels. (B) (1) Energy minimized geometry of a tetramer, hydrogen bonds: green dashed line. (2) Visualization of the intermolecular interactions of a dimer. C: gray, H: white, N: blue, and O: red. (D) Schematic representation of self-assembly of the nanosheet from gelators and β-CD. (A and B) Reprinted with permission from ref. 45. Copyright 2016, Royal Society of Chemistry. (C and D) Reprinted with permission from ref. 46. Copyright 2018, American Chemical Society. | ||
Fig. 4C gives an example of supramolecular nanosheets based on the host–guest interaction between the π-peptide dendron and β-cyclodextrin (β-CD).46 In the absence of cyclodextrin, only a helical nanofiber could be obtained. However, when β-CD was added, the host–guest interaction between them occurred, and the induction of β-CD to the gelator gave rise to the crystalline nature of the formed hydrogel. XRD results revealed that the maximum of d-spacing was estimated to be around 1.93 nm, which should be assigned to the bilayer length of the complex (Fig. 4D). Interestingly, due to the host–guest inclusion interaction, molecular level emission resulting from the pyrene moiety was observed. Moreover, this complex showed CPL exclusive from the monomer of pyrene with the formation of 2D nanosheets.46
2.3 Helical and twisted nanoarchitectures
It is interesting to note that chiral molecules are apt to form gels; thus chiral structures are frequently encountered in gels. Twisted or helical nanoarchitectures, such as α-helical proteins and double-helix DNA which are among the most fascinating structures found in nature, can be fabricated through artificial supramolecular gelation. Fig. 5A shows that the intermediate planar structure, which is usually self-assembled from an amphiphile as a bilayer or multi-bilayer unit, plays a very important role in the formation of twist or helical nanoarchitectures.47 Both the twisted and helical nanoarchitectures are generally derived from nanobelts or nanoribbons. However, it should be noted that twisted and helical structures are technically different. In terms of geometry differences, the helical ribbons have a cylindrical curvature and can be considered as precursors of tubules (Fig. 5B), while the twists exhibit a Gaussian or saddle-like curvature (Fig. 5C).47 On the other hand, twist structures may be thermodynamically stable, but helices are usually unstable and can eventually evolve into tubules. From the aspects of nanoarchitectonics, double helixes, triplexes, or a multi- or superhelix can be found in a higher-order structure. Furthermore, some of the twists can further develop into dendritic structures.48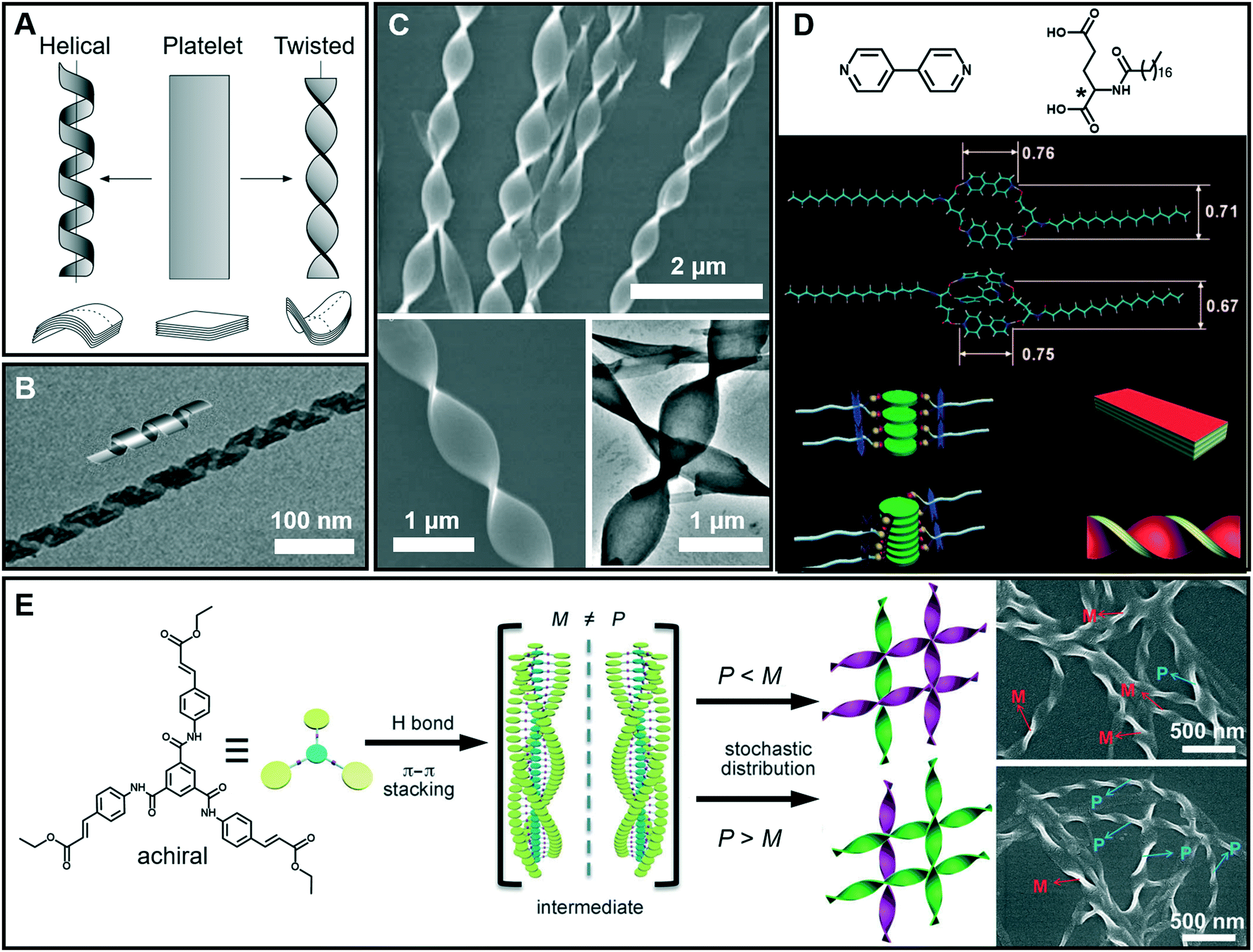 | ||
| Fig. 5 (A) Schematic illustration of twisted and helical ribbons. Reprinted with permission from ref. 47. Copyright 1999, Nature Publishing Group. (B) TEM image and cartoon representation of helical structures. Reprinted with permission from ref. 57. Copyright 2013, American Chemical Society. (C) SEM and TEM images of twisted ribbons. (D) Molecular structures and models for the formation mechanism of a co-assembled chiral twist in (C). Reprinted with permission from ref. 49. Copyright 2011, Wiley-VCH. (E) Formation of nanotwists by the hierarchical self-assembly of achiral building blocks via gelation. Reprinted with permission from ref. 56. Copyright 2014, Wiley-VCH. | ||
Fig. 5D shows the models for the formation mechanism of the chiral twist in Fig. 5C.49 Compared with the diverse hydrogen bonds between carboxylic acids, carboxylic acid–pyridyl hydrogen bonds were predominantly formed, indicating that a two component complex was produced as the basic building block for further assembly. It should be noted that the amide–amide hydrogen bonds were well preserved, and an ordered composite bilayer structure was formed due to these highly directional hydrogen bonds and the strong hydrophobic interactions between the alkyl chains. At an equal ratio of these two components, the carboxylic acid and bipyridine combined together and self-assembled into flat strips or ribbons. However, when bipyridine was in a slight excess, the flat ribbons started to twist because of the involvement of more π–π stacking interactions. The simulation model in Fig. 5D suggested that the additional bipyridine was inserted into the cavity and a sandwich supramolecular structure may be formed. In addition, a remarkable decrease in the distance between the two intermolecular carboxylic groups was observed. Consequently, the chiral centers of the C18 molecules are packed much more closely, similar to those in linear π-conjugated systems, and this eventually causes the nanoribbon to form a twist.49
Generally, chiral building blocks are likely to form twists and helical structures, because their chiral information can cause the evolution of planar structures as the result of the chiral interactions.50,51 However, achiral gelators could also form chiral architectures with an equal or unequal amount of enantiomers. The latter case can display macroscopic chirality52,53 or optical activity54 that resulted from spontaneous symmetry breaking,55 which is one of the most important approaches in obtaining chiral assemblies. As shown in Fig. 5E, a achiral C3-symmetric molecule can form supramolecular gels in a mixed solvent of N,N-dimethylformamide (DMF) and H2O.56 Left (M) and right-handed (P) twists simultaneously formed in an unequal number, resulting in macroscopic chirality without any chiral additives. At the early stages of gelation, one-dimensional helical aggregates with predominantly a P or M conformation were randomly formed via three-fold intermolecular hydrogen bonding and π–π stacking. Once the assemblies with a certain chiral bias start to form, the further growth follows the original chiral conformation, and larger twists with an unequal amount of conformations finally form (Fig. 5E).56
2.4 Tubular architectures
As mentioned above, tubular architectures can be obtained from the evolution of helical structures.47,58 1D nanotube architectures have been receiving increasing interest as functional materials and biomimetic systems because 1D hollow structures could provide a confined space for compound encapsulation, selected transportation, specified chemical reactions or sensing, ion transfer, and catalysis.59–63 Peptide-amphiphiles, glucolipids, amphiphilic metal complexes, phospholipids, cyclic peptides and bolaamphiphiles have been designed to form organic nanotubes.64–67 Here, as shown in Fig. 6A–D, some amphiphilic gelators based on glutamic acid are given to illustrate the diversity of tubular architectures during supramolecular gelation, including single-layer, multi-bilayer, trumpet-shaped and hexagonal tubes.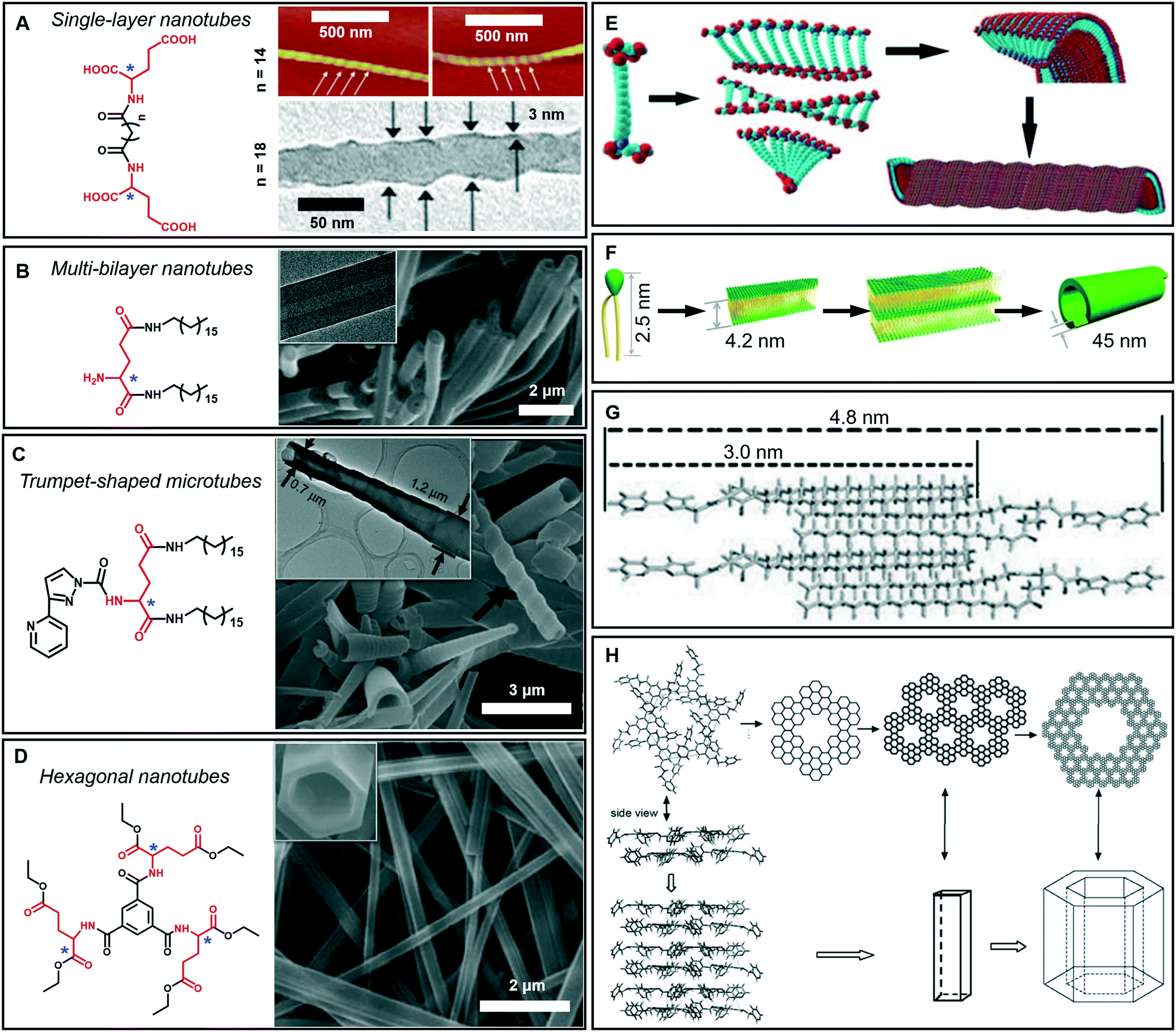 | ||
| Fig. 6 Tubular architectures based on glutamic acid-based amphiphilic gelators: from single-walled nanotubes to hexagonal nanotubes. (A) Single-layer nanotubes and (E) their possible formation mechanism. Reprinted with permission from ref. 68 (n = 18) and ref. 69 (n = 14). Copyright 2005 and 2010, Royal Society of Chemistry. (B) Multi-bilayer nanotubes and (F) their possible formation mechanism. Reprinted with permission from ref. 71. Copyright 2011, Wiley-VCH. (C) Trumpet-shaped microtubules and (G) the corresponding packing model of gelator molecules. Reprinted with permission from ref. 74. Copyright 2013, Wiley-VCH. (D) Hexagonal nanotubes. Reprinted with permission from ref. 75. Copyright 2012, Wiley-VCH. (H) Proposed formation process of the hexagonal nanotubes. Reprinted with permission from ref. 76 copyright 2008, Royal Society of Chemistry. | ||
Generally, bolaamphiphiles are able to form single-walled nanotubes due to the two hydrophilic head groups covalently linked by hydrophobic chains. For example, a bolaamphiphile is able to form a transparent gel in a mixture of ethanol and water during gelation (Fig. 6A).68,69 Both the TEM and AFM images indicate that the walls of the helical nanotubes have a single molecular-layer thickness. The simulation results revealed that the carboxylic acid units and amide groups are crucial for the formation of helical nanotubes (Fig. 6E). On the one hand, owing to the intermolecular H-bonds between carboxylic acid units, twisted dimer-like aggregates tended to form, which therefore yield the curved edges of the molecular assembly. On the other hand, the amide groups would allow the further aggregation of the twisted molecular assembly driven by the formation of the amide to amide H-bonds between the adjacent molecular assemblies and then form a curved helical ribbon and a subsequent helical spherical-nanotube (Fig. 6E).68 In contrast, traditional amphiphiles usually self-assemble into bilayer structures and further evolve into hollow tubes with double or multi-layered walls. For example, the wall of the helical tube fabricated from a dendritic glutarimide lipid is 4 nm, which corresponds to a bilayer thickness.70 The reason for the formation of the double-wall is that these multi hydrogen bonds are stabilized by the bilayer structures.
Another glutamic acid-based gelator was found to form ultra-long nanotubes with an aspect ratio higher than 1000, and the thickness of the nanotube walls was about 45 nm, indicating a multi-bilayer nanotube structure (Fig. 6B).71 As a result of the strong hydrogen bonds between the head groups as well as hydrophobic interactions among the tails, the lipid molecules packed into intermediate plates that consisted of many bilayers (Fig. 6F).71 Owing to the chiral nature of the formed layers, this multiple bilayer plate rolls up into a multi-layered thick-walled nanotube as shown in Fig. 6B. It should be noted that compared with the multilayer structures,72 the metal-coordinated single-layer nanotube is proved to be a more efficient catalyst for asymmetric reactions with high enantioselectivity (up to 97% ee),73 which is due to the alignment of the catalytic sites on the surface of the single-layer nanotube being much more efficient that that embedded in the layer walls for multilayer nanotubes.
In addition, some special tubular structures can also be formed during supramolecular gelation. For example, as shown in Fig. 6C, the introduction of heteroaromatic rings can be used to regulate the tubular nanoarchitectures.74 Similarly, this gelator tended to form a bilayer structure and served as the basic unit for the further stacking or rolling. Polar solvents, such as dimethyl sulfoxide, are suggested to form H-bonds with aromatic hydrogen atoms and the head-groups of gelators packed on the outside of the bilayer structure, as shown in Fig. 6G. Therefore, the π–π stacking of the pyridine and pyrazole rings was enhanced and favored the formation of nanotube architectures.74
Interestingly, a C3-symmetrical glutamic acid ethyl ester gelator was found to form hexagonal tubes in a wide range of mixed solvents through supramolecular gelation (Fig. 6D).75 The one-dimensional growth of the monomers into columnar-type supramolecular assemblies results in these special tubular structures, which is quite different from the formation of the amphiphilic peptide nanotubes. Previous reports suggested that six amide groups are firstly assembled into a cyclic basic unit, and then six basic units are further assembled to form a hexagonal 2D sheet, which is sequentially organized into a hexagonal tube (Fig. 6H).76,77 Due to the formation of multiple hydrogen bonds, the gelation capability of this C3-symmetrical gelator is significantly enhanced. Tubular structures can be always formed through instant gelation in many organic solvents. This excellent property permitted the encapsulation of various functional species, such as biomacromolecules, synthetic polymers, and low-molecular-mass molecules.75 In particular, achiral aggregation-induced emissive compounds (AIEgens) can be encapsulated into confined nanotubes via co-gelation, and emit tunable and controlled circularly polarized luminescence (CPL).78
Furthermore, these 1D nanoarchitectures including nanofibers,79 nanotwists80 and nanotubes69 can be used as templates to grow inorganic structures,81 leading to a wide range of inorganic nanoarchitectures based on supramolecular gelation.
2.5 Spherical architectures
Spherical and colloidal particles are of intense interest due to their applications in catalysis, separation science and chemical sensors.82–85 Supramolecular gelation is also applied to prepare spherical nanostructures.86 As shown in Fig. 7A, an amphiphilic gelator can self-assemble in aqueous media and result in a hydrogel with nanospherical morphologies.87 The nanosphere is solid with a diameter of 700 ± 100 nm. A two-step hierarchical self-assembly process was proposed for the formation of this nanosphere. Firstly, the gelators assembled into extended 2D sheets through intermolecular π–π stacking. As time proceeds, the nanosheets tend to minimize their contact with the solvent (H2O/MeOH) due to the hydrophobic effect. In contrast, the amide groups tend to be exposed to the polar medium due to H-bonding with the solvent. As a result, nanospheres whose exteriors are decorated with amide groups were formed. It should be noted that this spherical architecture with surface exposed amide groups can be utilized for catalysing the Knoevenagel condensation reaction.87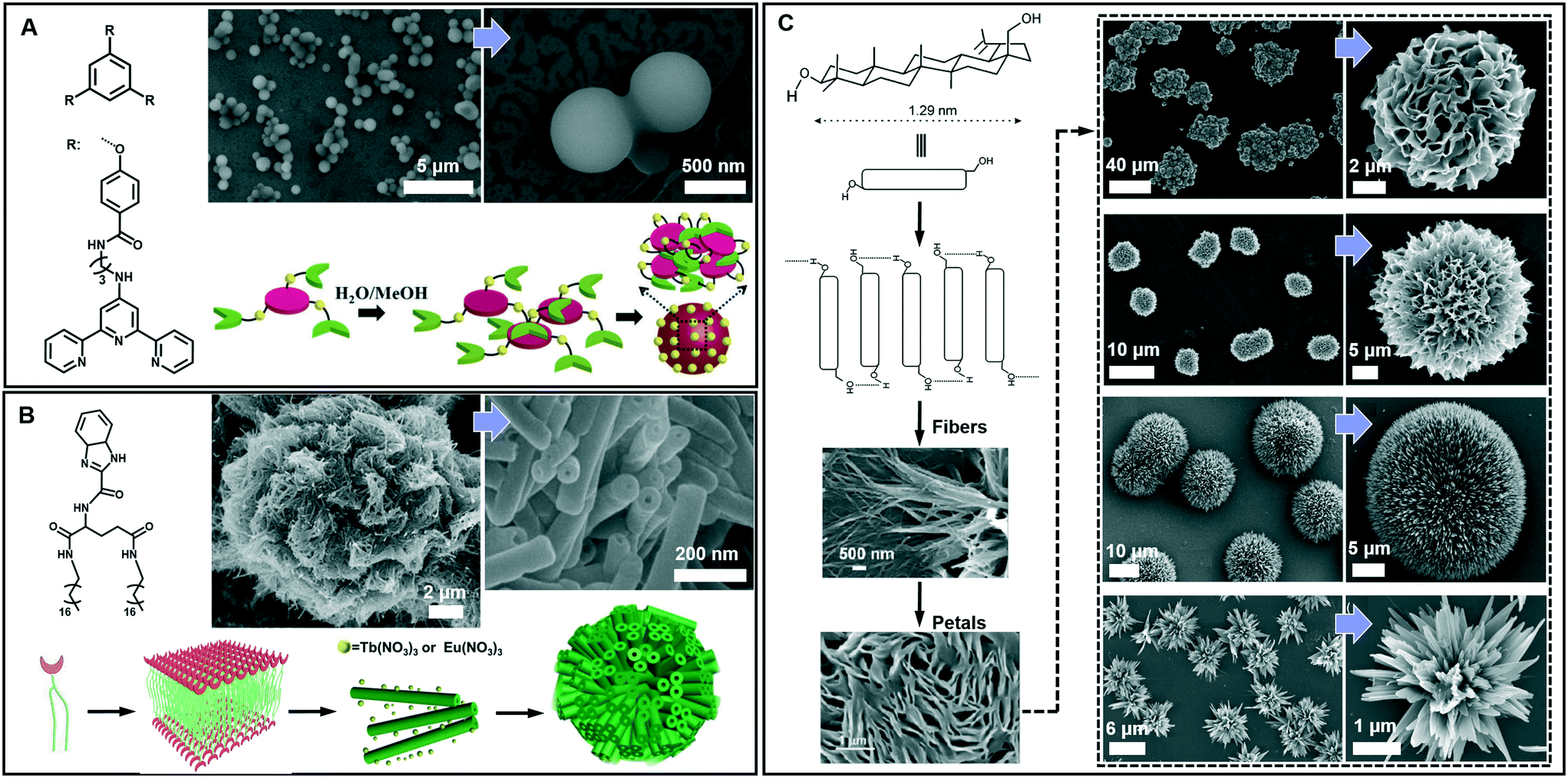 | ||
| Fig. 7 Diverse spherical architectures obtained in supramolecular gels. (A) The basic nanospheres and the self-assembly mechanism. Reprinted with permission from ref. 87. Copyright 2016, Royal Society of Chemistry. (B) Microtubular flower structures and the schematic illustration of the assembly process. Reprinted with permission from ref. 88. Copyright 2016, Wiley-VCH. (C) Schematic representation of the self-assembly of betulin affording hierarchical flower-like architectures consisting of petals and fibers. Reprinted with permission from ref. 89. Copyright 2015, American Chemical Society. | ||
From the viewpoint of nanoarchitectonics, the 1D and 2D nanostructures, such as nanotubes and nanosheets, can serve as basic units that may further evolve into various higher order nanostructures. Fig. 7B shows that microtubular flower structures can be formed by the coordination between benzimidazole and lanthanide ions.88 From the SEM observation, it is clearly shown that the microflower was hierarchical constructed from a huge number of nanotubes, whose outer diameter ranged from 50 to 80 nm. The gelators assembled into an interdigitated bilayer structure, serving as the basic building unit for further growth. However, after adding Eu(NO3)3 and Tb(NO3)3, the coordination between the gelator and Eu(III) or Tb(III) was hindered, because the O atom in NO3− showed a stronger coordination capability than the N atom. Therefore, the bilayer structure continuously turned into a nanotube, while Eu(NO3)3 and Tb(NO3)3 only attached on the surface. Due to the weak interaction, these nanotubes were aligned into 3D microflowers, as shown in Fig. 7B.88 It should be noted that the lanthanide ions not only assist the formation of this hierarchical architecture but also endow the system with special luminescence properties, resulting in chiroptical activities.88
Beside the microtubular flowers, other flower-like architectures can also be encountered in supramolecular gel systems. For example, as shown in Fig. 7C, betulin derivatives can self-assemble into various flower-like structures in different liquids.89 A lamellar self-assembly of the molecules was proposed, and the mechanism of the assembly process is illustrated in Fig. 7C. Although all the morphology characterization of dried assemblies showed flower-shaped superstructures of micrometer diameter, their shapes were not identical. The magnified SEM images revealed that the shape of the petals and their arrangement in these flower-like architectures are slightly different. Nanoflowers from inorganic substances have been reported, but flower-like self-assemblies resembling marigold, dahlia, and cadamba flowers obtained from the gelation process are rare.89
3. Transformation and switching of nanoarchitectures
In supramolecular gel systems, the self-assembly is a dynamic process and the structures sometimes form at an intermediate stage. More importantly, many gels are responsive that they change their gelation properties under external stimuli. During such a process, the transformation among different architectures is possible due to the dynamic feature of the supramolecular gel. Therefore, a full understanding of this transformation will provide a better understanding of nanoarchitectonics and its formation mechanism, and much assistance in developing new functional materials.31,90 Aside from molecular design,19 numerous exterior factors, such as solvent composition, ageing time, and physical stimuli (including sonication and photo-irradiation), can induce morphology transformation.24,33,34,91,92 Meanwhile, some specific nanostructures, such as dendritic nanotwists,48 nanotrumpets,93 feather-like dendrites70 and toruloid-like nanostructures,94,95 emerge as well during the transformation process. In this section, we will give several typical examples to show the switching of nanoarchitectures.3.1 Ageing time and temperature
Fig. 8 shows a typical work that was pioneered by Oda et al., in which the shape, size, and morphology transitions of twisted and helical membranes formed from non-chiral dicationic n-2-n gemini amphiphiles complexed with chiral tartrate anions are fine-tuned.96 At the beginning, the supramolecular gels consist of entangled fibrous structures, typical of physical gels. After 2 h, some initially ill-defined fibers were observed from the microscopy image (Fig. 8B). These structures evolved with time and became helical ribbons after around 3 h (Fig. 8C). It should be noted that the helicity of these ribbons depends on the molecular chirality of tartrate: right-handed ribbons were observed for L-tartrate, whereas left-handed ribbons were formed with D-tartrate. After about 36 h, the helical ribbons closed to form monodisperse tubules, and these tubules could be observed even after more than a year. Another important factor that can induce the morphology transformation in this system is the temperature. After complete solubilisation at 50 °C, the supramolecular gels were kept at several different temperatures below their melting temperature (∼43 °C) for a few months. The experiment results show that the gels kept between 38 and 40 °C displayed stable twisted ribbons even after 6 months. However, these twisted ribbons turn to tubular structures when the gels were kept at 35 °C for 1 month. After being kept at 40 °C for 2 months, these gels showed again twisted ribbons. These observations clearly indicated that the transition between twisted and helical morphologies of the ribbons is a reversible process. Below 35 °C, tubules are favoured, whereas at 40 °C twisted ribbons are favoured.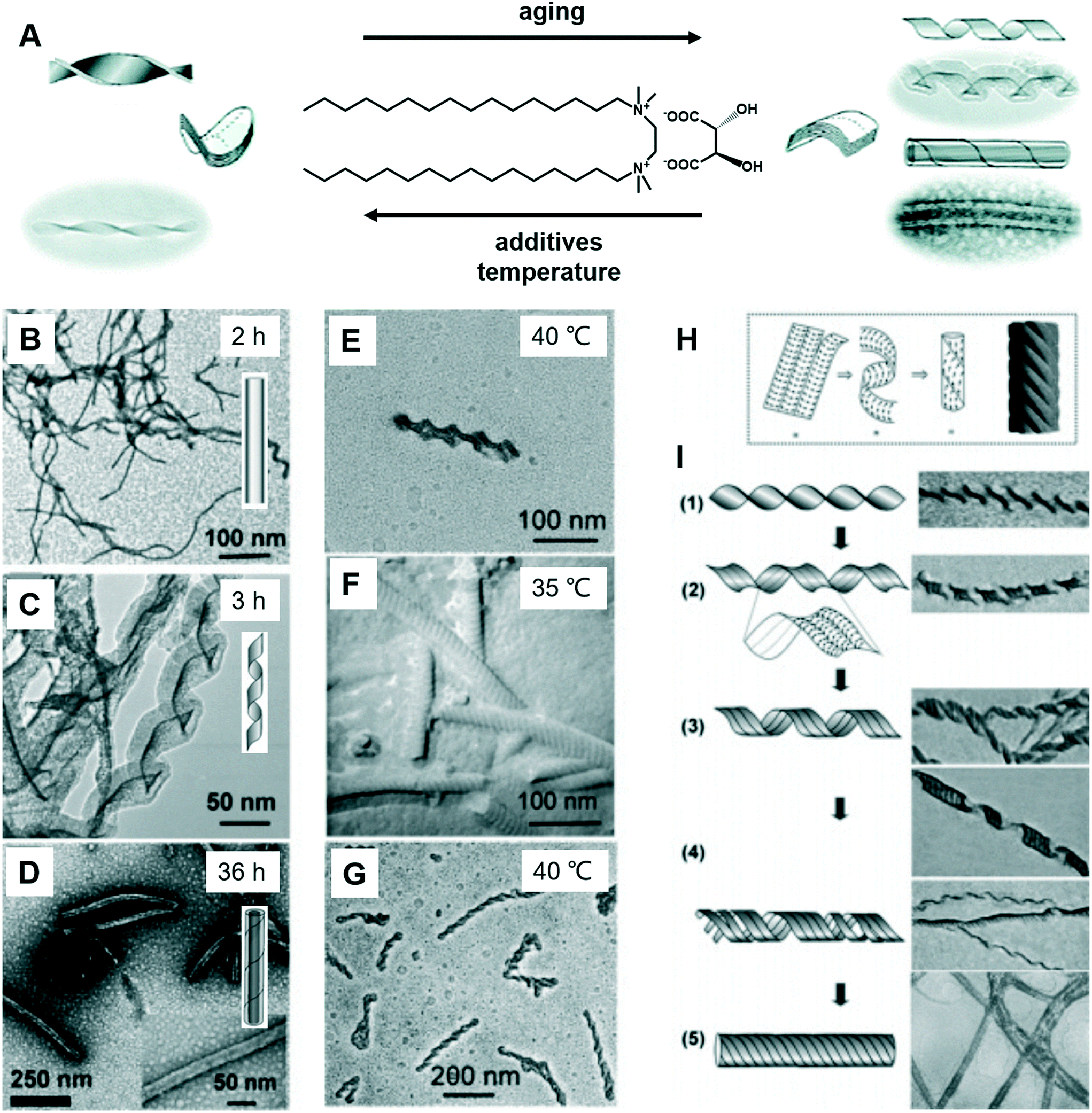 | ||
| Fig. 8 (A) Schematic illustration of the transformation between the twists, helical ribbons and tubules upon modifying the aging time, temperature and introducing additives. TEM micrographs and schematic representation of the evolution from fibrous nanostructures (B) to helical ribbons (C) and tubules (D). TEM micrographs of gels kept at 40 °C show twisted ribbons (E), whereas those kept at 35 °C or below shows a transition to tubules (F). If the gels were kept again at 40 °C, the tubules could be converted back to twisted ribbons (G). (H) The model proposed by Selinger et al.97 giving rise to ripples due to the modulation in the tilt direction separated by sharp domain walls. (I) Transition from twisted ribbons to helical ribbons, and then to tubules. Reprinted with permission from ref. 96. Copyright 2007, American Chemical Society. | ||
It was found that the “additive factor” is important during the architecture transformation.96 These likely transient twisted ribbons could not even be detected for the purest samples, which indicates that the formation of tubules is a cooperative process and that its kinetics is extremely sensitive to molecular composition. Some examples of the intermediate state of the ribbons are shown in Fig. 8I. From these images, some stripes could be observed at the same time when the twists transform to helical ribbons, which can be explained by a previous report by Selinger et al.97 It should be noted that these nanoarchitectures could be successfully ascribed to silica, leading to the formation of chiral silica nanoribbons, which show activity on stem cell adhesion.57
3.2 Second component
Since the ground-breaking work of Oda et al.,96 studies have increasingly focused on the self-assembly of two-component or multicomponent systems, wherein small amounts of additives can be used to tune the assembly behaviour.98,99 For chiral nanoarchitectures, these systems are very effective in regulating the chiral expression either by using chiral additives or achiral additives.100–103 Generally, it is easy to understand chiral additives due to the “sergeants and soldiers principle” and “majority rules”.104–106 Recently, achiral additives were found to induce morphology transformation and trigger chirality inversions through co-assembly with chiral compounds. As shown in Fig. 9, phenylalanine-based chiral enantiomers, named as DPF and LPF, are the basic units for constructing supramolecular chiral nanostructures.107 Two achiral derivatives (BPy1 and BPy2) were employed to regulate the nanoarchitectures. The handedness of the nanostructures could be inverted simply by exchanging the achiral molecule. Meanwhile, the helical fibers were transformed to twisted ribbons.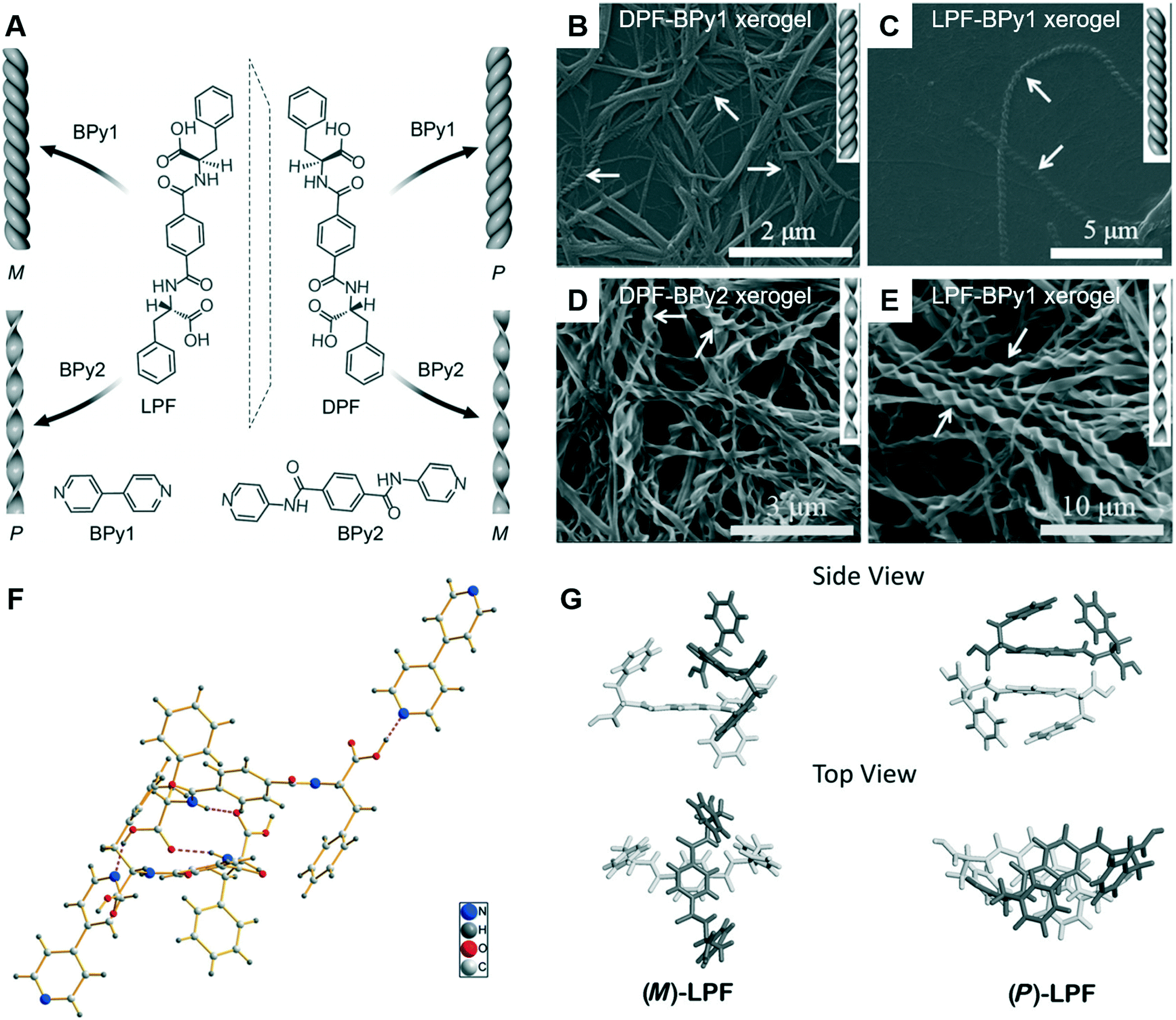 | ||
| Fig. 9 (A) Schematic illustration of helical inversion and morphology transformation. M and P denote left and right-handed helical nanofibers or twisted ribbons, respectively. LPF and DPF are enantiomeric gelators. BPy1 and BPy2 are achiral bis(pyridinyl) derivatives. (B–E) Corresponding SEM images of the helical fibers and twisted ribbons. (F) The packing structure of LPF–BPy1 stabilized by hydrogen bonds (pink dashed lines) in the cocrystal state. (G) Structures of the (M)-LPF dimer determined by crystal structure analysis and the (P)-LPF dimer optimized by simulation. Reprinted with permission from ref. 107. Copyright 2016, Wiley-VCH. | ||
The crystal structure and packing diagram in Fig. 9F revealed that two types of intermolecular hydrogen bonds, C–N⋯H–O and N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, are the main driving force for the co-assembly.107 Firstly, the one-dimensional self-assembly in a head-to-tail model formed via the hydrogen bonds between a pyridyl nitrogen atom and the HO group of carboxylic acid. Secondly, the hydrogen bonds between an amide N–H group and a carboxylic acid CO moiety generated the three-dimensional crystal networks. In addition, Fig. 9G suggested that the packing mode for the dimers was different. For the (M)-LPF dimer, two molecules were nearly perpendicular to each other and the two terminal phenyl groups were arranged in a trans fashion. However, for the (P)-LPF dimer, one molecule was nearly parallel to the other one and the two terminal phenyl groups are cis to each other.
C, are the main driving force for the co-assembly.107 Firstly, the one-dimensional self-assembly in a head-to-tail model formed via the hydrogen bonds between a pyridyl nitrogen atom and the HO group of carboxylic acid. Secondly, the hydrogen bonds between an amide N–H group and a carboxylic acid CO moiety generated the three-dimensional crystal networks. In addition, Fig. 9G suggested that the packing mode for the dimers was different. For the (M)-LPF dimer, two molecules were nearly perpendicular to each other and the two terminal phenyl groups were arranged in a trans fashion. However, for the (P)-LPF dimer, one molecule was nearly parallel to the other one and the two terminal phenyl groups are cis to each other.
An obvious difference between the two achiral additives is the hydrogen bonding site.107 BPy1 only has pyridine, whereas BPy2 has pyridine and amide. Therefore, BPy1 could interact with the chiral gelator via the COOH sites, while BPy2 interacts through both the COOH and amide groups. These two interactions result in a parallel arrangement, which might be the reason for the chirality inversion and morphology change.107 In addition, helicity inversion of the supramolecular assemblies based on LDF derivatives could be successfully achieved through inserting variable methylene units between the chiral centre and rigid aromatic core.108 Compared with the right-handed nanofibers, an increase in cell adhesion on the left-handed nanofibers is observed, even though both nanofibers were derived from L-phenylalanine derivatives.108
3.3 Solvent
A special nanotrumpet architecture was observed for an achiral C3-symmetric gelator when the solvent polarity was changed (Fig. 10).93 Supramolecular gels can be formed in the mixed solvent of DMF/H2O when their volume ratio ranged between 7 and 0.67. As shown in Fig. 10B–E, twisted and trumpet-like architectures were formed upon further increasing the amount of DMF. It should be noted that these nanotwists and nanotrumpets were not identical at different volume ratios of DMF and water. For example, the nanotrumpets were slender and smooth (Fig. 10C), then they became short. When the volume ratio was increased to 5, some wheel-like structures emerged on the surface of the nanotrumpets at a small aspect ratio. On the other hand, the nanotwists formed in a larger mixing range (Fig. 10E) were much more complex than those formed in a small volume ratio (Fig. 10B), which appeared to be twisted from a bunch of nanobelts.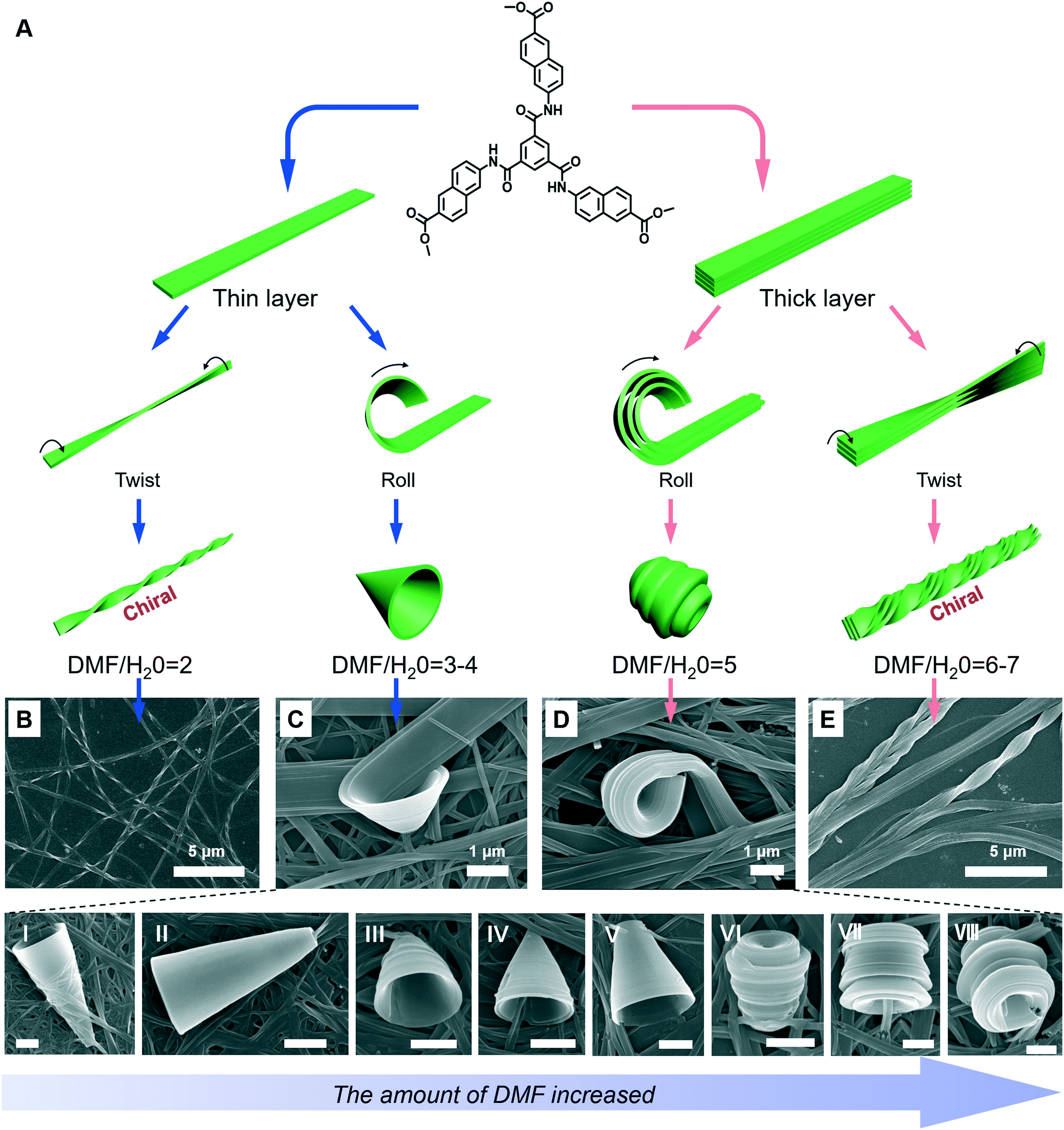 | ||
| Fig. 10 (A) Schematic illustration of the architecture transformation in various DMF/H2O mixtures. An achiral C3-symmetric gelator can form nanotwists (B), nanotrumpets (C and D) and hierarchical twisted nanostructures (E). I to VI show various types of trumpet-like nanostructures. Scale bar: 1 μm. (C and D) also show the rolling process of the nanobelts into nanotrumpets. Reprinted with permission from ref. 93. Copyright 2018, Royal Chemistry of Society. | ||
Fig. 10A shows the possible mechanism for the formation of various nanostructures when the solvent was changed.93 Owing to the intermolecular H-bonds and π–π stacking, the neighbouring molecules staked and formed a layer structure at the beginning. Depending on the DMF/H2O ratio, thinner or thicker 1D nanobelts could be further stacked. Such nanobelts serve as the bases for the subsequent hierarchical self-assembly. When the volume ratio was below 4, the nanobelts stacked as thinner 1D nanolayers and further twisted or rolled into nanotwists and nanotrumpets, respectively. However, when the volume ratio was beyond 4, the hydrogen bonding enhanced the molecular packing, resulting in thicker nanolayers. Therefore, the twists and trumpet-like architectures from thick and thin nanolayers were different. Moreover, such a rolling process was directly confirmed from Fig. 10C and D.93
3.4 pH
In the case of gelation, particularly for hydrogels, the pH value of the systems is important.109,110 To date, although the effects of pH on hydrogel formation and structure regulation have been reported, it is still a challenge to fabricate hydrogels with wide pH adaptability. As shown in Fig. 11A, an amino acid derivative has been found to form hydrogels in a range of pH 7.46–14.110 In phosphate buffer solution at pH 7.46, left-handed nanofibers with widths ranging from 30 nm to 55 nm were observed. When the pH value of this system turned from neutral to slightly basic (7.46–11.0), the gel fibers became helical in nature. Branching in nanofibers started from pH = 12 and feature-like morphologies were observed up to pH = 14. Experimental data suggested that the hydrogen-bonding interaction between carboxyl acid groups plays an important role in gel formation and morphology change.110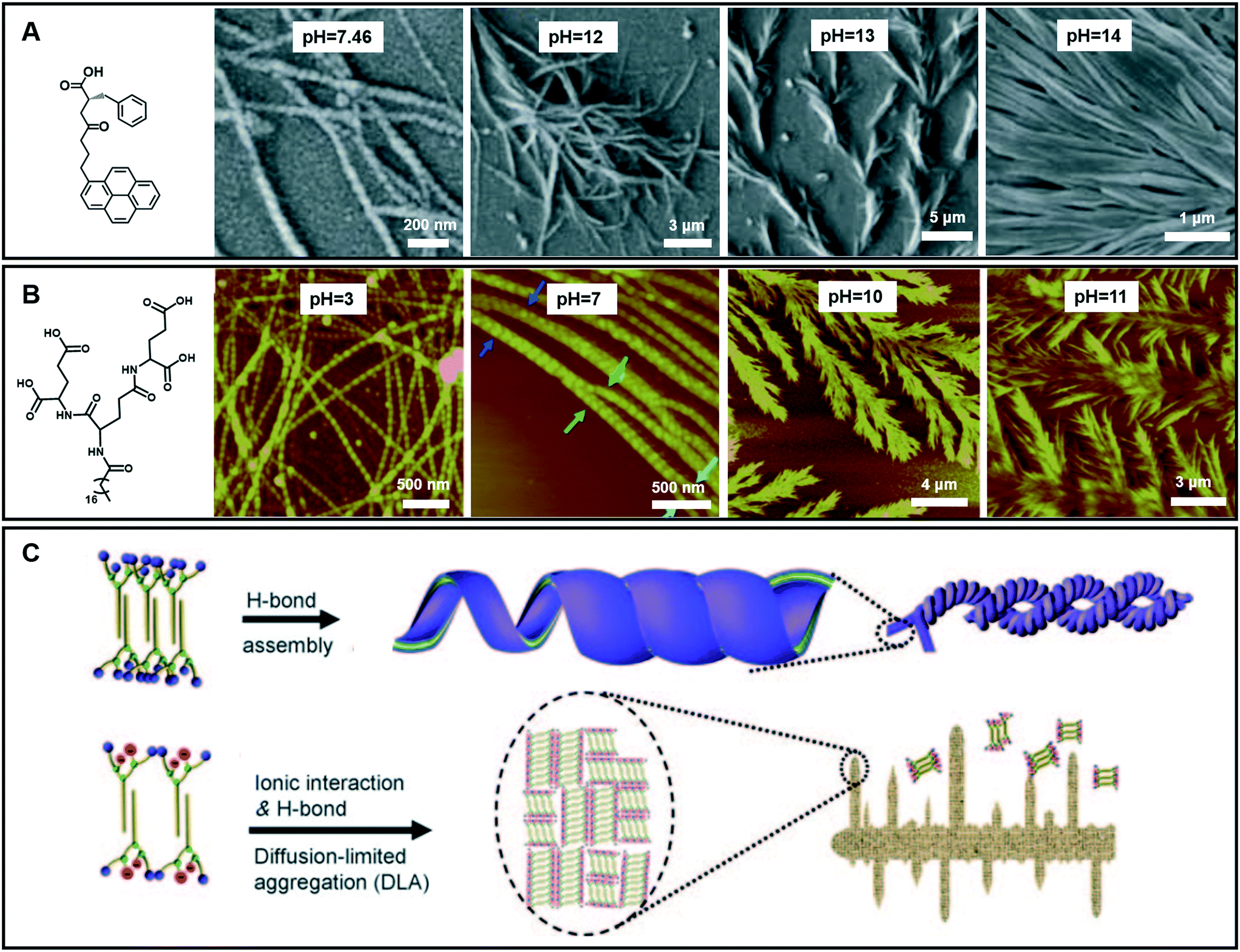 | ||
| Fig. 11 (A and B) Various nanoarchitectures of hydrogels accompanying the pH change. (C) Schematic illustration of architecture evolution in (B). (A) Reprinted with permission from ref. 110. Copyright 2013, Royal Chemistry of Society. (B and C) Reprinted with permission from ref. 70. Copyright 2011, Wiley-VCH. | ||
Fig. 11B gives an example of an amphiphilic dendron containing three dendrites (L-glutamic acid), which can form hydrogels over a wider pH range from 2 to 13.70 Accompanying the pH change, the compounds self-assembled into various nanostructures: from helical nanotubes with a string of beads, and coiled superhelixes to pine-like and feather-like dendrite nanostructures. Compared with the gelator in Fig. 11A, the wide pH adaptability and diverse structural changes lie in the special molecular structures, which have many hydrogen-bond sites, as shown in Fig. 11B.70 During the transformation, the four carboxylic acid groups and three amide groups are crucial. A bilayer unit was formed owing to the multiple-hydrogen bonds between the amide groups and the hydrophobic interaction between the alkyl chains. In a pH range of 2–9, the hydrogen bond between the carboxylic acid was dominant. This hydrogen bonding enables the bilayer structures to roll into helical nanotubes due to the chiral nature of the gelators. In this pH range, pH variation causes a subtle change in the hydrogen bond sites, resulting in different nanotube architectures. Meanwhile when the pH value is higher than 10, the ionic interaction becomes dominant because the hydrogen bond between the carboxylic acid was largely destroyed. However, the hydrogen bonds between the amide groups did not change much. Therefore, the bilayer structure is still stabilized and grows in a 2D manner to form a dendritic structure owing to the loss of the confinement at the head group, suggesting a diffusion-limited aggregation far from thermodynamic equilibrium.70 Interestingly, these dendrite structures changed from pine-like to feather-like structures depending on slight pH variations.
3.5 Ultrasound
Different from agitation, a physical medium is required for sonication in order to support their propagation. When such a kind of high frequency mechanical wave propagates in media like liquid or liquid–powder suspension, cavitation which can create extreme physical and chemical conditions usually occurs.111,112 This cavitation effect may provide high energy to induce architecture transformation.113,114 For example, as shown in Fig. 12, a gelator containing triphenylamine was found to form a stable gel in ethyl acetate with a solid microsphere structure.115 With the extended ultrasound time, most of the ball structures disappeared and some of them changed into fibers, then the fiber structures further changed into nanotube structures. Thus, the solid microsphere structure formed in the gel could be changed into a nanotube in the transition process of gel to gel via sonication. During the sonication process, the intermolecular hydrogen bonding of the self-assembly system was obviously enhanced, resulting in the decreased hydrophobicity of the xerogel film with a change of the contact angle from 142° to 129°.115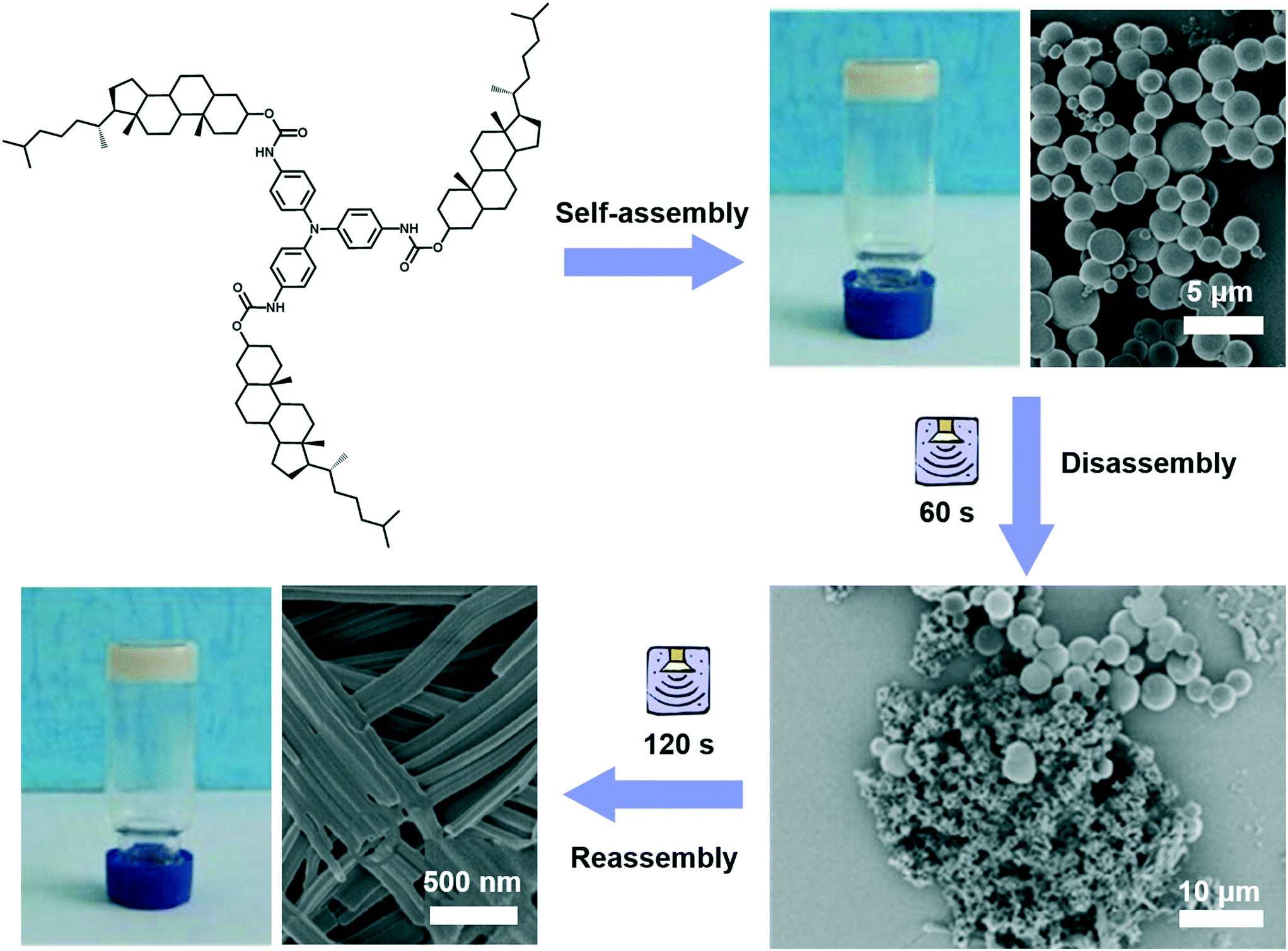 | ||
| Fig. 12 Schematic illustration of the architecture transformation from solid microsphere structures to nanotubes in the transition process of gel to gel via in situ sonication. Reprinted with permission from ref. 115. Copyright 2018, Taylor & Francis Group. | ||
3.6 Photo-irradiation
Most of the common physical stimuli, such as the examples in Fig. 12, will not change the chemical structure of the gelator but can modulate the molecular configuration or the self-assembly process except for the light stimulus.116,117 Light activated topochemical reactions, such as the [2 + 2] photocycloaddition of olefins and ring-opening reaction of spiropyran, can also be designed to realize architecture transformation.91,118,119 It was found that the initial white gel formed from an L-glutamide gelator with a β-naphthylacryl group (Fig. 13A) was transformed into a precipitate under light.94 The SEM image shows that long nanofibers were formed in the ethanol gel (Fig. 13B). With further irradiation, some wheel-like structures emerged on the surface of the fibers (Fig. 13C), and these wheels expanded in a concentric circular manner at the expense of the nanofibers (Fig. 13D). Thereafter, this kind of curved structure increased gradually, and the whole fiber changed into discrete a toruloid-like nanostructure (Fig. 13E). Enlarged images revealed that various types of nanoarchitectures appeared, such as cork-like (Fig. 13I), thumbtack-like (II), egg tart-like (III), spinning top-like (IV), screw-like (V), and Chinese artichoke-like (VI) structures. All of these structures had a hollow cavity from the lateral side. It should be noted that the nanofiber structures can be formed again when the precipitates were heated. These nanofibers could be changed into coaxial hollow toruloid-like nanostructures again under photo-irradiation. Thus, a reversible transformation between the nanofibers and toruloid-like nanostructures was achieved due to the topochemical [2 + 2] cycloaddition reaction.115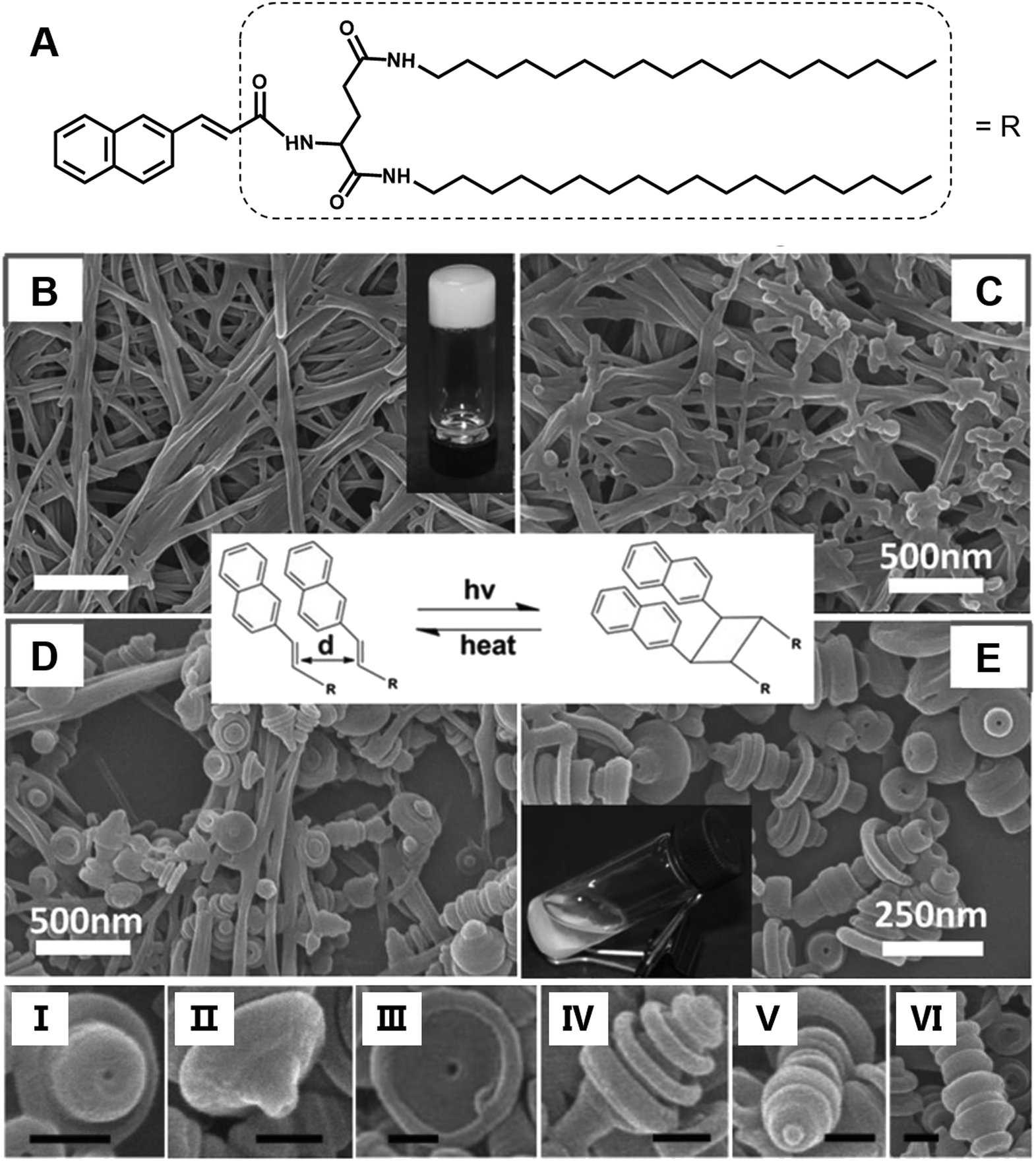 | ||
| Fig. 13 (A) Chemical structure and (B–E) the architecture transformation from nanofibers to coaxial toruloid-like nanostructures from a supramolecular gel. SEM images of (B) gels, intermediate states of the gels after irradiation for (C) 1 h and (D) 5 h, and the (D) precipitates. I to VI show various types of discrete coaxial hollow toruloid-like nanostructures; scale bar: 100 nm. Reprinted with permission from ref. 94. Copyright 2013, Wiley-VCH. | ||
4. Conclusion
Here, we summarized the various 0D, 1D, 2D and 3D architectures formed during supramolecular gelation. Using different kinds of molecules or building blocks, diverse nanostructures can be obtained. There is a strong relationship between the molecular structures and the nanostructures. However, the stacking of the molecules into nanostructures is affected by many factors such as the solvents, temperature, pH and physical factors including light and ultrasound. In addition, many of the structures are intermediate structures that can be further changed. However, if the gelation and further ageing process are stopped, all of these structures can be separated and solidified. Moreover, these organic nanostructures can serve as templates for growing inorganic nanostructures in many cases, thus providing an efficient way of obtaining or controlling nanostructures in a large quantity.We discussed the preformed architectures and their transformation here for the gel system. However, the gelator design and their properties or functions are the other two important areas. Among these three parts, gel architectures bridge them together. We showed some examples of interesting properties by different nanostructures in the gels. This should be more important in future research. Although the inherent molecular properties have a profound effect on their final properties, the assembled nanostructures also played an important role. Before fully performing the functions, it is important to better understand how a molecule can be tuned into various nanostructures. Thus, we hope that supramolecular gelation could provide a useful methodology to design and control functional nanomaterials under the concept of nanoarchitectonics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB12020200), Key Research Program of Frontier Sciences, CAS (QYZDJ-SSW-SLH044), and National Natural Science Foundation of China (91427302).References
- K. Ariga, J. B. Li, J. B. Fei, Q. M. Ji and J. P. Hill, Adv. Mater., 2016, 28, 1251–1286 CrossRef CAS PubMed.
- M. Aono and K. Ariga, Adv. Mater., 2016, 28, 989–992 CrossRef CAS PubMed.
- K. Ariga, Q. M. Ji, J. P. Hill, Y. Bando and M. Aono, NPG Asia Mater., 2012, 4, 11 CrossRef.
- B. Angelov, A. Angelova, S. K. Filippov, M. Drechsler, P. Stepanek and S. Lesieur, ACS Nano, 2014, 8, 5216–5226 CrossRef CAS PubMed.
- A. Angelova, B. Angelov, R. Mutafchieva, S. Lesieur and P. Couvreur, Acc. Chem. Res., 2011, 44, 147–156 CrossRef CAS PubMed.
- M. Ramanathan, K. Hong, Q. Ji, Y. Yonamine, J. P. Hill and K. Ariga, J. Nanosci. Nanotechnol., 2014, 14, 390–401 CrossRef CAS PubMed.
- M. Aono, Y. Bando and K. Ariga, Adv. Mater., 2012, 24, 150–151 CrossRef CAS PubMed.
- M. Komiyama, K. Yoshimoto, M. Sisido and K. Ariga, Bull. Chem. Soc. Jpn., 2017, 90, 967–1004 CrossRef.
- M. Komiyama, T. Mori and K. Ariga, Bull. Chem. Soc. Jpn., 2018, 91, 1075–1111 CrossRef CAS.
- K. Ariga, D. T. Leong and T. Mori, Adv. Funct. Mater., 2018, 28, 1702905 CrossRef.
- A. Angelova, B. Angelov, B. Papahadjopoulos-Sternberg, M. Ollivon and C. Bourgaux, Langmuir, 2005, 21, 4138–4143 CrossRef CAS PubMed.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef CAS PubMed.
- M. Liu, G. Ouyang, D. Niu and Y. Sang, Org. Chem. Front., 2018, 5, 2885–2900 RSC.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS PubMed.
- Z. Qi and C. A. Schalley, Acc. Chem. Res., 2014, 47, 2222–2233 CrossRef CAS PubMed.
- D. Yang, P. Duan and M. Liu, Angew. Chem., Int. Ed., 2018, 57, 9357–9361 CrossRef CAS PubMed.
- S. Cherumukkil, B. Vedhanarayanan, G. Das, V. K. Praveen and A. Ajayaghosh, Bull. Chem. Soc. Jpn., 2018, 91, 100–120 CrossRef CAS.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- L. Zhang, X. Wang, T. Wang and M. Liu, Small, 2015, 11, 1024–1024 CrossRef.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546–6596 RSC.
- J. K. H. Hui, Z. Yu and M. J. MacLachlan, Angew. Chem., Int. Ed., 2007, 46, 7980–7983 CrossRef CAS PubMed.
- R. N. Shah, N. A. Shah, M. M. D. R. Lim, C. Hsieh, G. Nuber and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 3293–3298 CrossRef CAS PubMed.
- E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041–6046 CrossRef CAS PubMed.
- T. Haino, M. Tanaka and Y. Fukazawa, Chem. Commun., 2008, 468–470 RSC.
- H. Engelkamp, S. Middelbeek and R. J. M. Nolte, Science, 1999, 284, 785–788 CrossRef CAS PubMed.
- A. Maity, M. Gangopadhyay, A. Basu, S. Aute, S. S. Babu and A. Das, J. Am. Chem. Soc., 2016, 138, 11113–11116 CrossRef CAS PubMed.
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 15727 CrossRef CAS PubMed.
- H. Jiang, L. Zhang and M. Liu, ChemNanoMat, 2018, 4, 720–729 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- R. Davis, R. Berger and R. Zentel, Adv. Mater., 2007, 19, 3878–3881 CrossRef CAS.
- M. Y. Yeh, C. W. Huang, J. W. Chang, Y. T. Huang, J. H. Lin, S. M. Hsu, S. C. Hung and H. C. Lin, Soft Matter, 2016, 12, 6347–6351 RSC.
- Y. Zhang, D. Yang, J. Han, J. Zhou, Q. Jin, M. Liu and P. Duan, Langmuir, 2018, 34, 5821–5830 CrossRef CAS PubMed.
- R. Oda, I. Huc, M. Schmutz, S. J. Candau and F. C. MacKintosh, Nature, 1999, 399, 566–569 CrossRef CAS PubMed.
- H. Cao, Q. Z. Yuan, X. F. Zhu, Y. P. Zhao and M. H. Liu, Langmuir, 2012, 28, 15410–15417 CrossRef CAS PubMed.
- X. F. Zhu, P. F. Duan, L. Zhang and M. H. Liu, Chem. – Eur. J., 2011, 17, 3429–3437 CrossRef CAS PubMed.
- L. Zhang, T. Wang, Z. Shen and M. Liu, Adv. Mater., 2016, 28, 1044–1059 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- T. Yamaguchi, T. Kimura, H. Matsuda and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 6350–6355 CrossRef CAS PubMed.
- J. Sun, Y. Li, F. Yan, C. Liu, Y. Sang, F. Tian, Q. Feng, P. Duan, L. Zhang, X. Shi, B. Ding and M. Liu, Nat. Commun., 2018, 9, 2599 CrossRef PubMed.
- J. M. Ribo, J. Crusats, F. Sagues, J. Claret and R. Rubires, Science, 2001, 292, 2063–2066 CrossRef CAS PubMed.
- L. Perez-Garcia and D. B. Amabilino, Chem. Soc. Rev., 2007, 36, 941–967 RSC.
- Z. Shen, T. Wang and M. Liu, Angew. Chem., Int. Ed., 2014, 53, 13424–13428 CrossRef CAS PubMed.
- R. K. Das, O. F. Zouani, C. Labrugere, R. Oda and M.-C. Durrieu, ACS Nano, 2013, 7, 3351–3361 CrossRef CAS PubMed.
- L. Ziserman, H.-Y. Lee, S. R. Raghavan, A. Mor and D. Danino, J. Am. Chem. Soc., 2011, 133, 2511–2517 CrossRef CAS PubMed.
- N. Kameta, H. Minamikawa and M. Masuda, Soft Matter, 2011, 7, 4539–4561 RSC.
- R. H. Baughman, A. A. Zakhidov and W. A. de Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- C. R. Martin and P. Kohli, Nat. Rev. Drug Discovery, 2003, 2, 29–37 CrossRef CAS PubMed.
- L. Zhang, Q. Jin and M. Liu, Chem. - Asian J., 2016, 11, 2642–2649 CrossRef CAS PubMed.
- J. Jiang, G. Ouyang, L. Zhang and M. Liu, Chemistry, 2017, 23, 9439–9450 CrossRef CAS PubMed.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1443 CrossRef CAS PubMed.
- R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe and S. Perrier, Chem. Soc. Rev., 2012, 41, 6023–6041 RSC.
- G. Chen and M. Jiang, Chem. Soc. Rev., 2011, 40, 2254–2266 RSC.
- T. Shimizu, Bull. Chem. Soc. Jpn., 2018, 91, 623–668 CrossRef CAS.
- C. L. Zhan, P. Gao and M. H. Liu, Chem. Commun., 2005, 462–464 RSC.
- J. Jiang, T. Wang and M. Liu, Chem. Commun., 2010, 46, 7178–7180 RSC.
- P. Duan, L. Qin, X. Zhu and M. Liu, Chem. – Eur. J., 2011, 17, 6389–6395 CrossRef CAS PubMed.
- X. F. Zhu, Y. G. Li, P. F. Duan and M. H. Liu, Chem. – Eur. J., 2010, 16, 8034–8040 CrossRef CAS PubMed.
- Q. X. Jin, L. Zhang, H. Cao, T. Y. Wang, X. F. Zhu, J. Jiang and M. H. Liu, Langmuir, 2011, 27, 13847–13853 CrossRef CAS PubMed.
- J. Jiang, Y. Meng, L. Zhang and M. Liu, J. Am. Chem. Soc., 2016, 138, 15629–15635 CrossRef CAS PubMed.
- Q. Jin, L. Zhang and M. Liu, Chem. – Eur. J., 2013, 19, 9234–9241 CrossRef CAS PubMed.
- H. Cao, P. F. Duan, X. F. Zhu, J. Jiang and M. H. Liu, Chem. – Eur. J., 2012, 18, 5546–5550 CrossRef CAS PubMed.
- N. Shi, G. Yin, H. Li, M. Han and Z. Xu, New J. Chem., 2008, 32, 2011–2015 RSC.
- X. Mu, W. Song, Y. Zhang, K. Ye, H. Zhang and Y. Wang, Adv. Mater., 2010, 22, 4905–4909 CrossRef CAS PubMed.
- J. Han, J. You, X. Li, P. Duan and M. Liu, Adv. Mater., 2017, 29, 1606503 CrossRef PubMed.
- Y. Qiao, Y. Wang, Z. Yang, Y. Lin and J. Huang, Chem. Mater., 2011, 23, 1182–1187 CrossRef CAS.
- M. Nakagawa and T. Kawai, J. Am. Chem. Soc., 2018, 140, 4991–4994 CrossRef CAS PubMed.
- K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980–999 CrossRef CAS PubMed.
- Y. Lu, A. Wittemann and M. Ballauff, Macromol. Rapid Commun., 2009, 30, 806–815 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer, S. S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS.
- L. Zerkoune, S. Lesieur, J.-L. Putaux, L. Choisnard, A. Geze, D. Wouessidjewe, B. Angelov, C. Vebert-Nardin, J. Doutch and A. Angelova, Soft Matter, 2016, 12, 7539–7550 RSC.
- B. G. Bag and R. Majumdar, Chem. Rec., 2017, 17, 841–873 CrossRef CAS PubMed.
- P. Sutar and T. K. Maji, Chem. Commun., 2016, 52, 13136–13139 RSC.
- X. Zhou, Q. Jin, L. Zhang, Z. Shen, L. Jiang and M. Liu, Small, 2016, 12, 4743–4752 CrossRef CAS PubMed.
- B. G. Bag and S. S. Dash, Langmuir, 2015, 31, 13664–13672 CrossRef CAS PubMed.
- L. Zhang, L. Qin, X. Wang, H. Cao and M. Liu, Adv. Mater., 2014, 26, 6959–6964 CrossRef CAS PubMed.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC.
- X. Wang and M. Liu, Chem. – Eur. J., 2014, 20, 10110–10116 CrossRef CAS PubMed.
- Y. Sang, P. Duan and M. Liu, Chem. Commun., 2018, 54, 4025–4028 RSC.
- X. Wang, P. Duan and M. Liu, Chem. – Eur. J., 2013, 19, 16072–16079 CrossRef CAS PubMed.
- X. Wang, F. Xie, P. Duan and M. Liu, Langmuir, 2016, 32, 12534–12541 CrossRef CAS PubMed.
- A. Brizard, C. Aime, T. Labrot, I. Huc, D. Berthier, F. Artzner, B. Desbat and R. Oda, J. Am. Chem. Soc., 2007, 129, 3754–3762 CrossRef CAS PubMed.
- J. V. Selinger, F. C. MacKintosh and J. M. Schnur, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 53, 3804–3818 CrossRef CAS.
- L. Ji, G. Ouyang and M. Liu, Langmuir, 2017, 33, 12419–12426 CrossRef CAS PubMed.
- D. Niu, L. Ji, G. Ouyang and M. Liu, Chem. Commun., 2018, 54, 1137–1140 RSC.
- J. Chen, T. Wang and M. Liu, Chem. Commun., 2016, 52, 6123–6126 RSC.
- D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC.
- M. Deng, L. Zhang, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2016, 55, 15062–15066 CrossRef CAS PubMed.
- Y. Liu, C. Chen, T. Wang and M. Liu, Langmuir, 2016, 32, 322–328 CrossRef CAS PubMed.
- S. Lifson, C. Andreola, N. C. Peterson and M. M. Green, J. Am. Chem. Soc., 1989, 111, 8850–8858 CrossRef CAS.
- A. J. Wilson, J. van Gestel, R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2006, 4404–4406 RSC.
- J. J. van Gorp, J. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef CAS PubMed.
- G. F. Liu, L. Y. Zhu, W. Ji, C. L. Feng and Z. X. Wei, Angew. Chem., Int. Ed., 2016, 55, 2411–2415 CrossRef CAS PubMed.
- J. Liu, F. Yuan, X. Ma, D.-I. Y. Auphedeous, C. Zhao, C. Liu, C. Shen and C. Feng, Angew. Chem., Int. Ed., 2018, 57, 6475–6479 CrossRef CAS PubMed.
- D. Kurouski, X. Lu, L. Popova, W. Wan, M. Shanmugasundaram, G. Stubbs, R. K. Dukor, I. K. Lednev and L. A. Nafie, J. Am. Chem. Soc., 2014, 136, 2302–2312 CrossRef CAS PubMed.
- J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198 RSC.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- N. Komiya, T. Muraoka, M. Iida, M. Miyanaga, K. Takahashi and T. Naota, J. Am. Chem. Soc., 2011, 133, 16054–16061 CrossRef CAS PubMed.
- X. Yu, Q. Liu, J. Wu, M. Zhang, X. Cao, S. Zhang, Q. Wang, L. Chen and T. Yi, Chem. – Eur. J., 2010, 16, 9099–9106 CrossRef CAS PubMed.
- Y. Wang, C. Zhan, H. Fu, X. Li, X. Sheng, Y. Zhao, D. Xiao, Y. Ma, J. S. Ma and J. Yao, Langmuir, 2008, 24, 7635–7638 CrossRef CAS PubMed.
- X. Cao, A. Gao, D. Liu, N. Zhao, Q. Ding and Y. Li, Supramol. Chem., 2018, 30, 674–680 CrossRef CAS.
- X. Cao, A. Gao, H. Lv, Y. Wu, X. Wang and Y. Fan, Org. Biomol. Chem., 2013, 11, 7931–7937 RSC.
- J. H. Kim, M. Seo, Y. J. Kim and S. Y. Kim, Langmuir, 2009, 25, 1761–1766 CrossRef CAS PubMed.
- C. Liu, D. Yang, Q. Jin, L. Zhang and M. Liu, Adv. Mater., 2016, 28, 1644–1649 CrossRef CAS PubMed.
- W. Miao, S. Wang and M. Liu, Adv. Funct. Mater., 2017, 27, 1701368 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |