
Sticky ends in a self-assembling ABA triblock copolymer: the role of ureas in stimuli-responsive hydrogels†
Ryan T.
Shafranek
 a,
Joel D.
Leger
a,
Song
Zhang
b,
Munira
Khalil
a,
Xiaodan
Gu
b and
Alshakim
Nelson
*a
a,
Joel D.
Leger
a,
Song
Zhang
b,
Munira
Khalil
a,
Xiaodan
Gu
b and
Alshakim
Nelson
*a
aDepartment of Chemistry, University of Washington, Seattle, WA 98105, USA. E-mail: alshakim@uw.edu
bSchool of Polymer Science and Engineering, University of Southern Mississippi, Hattiesburg, MS 39406, USA
First published on 21st November 2018
Abstract
Directing polymer self-assembly through noncovalent interactions is a powerful way to control the structure and function of nanoengineered materials. Dynamic hydrogen bonds are particularly useful for materials with structures that change over time or in response to specific stimuli. In the present work, we use the supramolecular association of urea moieties to manipulate the morphology, thermal response, and mechanical properties of soft polymeric hydrogels. Urea-terminated poly(isopropyl glycidyl ether)-b-poly(ethylene oxide)-b-poly(isopropyl glycidyl ether) ABA triblock copolymers were synthesized using controlled, anionic ring-opening polymerization and subsequent chain-end functionalization. Triblock copolymers with hydroxy end-groups were incapable of hydrogelation, while polymers terminated with meta-bis-urea motifs formed robust gels at room temperature. Rheometric analysis of the bulk gels, variable-temperature infrared spectroscopy (VT-IR), differential scanning calorimetry (DSC), and small-angle X-ray scattering (SAXS) confirmed the formation of structured hydrogels via association of the meta-bis-urea end-groups. Monourea end-groups did not result in the same regular structure as the meta-bis-urea. In future, the reported hydrogels could be useful for elastomeric, shape-morphing 3D-printed constructs, or as biomimetic scaffolds with precisely tailored porosity and mechanical properties.
Design, System, ApplicationWe report a strategy to prepare hierarchically self-assembled polymeric hydrogels based on amphiphilic ABA triblock copolymers with hydrogen-bonding end-groups. The polymers were designed with a central hydrophilic poly(ethylene oxide) ‘B’ block that is flanked with hydrophobic poly(isopropyl glycidyl ether) ‘A’ blocks and terminated with aryl urea chain ends. The localization of the hydrogen-bonding groups onto the terminal end of the low-Tg ‘A’ blocks with temperature-dependent aqueous solubility (LCST behavior) allowed the ureas to self-assemble despite the aqueous medium of the gel. Moreover, the resulting hydrogels exhibited a temperature-dependent sol–gel transition and a shear-responsive behavior. The stimuli-responsive viscoelastic properties of these gels make them ideal for direct-write 3D printing, soft robotics, and tissue engineering applications. |
Introduction
Hydrogel-forming biopolymers, like collagen and gelatin, are exquisite examples of biopolymers that possess a defined composition, length, and sequence. These linear polymers fold or self-assemble into well-defined higher-order structures that can bind molecules and create three-dimensional scaffolds for cells. In the case of collagen, linear protein strands hierarchically assemble to form fibrous bundles that not only provide mechanical structure, but also present recognition sites that promote cellular adhesion.1 Non-covalent interactions,2,3 including hydrogen bonding, ionic interactions, and van der Waals forces—introduced at precise locations along the polymer chain—drive the self-assembly of biopolymers. Wholly synthetic polymers that show the same level of compositional precision are not possible yet, even using controlled polymerization techniques. Block copolymers that microphase separate have received significant attention in the polymer sciences based on their ability to organize into nano- and microscale morphologies.4–7 However, these polymers do not self-assemble with the same level of complexity observed in proteins or DNA. While the sequence of polymer blocks can be controlled in a segmented block copolymer, the individual monomer sequence cannot. The design rules that govern the self-assembly of synthetic polymers with precisely defined monomer sequences is likely to be incredibly complex.8 Accordingly, there is a need to develop a strategy to afford hierarchically self-assembled synthetic polymers that does not rely on precise monomer sequences. These alternative strategies could complement existing efforts to create sequence-controlled polymers.9The precision molecular engineering of polymers to control nanoscale architecture remains non-trivial.10–14 However, the molecular-level design of nanostructured polymers and macromolecular materials (also referred to as nanoarchitectonics15–17) has led to the emergence of precisely self-assembled materials with catalytic behavior,18,19 dynamic mechanical properties,20 and electromechanical actuation.21 Nanoarchitectonic-based approaches to polymer design have recently been applied toward the fabrication of stimuli-responsive hydrogels.22–25 Stimuli-responsive hydrogels26 adapt to changes in their environments such as temperature,27–30 pH and ion concentration,31–35 and shear stress.29,36–39 These “smart” hydrogels are ideal for biomimetic tissues and tissue culture,40–45 as well as drug-delivery systems.46–48 In particular, shear-thinning hydrogels have potential as injectable therapeutics36,37 and are excellent inks for direct-write 3D printing.29,38,43,49 The manner by which polymeric hydrogels self-assemble and react to stimuli can be tuned with small molecular modifications to the main-chain, side-chain or end-group functionality. Despite the small fraction of the polymer chain that the end-groups represent, supramolecular functionalities at the chain termini can have dramatic effects on the characteristics of the resulting self-assembled networks.50–52 Supramolecular hydrogen-bonding molecules can form ordered assemblies in solution and bulk polymer and can be incorporated into the design of a polymer.53–58 For example, Sijbesma and Meijer demonstrated that the incorporation of the ureidopyrimidinone (UPy) motif36,59–61 can have a profound, measurable effect on polymer morphology in both solution and the bulk phase. For hydrogels, the use of hydrogen-bonding groups influences not only morphology, but also moduli, shear-thinning characteristics, and thermoresponsive behavior.36,37,62,63
Herein, we report temperature and shear-responsive hydrogels composed of poly(isopropyl glycidyl ether)-b-poly(ethylene oxide)-b-poly(isopropyl glycidyl ether) (PiPGE-b-PEO-b-PiPGE). Our group has developed ABA triblock copolymers based on poly(alkyl glycidyl ethers) as platforms for stimuli-responsive hydrogels,29,64 wherein ‘A’ is a poly(alkyl glycidyl ether) and ‘B’ is poly(ethylene oxide). These block copolymers self-assemble into flower-like micelles65 (Fig. 1) in aqueous media and form physically entangled micelle networks as the polymer concentration increases.29 Micellization is driven by the hydrophobic aggregation of the poly(alkyl glycidyl ether) blocks. Some poly(alkyl glycidyl ethers), such as poly(ethyl glycidyl ether), have lower critical solution temperatures (LCSTs) as homopolymers.66–68 Others, such as poly(isopropyl glycidyl ether), are insoluble at all temperatures and concentrations. However, when either of these alkyl glycidyl ethers is the ‘A’ component in a PEO-based ABA triblock copolymer, the copolymer has an LCST due to entropically-driven hydrophobic aggregation of the ‘A’ blocks. This phase transition results in self-supporting hydrogels only when the ‘A’ blocks constitute a volume fraction ≥20 wt%.
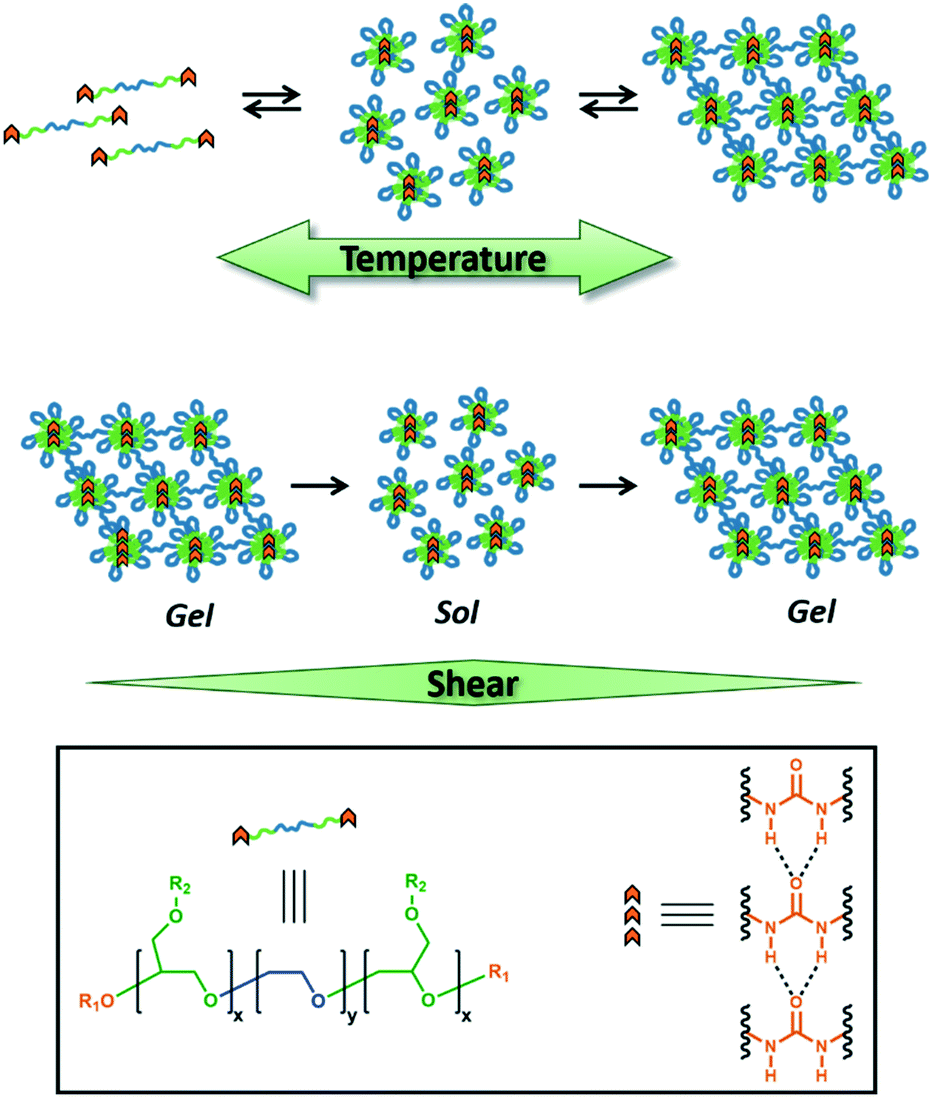 | ||
| Fig. 1 Idealized representation of dynamic, reversible self-assembly of amphiphilic triblock copolymers in aqueous media. The equilibrium distribution of solvated unimers, flowerlike micelles, and physically entangled micelle networks changes with temperature (top). The hydrogel network can also be temporarily disrupted with shear stress (middle). Hydrogen-bonding end groups (orange chevrons) self-assemble in the hydrophobic domains, enhancing mechanical strength. | ||
Ureas were chosen as the hydrogen-bonding group of choice for the present study due to their strong self-association and ability to form extended supramolecular arrays both as small molecules69–76 as well as in segmented and block copolymers.56,58,62,77–81 The introduction of urea motifs into block copolymers results in crystalline or semi-crystalline domains due to the regular pattern of intermolecular hydrogen bonds. The geometric structure of the array, as well as the strength of the hydrogen-bonding association, varies depending on the placement of the ureas within the block copolymer, as well as the nature of the substituents adjacent to the urea groups.62,72,73,76,82–84
In the case of stimuli-responsive hydrogels, ureas play an influential role in polymer self-assembly. In most cases, ureas are placed in a hydrophobic domain to minimize competing hydrogen-bonding interactions with water.10,31,32 This placement allows the formation of supramolecular hydrogen-bonding arrays. Ideally, these hydrogen bonds act as physical crosslinks between polymer chains, enhancing the mechanical strength of the resulting hydrogels while retaining shear-thinning, self-healing, and temperature-responsive characteristics.
The polymers in the present study demonstrate that the inclusion of urea-containing hydrogen bonding groups at the chain ends can significantly alter the viscoelastic behavior of multi-stimuli responsive hydrogels. Changes in morphology, mechanical properties, and thermal response demonstrate the enormous impact of relatively minor synthetic modifications and imply that hydrogen-bonding groups could be used to precisely tune the thermo- and mechano-responsive behavior of polymeric hydrogels.
Experimental
Materials
All solvents and reagents were purchased from Sigma-Aldrich, Fisher Scientific, or TCI America and used as received, unless noted otherwise. Dichloromethane (DCM, HPLC grade) and tetrahydrofuran (THF, Optima) were dried with neutral alumina using a Pure Process Technology Glass Contour solvent purification system and were dispensed under argon. Diethyl ether (Fisher, Certified ACS, Anhydrous, BHT stabilized, 99.9%) was dried over anhydrous magnesium sulfate (Fisher, Certified Powder) immediately prior to use. Acetonitrile was purchased from Fisher (HPLC Grade, 99.9%).Instrumentation
All NMR spectra were collected using a Bruker AV 500 MHz spectrometer equipped with a double resonance broadband (BBI) probe and Oxford narrow bore cryomagnet, with 24 scans and 5 or 10 s pulse delay times (d1) at 298 K. Thin-film IR spectra were collected on a Bruker Vector 33 FT-IR spectrophotometer using KBr salt plates. Variable-temperature IR experiments were performed using a JASCO Model FT/IR-4100A spectrophotometer at 1 cm−1 resolution, with a sealed brass sample cell. The cell temperature was controlled using inlet/outlet tubing connected to an ethylene glycol-based heat exchanger. The sample chamber was purged with N2 to remove CO2 and traces of water vapor. Sol–gels were prepared at 23 wt% in D2O to better observe urea stretching and bending modes between 1500–1800 cm−1. Heating/cooling rates were maintained at approximately 0.5 °C min−1 to ensure complete equilibration and self-assembly. Gel permeation chromatography was performed using a Waters chromatograph (Waters 1525 Binary HPLC Pump with in-line degasser) equipped with two 10 μm Malvern columns (300 mm × 7.8 mm) connected in series with increasing pore size (1000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å), using chloroform (Optima, 0.1% v/v triethylamine, 1.0 mL min−1 flow rate) as the eluent, and calibrated with poly(ethylene oxide) standards (Fluka Analytical, 400 to 40000 g mol−1). The relative molecular weights were measured using a refractive index detector (Waters 2414). All rheological measurements were performed on a TA Instruments Discovery HR-2 hybrid rheometer equipped with Peltier temperature control accessory. Each rheological experiment was performed with a 20 mm flat-plate geometry, except for cyclic oscillatory strain experiments, which utilized a 40 mm cone-and-plate geometry and solvent trap. All tests were performed in duplicate to ensure reproducibility. Differential scanning calorimetry was performed using a TA Instruments DSC 250 calorimeter equipped with a TA RCS90 cooling system, using Tzero standard aluminum pans (TA Instruments) and calibrated with an indium standard. The heating and cooling rate for all samples was 2 °C min−1, except for the initial heating cycle, which was run at a rate of 10 °C min−1. SAXS measurements were performed using a Xeuss 2.0 laboratory beamline (Xenocs Inc.) with an X-ray wavelength of 1.54 Å and a sample-to-detector distance of 2.5 m. Diffraction images were recorded on a Pilatus 1M Detector (Dectris Inc.) with an exposure time of 2 h, then analyzed using the Nika software package.85 Measurements were collected in both the hydrated (25 wt%) and dry states for each polymer composition.
000 Å), using chloroform (Optima, 0.1% v/v triethylamine, 1.0 mL min−1 flow rate) as the eluent, and calibrated with poly(ethylene oxide) standards (Fluka Analytical, 400 to 40000 g mol−1). The relative molecular weights were measured using a refractive index detector (Waters 2414). All rheological measurements were performed on a TA Instruments Discovery HR-2 hybrid rheometer equipped with Peltier temperature control accessory. Each rheological experiment was performed with a 20 mm flat-plate geometry, except for cyclic oscillatory strain experiments, which utilized a 40 mm cone-and-plate geometry and solvent trap. All tests were performed in duplicate to ensure reproducibility. Differential scanning calorimetry was performed using a TA Instruments DSC 250 calorimeter equipped with a TA RCS90 cooling system, using Tzero standard aluminum pans (TA Instruments) and calibrated with an indium standard. The heating and cooling rate for all samples was 2 °C min−1, except for the initial heating cycle, which was run at a rate of 10 °C min−1. SAXS measurements were performed using a Xeuss 2.0 laboratory beamline (Xenocs Inc.) with an X-ray wavelength of 1.54 Å and a sample-to-detector distance of 2.5 m. Diffraction images were recorded on a Pilatus 1M Detector (Dectris Inc.) with an exposure time of 2 h, then analyzed using the Nika software package.85 Measurements were collected in both the hydrated (25 wt%) and dry states for each polymer composition.
Monomer distillation
Isopropyl glycidyl ether (iPGE, Aldrich 98%) was dried over CaH2 (Fisher, Laboratory Grade) for 24 h under N2 atmosphere. Next, iPGE was distilled under reduced pressure into a flask containing ∼1 mL butyl magnesium chloride (dried 2.0 M THF solution, Aldrich). Finally, iPGE was distilled into a clean, dry flask containing activated 4 Å molecular sieves (Fisher, Grade 514, 8–12 mesh beads). Immediately prior to use, distilled iPGE was degassed using three full freeze–pump–thaw cycles.Potassium naphthalenide preparation
Potassium naphthalenide was prepared as a 1.0 M solution in THF. Typical preparation proceeded as follows: naphthalene (2.57 g, Fisher Certified Crystalline) was added to an oven-dried 100 mL Schlenk flask, then dissolved in 20 mL anhydrous THF under N2 atmosphere. Potassium (0.783 g, Aldrich 99.5%) was transferred from mineral oil to hexanes, then added quickly to the dissolved naphthalene. The resulting dark green solution was stirred under N2 atmosphere for 6 h.Synthesis of polymer 1
An oven-dried 5-neck reactor flask was charged with poly(ethylene oxide) (PEO, 20.2 g, Sigma BioUltra Mn = 8000 g mol−1) and a glass-coated stir bar. The flask was evacuated and purged with argon three times, and PEO was then dried under reduced pressure at 40 °C for 24 h. Dried PEO was dissolved in 200 mL anhydrous THF and warmed to 50 °C under argon atmosphere. Potassium naphthalenide 1.0 M THF solution (7 mL) was slowly added via syringe to the dissolved PEO until a yellow-green color persisted. Degassed iPGE (6.29 g) was added quickly via syringe. After 48 h of polymerization at 50 °C under argon atmosphere, the reaction was quenched by addition of degassed 1% (v/v) acetic acid:MeOH (5 mL). The reaction mixture was concentrated in vacuo, then precipitated in large excess (800 mL) of anhydrous Et2O. The slurry was centrifuged at 4400 rpm, the supernatant was decanted, and the precipitated polymer was rinsed three times with fresh Et2O. Recovered white solid was dried in air at room temperature for 24 h, then dried under reduced pressure at 40 °C. To remove traces of potassium ion, dried polymer was reconstituted in 150 mL DCM, then stirred with 1 g Dowex 50WX8 cation exchange resin (Sigma-Aldrich, hydrogen form, 100–200 mesh) for 30 min. Following filtration, concentration, and a final precipitation from Et2O, polymer was dried under reduced pressure at 50 °C, then stored in an amber bottle at 5 °C (21.6 g, 82% yield). Degree of polymerization (DP) for the poly(isopropyl glycidyl ether) blocks was estimated by 1H NMR spectroscopy to be DP = 8 (16 total, approximately 1900 g mol−1) as described in the ESI.† Gel permeation chromatography (GPC) indicated narrow dispersity, Đ = 1.10, suggesting little to no chain transfer or termination, in agreement with the living nature of the polymerization.
Steglich esterification with Boc-protected glycine
Polymer 1 and Boc-protected glycine (Boc-gly, 25 eq., Aldrich ≥99%) were dried under reduced pressure at 50 °C for 24 h. Dried Boc-gly was dissolved in anhydrous DCM under N2 atmosphere, while dried polymer was dissolved in DCM in a separate flask. 4-Dimethylaminopyridine (DMAP, 0.25 eq., Aldrich ReagentPlus ≥99%) was dissolved in a minimal amount of DCM, then mixed with dissolved polymer. The polymer/DMAP solution was added dropwise to dissolved Boc-gly. Dicyclohexylcarbodiimide (DCC, 25 eq., Aldrich 99%) was melted for easy handling, then dissolved in anhydrous DCM and added dropwise to the rest of the reaction mixture. The final reaction mixture (approximately 10 wt% polymer) was brought to reflux (55 °C) and stirred under N2 for 24 h. The reaction mixture was cooled and vacuum-filtered. Solvent was removed in vacuo, then residue was reconstituted in CH3CN (Fisher, HPLC Grade, 99.9%) and stirred for 15 min. The solution was centrifuged, resulting in a small amount of settled white solid. Supernatant was filtered through a fritted funnel (medium porosity), and transparent filtrate was concentrated in vacuo. The residue was stirred in excess Et2O, centrifuged, and rinsed twice with fresh Et2O. The Boc-functionalized polymer was isolated as an off-white solid and dried in air at room temperature for 24 h.TFA-mediated Boc cleavage
The Boc-functionalized polymer was stirred in a 1:1 (v/v) mixture of DCM and trifluoroacetic acid (TFA, Sigma-Aldrich ReagentPlus, 99%) for 18 h at room temperature. Solvent was removed in vacuo, concentrate was reconstituted in fresh DCM, and this process was repeated 3 times. The resulting residue was precipitated in excess Et2O, centrifuged, then rinsed once with fresh Et2O. The recovered off-white solid was dried under reduced pressure at 40 °C to afford the amine-functionalized polymer.
Synthesis of polymer 1-U
The amine-functionalized polymer was dried under reduced pressure at 40 °C prior to synthesis. para-Tolyl isocyanate (10 eq., Aldrich 99%) was removed from cold storage and brought to room temperature in a sealed bottle. The isocyanate was dissolved in anhydrous DCM in a flask containing activated 4 Å molecular sieves. Dried polymer was dissolved in DCM along with triethylamine (NEt3, 2.5 eq., TCI America >99%, distilled from CaH2). Dissolved polymer was added dropwise to the isocyanate, and the reaction mixture was stirred under N2 atmosphere for 18 h. Next, the reaction mixture was vacuum-filtered. The slightly turbid, golden filtrate was centrifuged at 4400 rpm. The supernatant was syringe-filtered (0.7 μm glass microfiber, Whatman) and solvent was removed in vacuo. The concentrated residue was stirred in excess Et2O, centrifuged, then rinsed twice with fresh Et2O and dried in air at room temperature for 24 h to afford polymer 1-U. Degree of functionalization (fn) was estimated by 1H NMR spectroscopy as described in the ESI.†fn = 82–91%.Synthesis of polymer 1-bisU
The amine-functionalized polymer was dried under reduced pressure at 40 °C prior to synthesis. Tolylene-2,4-diisocyanate (100 eq., Aldrich 95%, 4% as 2,6-isomer) was dissolved in anhydrous DCM in a flask containing activated 4 Å molecular sieves. Dried polymer was dissolved in DCM along with NEt3 (2.5 eq., TCI America >99%, distilled from CaH2.) Dissolved polymer was added dropwise to the diisocyanate over 1 h. Reaction mixture was stirred under N2 atmosphere for 16 h, then vacuum-filtered. The slightly turbid, golden filtrate was syringe-filtered (0.7 μm glass microfiber) then concentrated in vacuo. The residue was stirred in a large excess of anhydrous Et2O, centrifuged, then rinsed three times with fresh Et2O. Recovered polymer was dried under reduced pressure at RT for 3 h. Aniline (100 eq., Fisher, Certified ACS, 99.9%) was dried over activated 4 Å molecular sieves for 2 h, then dissolved in dry DCM in a flask containing molecular sieves. The partially dried polymer was reconstituted in dry DCM, then added dropwise to the aniline over 1 h. The reaction mixture was stirred under N2 atmosphere for 21 h, then vacuum-filtered and centrifuged. The supernatant was syringe-filtered, yielding a clear, golden solution which was subsequently concentrated. The residue was stirred in a large excess of Et2O, centrifuged, and rinsed three times with fresh Et2O. Recovered polymer was dried under reduced pressure at 30 °C to afford polymer 1-bisU. Degree of functionalization (fn) was estimated by 1H NMR spectroscopy as described in the ESI.†fn = 90–93%.Results and discussion
We developed a modular route (Scheme 1) to the syntheses of the polymers in this study. Polymer 1 was synthesized via anionic ring-opening polymerization of isopropyl glycidyl ether initiated from poly(ethylene oxide). Molecular weight was controlled by changing the ratio of isopropyl glycidyl ether monomer to PEO initiator, and dispersity indices were consistently low (Đ < 1.2). Polymer 1 was derivatized with Boc-glycine, and the protecting groups were removed under acidic conditions to afford an amine-functionalized polymer. Further reactions with aryl isocyanates afforded the urea-terminated polymer 1-U and polymer 1-bisU. GPC traces of the urea-terminated polymers indicated lower apparent molecular weights than the hydroxy-terminated polymer (Fig. S5†). This effect may have been due to the formation of compact aggregates by the urea-derivatized polymers in chloroform, which would have decreased the hydrodynamic radius of the polymer chains. Alternatively, the lower detected molecular weights could have been due to secondary retention, or noncovalent interactions between the polymers and the column material. Polymer 1-bisU, in addition to having a smaller apparent molecular weight, showed a broader GPC trace and higher dispersity (Đ = 1.68) than polymer 1 (Đ = 1.10) and polymer 1-U (Đ = 1.09). The presence of a small, high molecular weight shoulder in the bis-urea polymer trace suggested that a small degree of step-growth polymerization may have occurred, and accounts for the increased dispersity. No such shoulders were present for polymer 1-U, which was incapable of step-growth polymerization.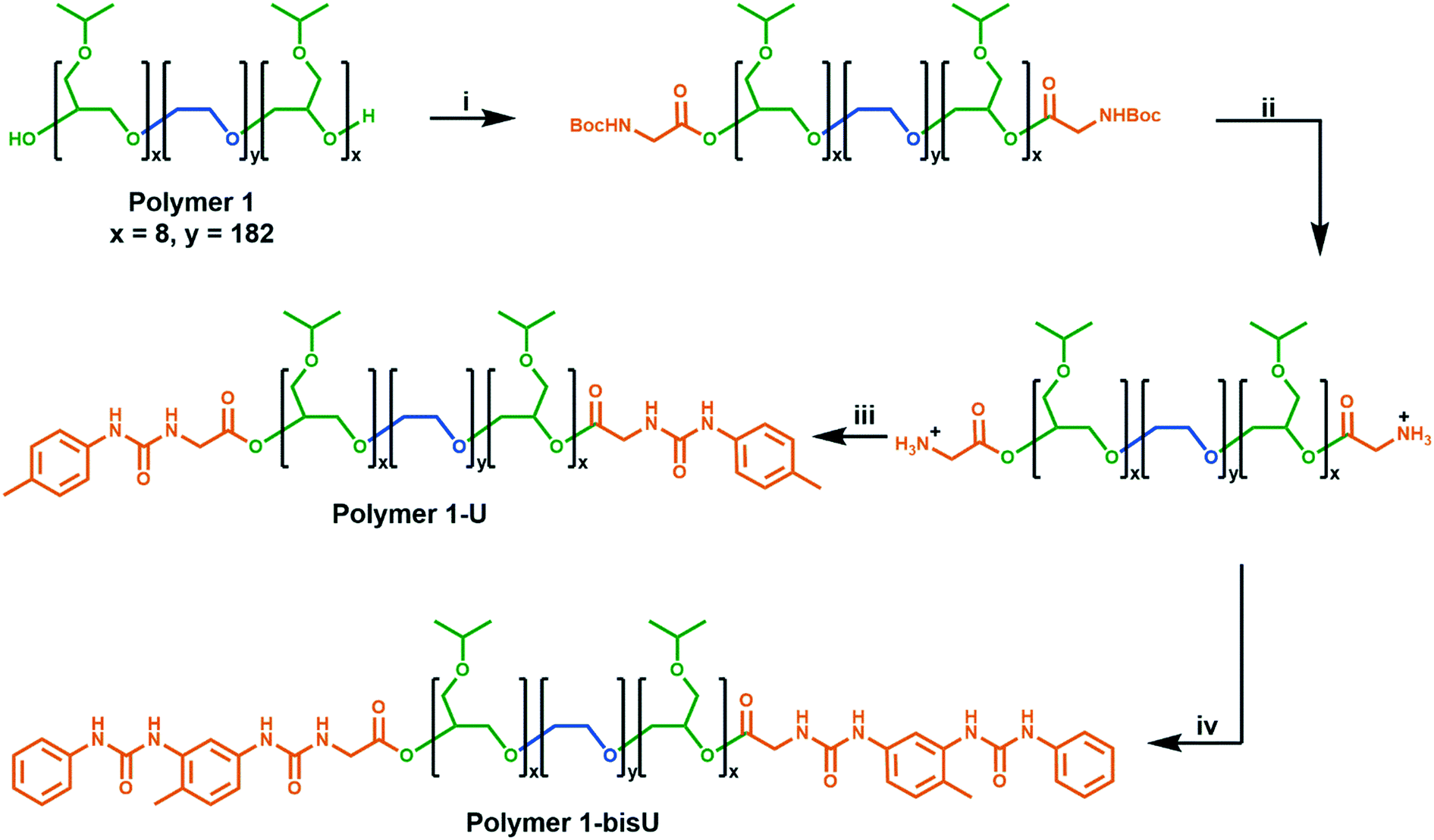 | ||
| Scheme 1 Chain-end functionalization of PiPGE-b-PEO-b-PiPGE triblock copolymer. For each of the polymers, x ≈ 8 and y ≈ 182 (Mn ≈ 9900 g mol−1). (i) DCC/Boc-Gly-OH (excess); DMAP (cat.); DCM, 55 °C, 24 h. (ii) 1:1 v/v TFA:DCM; RT, 18 h. (iii) p-Tolyl isocyanate/NEt3; DCM, RT, 18 h. (iv) (1) Tolylene-2,4-diisocyanate (excess)/NEt3; DCM, RT, 16 h. (2) Aniline (excess); DCM, RT, 21 h. | ||
Phase diagrams and rheology
The incorporation of the single- and bis-urea motifs to the chain ends of polymer 1 (to afford polymer 1-U and polymer 1-bisU) altered the viscoelastic behaviors of the corresponding aqueous solutions or gels. Phase diagrams were constructed by preparing a series of polymer solutions as described in the ESI† at different concentrations (2–30 wt% in DI water) and performing rheological temperature-ramp experiments (5–50 °C at 2 °C min−1, Fig. S6–S8†) for each concentration. Relevant data from the temperature-ramp experiments are summarized in Table 1. At the end of the temperature-ramp tests, the urea-functionalized polymer hydrogels were stiffer than the unfunctionalized gels, as indicated by greater G′ values. As expected, the introduction of chain-end ureas lowered both the gelation temperature (Tgel) for a particular concentration, as well as the minimum polymer concentration (Cmin) required for gelation at a particular temperature (Fig. 2). The reduction of Tgel in polymer 1-bisU gels was due to both hydrophobic and hydrogen-bonding interactions. The aryl bis-urea chain ends were hydrophobic, as evidenced by qualitative solubility tests of small-molecule aryl urea analogs (Table S1†). The introduction of aryl bis-urea chain ends therefore increased the overall hydrophobicity of the PiPGE ‘A’ blocks in polymer 1-bisU, driving micellization at lower temperatures. Strong hydrogen-bonding interactions between the bis-urea chain-ends resulted in microphase separation of crystalline domains, as supported by DSC and SAXS, to be discussed in a later section. The presence of these hard domains likely were responsible for imparting gel-like mechanical properties to the polymer 1-bisU gel. Therefore, the increased hydrophobicity of polymer 1-bisU shifted the equilibrium distribution of solvated polymer chains and bridged/unbridged flowerlike micelles, while the strong hydrogen-bonding interactions slowed exchange dynamics between micellized and water-solvated polymer chains. As a result, the Tgel decreased with the introduction of hydrogen-bonding domains. Surprisingly, the monourea end-group in polymer 1-U had a subtle, almost imperceptible effect on the thermal response of the hydrogels, as depicted in the phase diagrams (Fig. 2). While a single urea lowered the Tgel for each solution slightly, the most noticeable effect of monourea chain ends was the appearance of a viscous regime at higher concentrations and temperatures (≥20 wt% and ≥20 °C). While polymer 1 underwent a transition directly from free-flowing solution to self-supporting gel, polymer 1-U first became thick and viscous before becoming a free-standing gel. The increased viscosity and slightly lowered gelation temperature of polymer 1-U solutions was likely the result of hydrophobic aggregation of the terminal ureas. Polymer 1-bisU exhibited a much broader “viscous” range and gelled at a much lower temperature and concentration than polymers 1 and 1-U. It is likely that the bis-urea domains never became fully solvated, except perhaps at polymer concentrations <10 wt%. This hypothesis was supported by VT-IR experiments (discussed later) and by the extreme hydrophobicity of a small-molecule bis-urea analog (Table S1†), which would not dissolve in water at any temperature, even at concentrations <0.5 mg mL−1.| Polymer | T gel (°C) | Crossover modulus (Pa) | Modulus at 50 °C (G′, Pa) |
|---|---|---|---|
| Polymer 1 | 39.6 | 6.25 | 2460 |
| Polymer 1-U | 37.2 | 62.2 | 2480 |
| Polymer 1-bisU | 14.9 | 765 | 10100 |
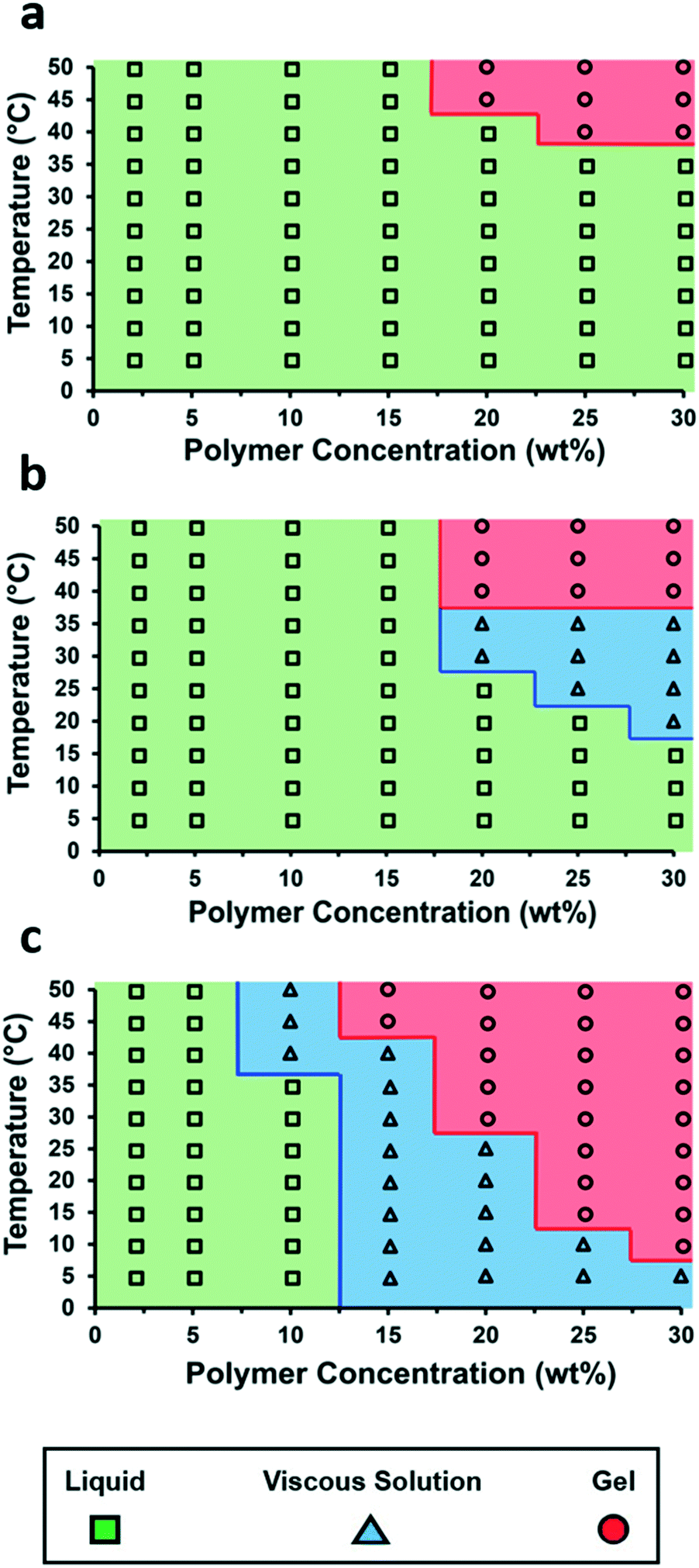 | ||
| Fig. 2 Temperature–concentration phase diagrams for (a) polymer 1, (b) polymer 1-U, and (c) polymer 1-bisU. | ||
Based on comparisons of the phase diagrams for each polymer, 25 wt% was chosen as the concentration at which to compare rheological properties. All tests were conducted at 25 °C. As the phase diagrams suggest, polymer 1-bisU was a gel at this temperature, polymer 1-U was a viscous solution, and polymer 1 was a free-flowing solution. Indeed, polymer 1-bisU showed the self-healing (Fig. 3b) and shear-thinning (Fig. 3d) properties expected of these stimuli-responsive hydrogels. The dynamic oscillatory strain test, in which the material was subjected to alternating periods of high (100%) and low (1%) strain amplitude, indicated that at 25 wt% and 25 °C, polymer 1-bisU behaved as a solid gel at low strain and as a viscous fluid at high strain. Importantly, the material yielded and recovered almost instantaneously, with minimal mechanical hysteresis between strain cycles. With increasing shear rate, the viscosity of the polymer 1-bisU gel decreased over four orders of magnitude. This shear-thinning behavior confirmed the gelled nature of polymer 1-bisU and demonstrated its suitability as an injectable or extrudable gel. By contrast, the viscosities of polymer 1 (Fig. S9†) and polymer 1-U (Fig. 3c) solutions were largely independent of shear rate, suggesting that they were more like viscous liquids than elastic solids. Note that the viscosity of the polymer 1-U solution was consistently greater than that of polymer 1 at most shear rates, reflecting the thicker nature of the polymer 1-U solution.
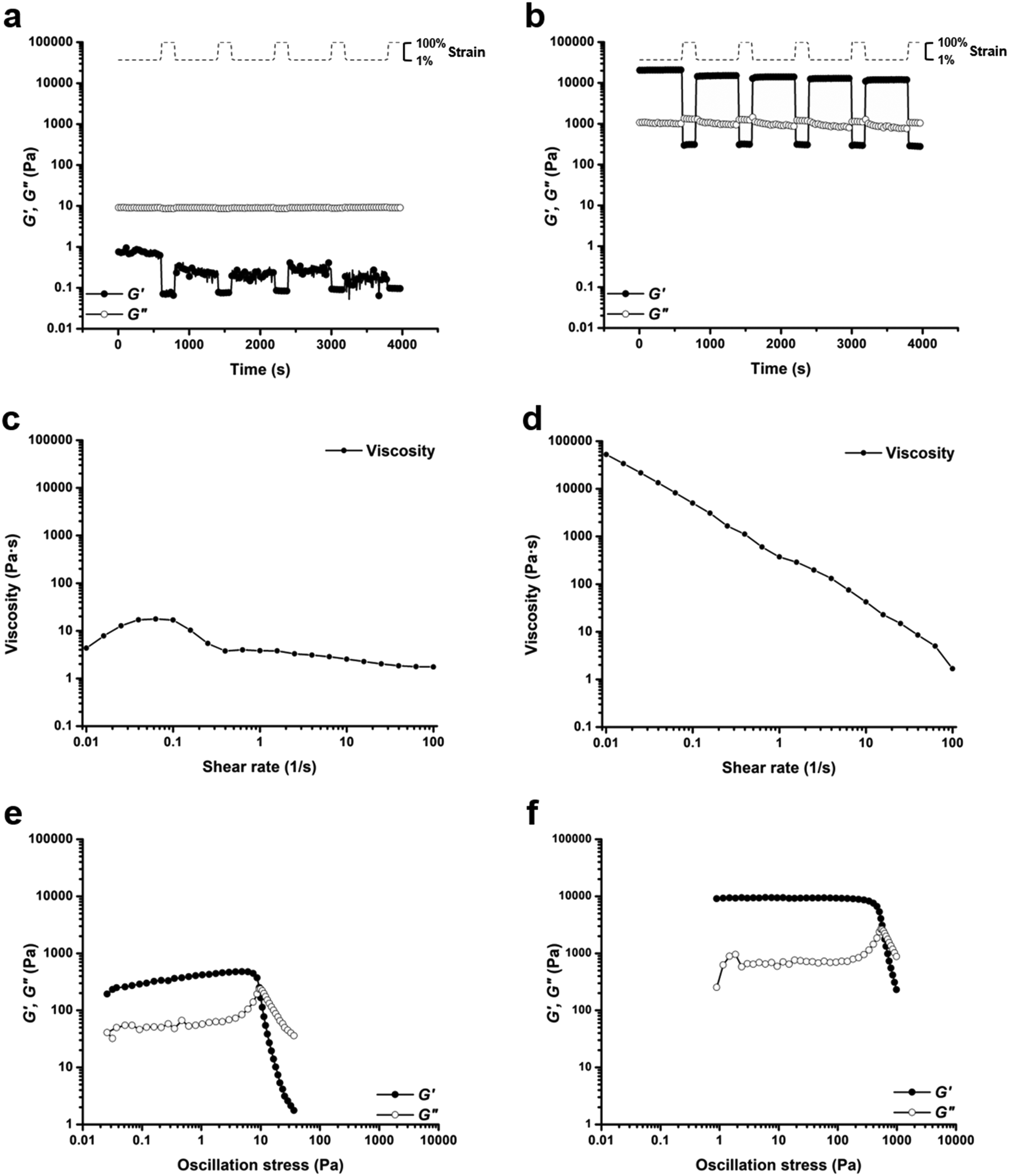 | ||
| Fig. 3 Rheological experiments for polymer 1-U and polymer 1-bisU conducted at 25 wt% and 25 °C. Cyclic oscillatory strain tests (angular frequency, ω = 6 rad s−1) for (a) polymer 1-U and (b) polymer 1-bisU. Viscosity vs. shear rate plots for (c) polymer 1-U and (d) polymer 1-bisU. Oscillatory stress sweep experiments (angular frequency, ω = 6 rad s−1) for (e) polymer 1-U and (f) polymer 1-bisU. The stress sweep experiments were used to determine static yield stress (σstat), reported as the onset of G′ decrease. For polymer 1-U, σstat = 7.58 Pa. For polymer 1-bisU, σstat = 447 Pa. | ||
The dynamic oscillatory strain test for a 25 wt% polymer 1 solution (Fig. S10†) showed an interesting phenomenon: the material was initially a fluid with G′′ > G′, but following each cycle of 100% strain amplitude, the storage modulus increased and eventually crossed over the loss modulus. This strange measurement was attributed to the frequency dependence of the moduli for polymer 1 (Fig. S11†). Frequency-sweep measurements for each of the polymers (Fig. S11–S13†) allowed the estimation of average terminal relaxation time, τ, defined as the inverse of the radial frequency ω at the point of modulus crossover.86,87 For polymer 1, τ = 0.147 s, while for polymer 1-U τ = 4.51 s, and for polymer 1-bisU there was no modulus crossover in the tested frequency range. If the mechanism of stress relaxation is assumed to be disruption of bridging interactions between micelles or disruption of individual micelle structure, then this trend is logical. Because polymer 1 was not hydrophobic enough to form self-supporting hydrogels at 25 wt% and 25 °C, the exchange dynamics between chain-ends in micelle cores and water-solvated chain-ends was fast. The introduction of hydrophobic, hydrogen-bonding groups slowed the exchange dynamics for polymer 1-U, resulting in a longer relaxation time, while for polymer 1-bisU the exchange dynamics were too slow to be measured. The frequency-sweep measurements reflected the physical nature of each solution or gel: polymer 1 was a free-flowing liquid, polymer 1-U was a viscous solution, and polymer 1-bisU was a free-standing hydrogel. The angular frequency for the cyclic strain measurements was 6 rad s−1 (1 Hz), which was near the fluidlike-solidlike transition observed in the frequency-sweep test for polymer 1.
Oscillatory strain sweep tests were conducted for 25 wt% solutions/gels of all polymers, in which solutions/gels were subjected to increasing oscillatory strain amplitudes (0.01 to 100%) at a fixed angular frequency (6 rad s−1). From this data, plots of storage and loss moduli (G′ and G′′) against oscillatory stress (σ) were made (Fig. 3e and f, S14†). These plots were used to determine the static yield stress (σstat) for each gel, or the minimum amount of pressure needed for the gel to yield and to begin flowing, a crucial factor for extrusion-based applications. The polymer 1 solution was an unstructured fluid, and therefore did not possess a measurable yield stress. Under the conditions of the test, polymer 1-U was a weak gel, with a yield stress of σstat = 7.58 Pa (Fig. 3e). As expected, the polymer 1-bisU gel had the highest yield stress of σstat = 447 Pa (Fig. 3f). This yield stress was more than 5 times the value for a 20 wt% gel based on Pluronic F127,29 but several orders of magnitude less than a similar bis-urea-containing, PEO-based gel.37 While the F127 gel exhibited poor mechanical stability, the PEO-based gel was both mechanically robust and extrudable from a 29-gauge syringe needle. Polymer 1-bisU gels therefore had a high enough yield stress to maintain structural integrity in the absence of applied pressure, but a low enough yield stress that they could be easily extruded from a nozzle.
Differential scanning calorimetry
While rheology and phase diagrams clearly demonstrated that the presence of the urea and bis-urea end-groups affected the viscoelastic behavior of the hydrogels, we turned to other techniques to further probe the assembly of the hydrogen-bonding assemblies. Urea-based assemblies are known37,80 to form anisotropic crystalline domains which can be represented by a melting transition in a DSC thermogram at temperatures above 170 °C. As shown in Fig. S15 and S16,† polymer 1-bisU exhibited a melting exotherm that suggested that the bis-ureas formed hard domains within the hydrogel assemblies. Replicate measurements demonstrated that the melting transition occurred closer to 170 °C when the sample was cooled to 0 °C with no additional equilibration time, and closer to 193 °C when the sample was equilibrated for 90 min at 0 °C (Table 2). This difference in melting transition may reflect differences in the crystalline packing of the asymmetric bis-urea.76,84,88,89 The single urea chain-end of polymer 1-U did not exhibit a melting transition, which suggested that the ureas were not strongly associating into crystalline domains. We, and others,90–93 have previously reported glass transition temperatures for poly(alkyl glycidyl ethers) that range from −70 to −60 °C. However, we did not observe a measurable glass transition in this temperature range. Polymer 1, polymer 1-U, and polymer 1-bisU exhibited PEO melting and crystallization transitions at 50–54 °C and 35–40 °C, respectively. Overall, the DSC data demonstrated phase separation of the urea end-groups only in polymer 1-bisU, which was consistent with the SAXS data.| Polymer | Range (°C) | Transition 1 | Transition 2 | ||
|---|---|---|---|---|---|
| T (°C) | ΔH (J g−1) | T (°C) | ΔH (J g−1) | ||
| a No equilibration time at 0 °C. b Allowed to equilibrate at 0 °C for 90 min following initial heating/cooling cycle. | |||||
| Polymer 1 | −90 ↔ 200 | 53.9 | 126 | — | — |
| Polymer 1-U | 0a ↔ 200 | 53.1 | 112 | — | — |
| Polymer 1-bisU | 0a ↔ 200 | 51.7 | 99.7 | 170 | 0.621 |
| Polymer 1-bisU | 0b ↔ 200 | 52.0 | 95.6 | 193 | 0.557 |
Infrared spectroscopy
Thin-film and solution-phase IR spectroscopic experiments were performed to further probe the association of the hydrogen bonding end-groups. Thin-film IR spectra of polymer 1-U and polymer 1-bisU exhibited peaks in the range of 1500–1800 cm−1 that corresponded to the carbonyl stretching frequencies. For both polymer 1-U and polymer 1-bisU, the chain-end ester carbonyl stretching mode was present at 1751 cm−1. More complex amide I, amide II, and aromatic carbon–carbon stretching vibrations appeared between 1500–1700 cm−1. The amide I mode (carbonyl stretch) appeared as a mixture of strongly hydrogen-bonded (1640 cm−1) and weakly-hydrogen-bonded (1700 cm−1) populations in the thin film. Unfortunately, these stretches were overlapped by the presence of a water bending mode at 1640–1650 cm−1, which complicated the analysis. However, these peak assignments are in agreement with previously reported IR spectra for similar urea motifs.72,80,89,94,95To probe the association and dissociation of the urea groups, solution-phase variable-temperature IR (VT-IR) experiments were performed. In these experiments, 23 wt% gels of polymer 1-U and polymer 1-bisU were prepared in D2O to observe the carbonyl stretching modes between 1600–1800 cm−1. As previously noted, these ABA triblock copolymers exhibit a temperature response in which they undergo a reversible sol–gel transition in water above a critical temperature. In the ‘sol’ state, the entire triblock copolymer is water soluble, whereas in the ‘gel’ state, the PiPGE block is hydrophobic and drives the micellization of the polymers.29 Thus, we hypothesized that, as the PiPGE blocks transitioned from a solvated aqueous state to a hydrophobically collapsed micelle core, the chain-end ureas would transition from a partially-solvated state to a self-associated supramolecular array within the hydrophobic domain of the glycidyl ether blocks.
The VT-IR spectra suggested that the association of the hydrogen-bonded ureas remained relatively constant with temperature, even as the polymer chains underwent nanoscale rearrangement. Baseline-corrected spectra of polymer 1-bisU in the range of 1615–1715 cm−1 (Fig. S19†) indicated a slight change to the peak-shape for the carbonyl stretching frequencies as temperature decreased from 50 to 5 °C. This subtle, if not minimal, change suggested that at low temperatures, when the PiPGE blocks were more water-soluble, the urea end-groups were still hydrogen-bonded. The VT-IR tests therefore implied that the effects of water solvation on urea hydrogen-bonding in this system were minimal, and that these interactions persisted despite the solubility changes in the polymer.
We also observed that IR spectroscopy could be used to track the assembly/disassembly of the PiPGE chains. The peak at approximately 1348 cm−1 underwent measurable changes in intensity with varying temperature, increasing in area continuously as temperature decreased from 50 to 5 °C (Fig. 4). This peak likely corresponded to a C–H bending mode of the gem-dimethyl moiety in the PiPGE blocks. As these glycidyl ether blocks underwent hydrophobic collapse at higher temperatures, their effective concentration in D2O decreased and their rotational freedom was restricted. As a result, CH3 bending modes decreased in intensity. Conversely, disassembly of micelles upon cooling and subsequent solvation of the glycidyl ether blocks resulted in an increase in intensity for the peak at 1348 cm−1. While these changes in the IR spectra are not directly related to the urea hydrogen-bonding motifs, they nevertheless reveal a useful protocol to track the self-assembly and disassembly of polymer chains with respect to temperature. In future experiments, deuteration of the isopropyl groups might be an easier way to track these bending modes in IR spectra.
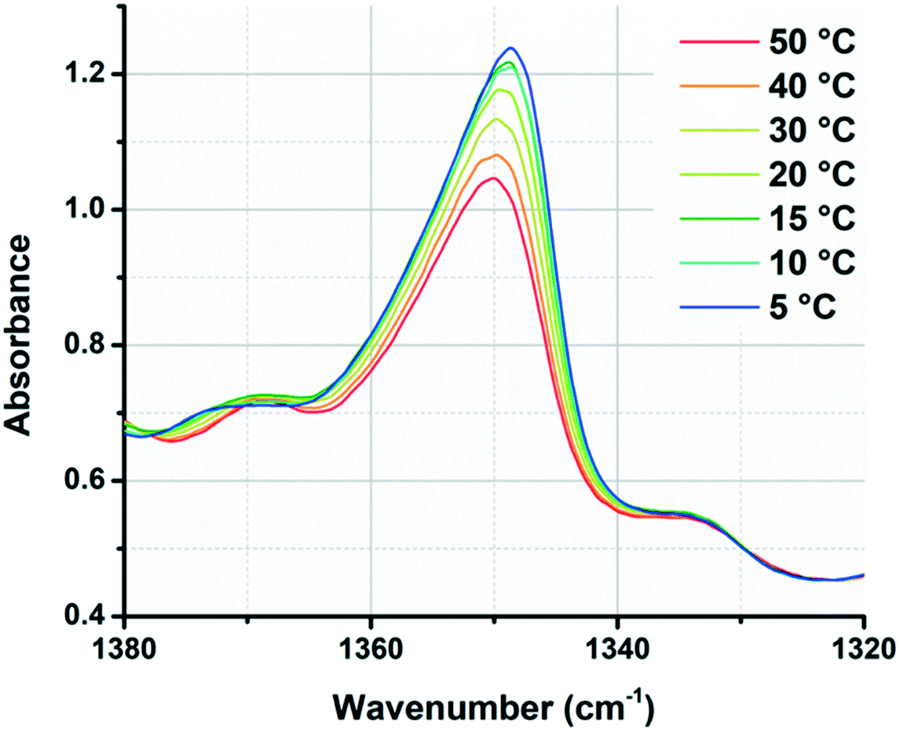 | ||
| Fig. 4 Overlaid spectra of polymer 1-bisU 23 wt% in D2O, showing continuous changes in gem-dimethyl (1348 cm−1) peak intensity with increasing/decreasing temperature. | ||
Small-angle X-ray scattering
Small-angle X-ray scattering (SAXS) was used to characterize the morphology of the assembled polymers (Fig. 5). Previously, SAXS has been used to characterize the morphology of hydrogen-bonding polymers in hydrogels, organogels, and in the solid state.25,37,53,61,80,96–99 Each polymer was examined in both the hydrated state (at 25 wt% polymer concentration) and the dry state. For the base polymer, polymer 1, the volume fraction of the PiPGE blocks was insufficient to induce phase separation as demonstrated by the absence of any correlation peaks. The same was also true for polymer 1-U at this concentration, which is consistent with the data we have discussed thus far. On the other hand, polymer 1-bisU exhibited a well-defined, phase-separated morphology in both the hydrated (25 wt%) and dry states. A sharp correlation peak at q = 0.494 nm−1 was observed, which corresponded to a characteristic spacing (periodicity, d) of 12.7 nm, according to the equation d = 2π/q. Smaller scattering peaks were also present at q = 0.694 and q = 0.856 nm−1. These peaks corresponded to the peak-position ratio of 1,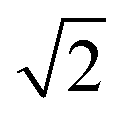 , and
, and 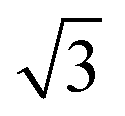 , which suggested a morphology of spherical micelles in a body-centered cubic (bcc) arrangement.100,101 Indeed, for an ABA triblock copolymer with weight fractions A ≈ 0.2 and B ≈ 0.8, cubic morphology was expected.102–104 This result suggested that polymer 1-bisU was the only polymer with a well-defined, structured morphology at 25 wt%.
, which suggested a morphology of spherical micelles in a body-centered cubic (bcc) arrangement.100,101 Indeed, for an ABA triblock copolymer with weight fractions A ≈ 0.2 and B ≈ 0.8, cubic morphology was expected.102–104 This result suggested that polymer 1-bisU was the only polymer with a well-defined, structured morphology at 25 wt%.
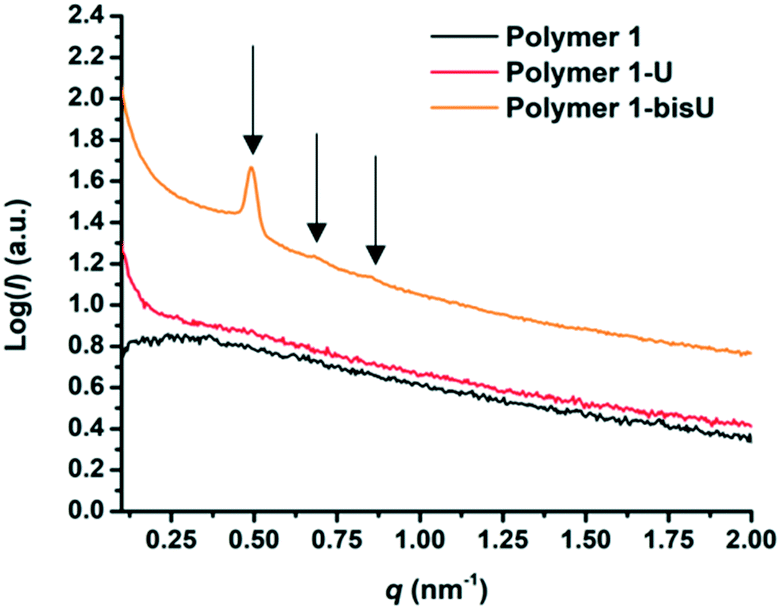 | ||
| Fig. 5 SAXS plots of intensity (I) vs. scattering vector (q) for each of the three polymer compositions in the hydrated state (25 wt%). | ||
Conclusions
In conclusion, end-groups capable of strong hydrogen bonding interactions can drastically influence both the nano- and macroscale properties of stimuli-responsive polymeric hydrogels. An ABA triblock copolymer of PEO (‘B’ block; DP = 182) and PiPGE (‘A’ blocks; DP = 8), which could not form hydrogels at room temperature, could be transformed into a gel when its end-groups were changed to aryl bis-ureas. As expected, bis-urea end-groups self-associated more strongly than monourea end-groups, resulting in more dramatic changes at the molecular and macroscopic levels. The changes in viscoelastic behavior induced by bis-urea motifs were the result of both the hydrophobicity of the two phenyl groups, as well as the strong hydrogen-bonding between ureas. The effects of hydrophobicity and hydrogen-bonding were complementary: additional hydrophobicity led to polymer chains that micellized more readily, which in turn allowed for higher local concentrations of ureas in the micelle core. The ureas localized within these hydrophobic domains formed hydrogen-bonding supramolecular assemblies.The introduction of bis-urea motifs also altered the processing conditions required for the gels. Polymer 1 (hydroxy-terminated) solutions were free-flowing and easy to manipulate at low temperatures, while polymer 1-bisU gels were extremely viscous at low temperatures. DSC and SAXS experiments suggested that these changes in the bulk material were due to nanoscale organization and microphase separation of the urea groups. Overall, the data demonstrate that end-group composition is a profoundly important parameter in block copolymer self-assembly. In future work, changing the composition of the ‘A’ blocks – for example, to statistical copolymers of isopropyl glycidyl and ethyl glycidyl ether – could help to optimize the thermoresponsive properties of urea-containing polymers. We anticipate that alternative polymer compositions and hydrogen-bonding end-groups will result in materials with precisely tunable mechanical properties and stimuli-responsive behaviors. With further optimization, the soft gels could find applications in shape-morphing 3D-printed materials and tissue engineering scaffolds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support of this research by the NSF CAREER Grant 1752972. We also acknowledge Dylan G. Karis, Christopher R. Fellin, and Robert J. Ono for synthetic guidance.Notes and references
- E. E. Antoine, P. P. Vlachos and M. N. Rylander, Tissue Eng., Part B, 2014, 20, 683–696 CrossRef CAS PubMed.
- L. Voorhaar and R. Hoogenboom, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2015, 15, 13–26 CrossRef PubMed.
- M. Park, C. Harrison, P. M. Chaikin, R. A. Register and D. H. Adamson, Science, 1997, 276, 1401–1404 CrossRef CAS.
- B. S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef.
- A. V. Ruzette and L. Leibler, Nat. Mater., 2005, 4, 19–31 CrossRef CAS PubMed.
- G. S. Doerk and K. G. Yager, Mol. Syst. Des. Eng., 2017, 2, 518–538 RSC.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 1238149 CrossRef PubMed.
- J.-F. Lutz, T. Y. Meyer, M. Ouchi and M. Sawamoto, Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties, American Chemical Society, Washington, D.C., 2014 Search PubMed.
- K. Ariga and M. Aono, Jpn. J. Appl. Phys., 2016, 55, 1102A6 CrossRef.
- K. Ariga, T. Mori and J. Li, Langmuir, 2018 DOI:10.1021/acs.langmuir.8b01434 , Article ASAP.
- Y. Wakayama, Jpn. J. Appl. Phys., 2016, 55, 1102AA CrossRef.
- P. Kujawa and F. M. Winnik, Polym. Int., 2014, 63, 377–380 CrossRef CAS.
- K. Ariga, M. V. Lee, T. Mori, X. Y. Yu and J. P. Hill, Adv. Colloid Interface Sci., 2010, 154, 20–29 CrossRef CAS PubMed.
- K. Ariga, D. T. Leong and T. Mori, Adv. Funct. Mater., 2018, 28, 1702905 CrossRef.
- K. Ariga, Mater. Chem. Front., 2017, 1, 208–211 RSC.
- E. Stulz, Acc. Chem. Res., 2017, 50, 823–831 CrossRef CAS PubMed.
- M. L. Cortez, A. Lorenzo, W. A. Marmisollé, C. Von Bilderling, E. Maza, L. Pietrasanta, F. Battaglini, M. Ceolín and O. Azzaroni, Soft Matter, 2018, 14, 1939–1952 RSC.
- A. Lorenzo, W. A. Marmisollé, E. M. Maza, M. Ceolín and O. Azzaroni, Phys. Chem. Chem. Phys., 2018, 20, 7570–7578 RSC.
- E. Maza, C. Von Bilderling, M. L. Cortez, G. Díaz, M. Bianchi, L. I. Pietrasanta, J. M. Giussi and O. Azzaroni, Langmuir, 2018, 34, 3711–3719 CrossRef CAS PubMed.
- B. Kim, Y. D. Park, K. Min, J. H. Lee, S. S. Hwang, S. M. Hong, B. H. Kim, S. O. Kim and C. M. Koo, Adv. Funct. Mater., 2011, 21, 3242–3249 CrossRef CAS.
- H. Sawada and M. Yamanaka, Chem. – Asian J., 2018, 13, 929–933 CrossRef CAS PubMed.
- L. Li, P. Zhang, Z. Zhang, Q. Lin, Y. Wu, A. Cheng, Y. Lin, C. M. Thompson, R. A. Smaldone and C. Ke, Angew. Chem., Int. Ed., 2018, 57, 5105–5109 CrossRef CAS PubMed.
- G. Zhang, Y. Chen, Y. Deng, T. Ngai and C. Wang, ACS Macro Lett., 2017, 6, 641–646 CrossRef CAS.
- H. Yuan, J. Xu, E. P. Van Dam, G. Giubertoni, Y. L. A. Rezus, R. Hammink, H. J. Bakker, Y. Zhan, A. E. Rowan, C. Xing and P. H. J. Kouwer, Macromolecules, 2017, 50, 9058–9065 CrossRef CAS PubMed.
- C. Tsitsilianis, Soft Matter, 2010, 6, 2372–2388 RSC.
- M. D. C. Topp, P. J. Dijkstra, H. Talsma and J. Feijen, Macromolecules, 1997, 30, 8518–8520 CrossRef CAS.
- M. S. Kim, H. Hyun, K. S. Seo, Y. H. Cho, J. W. Lee, C. R. Lee, G. Khang and H. B. Lee, J. Polym. Sci. Part A Polym. Chem., 2006, 44, 5413–5423 CrossRef CAS.
- M. Zhang, A. Vora, W. Han, R. J. Wojtecki, H. Maune, A. B. A. Le, L. E. Thompson, G. M. McClelland, F. Ribet, A. C. Engler and A. Nelson, Macromolecules, 2015, 48, 6482–6488 CrossRef CAS.
- A. P. Constantinou and T. K. Georgiou, Polym. Chem., 2016, 7, 2045–2056 RSC.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- H. Gao, N. Wang, X. Hu, W. Nan, Y. Han and W. Liu, Macromol. Rapid Commun., 2013, 34, 63–68 CrossRef CAS PubMed.
- S. Zhang, A. M. Bellinger, D. L. Glettig, R. Barman, Y. A. L. Lee, J. Zhu, C. Cleveland, V. A. Montgomery, L. Gu, L. D. Nash, D. J. Maitland, R. Langer and G. Traverso, Nat. Mater., 2015, 14, 1065–1071 CrossRef CAS PubMed.
- T. G. O'Lenick, X. Jiang and B. Zhao, Langmuir, 2010, 26, 8787–8796 CrossRef PubMed.
- P. Y. W. Dankers, T. M. Hermans, T. W. Baughman, Y. Kamikawa, R. E. Kieltyka, M. M. C. Bastings, H. M. Janssen, N. A. J. M. Sommerdijk, A. Larsen, M. J. A. Van Luyn, A. W. Bosman, E. R. Popa, G. Fytas and E. W. Meijer, Adv. Mater., 2012, 24, 2703–2709 CrossRef CAS PubMed.
- G. M. Pawar, M. Koenigs, Z. Fahimi, M. Cox, I. K. Voets, H. M. Wyss and R. P. Sijbesma, Biomacromolecules, 2012, 13, 3966–3976 CrossRef CAS PubMed.
- C. B. Highley, C. B. Rodell and J. A. Burdick, Adv. Mater., 2015, 27, 5075–5079 CrossRef CAS PubMed.
- L. Li, B. Yan, J. Yang, L. Chen and H. Zeng, Adv. Mater., 2015, 27, 1294–1299 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, 41–56 CrossRef PubMed.
- S. Khetan and J. A. Burdick, Soft Matter, 2011, 7, 830–838 RSC.
- D. B. Kolesky, R. L. Truby, A. S. Gladman, T. A. Busbee, K. A. Homan and J. A. Lewis, Adv. Mater., 2014, 26, 3124–3130 CrossRef CAS PubMed.
- H. Wang and S. C. Heilshorn, Adv. Mater., 2015, 27, 3717–3736 CrossRef CAS PubMed.
- A. M. Rosales and K. S. Anseth, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- S. R. Sershen, S. L. Westcott, N. J. Halas and J. L. West, J. Biomed. Mater. Res., 2000, 51, 293–298 CrossRef CAS PubMed.
- T. Yamagata, M. Morishita, N. J. Kavimandan, K. Nakamura, Y. Fukuoka, K. Takayama and N. A. Peppas, J. Controlled Release, 2006, 112, 343–349 CrossRef CAS PubMed.
- M. J. Webber, J. B. Matson, V. K. Tamboli and S. I. Stupp, Biomaterials, 2012, 33, 6823–6832 CrossRef CAS PubMed.
- R. A. Barry, R. F. Shepherd, J. N. Hanson, R. G. Nuzzo, P. Wiltzius and J. A. Lewis, Adv. Mater., 2009, 21, 2407–2410 CrossRef CAS.
- R. J. Wojtecki and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 457–472 CrossRef CAS.
- L. Yu, G. Chang, H. Zhang and J. Ding, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1122–1133 CrossRef CAS.
- M. Popescu, G. Liontos, A. Avgeropoulos, E. Voulgari, K. Avgoustakis and C. Tsitsilianis, ACS Appl. Mater. Interfaces, 2016, 8, 17539–17548 CrossRef CAS PubMed.
- W. H. Binder, L. Petraru, T. Roth, P. W. Groh, V. Pálfi, S. Keki and B. Ivan, Adv. Funct. Mater., 2007, 17, 1317–1326 CrossRef CAS.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- J. H. K. K. Hirschberg, L. Brunsveid, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167–170 CrossRef CAS PubMed.
- R. A. Koevoets, R. M. Versteegen, H. Kooijman, A. L. Spek, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 2999–3003 CrossRef CAS PubMed.
- W. H. Binder, M. J. Kunz, C. Kluger, G. Hayn and R. Saf, Macromolecules, 2004, 37, 1749–1759 CrossRef CAS.
- A. Vora, B. Zhao, D. To, J. Y. Cheng and A. Nelson, Macromolecules, 2010, 43, 1199–1202 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761–6769 CrossRef CAS.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- N. Chebotareva, P. H. H. Bomans, P. M. Frederik, N. a J. M. Sommerdijk and R. P. Sijbesma, Chem. Commun., 2005, 4967–4969 RSC.
- M. Tharcis, T. Breiner, J. Belleney, F. Boué and L. Bouteiller, Polym. Chem., 2012, 3, 3093 RSC.
- D. G. Karis, R. J. Ono, M. Zhang, A. Vora, D. Storti, M. A. Ganter and A. Nelson, Polym. Chem., 2017, 8, 4199–4206 RSC.
- T. Liu, Z. Zhou, C. Wu, B. Chu, D. K. Schneider and V. M. Nace, J. Phys. Chem. B, 1997, 101, 8808–8815 CrossRef CAS.
- S. Aoki, A. Koide, S. Imabayashi and M. Watanabe, Chem. Lett., 2002, 31, 1128–1129 CrossRef.
- T. Isono, K. Miyachi, Y. Satoh, S. Sato, T. Kakuchi and T. Satoh, Polym. Chem., 2017, 8, 5698–5707 RSC.
- S. Heinen, S. Rackow, A. Schäfer and M. Weinhart, Macromolecules, 2017, 50, 44–53 CrossRef CAS.
- Y. L. Chang, M. A. West, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 1993, 115, 5991–6000 CrossRef CAS.
- J. Van Esch, F. Schoonbeek, M. De Loos, H. Kooijman, A. L. Spek, R. M. Kellogg and B. L. Feringa, Chem. – Eur. J., 1999, 5, 937–950 CrossRef CAS.
- F. Lortie, S. Boileau, L. Bouteiller, C. Chassenieux, B. Demé, G. Ducouret, M. Jalabert, F. Lauprêtre and P. Terech, Langmuir, 2002, 18, 7218–7222 CrossRef CAS.
- F. Lortie, S. Boileau and L. Bouteiller, Chem. – Eur. J., 2003, 9, 3008–3014 CrossRef CAS PubMed.
- V. Simic, L. Bouteiller and M. Jalabert, J. Am. Chem. Soc., 2003, 125, 13148–13154 CrossRef CAS PubMed.
- L. Bouteiller, O. Colombani, F. Lortie and P. Terech, J. Am. Chem. Soc., 2005, 127, 8893–8898 CrossRef CAS PubMed.
- M. Bellot and L. Bouteiller, Langmuir, 2008, 24, 14176–14182 CrossRef CAS PubMed.
- P. Brocorens, M. Linares, C. Guyard-Duhayon, R. Guillot, B. Andrioletti, D. Suhr, B. Isare, R. Lazzaroni and L. Bouteiller, J. Phys. Chem. B, 2013, 117, 5379–5386 CrossRef CAS PubMed.
- R. A. Koevoets, S. Karthikeyan, P. C. M. M. Magusin, E. W. Meijer and R. P. Sijbesma, Macromolecules, 2009, 42, 2609–2617 CrossRef CAS.
- L. Ning, W. De-Ning and Y. Sheng-Kang, Polymer, 1996, 37, 3577–3583 CrossRef CAS.
- O. Colombani, C. Barioz, L. Bouteiller, C. Chanéac, L. Fompérie, F. Lortie and H. Montés, Macromolecules, 2005, 38, 1752–1759 CrossRef CAS.
- R. M. Versteegen, R. Kleppinger, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 772–783 CrossRef CAS.
- R. M. Versteegen, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2005, 38, 3176–3184 CrossRef CAS.
- S. Boileau, L. Bouteiller, F. Lauprêtre and F. Lortie, New J. Chem., 2000, 24, 845–848 RSC.
- J. P. Sheth, D. B. Klinedinst, T. W. Pechar, G. L. Wilkes, E. Yilgor and I. Yilgor, Macromolecules, 2005, 38, 10074–10079 CrossRef CAS.
- F. Piana, D. H. Case, S. M. Ramalhete, G. Pileio, M. Facciotti, G. M. Day, Y. Z. Khimyak, J. Angulo, R. C. D. Brown and P. A. Gale, Soft Matter, 2016, 12, 5489–5489 RSC.
- J. Ilavsky, J. Appl. Crystallogr., 2012, 45, 324–328 CrossRef CAS.
- C. Chassenieux, O. Colombani and T. Nicolai, Macromolecules, 2011, 44, 4487–4495 CrossRef.
- C. Tsitsilianis, G. Serras, C. Ko, F. Jung, C. M. Papadakis, M. Rikkou-Kalourkoti, C. S. Patrickios, R. Schweins and C. Chassenieux, Macromolecules, 2018, 51, 2169–2179 CrossRef CAS.
- S. Sami, E. Yildirim, M. Yurtsever, E. Yurtsever, E. Yilgor, I. Yilgor and G. L. Wilkes, Polymer, 2014, 55, 4563–4576 CrossRef CAS.
- S. Baddi, S. S. Madugula, D. S. Sarma, Y. Soujanya and A. Palanisamy, Langmuir, 2016, 32, 889–899 CrossRef CAS PubMed.
- A. Labbé, S. Carlotti, A. Deffieux and A. Hirao, Macromol. Symp., 2007, 249–250, 392–397 CrossRef.
- M. Schömer and H. Frey, Macromolecules, 2012, 45, 3039–3046 CrossRef.
- K. P. Barteau, M. Wolffs, N. A. Lynd, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2013, 46, 8988–8994 CrossRef CAS.
- A. Lee, P. Lundberg, D. Klinger, B. F. Lee, C. J. Hawker and N. A. Lynd, Polym. Chem., 2013, 4, 5735–5742 RSC.
- I. Yilgor, E. Yilgor, S. Das and G. L. Wilkes, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 471–483 CrossRef CAS.
- E. Yilgor, M. Isik and I. Yilgor, Macromolecules, 2010, 43, 8588–8593 CrossRef CAS.
- L. Montero De Espinosa, S. Balog and C. Weder, ACS Macro Lett., 2014, 3, 540–543 CrossRef CAS.
- J. S. Nambam and J. Philip, Langmuir, 2012, 28, 12044–12053 CrossRef CAS PubMed.
- C. Y. Liaw, K. J. Henderson, W. R. Burghardt, J. Wang and K. R. Shull, Macromolecules, 2015, 48, 173–183 CrossRef CAS.
- A. H. Gröschel and A. Walther, Angew. Chem., Int. Ed., 2017, 56, 10992–10994 CrossRef PubMed.
- B. Chu and B. S. Hsiao, Chem. Rev., 2001, 101, 1727–1761 CrossRef CAS PubMed.
- M. Li, Y. Liu, H. Nie, R. Bansil and M. Steinhart, Macromolecules, 2007, 40, 9491–9502 CrossRef CAS.
- M. W. Matsen and M. Schick, Phys. Rev. Lett., 1994, 72, 2660–2663 CrossRef CAS PubMed.
- C. A. Tyler and D. C. Morse, Phys. Rev. Lett., 2005, 94, 1–4 CrossRef PubMed.
- M. W. Matsen, Macromolecules, 2012, 45, 2161–2165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8me00063h |
| This journal is © The Royal Society of Chemistry 2019 |