
A successful collaboration between academia, biotech and pharma led to discovery of erdafitinib, a selective FGFR inhibitor recently approved by the FDA
Christopher W.
Murray
a,
David R.
Newell
b and
Patrick
Angibaud
c
aAstex Pharmaceuticals, 436 Cambridge Science Park, Milton Road, Cambridge, CB4 0QA, UK. E-mail: chris.murray@astx.com
bNorthern Institute of Cancer Research, Newcastle University, Newcastle, NE2 4AD, UK. E-mail: herbie.newell@newcastle.ac.uk
cJanssen Cilag, CS10615, Chausée du Vexin, 27106 Val de Reuil, France. E-mail: pangibau@its.jnj.com
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year.1 Patients with metastatic bladder cancer have a poor prognosis and 15–20% of these patients exhibit FGFR alterations.
000 deaths per year.1 Patients with metastatic bladder cancer have a poor prognosis and 15–20% of these patients exhibit FGFR alterations.
Through a collaboration between the Northern Institute of Cancer Research (NICR) at Newcastle University and Astex, early FGFR inhibitors were identified using a structure-based approach. These early compounds were further optimized and developed clinically in a collaboration between Astex and Janssen which led to erdafitinib, a selective pan-FGFR inhibitor. The compound shows selectivity for FGFR1–4 over highly homologous receptor tyrosine kinases such as VEGFR2. In April 2019, BALVERSA™ (erdafitinib) was the first FGFR kinase inhibitor to receive FDA accelerated approval against locally advanced or metastatic urothelial carcinoma that has susceptible FGFR3 or FGFR2 genetic alterations and progressed during or following at least one line of prior platinum-containing chemotherapy.1 This accelerated approval followed breakthrough therapy designation by the FDA in March 2018. The phase 2 study in this patient population showed an overall response rate of 40% for patients whose tumours harboured actionable FGFR alterations. Further details of these results were recently published in The New England Journal of Medicine.2
In 2006, at the time of the collaboration initiation between NICR and Astex, most FGFR inhibitors in clinical development also inhibited VEGFR2, and VEGFR2 inhibition was believed to be linked to hypertension, limiting the ability to reach the exposure required for therapeutic inhibition of the FGFR pathway during dose escalation. Based on this hypothesis, a fragment-based drug discovery screen was initiated with the goal of identifying novel and selective FGFR inhibitors. In early 2008, a fragment-based screening approach followed by combined exploratory chemistry had led to the identification of highly potent and selective FGFR inhibitors from an imidazopyridine series. The compounds from this imidazopyridine series showed promising oral, in vivo efficacy against a range of FGFR-dependent human xenograft tumour models and unlike non-selective inhibitors, showed absence of activity against FGFR-independent xenografts models, demonstrating the FGFR selectivity of these compounds.3 In June 2008, Astex initiated a collaboration with Janssen Pharmaceutica and while extensive lead optimisation efforts were applied to this series, it was also decided to investigate other chemical series.
Fortunately, quinoxaline fragments (compounds 1 and 2) were previously identified and through virtual screening efforts based on their crystal structures with FGFR1, a related analogue, compound 3, was identified (see Fig. 1). Compound 3 showed FGFR activity (330 nM IC50 against FGFR3) and selectivity against VEGFR2 (∼15 fold). Replacement of the benzyl group in compound 3 by an aniline (compound 4) led to a 30-fold improvement in potency. Superposition of the crystal structure of compound 4 and that of the earlier imidazopyridine series (Fig. 2) inspired further substitutions of the aniline which led to increased affinity and favourable physicochemical and pharmacokinetic properties. Collaborative lead optimisation culminated in the synthesis of compound 5 (erdafitinib) which was extensively characterized by both teams and advanced by Janssen into clinical development.
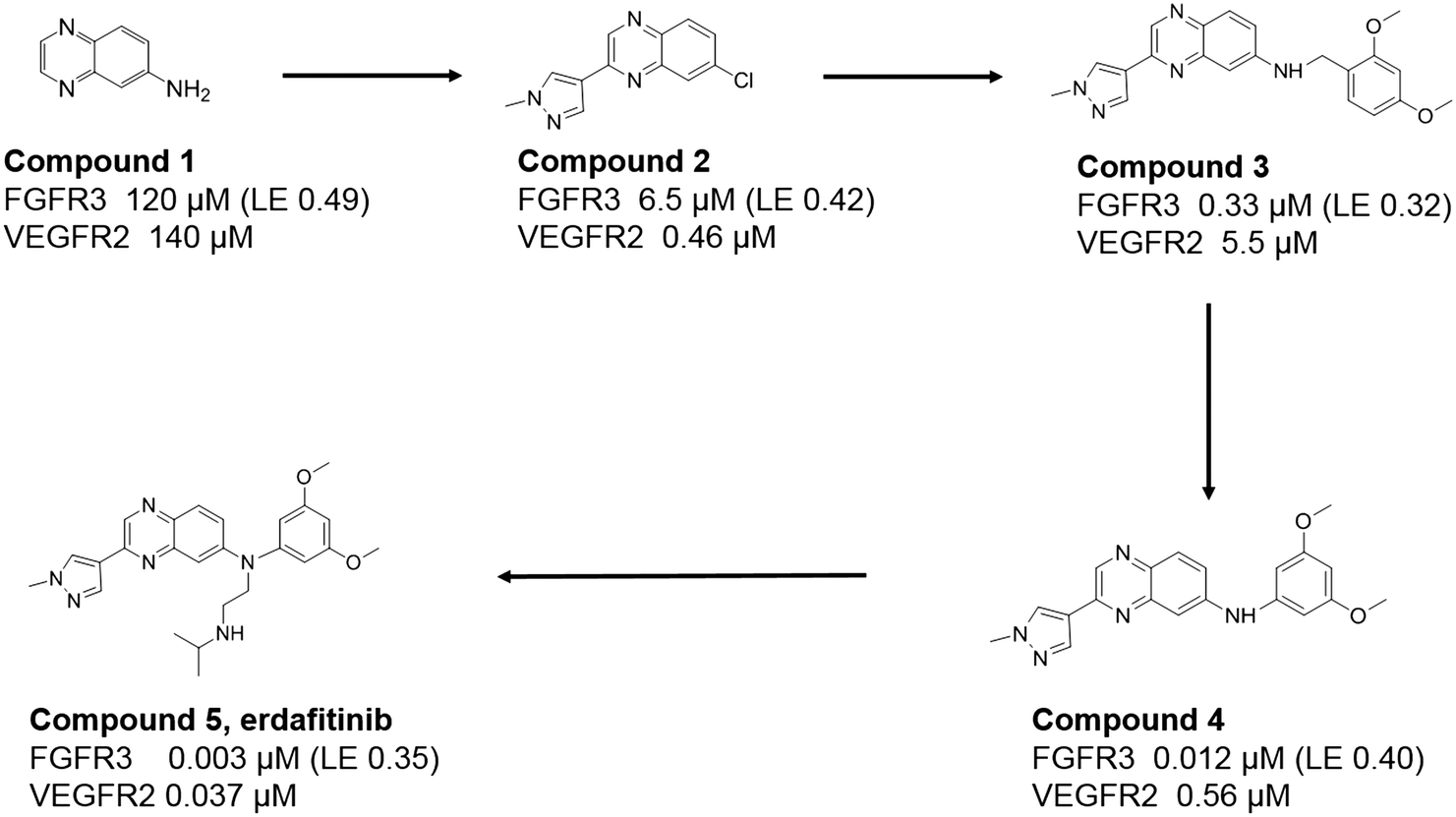 | ||
| Fig. 1 Summary of the discovery of erdafitinib starting from fragments. Potencies versus FGFR3 and VEGFR2 are specified together with ligand efficiency (LE) versus FGFR3 given in kcal mol−1 per heavy atom. | ||
 | ||
| Fig. 2 Left side shows overlay of the crystal structure of compound 4 in FGFR1 with the crystal structure of an imidazopyridine. Right side shows crystal structure of erdafitinib in FGFR1. The optimisation strategy is annotated. Further details on the design strategy were presented by Angibaud et al., at the 2014 American Association for Cancer Research (AACR) meeting. | ||
The pharmacological profile of erdafitinib has recently been published.4 Erdafitinib demonstrated high potency against cancer cell lines with FGFR alterations (ranging from 0.1–130 nM), while demonstrating as intended high selectivity against VEGFR2 inhibition (>50 fold selectivity over VEGFR2 in BaF3 cells). High cellular potency and favourable pharmacokinetic properties in multiple animal species led to strong anti-tumoral efficacy in multiple FGFR-driven human xenograft models.4 These preclinical properties combined with a favourable toxicology profile led to evaluation of erdafitinib in human clinical trials2,5 and its recent FDA approval.1
Collaboration and open innovation between multiple teams, biotech (Astex), academia (UK University) and pharma (Janssen), led to the discovery of erdafitinib. Each team brought their specific expertise and most importantly, wisely and collaboratively capitalized on each other’s strengths; from fragment/structure-based technology to chemistry optimization and finally clinical development, all with the focus to benefit patients.
A large number of researchers at the NICR, Astex and Janssen contributed to the FGFR program that ultimately led to discovery of erdafitinib and the authors, representatives of these discovery teams, express their appreciation to all contributors, and gratefully acknowledge the scientists, clinicians and patients who enabled the successful development of BALVERSA™.
References
- Johnson & Johnson press releases (accessed August 16, 2019), (https://www.jnj.com/media-center/press-releases/janssen-announces-us-fda-breakthrough-therapy-designation-for-erdafitinib-in-the-treatment-of-metastatic-urothelial-cancer), (https://www.jnj.com/balversa-erdafitinib-receives-u-s-fda-approval-for-the-treatment-of-patients-with-locally-advanced-or-metastatic-urothelial-carcinoma-with-certain-fgfr-genetic-alterations).
- Y. Loriot, A. Necchi, S. H. Park, J. Garcia-Donas, R. Huddart, E. Burgess, M. Fleming, A. Rezazadeh, B. Mellado, S. Varlamov, M. Joshi, I. Duran, S. T. Tagawa, Y. Zakharia, B. Zhong, K. Stuyckens, A. Santiago-Walker, P. De Porre, A. O'Hagan, A. Avadhani and A. O. Siefker-Radtke, Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma, N. Engl. J. Med., 2019, 381, 338–348 CrossRef.
- M. Squires, G. Ward, G. Saxty, V. Berdini, A. Cleasby, P. King, P. Angibaud, T. Perera, L. Fazal, D. Ross, C. Griffiths-Jones, A. Madin, R. K. Benning, E. Vickerstaffe, A. O'Brien, M. Frederickson, M. Reader, C. Hamlett, M. A. Batey, S. Rich, M. Carr, D. Miller, R. Feltell, A. Thiru, S. Bethell, L. A. Devine, B. L. Graham, A. Pike, J. Cosme, E. J. Lewis, E. Freyne, J. Lyons, J. Irving, C. Murray, D. R. Newell and N. T. Thompson, Potent, selective inhibitors of fibroblast growth factor receptor define fibroblast growth factor dependence in preclinical cancer models, Mol. Cancer Ther., 2011, 10, 1542–1552 CrossRef CAS.
- T. P. S. Perera, E. Jovcheva, L. Mevellec, J. Vialard, D. De Lange, T. Verhulst, C. Paulussen, K. Van De Ven, P. King, E. Freyne, D. C. Rees, M. Squires, G. Saxty, M. Page, C. W. Murray, R. Gilissen, G. Ward, N. T. Thompson, D. R. Newell, N. Cheng, L. Xie, J. Yang, S. J. Platero, J. D. Karkera, C. Moy, P. Angibaud, S. Laquerre and M. V. Lorenzi, Discovery and pharmacological characterization of JNJ-42765493 (erdafitinib), a functionally selective small-molecule FGFR family inhibitor, Mol. Cancer Ther., 2017, 16, 1010–1020 CrossRef CAS.
- J. Tabernero, R. Bahleda, R. Dienstmann, J. R. Infante, A. Mita, A. Italiano, E. Calvo, V. Moreno, B. Adamo, A. Gazzah, B. Zhong, S. J. Platero, J. W. Smit, K. Stuyckens, M. Chatterjee-Kishore, J. Rodon, V. Peddareddigari, F. R. Luo and J.-C. Soria, Phase 1 Dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors, J. Clin. Oncol., 2015, 33, 3401–3408 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |