
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMethyl sulfonamide substituents improve the pharmacokinetic properties of bicyclic 2-pyridone based Chlamydia trachomatis inhibitors
Martina
Kulén†
ab,
Carlos
Núñez-Otero†
 bde,
Andrew G.
Cairns
ab,
Jim
Silver
bcd,
Anders E. G.
Lindgren
ab,
Emma
Wede
bcd,
Pardeep
Singh
ab,
Katarina
Vielfort
bcd,
Wael
Bahnan‡
bcd,
James A. D.
Good§
ab,
Richard
Svensson
fg,
Sven
Bergström
*bcd,
Åsa
Gylfe
*bde and
Fredrik
Almqvist
*ab
bde,
Andrew G.
Cairns
ab,
Jim
Silver
bcd,
Anders E. G.
Lindgren
ab,
Emma
Wede
bcd,
Pardeep
Singh
ab,
Katarina
Vielfort
bcd,
Wael
Bahnan‡
bcd,
James A. D.
Good§
ab,
Richard
Svensson
fg,
Sven
Bergström
*bcd,
Åsa
Gylfe
*bde and
Fredrik
Almqvist
*ab
aDepartment of Chemistry, Umeå University, 901 87 Umeå, Sweden. E-mail: fredrik.almqvist@umu.se
bUmeå Centre for Microbial Research, Umeå University, 901 87 Umeå, Sweden. E-mail: sven.bergstrom@umu.se; asa.gylfe@umu.se
cDepartment of Molecular Biology, Umeå University, 901 87 Umeå, Sweden
dLaboratory for Molecular Infection Medicine Sweden (MIMS), Umeå University, 901 87 Umeå, Sweden
eDepartment of Clinical microbiology, Umeå University, 901 85 Umeå, Sweden
fThe Uppsala University Drug Optimization and Pharmaceutical Profiling Platform, Department of Pharmacy, Uppsala University, SE-751 23 Uppsala, Sweden
gSciLifeLab Drug Discovery and Development Platform, ADME of Therapeutics Facility, Uppsala University, SE-751 23 Uppsala, Sweden
First published on 17th October 2019
Abstract
Chlamydia trachomatis infections are a global health problem and new approaches to treat C. trachomatis with drugs of high specificity would be valuable. A library of substituted ring fused 2-pyridones has been synthesized and evaluated for their ability to attenuate C. trachomatis infectivity. In vivo pharmacokinetic studies were performed, with the best candidates demonstrating that a C8-methylsulfonamide substituent improved pharmacokinetic properties important for oral administration. C8-Methyl sulfonamide analogue 30 inhibited C. trachomatis infectivity in low micromolar concentrations. Further pharmacokinetic evaluation at an oral dose of 10 mg kg−1 showed an apparent bioavailability of 41%, compared to C8-cyclopropyl and -methoxy analogues which had negligible oral uptake. In vitro ADME (absorption, distribution, metabolism and excretion) testing of solubility and Caco-2 cell permeability revealed that both solubility and permeability is greatly improved with the C8-methyl sulfonamide 30, effectively moving it from BCS (Biopharmaceutical Classification System) class IV to II.
Introduction
Chlamydia trachomatis is the most common cause of sexually transmitted disease and infectious blinding, infecting over one hundred million individuals globally every year.1 Chronic genital infections can cause infertility in women and inflammatory arthritis in both genders.2Chlamydia is a Gram negative obligate intracellular bacterium with a unique developmental cycle. The extracellular form, elementary body (EB), attaches and enters the host cell.3 Once intracellular, Chlamydia differentiates to the non-infectious replicative intracellular form, reticulate body (RB).4 RBs replicate within a vacuole-like structure termed inclusion for 36 to 72 hours. After replication, RBs differentiate back to EBs, which abandon the host cell by lysis or extrusion.5Infections with C. trachomatis are routinely treated with doxycycline or azithromycin, with high efficacy.6 However, the treatment of uncomplicated infections with broad-spectrum antibiotics disturbs the commensal flora in both the short and long term.7 Moreover, exposure to antibiotics contributes to the overall selective pressure on bacterial resistance.8 Anti-virulence compounds with selective effect on Chlamydia would not only reduce the use of important broad-spectrum antibiotics but also reduce side effects on the normal flora and the resulting selection for antibiotic resistant strains.
In search for potent anti-virulence compounds targeting C. trachomatis we previously identified compound 1 (Fig. 1) as a possible candidate.9 When C. trachomatis was treated with this compound at 2.5 μM in vitro, progeny was produced but their ability to infect new cells was attenuated. It was observed that inhibition of the pathogenic ability of C. trachomatis was partly due to an effect on glucose uptake.9 Introduction of an amine substituent in the C6-position and saturation of the C2–C3 double bond resulted in compound 2, with higher activity and better physiochemical properties than its precursor.10 Further development by exchanging the hydrolysable C3-phenyl amide to non-hydrolysable amide isosteres resulted in the potent 1,2,3-triazole analogue 3 (Fig. 1).11 The ring-fused 2-pyridone analogues 2 and 3 inhibit Chlamydial infectivity (EC50 < 60 nM) in a cell based assay without effecting host microbiota and showed no mutagenic potential when assessed with the Ames test.11,12 Ideally a drug for the genital C. trachomatis infection would be administered orally. In vitro ADME (absorption, distribution, metabolism, excretion) testing regarding solubility and permeability was performed on 2 (not shown), yielding poor solubility and moderate permeability. Both compounds were also tested in vivo, via intravenous (IV) and per oral (PO) administration. The data showed very low blood concentrations especially of 2, while 3 had high enough blood concentrations to enable calculation of intravenous pharmacokinetic parameters indicating a high steady state volume of distribution (Vss) (Table 4, Fig. 2). Intending to improve pharmacokinetic properties, we decided to explore the possibility of modifying the C8-position of the central fragment. In our previous structure–activity relationship investigations, our focus was on ring-fused 2-pyridone analogues with structural changes at the C7-, C6-, and C3-positions, while the C8-cyclopropyl substituent remained unmodified. In this study, we have instead synthesized and evaluated a small library of C8-oxygen and nitrogen analogues with the overall aim to maintain anti-Chlamydial activity and improve oral availability. An earlier study10 showed that there was no discernable difference between the two enantiomers of compound 24 and therefore all compounds presented here were prepared and tested as racemic mixtures.
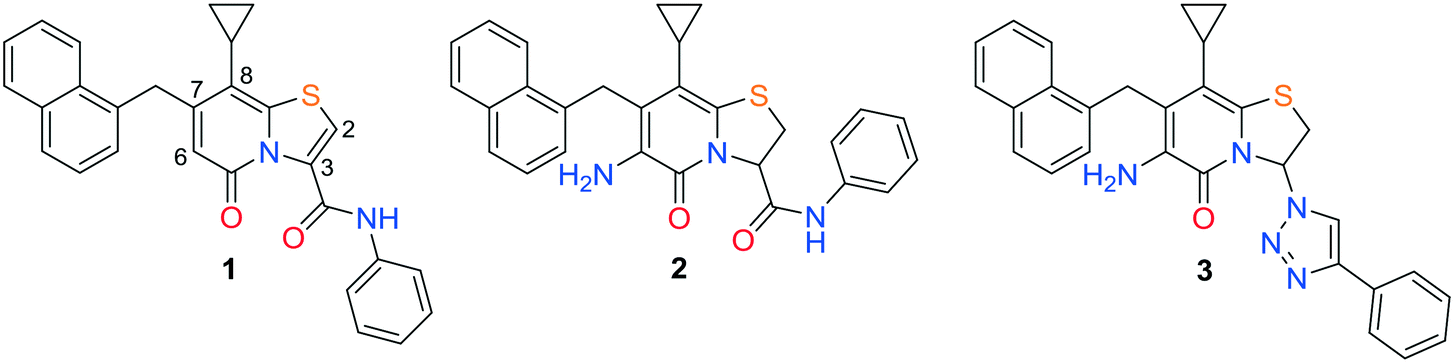 | ||
| Fig. 1 Chemical structures of compounds 1, 2, and 3. | ||
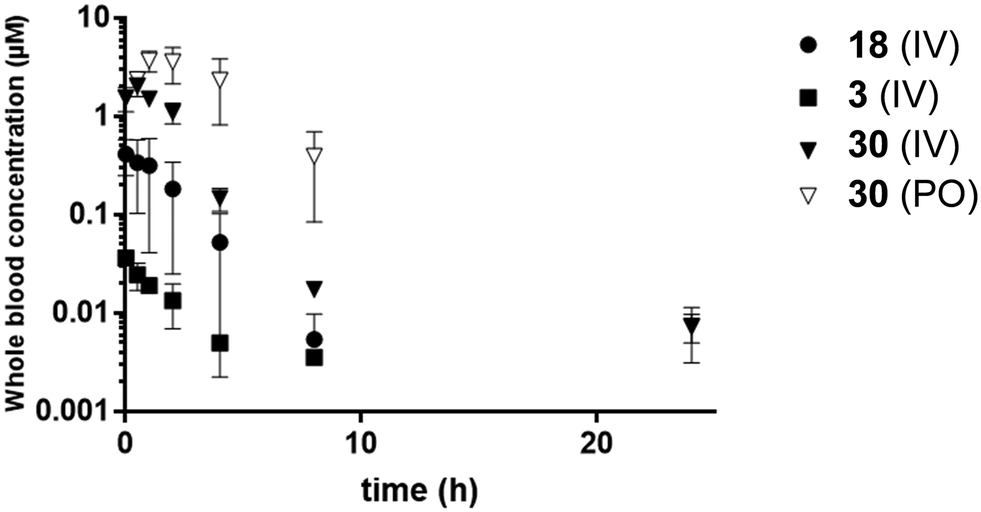 | ||
| Fig. 2 Blood concentration versus time of 3 (1.2 mg kg−1), 18 (0.9 mg kg−1), and 30 (1.0 mg kg−1 IV, 10 mg kg−1 PO). Error bars indicate SEM. | ||
Results & discussion
By introducing heteroatom containing substituents in the C8-position we aimed to reduce the lipophilicity and improve the drug-like properties of the compounds. First a set of C8-hydroxyl or -methoxy substituted analogues were designed and for these the previously published carboxylic acid 4 (ref. 13) served as a key intermediate (Scheme 1). In our previous study10 the phenyl amide and p-methyl phenyl amide at the C3-position gave similar biological results. We therefore prepared C8-methoxy, C3-amide analogues 5 and 6 by amide coupling under basic conditions with the corresponding amine and propylphosphonic anhydride as coupling reagent.14 C3-Phenyl amide 7 was generated by demethylation of analogue 5 with boron tribromide (Scheme 1). Nitration and subsequent reduction with zinc in acetic acid15 starting from amides 5 and 6 gave the desired C6-amines 8 and 9 (Scheme 1).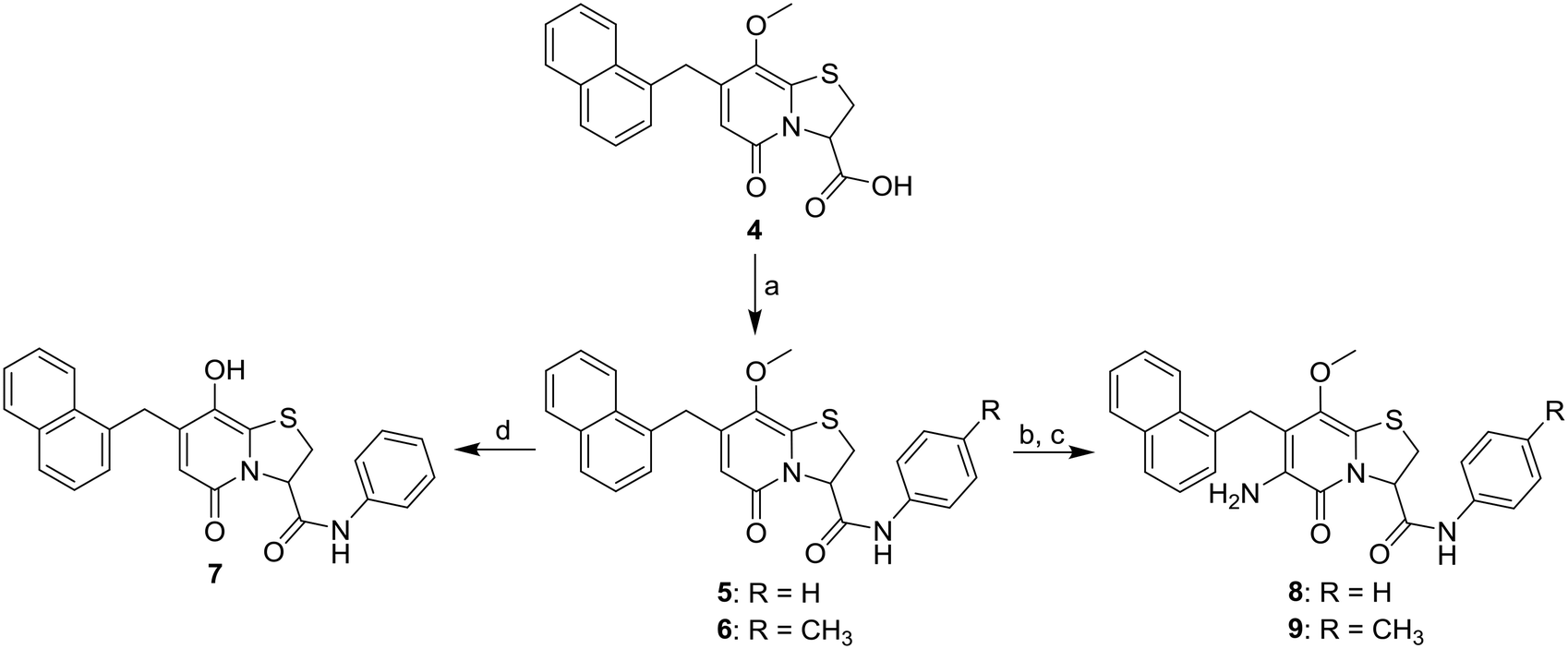 | ||
Scheme 1 Synthesis of C8-methoxy amide analogues 5–7. Reagents and conditions: a) aniline or 4-methylaniline, propylphosphonic anhydride (50% in EtOAc), pyridine, MeCN/EtOAc (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), −10 °C to rt, 24–43 h, 5: 58%, 6: 38%; b) NaNO2, TFA, DCM, O2 atmosphere, rt, 1–7 h; c) activated Zn dust, AcOH, rt, 31–48 h, 8: 30% over two steps; 9: 35% over two steps; d) BBr3, DCM, −40 °C to rt, 6 h, 10%. :1), −10 °C to rt, 24–43 h, 5: 58%, 6: 38%; b) NaNO2, TFA, DCM, O2 atmosphere, rt, 1–7 h; c) activated Zn dust, AcOH, rt, 31–48 h, 8: 30% over two steps; 9: 35% over two steps; d) BBr3, DCM, −40 °C to rt, 6 h, 10%. | ||
The previously observed potency of 3 (Fig. 1) also motivated us to synthesize the 1,2,3-triazole analogue. Thus, the carboxylic acid intermediate 4 underwent Curtius rearrangement in tert-butyl alcohol to acquire the BOC-protected amine 10, which upon acidic deprotection resulted in the primary amine 11 (Scheme 2). The final 1,2,3-triazole 12 was obtained by diazo transfer followed by Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) (Scheme 2).
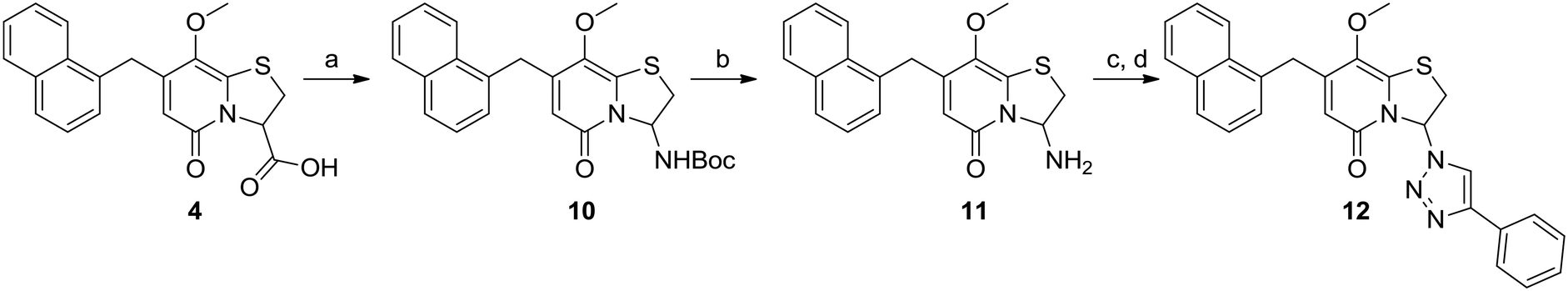 | ||
| Scheme 2 Synthesis of C8-methoxy 1,2,3-triazole 12. Reagents and conditions: a) DPPA, Et3N, t-BuOH, 2 h, 85 °C, 60%; b) TFA, DCM, 2 h, rt, 32%; c) imidazole-1-sulfonyl azide H2SO4, CuSO4, K2CO3, MeOH, 40 h, 50 °C; d) phenyl acetylene, CuSO4, sodium ascorbate, DMF, H2O, rt, 4 h, 4% from 11. | ||
Our previous study also revealed that the 1-naphthyl substituent could be exchanged for a 2,3-dimethyl phenyl substituent without loss of potency.10 This C7-substituent was incorporated into the ring-fused 2-pyridone scaffold using 2,3-dimethyl phenyl substituted Meldrum's acid 14 to give ring-fused 2-pyridone 15 (Scheme 3). The three C3-amides (16, 17, and 18) were obtained through methyl ester hydrolysis and then subsequent amide coupling of the generated acid using the corresponding aniline derivative. We have previously prepared and tested 1,2,4-oxadiazole substituted compounds which are easily accessible directly from the corresponding carboxylic acids.11 To further evaluate these moieties together with other C7- and C8-substituents we prepared two more of these amide isosteres via hydrolysis of methyl ester 15 and then amide coupling with benzamide oxime and TBTU followed by cyclization16 to generate the 1,2,4-oxadiazole 19. These analogues were C6-aminated via the established nitration-reduction route resulting in four C6-amine analogues 20, 21, 22, and 23 (Scheme 3).
 | ||
| Scheme 3 Synthesis of C8-methoxy analogues 16–23 with a C7-2,3-dimethyl phenyl substituent. Reagents and conditions: a) TFA, DCE, MWI, 120 °C, 3 min, 80%; b) 1 M LiOH(aq), THF, rt, 15 h; c) for 16 and 17: aniline, 4-methylaniline, propylphosphonic anhydride (50% in EtOAc), pyridine, MeCN/EtOAc (1:1), −10 °C to rt, 24 h, 16: 82%, 17: 39%; for 18: 3-fluoro-5-methylaniline, HATU, DIPEA, DCM, 2 h, 95%; d) NaNO2, TFA, DCM, O2 atmosphere, rt, 2.5–6 h; e) activated Zn dust, AcOH, rt, 20–23 h, 21: 30%, 22: 17%, 23: 38% over two steps; f) 1 M LiOH(aq), THF, rt, 15 h; g) benzamidoxime, TBTU, DIPEA, DMF, MWI, 170 °C,12 min, 46%; h) NaNO2, TFA, DCM, O2 atmosphere, rt, 1 h; i) activated Zn dust, AcOH, rt, 19 h, 60%. | ||
The fourteen C8-oxygen analogues were evaluated for their ability to inhibit C. trachomatis infectivity by the previously described reinfect assay (Table 1). Reinfect values show inhibition of Chlamydia infectivity by the tested compound in relation to DMSO treated control infections.10,11 The reinfect assay is cell based and also provides preliminary information about the abilities of the compounds to penetrate biological membranes, attach to their target and exert their function on the intracellular bacteria. In brief, HeLa cells infected with C. trachomatis serovar L2 were treated with a compound (2.5 μM or 1 μM) and incubated for 48 h before Chlamydia was harvested by cell lysis. The cell lysate was used to reinfect fresh HeLa cells and the number of Chlamydia inclusion forming units (IFU) formed during the reinfection were enumerated after 48 hours incubation. The reinfect value is the reduction in percent of IFUs in a compound treated infection compared to DMSO treated controls. The cytotoxicity of the compounds at 10 μM was tested in a resazurin cell toxicity assay (HeLa cells) to assess that the anti-chlamydial effect of the compounds was not due to host cell toxicity.10,11
P is the logarithm of the partition coefficient between n-octanol and water
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
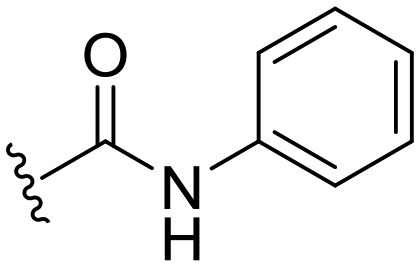
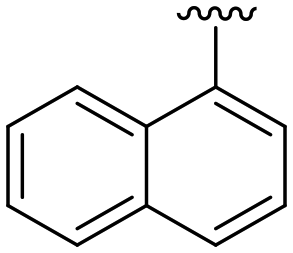
| ID | R3 | R6 | R7 | R8 | Reinfect 2,5 μM (%) | Reinfect 1 μM (%) | HeLa viability 10 μM (%) | Calculated logP (ref. 17) |
| n.d. not determined. a Data provided from previous studies.10,11 | ||||||||
| 24 |
|
H |
|
|
87 ± 6 | 67 ± 8 | 100 ± 4a | 5.4 |
| 2 |
|
NH2 |
|
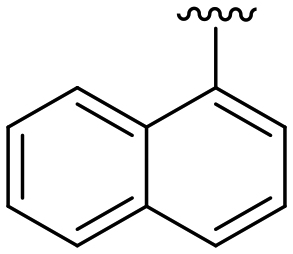
|
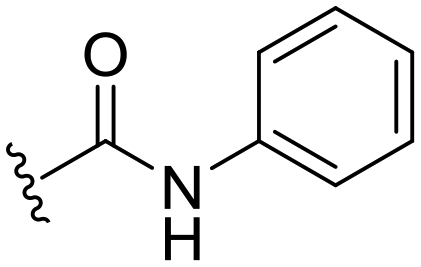
98 ± 2 | 94 ± 5 | 101 ± 3a | 4.9 |
| 3 |
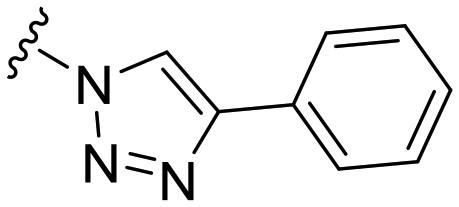
|
NH2 |
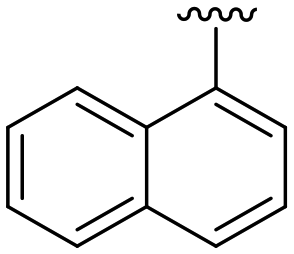
|
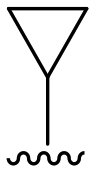
|
91 ± 4 | 90 ± 4 | 105 ± 4a | 5.1 |
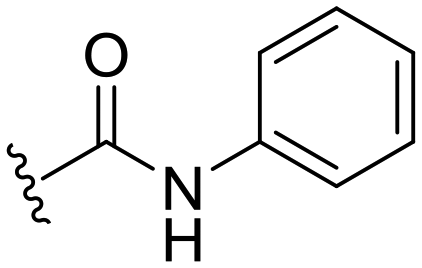
| 7 |
|
H |

|
OH | 21 ± 14 | n.d. | 93 ± 6 | 4.8 |
| 5 |
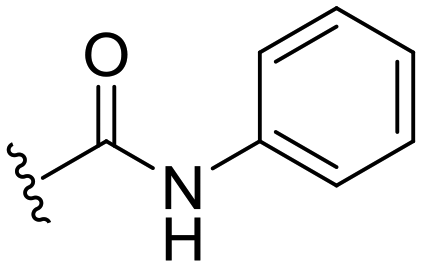
|
H |
|
OCH3 | 24 ± 8 | n.d. | 91 ± 6 | 5.0 |
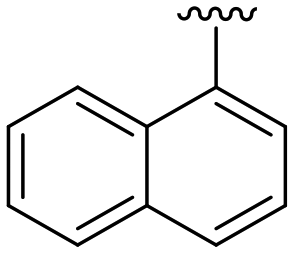
| 8 | NH2 | 25 ± 14 | n.d. | 94 ± 2 | 4.5 | |||
| 6 |
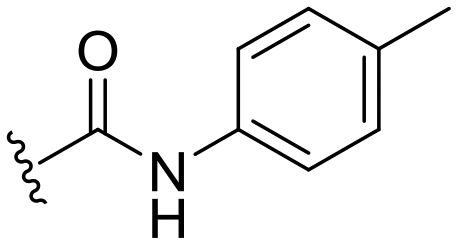
|
H |
|
OCH3 | 94 ± 0 | 62 ± 1 | 89 ± 4 | 5.3 |
| 9 | NH2 | 51 ± 16 | n.d. | 91 ± 2 | 4.8 | |||
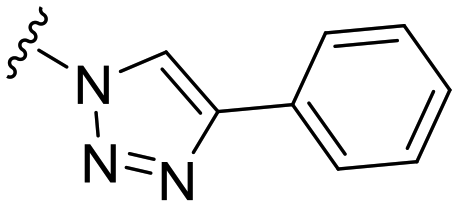
| 12 |
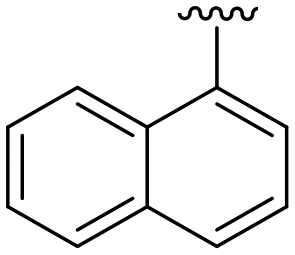
|
H |
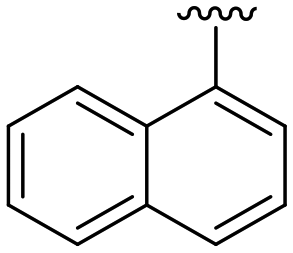
|
OCH3 | 81 ± 9 | n.d. | 87 ± 3 | 5.2 |
| 16 |
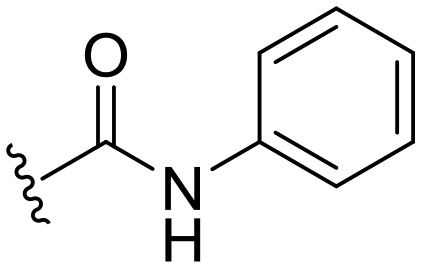
|
H |
|
OCH3 | 51 ± 5 | 22 ± 2 | 89 ± 2 | 4.4 |
| 21 | NH2 | 25 ± 14 | n.d. | 94 ± 1 | 3.9 | |||
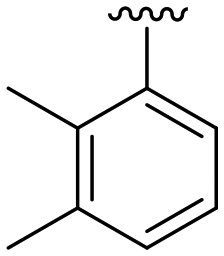
| 17 |
|
H |

|
OCH3 | 98 ± 1 | 71 ± 4 | 88 ± 3 | 4.7 |
| NH2 | ||||||||
| 22 | ||||||||
| 56 ± 13 | n.d. | 93 ± 1 | 4.2 | |||||
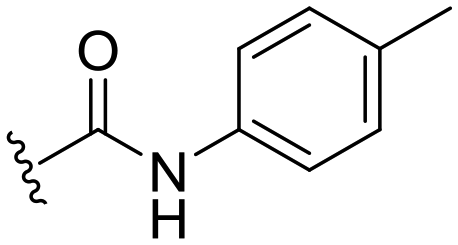
| 18 |
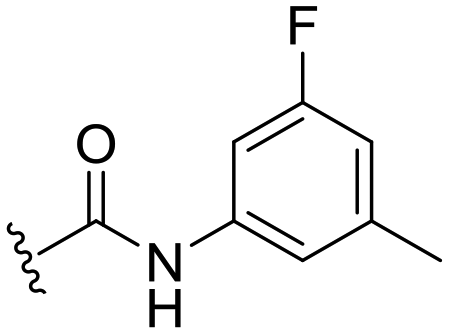
|
H |
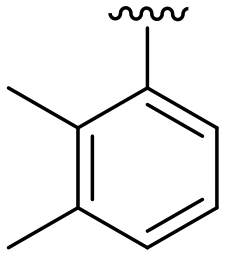
|
OCH3 | 95 ± 2 | 77 ± 4 | 90 ± 2 | 5.0 |
| NH2 | ||||||||
| 23 | ||||||||
| 47 ± 16 | n.d. | 92 ± 2 | 4.4 | |||||
| 19 |
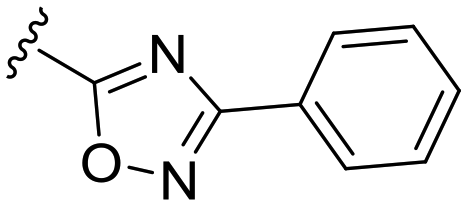
|
H |

|
OCH3 | 1 ± 4 | n.d. | 91 ± 3 | 4.8 |
| 20 | NH2 | 17 ± 8 | n.d. | 94 ± 2 | 4.3 | |||
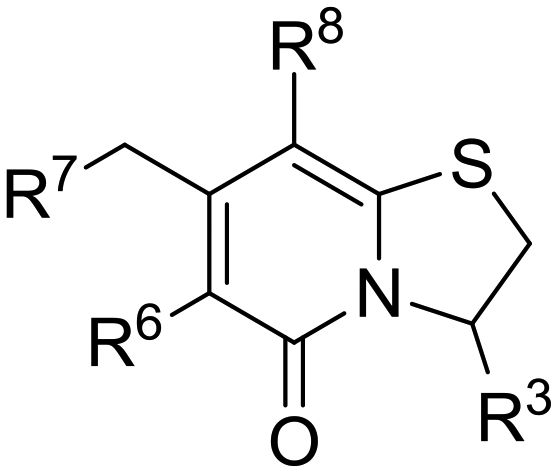
The C8-hydroxy (7) and -methoxy (5) analogues, both had slightly lower calculated logP (ref. 17) value than the previously reported cyclopropyl analogue 24,10 but proved less active (Table 1). In addition, the synthesis of the hydroxyl analog required additional low yielding steps and therefore we decided not to continue with this series. In previous studies introduction of the C6-amine has been shown to improve activity10,11 but the corresponding C6-amine analogue 8 showed only 25% inhibition at 2.5 μM. Exchanging the 1-naphthyl group to a 2,3-dimethyl phenyl substituent at the C7-position resulted in a decreased logP value as seen when comparing analogues 5 (5.0) and 13 (4.4), together with a small improvement in potency. However, the p-methyl phenyl amide 6 and the analogue 17 with a 2,3-dimethyl phenyl group at the C7-position demonstrated comparable anti-infective activity to phenyl amide 24 at 2.5 μM. Unfortunately, their effect dropped at 1 μM to 62% and 71% inhibition respectively. A more promising result was obtained with the 3-fluoro-5-methylaniline analogue 18, although the potency at 1 μM was still lower than 2 and 3. The 1,2,4-oxadiazole 19 and the 1,2,3-triazole 12 analogues were evaluated but proved less active than analogue 2 and the best C8 methoxy analogue 18. In contrast to previous studies, introduction of a C6-amine gave little benefit with a C8-methoxy substituent (Table 1). All the tested compounds showed low toxicity towards HeLa cells at 10 μM.
A general drop in potency of the C8-oxygen analogues compared with the cyclopropyl analogues was observed, with all C8-oxygen compounds losing activity at concentrations below 1 μM compared to the cyclopropyl analogues 2 and 3. However, analogue 18 was one of the most potent among the oxygen analogues and its synthesis in larger scale also proved straightforward resulting in a crystalline product suitable for various formulations. Therefore, this analogue was chosen for further evaluation together with the previously identified compounds 2 and 3. A formulation in lipid emulsion (for details about the vehicle see the Experimental section) enabled an oral dose of 10 mg kg−1. This formulation was used in an in vivo pharmacokinetic evaluation (Table 4, Fig. 2). Although the peak blood concentrations (Cmax) and AUC of compound 18 were higher compared to compound 3 after the intravenous administration, the oral uptake was unfortunately too low to allow for proper determination of pharmacokinetic parameters after oral administration. The concentrations of compound 2 after both intravenous and oral administration were too low to allow calculation of pharmacokinetic parameters. It was therefore clear that the oral bioavailability of these compounds was extremely poor.
We next considered that introduction of nitrogen based C8-substituents would potentially have a greater influence on drug-like properties. Therefore, a set of five analogues was designed for evaluation. The previously published primary amine intermediate 25 (ref. 13) served as the starting point and a collection of five compounds with different C8-nitrogen substituents were obtained (Scheme 4A). Methylation of the C8-primary amine, hydrolysis of the C3-methyl ester and following amide coupling using aniline resulted in dimethylamine 27. The secondary amine 26 was obtained through reductive alkylation of the C8-primary amine, followed by ester hydrolysis and amide coupling. Compounds 28 and 29 were synthesized by reaction with methyl chloroformate and acetyl chloride, respectively, followed by C3-methyl ester hydrolysis and amide coupling. The final methyl sulfonamide analogue was prepared by allowing the C8-amine intermediate 25 to react with methane sulfonyl chloride followed by hydrolysis and amide coupling using aniline to generate the C8-methyl sulfonamide 30 (Scheme 4A). Evaluation of their biological activity again showed a drop in potency compared to the cyclopropyl analogues but both 27 and 30 showed about 90% inhibition at 2.5 μM (Table 2). However, the calculated logP was significantly lower for the methyl sulfonamide analogue 30 compared to the dimethylamine 27. To improve the potency a set of four additional methyl sulfonamide analogues were synthesized. Two of these were substituted phenyl amides (32 and 33, Scheme 4B and Table 2) chosen based on results of our previous study10 and prepared by amide coupling from the carboxylic acid 31.
 | ||
| Scheme 4 A. Synthesis of C8-nitrogen analogues 26–30. B. Synthesis of C8-methyl sulfonamide C3-amide derivatives 32 and 33. Reagents and conditions: a) CH3I, NaHCO3, DMF, 70 °C, 10 h; b) 1 M LiOH(aq), THF:MeOH (5:1), rt; c) aniline, HATU, DIPEA, DMF, 1 h, 55% from 25; d) NaBH(OAc)3, TFA, DCM, acetone, −15 °C to rt, overnight; e) 1 M LiOH(aq), THF:MeOH (4:1), rt, 1 h; f) suspended in DCM and dripped in to ethereal HCl, −20 °C, overnight; g) aniline, HATU, DIPEA, DMF, 1 h, 43% from 25; h) methyl chloroformate, pyridine, DCM, rt, 2 h; i) 1 M LiOH(aq), THF:MeOH (4:1), rt; j) aniline, HATU, DIPEA, DMF, 1 h, 38% from 25; k) methanesulfonyl chloride, pyridine, THF, rt, overnight; then: 1 M LiOH(aq), MeOH or THF:MeOH (3:1), rt; l) aniline, HATU, DIPEA, DMF, 1 h, 30: 52% from 25; m) acetyl chloride, pyridine, DCM, rt, overnight; n) 1 M LiOH(aq), THF:MeOH (5:1), rt, 2 h; o) aniline, HATU, DIPEA, DMF, 1 h, 50% from 25; p) 3-methylaniline or 3-fluoro-5-methylaniline, HATU, DIPEA, DMF, 4 h, 32: 49%, 33: 78%. The acid intermediates as isolated compounds are published previously.13 | ||
P is the logarithm of the partition coefficient between n-octanol and water
|
|
||||||
|---|---|---|---|---|---|---|
| ID | R3 | R8 | Reinfect 2.5 μM (%) | Reinfect 1 μM (%) | HeLa viability 10 μM (%) | Calculated logP (ref. 17) |
| n.d. not determined. a Variability observed between experimental replicates was a sign of low compound solubility. | ||||||
| 26 |
|
|
79 ± 9 | 25 ± 4 | 91 ± 4 | 5.5 |
| 27 |
|
|
94 ± 0 | 78 ± 20 | 92 ± 3 | 5.0 |
| 28 |
|
|
12 ± 15 | −19 ± 21 | 88 ± 3 | 4.6 |
| 29 |
|
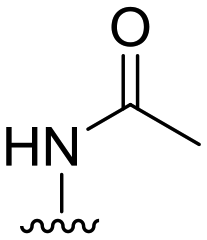
|
3 ± 9 | 44 ± 47 | 90 ± 5 | 4.4 |
| 30 |
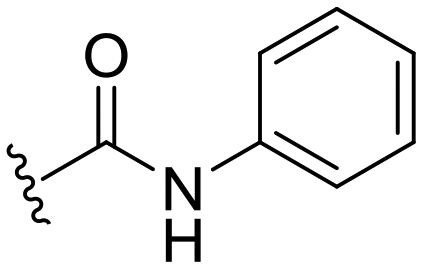
|
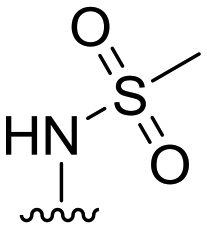
|
89 ± 4 | 23 ± 12 | 94 ± 2 | 4.2 |
| 32 |
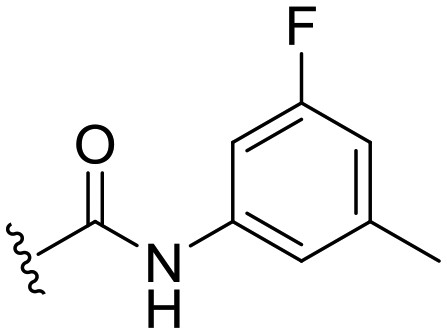
|
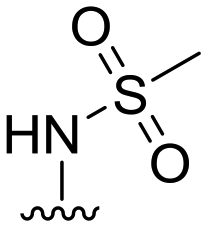
|
6 ± 17 | 6 ± 6 | 91 ± 2 | 4.7 |
| 33 |
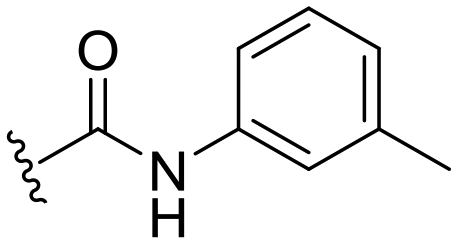
|
|
−2 ± 5 | n.d. | 92 ± 2 | 4.5 |
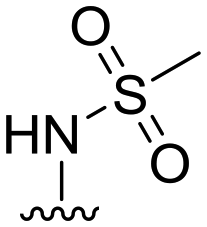
| 38 |
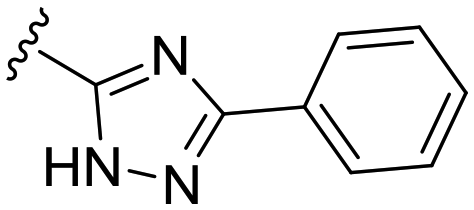
|

|
20 ± 4 | n.d. | 93 ± 1 | 4.7 |
| 46 |
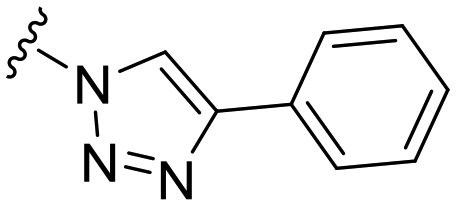
|
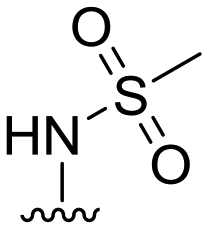
|
88 ± 7 | n.d. | 91 ± 0 | 4.3 |
In addition, two methyl sulfonamide analogues with C3-amide isosteres instead of phenyl amides were synthesized. The first of these two, the 1,2,4-triazole 38 easily accessible from the corresponding methyl ester, was chosen based on results from our previous study11 and obtained as described in Scheme 5. A one-pot reaction18 was performed on the N-carboxybenzyl (Cbz) protected methyl ester 36 to give the 1,2,4-triazole 37. The desired methyl sulfonamide 38 was then prepared by acidic Cbz-deprotection followed by installment of the methyl sulfonamide functionality via a reaction with methane sulfonyl chloride (Scheme 5).
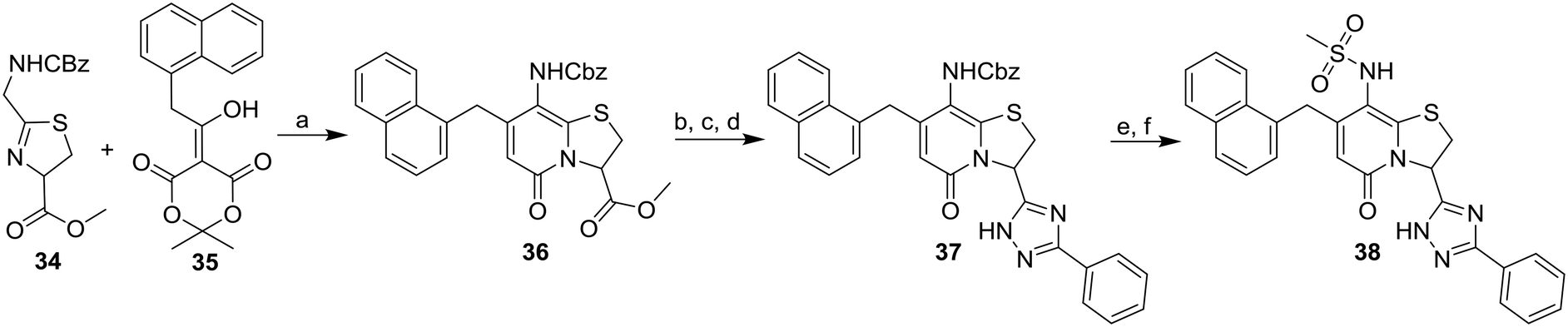 | ||
| Scheme 5 Synthesis of C8-methyl sulfonamide 1,2,4-triazole 38. Reagents and conditions: a) 35, TFA, DCM, 70 °C, 20 h, 85%; b) 1 M LiOH(aq), THF, rt, 2 h; c) benzamidine·HCl, HATU, DIPEA, DMF, 3.5 h; then (4-methoxybenzyl)hydrazine·HCl, AcOH, 80 °C, 20 h; d) TFA, DCM, 50 °C, 22 h, 39% from 36; e) 33% HBr/AcOH, DCM, rt, 2 h; f) methane sulfonyl chloride, pyridine, rt, 18 h; then: 1 M LiOH(aq), THF:MeOH (3:1), rt, 53% from 37. | ||
Earlier studies showed that the 1,2,3-triazoles were the most potent amide bioisosteres and the 1,2,3-triazole 3 has previously successfully been obtained via diazo transfer with imidazole-1-sulfonyl-azide of the C3-primary amine.11 However, with a N-carboxybenzyl-protected amine at the C8-position, the diazo transfer reaction proved unsuccessful and was exchanged for a phthalimide as protecting group instead. The phthalimide substituted thiazoline derivative 40 was synthesized from 2-(1,3-dioxoisoindolin-2-yl)acetonitrile via the ethyl imidate 39 (Scheme 6). The ring-fused 2-pyridone methyl ester 41 was then obtained from 40 and 35 after 4 days at 80 °C and the following hydrolysis and re-closure of phthalimide gave the carboxylic acid 42. The BOC-protected 43 was generated by Curtius rearrangement in tert-butyl alcohol of intermediate 42 and subsequent acidic deprotection of 43 resulted in the C3-amine 44. Diazo transfer of intermediate 44 using imidazole-1-sulfonyl azide followed Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) with phenyl acetylene resulted in the 1,2,3-triazole 45. The final compound 46 was obtained via deprotection, followed by reaction with methane sulfonyl chloride to generate the C8-methyl sulfonamide moiety.
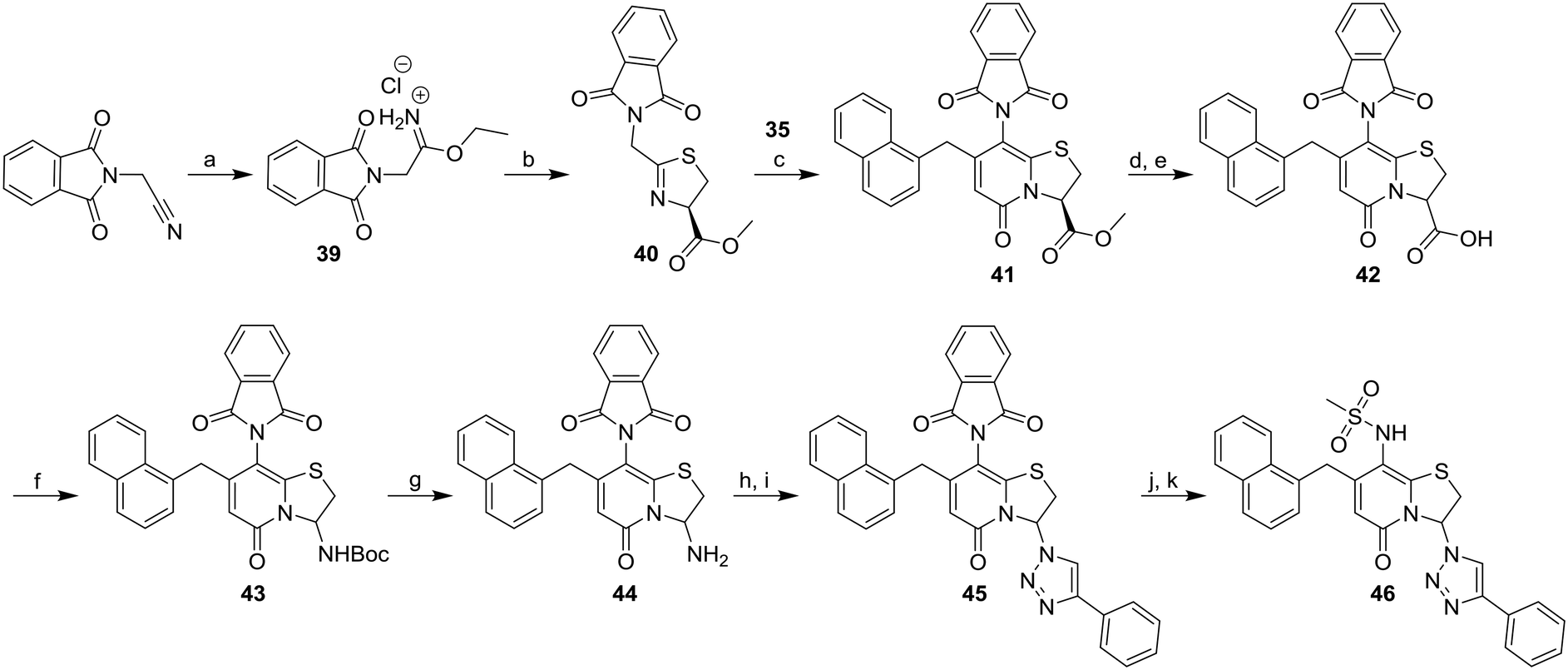 | ||
| Scheme 6 Synthesis of C8-methyl sulfonamide 1,2,3-triazole 46. Reagents and conditions: a) AcCl, EtOH/EtOAc (1:1), 0 °C to rt, 17 h, (see procedure); b) L-cysteine methyl ester·HCl, Et3N, DCM, rt, 3 days, 64%; c) 35, TFA, DCM, 80 °C, 4 days, 62%; d) 1 M LiOH(aq), THF, rt, 4 h; e) AcOH, DMF, 150 °C, 20 h, 78% from 41; f) DPPA, Et3N, t-BuOH, 2 h, 85 °C, 57%; g) TFA, DCM, rt, 2 h, 88%, h) imidazole-1-sulfonyl azide·H2SO4, CuSO4, K2CO3, MeOH, 24 h, 50 °C; i) phenyl acetylene, CuSO4, sodium ascorbate, DMF, H2O, rt, 3.5 h, 1% from 44; j) methyl amine, EtOH, rt, 20 h; k) methanesulfonyl chloride, pyridine, rt, 26 h; then: 1 M LiOH(aq), THF:MeOH (3:1), rt, 2 h, 99% from 45. | ||
The four additional methyl sulfonamide analogues were evaluated in the cell-based infection assay but none of the new analogues showed any improvement compared to compound 30. Since the activity of 30 was still in the low μM range, with low toxicity and a lower calculated logP compared to the analogues previously evaluated for pharmacokinetic properties, the methyl sulfonamide 30 was chosen together with analogues 3 and 18 for in vitro ADME determination (Table 3). Compounds 3 and 18 showed similar properties: very low solubility; low to moderate permeability with low or no efflux and low metabolic stability in both human and mouse liver microsomes (HLM, MLM). Analogue 30 on the other hand distinguished itself from 3 and 18 by improved solubility, much higher permeability and higher metabolic stability in both species. The mouse plasma protein binding was high, especially for analogues 3 and 18, which influences scaling (static well-stirred model) to hepatic clearance (CL) when using the plasma protein binding as a surrogate for the whole blood free fraction. Another limitation is that the blood to plasma ratio for these compounds are unknown, and it is known that in particular for high CL molecules the impact of the blood to plasma ratio may be large for these extrapolations.19 Thus, due to the high protein binding, the predicted unbound hepatic extraction (Eu) in mouse was low (Table 3).
| Assay | 3 | 18 | 30 |
|---|---|---|---|
| Solubility (μM ± SD) | 0.31 ± 0.16 | 0.32 ± 0.04 | 1.3 ± 0.8 |
| Caco-2 A-B Papp (×106 cm s−1 ± SD) | 0.33 ± 0.05 | 2.1 ± 0.04 | 30 ± 2.6 |
| Caco-2 efflux ratio | 0.6 | 3.2 | 2.0 |
| Mouse plasma protein binding (fraction unbound, %) | <0.1 | 0.1 | 0.39 |
| Intrinsic clearance (HLM, μl min−1 mg−1) | 187 | 184 | 39 |
| Intrinsic clearance (MLM, μl min−1 mg−1) | 135 | 440 | 59 |
| E u (mouse unbound hepatic extraction) | 0.01 | 0.09 | 0.001 |
An in vivo pharmacokinetic validation to investigate the in vitro–in vivo correlation was then performed. The compounds were administered both intravenously and orally with the same formulation as previously used. The oral data for compound 3 and 18 was too low to be able to be quantified, therefore only intravenous is shown (Table 4, Fig. 2). Interestingly, a remarkable improvement was observed for analogue 30 compared to 3, 18 and the earlier evaluated candidates. Compound 30 yielded oral blood concentrations in the μM range, lower CL and an oral bioavailability of 41%. Analogue 18 showed very low AUC in comparison to 3 despite the similarity in in vitro ADME properties and it is likely that the increased membrane permeation contributed to the higher CL of compound 3. In comparison to the obtained in vitro data, the predicted CL is underestimated indicating erroneous assumptions in models or large extrahepatic contributions. Taken together, incorporation of a sulfonamide contributes to a lowered CL in vitro and in vivo, resulting in a longer in vivo half-life (t1/2). Further, the sulfonamide effectively shifted compound 30 into a Biopharmaceutical Classification System (BCS) category II compound (high permeability, low solubility), while compounds 3 and 18 were categorized as class IV compounds (low permeability, low solubility), which are difficult to handle in further drug development.
| 3 | 18 | 30 | 30 | |
|---|---|---|---|---|
| a Variability in the compounds dose was caused by the different weight of mice between experiments. | ||||
| Administration | IV | IV | IV | PO |
| Dose (mg kg−1)a | 1.2 | 0.90 | 1.0 | 10 |
| C 0/Cmax (MM) | 0.040 | 0.41 | 1.7 | 3.7 |
| T max (h) | — | — | — | 1 |
| AUC0−∞ (μM × h) | 0.10 ± 0.01 | 0.95 ± 0.28 | 4.8 ± 0.4 | 20.3 ± 4.5 |
| t 1/2 (h) | 3.7 | 1.2 | 5.3 | 2.6 |
| CL (L h−1) | 0.51 ± 0.05 | 0.040 ± 0.01 | 0.010 ± 0.001 | — |
| V ss (L kg−1) | 27 ± 22 | 1.1 ± 0.6 | 0.62 ± 0.16 | — |
| F (%) | — | — | — | 41 |
With these encouraging results in hand, a first treatment study was conducted. Although the solubility had improved slightly it was not possible to dissolve analogue 30 in the vehicle at a higher dose than 10 mg kg−1, which was given twice a day via oral gavage to nine C. trachomatis-infected mice. As a control, vehicle without active compound was given to nine mice, while six mice received doxycycline. The mice showed no negative vital signs from the treatment, but unfortunately the course of infection was not improved at this dose (Fig. 3).
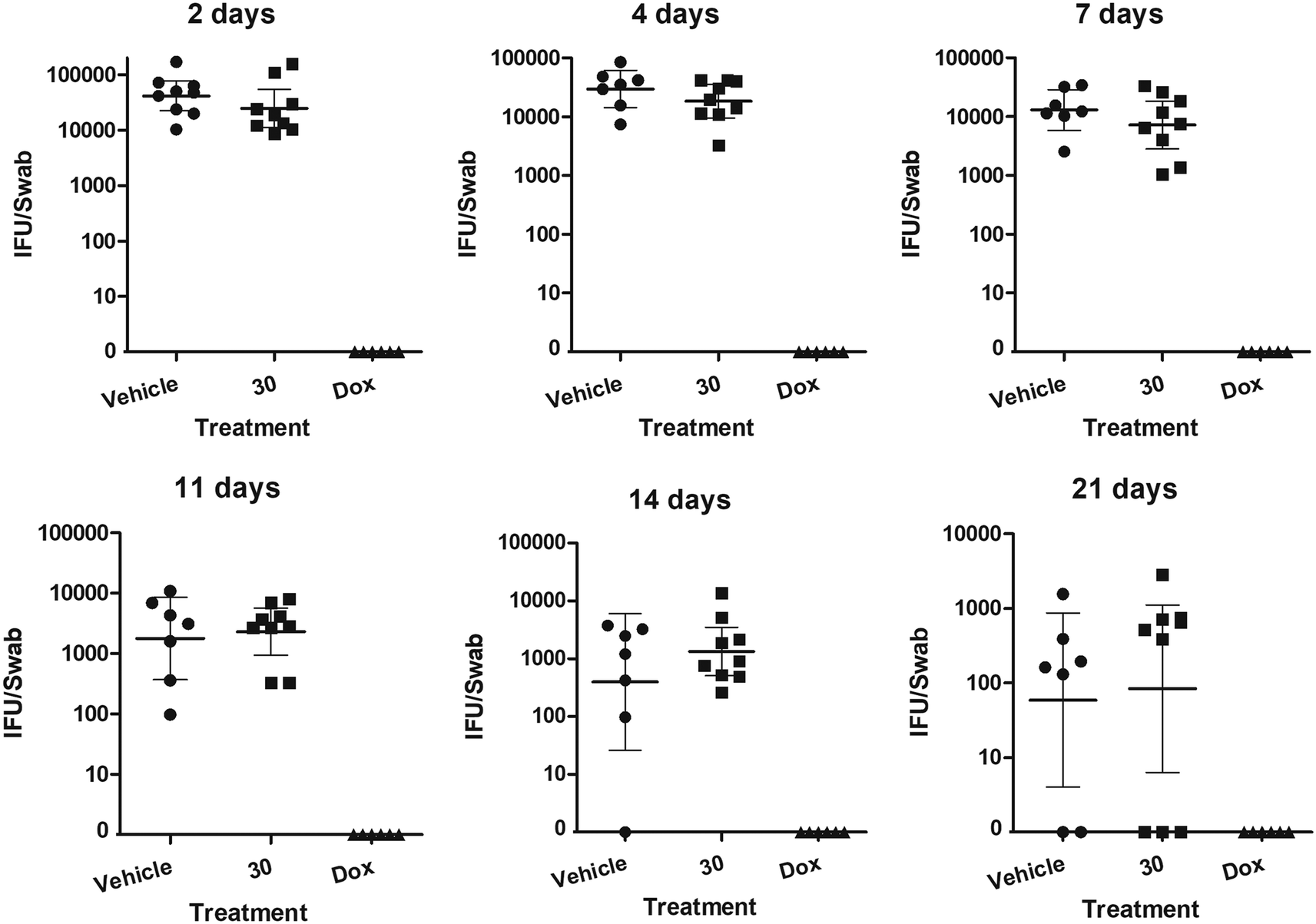 | ||
| Fig. 3 Oral treatment with compound 30 did not affect vaginal C. trachomatis infection in mice. The number of C. trachomatis IFUs per vaginal swab over time during the 7 days treatment and afterwards. C3H/HeJ mice were vaginally infected with C. trachomatis and treated with 10 mg kg−1 of compound 30, vehicle alone or 2 mg kg−1 doxycycline. | ||
The pharmacokinetic improvements observed represent a great advance towards a proof of concept for an orally available drug to treat C. trachomatis infection. The finding that C8-methyl sulfonamides so strongly can influence important pharmacokinetic parameters in favor of oral administration is an important step forward. Although no treatment effect was observed at the tested dose, the compounds show very low toxicity and therefore higher doses of the active compound could be utilized in future trials. To accomplish this, improved vehicles will be required to deliver these analogues in higher dose. In parallel, additional fine tuning of the substitution pattern to improve the potency while retaining the important pharmacokinetic properties observed with the methyl sulfonamide 30 is ongoing in our laboratories.
Materials and methods
Biology
:1000), followed by secondary donkey anti-rabbit FITC-labeled antibody (1:1000) (Jackson ImmunoResearch). Cell nuclei were stained with DAPI (1:1000). Infected cells were observed and measured by Arrayscan automated microscopy (ArrayScan VTI HCS, Thermo Scientific). Reinfect value was calculated as the percentage of IFUs compared to a DMSO treated control. The presented value is the average of three independent experiments with duplicates for each condition.
The thermodynamic solubility assay utilized solid weighed compound (0.2–0.5 mg) in a HPLC vial and PBS was added to yield a saturated solution. The vial was sealed and shaken (900 rpm) at room temperature for 48 h. After the incubation the suspension was transferred to conical glass inserts and centrifuged for 30 min at 10000×g, at 23 °C. The supernatant was then diluted in acetonitrile/H2O (50/50) 10–1000× for LC-MS/MS analysis, in two steps.
The Caco-2 study was performed in accordance with published protocols.22 Caco-2 cell monolayers (passage 94–105) were grown on permeable filter support and used for transport study on day 21 after seeding. Prior to the experiment a drug solution of 1 μM was prepared and warmed to 37 °C. The Caco-2 filters were washed with pre-warmed HBSS prior to the experiment, and thereafter the experiment was started by applying the donor solution on the apical or basolateral side. The transport experiments were carried out at pH 7.4 in both the apical and basolateral chamber. The experiments were performed at 37 °C and with a stirring rate of 500 rpm. The receiver compartment was sampled at 15, 30 and 60 minutes, and at 60 minutes also a final sample from the donor chamber was taken in order to calculate the mass balance of the compound. The samples (100 μl) were transferred to a 96-well plate containing 100 μl acetonitrile and Warfarin as internal standard and was sealed until LC-MS/MS analysis.
The metabolic stability assay was determined in 0.5 mg ml−1 human or animal liver microsomes (XenoTech LLC, KS, USA) at a compound concentration of 1 μM in 100 mM phosphate buffer pH 7.4 in a total incubation volume of 500 μl. The reaction was initiated by addition of 1 mM NADPH (final concentration). At various incubation times, i.e. at 0, 5, 10, 20, 40 and 60 min, a sample was withdrawn from the incubation and the reaction was terminated by addition to 1.5 volumes of cold acetonitrile. After centrifugation the supernatant was analyzed for the amount of parent compound remaining by LC-MS/MS. To determine the in vitro half-life, the percent remaining compound (analyte area/IS area) was calculated using time 0 min sample as 100%. The natural logarithm of the % remaining compound was plotted against time followed by linear regression.25
The plasma protein binding assay utilized rapid equilibrium dialysis (RED, Thermo Scientific Inc., USA) device inserts which allow for short dialysis times (2–4 h) as compared to traditional methods (>8 h) and minimal drug consumption in a 96-well format. Pooled human plasma with citric acid as anti-coagulant from two healthy non-smoking blood donors was provided by Uppsala Academic Hospital. Human plasma was obtained from volunteered, healthy, regular blood donors and no ethical improvement was required. Informed consent was obtained from human donors. Plasma from CD-1 mice with EDTA or Heparin as anti-coagulant was purchased from Novakemi AB, Sweden. In brief, 0.2 ml 10% plasma, 90% isotonic buffer test solution was spiked 10 μM compound and 1% final DMSO concentration and transferred to the membrane tube in the RED insert. 0.35 ml isotonic phosphate buffer pH 7.4 was added to the other side of the membrane. The 96-well base plate was then sealed with an adhesive aluminum foil film to prevent evaporation. The sample was incubated with rapid rotation (900 rpm) on a Kisker rotational incubator (Kisker Biotech Gmbh & Co. KG, Germany) at 37 °C for 4 h to achieve equilibrium. A stability test of the test solution was also prepared to allow detection of drug degradation, 100 μL of the spiked plasma test solution (in a plastic vial or on a sealed plate) was incubated at 37 °C for 4 h (or as long as the dialysis time). The plasma test solution was frozen directly after the administration to prevent any degradation. Prior to LC-MS/MS analysis the plasma and buffer sample were treated with the addition of methanol (1:3) containing Warfarin as analytical internal standard to precipitate proteins. The standard curve was created using the previously frozen plasma standard. The volume in each well consisted of 25 μl plasma and 25 μl buffer to minimize any matrix related effects in interpretation. The plate was then sealed, centrifuged and the supernatant was analyzed by mass spectrometry (LC-MS/MS). To adjust the fraction unbound (fu) for the diluted plasma the following equation was used: undiluted fu = 1/DF/{[(1/fu) − 1] + 1/DF}, DF = dilution factor.23
:4) followed by centrifugation at 3500rpm prior to analysis. Standard curves were constructed using blank mouse blood matrix spiked with compound from 10 mM DMSO stocks. Calculations of non-compartmental pharmacokinetic properties utilized Microsoft Excel and Graphpad Prism 7. Errors are standard deviations of the AUC and nonlinear regression fitting.
Animal experiments were performed according with the Guidelines for Care and Use of Laboratory Animals of Umeå University and approved by Umeå Animal Research Ethical Committee, the Swedish Board of Agriculture (A4-14, A75-15).
:2, 1:20, 1:200 and 1:2000. Thirty μL of vaginal swab dilution was inoculated in duplicates in a 96 well plate with 20000 HeLa cells per well. The plate was centrifuged at 900×g for 1 h at 37 °C. The inoculum was replaced by 100 μL preheated RPMI supplemented as above and with gentamicin (10 μg mL−1) per well and incubated for 42–46 h prior to methanol fixation, immunostaining and Chlamydia CFU quantification by Array Scan as described above.
Comp log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P
P
Computed logP values were calculated in MOE.17
Chemistry
:10 MeCN:H2O. HPLC purifications were performed on a system (Gilson) equipped with a 250 × 21.5 mm Nucleodur® C18 HTEC (particle size 5 μM) column using a flow rate of 18–20 mL min−1 and detection at 214 nm. 1H and 13C NMR spectra were recorded on a Bruker Avance III 400 MHz or a Bruker Avance III HD 600 MHz spectrometer at 298 K unless otherwise stated and calibrated by using the residual peak of the solvent as the internal standard (CDCl3: δH = 7.26 ppm; δC = 77.16 ppm; DMSO-d6: δH = 2.50 ppm; δC = 39.52 ppm; CD3OD: δH = 3.31 ppm; δC = 49.00 ppm). 19F NMR spectra were recorded with proton decoupling on a 400 MHz spectrometer at 298 K with CF3CO2H as an internal standard (δF = −76.5 ppm). The asterisk * indicates peaks determined with two-dimensional proton–carbon correlation techniques (HSQC and HMBC), which were used to supplement and complete the 13C NMR spectra where signals were otherwise broadened or weak. Amide coupling reactions with propylphosphonic anhydride (T3P) were prepared by adaptation of the procedure reported by Dunetz et al.14 The following compounds were synthesized and characterized as described previously: 1,92,103,114,1313, 14, 24,1025,1331,1334,1335.24
Experimental
:1, 6 mL) and after cooling the suspension to ≃10 °C (NaCl/ice), pyridine (130 μL, 1.61 mmol) was added. The reaction mixture was stirred for 5 min, and aniline (71 μL, 0.79 mmol) was added, followed by dropwise addition of T3P (50% in EtOAc; 0.62 mL, 1.0 mmol). The reaction was stirred for 3 h, then allowed to warm to room temperature and stirred for 16.5 h. Additional aniline (36 μL, 0.40 mmol) was added and after 45 min additional pyridine (42 μL, 0.52 mmol) was added and stirred at room temperature for 23 h. The reaction mixture was cooled to 0 °C, quenched with 1 M HCl(aq) and extracted with DCM (×3). The organic extracts were washed successively with water and brine, dried (Na2SO4) and the solvent removed under reduced pressure. Purification by flash chromatography (SiO2, 15–100% EtOAc in heptane) and freeze-drying from H2O:MeCN (6:1) afforded the amide 5 as a white powder (134 mg, 58%). 1H-NMR (400 MHz, CDCl3) δ 10.35 (s, 1H), 7.91–7.79 (m, 3H), 7.52–7.41 (m, 5H), 7.37–7.32 (m, 1H), 7.24–7.17 (m, 2H), 7.06–6.99 (m, 1H), 5.78–5.74 (m, 1H), 5.71–5.66 (m, 1H), 4.36, 4.22 (ABq, JAB = 17.0 Hz, 2H), 4.04–3.97 (m, 1H), 3.77 (s, 3H), 3.54 (dd, J = 8.2, 11.4 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 164.7, 161.3, 152.3, 140.5, 137.9, 137.8, 134.0, 133.1, 131.8, 128.9, 128.8, 128.04, 128.02, 126.5, 125.9, 125.6, 124.2, 123.8, 119.8, 114.4, 65.1, 60.8, 32.7, 30.5. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C26H23N2O3S, 443.1423; found, 443.1432.
:1, 6 mL) and after cooling the suspension to ≃10 °C (NaCl/ice), pyridine (130 μL, 1.61 mmol) was added. After stirring for 5 min, p-methylaniline (70 mg, 0.65 mmol) was added, followed by dropwise addition of T3P (50% in EtOAc; 0.63 mL, 1.1 mmol). Stirred at −10 °C for 3 h and then allowed to return to rt and stirred for 16.5 h. Additional p-methylaniline (47 mg, 0.44 mmol) was added and stirred for 45 min and then pyridine (42 μL, 0.52 mmol) was added and stirred for another 4 h. The reaction mixture was cooled to 0 °C and quenched with 1 M HCl(aq) (10 mL), diluted with EtOAc and extracted with EtOAc (×3). The combined organic layers were washed successively with water and brine, then the precipitate was filtered off and the filtrate was dried (Na2SO4) and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 30–100% EtOAc in heptane) and freeze-drying from H2O:MeCN (6:1) afforded the amide 6 as a white powder (107 mg, 44%). 1H-NMR (400 MHz, CDCl3) δ 10.23 (s, 1H), 7.91–7.77 (m, 3H), 7.51–7.37 (m, 5H), 7.36–7.32 (m, 1H), 7.08–7.03 (m, 2H), 5.79–5.77 (m, 1H), 5.73 (d, J = 7.9 Hz, 1H), 4.40–4.17 (m, 3H), 3.76 (s, 3H), 3.62 (dd, J = 7.9, 11.2 Hz, 1H), 2.28 (s, 3H). 13C-NMR (100 MHz, CDCl3) δ 164.3, 161.5, 152.3, 140.3, 138.1, 135.4, 134.1, 134.0, 133.1, 131.9, 129.5, 129.0, 128.12, 128.07, 126.6, 126.0, 125.6, 123.9, 119.9, 114.8, 65.2, 60.9, 32.8, 30.3, 21.0. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H25N2O3S, 457.1579; found, 457.1590.
:MeCN afforded the amide 7 as a white powder (5.8 mg). A white precipitate in the combined aqueous layers was filtered off and after freeze-drying from H2O:MeCN afforded the amide 7 (8.0 mg). The total yield of the reaction: 14 mg, 10%. 1H-NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.56 (s, 1H), 8.00–7.84 (m, 3H), 7.59–7.47 (m, 5H), 7.42 (d, J = 6.9 Hz, 1H), 7.35–7.26 (m, 2H), 7.10–7.01 (m, 1H), 5.50–5.44 (m, 1H), 5.27 (s, 1H), 4.32, 4.23 (ABq, JAB = 16.8 Hz, 2H), 3.91 (dd, J = 11.8, 9.0 Hz, 1H), 3.62–3.54 (m, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 166.0, 158.7, 149.8, 138.7, 136.2, 134.3, 133.5, 131.5, 131.4, 128.8, 128.6, 127.6, 127.3, 126.3, 125.8, 125.7, 124.0, 123.6, 119.0, 112.7, 64.3, 32.4, 32.2. HRMS (ESI+) (m/z): [M + Na+]+ calcd. for C25H21N2O3S, 429.1267; found, 429.1269.
:MeCN (10:1) afforded the amide 8 as a grey powder (30 mg, 30%). 1H-NMR (400 MHz, CDCl3) δ 10.40 (s, br, 1H), 8.17 (d, J = 8.4 Hz, 1H), 7.93–7.88 (m, 1H), 7.77 (d, J = 8.3 Hz, 1H), 7.64–7.52 (m, 4H), 7.39–7.28 (m, 3H), 7.17–7.07 (m, 2H), 5.85 (d, J = 7.5 Hz, 1H), 4.38, 4.31 (ABq, JAB = 16.5 Hz, 2H), 4.20 (d, J = 11.3 Hz, 1H), 3.99 (s br, 2H), 3.68 (dd, J = 7.7, 11.3 Hz, 1H), 3.58 (s, 3H). 13C-NMR (100 MHz, CDCl3) δ 164.7, 156.3, 139.1, 138.1, 134.1, 133.2, 132.1, 131.8, 129.1*, 127.8, 126.6, 126.1, 125.7, 124.7, 124.6, 124.5, 124.1, 123.4, 120.1, 65.5, 61.4, 30.3, 28.2. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C26H24N3O3S, 458.1532; found, 458.1523. *Note: overlap of two non-identical aromatic carbons, can be seen in HSQC.
:MeCN afforded the amine 9 as a grey powder (28 mg, 35%). 1H-NMR (400 MHz, CDCl3) δ 10.29 (br s, 1H), 8.17 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.77 (d, J = 8.2 Hz, 1H), 7.64–7.52 (m, 2H), 7.50–7.45 (m, 2H), 7.40–7.33 (m, 1H), 7.16–7.08 (m, 3H), 5.84 (d, J = 7.6 Hz, 1H), 4.38, 4.30 (ABq, JAB = 16.5 Hz, 2H), 4.20 (d, J = 11.2 Hz, 1H), 3.98 (br s, 2H), 3.67 (dd, J = 7.6, 11.2 Hz, 1H), 3.58 (s, 3H), 2.31 (s, 3H). 13C-NMR (100 MHz, CDCl3) δ 164.6, 156.3, 139.1, 135.5, 134.0, 133.1, 132.1, 131.8, 129.5, 129.0, 127.8, 126.6, 126.1, 125.7, 124.6, 124.1, 123.4, 120.1, 65.5, 61.4, 30.3, 28.2, 21.0. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H26N3O3S, 472.1688; found, 472.1687.
:1, 4 mL) and after cooling the suspension to ≃10 °C (NaCl/ice), pyridine (0.11 mL, 1.4 mmol) was added. After stirring for 5 min, aniline (60 μL, 0.66 mmol) was added, followed by dropwise addition of T3P (50% in EtOAc; 0.52 mL, 1.7 mmol). The reaction mixture was stirred at −10 °C for 1 h, then allowed to return to rt and stirred for 23 h. The reaction was cooled to 0 °C, then quenched with 1 M HCl(aq), diluted with EtOAc and extracted with EtOAc (×3). The combined organic layers were washed successively with H2O (1 mL 1 M HCl(aq) was added to aid separation) and brine, dried (Na2SO4), and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 25–100% EtOAc in heptane) and freeze-drying from H2O:MeCN (6:1) afforded the amide 16 as a white powder (101 mg, 82%). 1H-NMR (600 MHz, CDCl3) δ 10.43 (s, 1H), 7.57–7.53 (m, 2H), 7.30–7.25 (m, 2H), 7.11–7.04 (m, 3H), 6.95 (d, J = 7.3 Hz, 1H), 5.81 (d, J = 8.0 Hz, 1H), 5.78 (s, 1H), 4.21 (d, J = 11.2 Hz, 1H), 3.90, 3.82 (ABq, JAB = 17.3 Hz, 2H), 3.71 (s, 3H), 3.66 (dd, J = 11.2, 8.0 Hz, 1H), 2.29 (s, 3H), 2.10 (s, 3H). 13C-NMR (151 MHz, CDCl3) δ 164.5, 161.6, 152.9, 140.3, 138.4, 138.0, 137.5, 135.3, 135.0, 129.2, 129.0, 128.4, 125.9, 124.5, 120.0, 114.2, 65.3, 60.7, 33.9, 30.4, 20.8, 15.6. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C24H25N2O3S, 421.1580; found, 421.1589.
:1, 4 mL) and after cooling the suspension to ≃10 °C (NaCl/ice), pyridine (0.11 mL, 1.4 mmol) was added. After stirring for 15 min, p-methylaniline (70 mg, 0.65 mmol) was added and followed by dropwise addition of T3P (50% in EtOAc; 0.51 mL, 0.86 mmol). Stirred at −10 °C for 1 h and then allowed to return to rt and stirred for 23 h. The reaction mixture was cooled to 0 °C and quenched with 1 M HCl(aq), diluted with EtOAc and extracted with EtOAc (×3). The combined organic layers were washed successively with water and brine, dried (Na2SO4) and concentrated under reduced pressure. Purification by flash chromatography (SiO2, 25–100% EtOAc in heptane) and freeze-drying from H2O:MeCN afforded the amide 17 as an off-white powder (49 mg, 39%). 1H-NMR (400 MHz, CDCl3) δ 10.28 (s br, 1H), 7.45–7.40 (m, 2H), 7.12–7.03 (m, 4H), 6.98–6.93 (m, 1H), 5.80–5.73 (m, 2H), 4.21 (d, J = 11.2 Hz, 1H), 3.90, 3.82 (ABq, JAB = 17.3 Hz, 2H), 3.72 (s, 3H), 3.64 (dd, J = 7.9, 11.2 Hz, 1H), 2.31–2.27 (m, 6H), 2.10 (s, 3H). 13C-NMR (100 MHz, CDCl3) δ 164.3, 161.7, 152.6, 140.0, 138.2, 137.5, 135.4, 135.3, 135.0, 134.0, 129.5 (2C), 129.1, 128.4, 125.9, 120.0 (2C), 114.3, 65.1, 60.7, 33.9, 30.3, 20.9, 20.8, 15.6. HRMS (ESI+) (m/z): [M + Na+]+ calcd. for C25H26N2NaO3S, 457.1555; found, 457.1572.
:MeCN (6:1) afforded the 1,2,4-oxadiazole 19 as a yellow powder (139 mg, 46%). 1H-NMR (400 MHz, CDCl3) δ 8.06–8.01 (m, 2H), 7.53–7.43 (m, 3H), 7.11–7.04 (m, 2H), 7.02–6.97 (m, 1H), 6.40 (dd, J = 1.7, 7.8 Hz, 1H), 5.76–5.74 (m, 1H), 3.97 (dd, J = 7.8, 11.8 Hz, 1H), 3.94–3.83 (m, 2H), 3.77–3.71 (m, 4H), 2.30 (s, 3H), 2.13 (s, 3H). 13C-NMR (100 MHz, CDCl3) δ 175.1, 168.9, 160.4, 152.7, 137.6, 137.52, 137.49, 135.3, 135.2, 131.6, 129.2, 129.0, 128.4, 127.8, 126.4, 125.9, 115.4, 60.9, 58.3, 34.2, 34.1, 20.8, 15.6. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C25H24N3O3S, 446.1532; found, 446.1531.
:MeCN (10:1) afforded the amine 20 as a grey powder (68 mg, 60%). 1H-NMR (400 MHz, CDCl3) δ 8.09–8.04 (m, 2H), 7.53–7.43 (m, 3H), 7.09–6.98 (m, 2H), 6.86 (d, J = 7.4, 1H), 6.47 (dd, J = 1.8, 7.5 Hz, 1H), 4.07–3.81 (m, 5H), 3.73 (dd, J = 1.8, 11.8 Hz, 1H), 3.59 (s, 3H), 2.33 (s, 3H), 2.30 (s, 3H). 13C-NMR (100 MHz, CDCl3) δ 175.3, 168.8, 155.7, 138.4, 137.3, 135.4, 134.4, 133.2, 131.5, 128.9 (2C), 128.7, 127.7 (2C), 126.4, 125.9, 124.5, 124.0, 122.0, 61.3, 58.6, 34.1, 29.3, 20.8, 15.3. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C25H25N4O3S, 461.1641; found, 461.1639.
:MeCN (10:1) afforded the amine 21 as a grey powder (25 mg, 30%). 1H-NMR (400 MHz, CDCl3) δ 10.41 (bs, 1H) 7.61–7.55 (m, 2H), 7.34–7.28 (m, 2H), 7.12–7.04 (m, 2H), 7.03–6.97 (m, 1H), 6.80 (d, J = 7.5 Hz, 1H), 5.85 (d, J = 7.6 Hz, 1H), 4.19 (d, J = 11.2 Hz, 1H), 4.01–3.78 (m, 4H), 3.66 (dd, J = 7.6, 11.2 Hz, 1H), 3.57 (s, 3H), 2.33 (s, 3H), 2.29 (s, 3H). 13C-NMR (100 MHz, CDCl3) δ 164.8, 156.3, 139.1, 138.1, 137.4, 135.4, 134.3, 132.9, 129.0, 128.8, 126.0, 125.6, 124.5, 124.4, 120.1, 65.5, 61.3, 30.3, 29.3, 20.8, 15.3. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C24H26N3O3S, 436.1689; found, 436.1691.
:MeCN (10:1) afforded the amine 22 as an off-white powder (5.8 mg, 17%). 1H-NMR (400 MHz, CDCl3) δ 10.29 (br s, 1H), 7.49–7.43 (m, 2H), 7.14–7.08 (m, 2H), 7.08–7.03 (m, 1H), 7.03–6.96 (m, 1H), 6.79 (d, J = 7.5 Hz, 1H), 5.84 (d, J = 7.6 Hz, 1H), 4.20 (d, J = 11.2 Hz, 1H), 4.05–3.77 (m, 4H), 3.66 (dd, J = 7.6, 11.2 Hz, 1H), 3.57 (s, 3H), 2.34–2.27 (9H). 13C-NMR (100 MHz, CDCl3) δ 164.6, 156.4, 139.1, 137.4, 135.5, 135.4, 134.3, 134.1, 132.9, 129.6, 128.8, 126.0, 125.6, 124.61, 124.57, 120.1, 65.5, 61.3, 30.3, 29.4, 21.0, 20.9, 15.4. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C25H28N3O3S, 450.1845; found, 450.1848.
:MeCN (10:1) afforded the amine 23 as a brown powder (20 mg, 39%). 1H-NMR (400 MHz, CDCl3) δ 10.48 (br s, 1H), 7.36–7.30 (m, 1H), 7.08–6.96 (m, 3H), 6.81–6.77 (m, 1H), 6.65–6.58 (m, 1H), 5.82 (d, J = 7.7 Hz, 1H), 4.17 (d, J = 11.3 Hz, 1H), 4.02–3.77 (m, 4H), 3.66 (dd, J = 7.7, 11.3 Hz, 1H), 3.57 (s, 3H), 2.32 (s, 3H), 2.31–2.28 (m, 6H). 13C-NMR (100 MHz, CDCl3) δ 164.9, 163.0 (d, JCF = 244.1 Hz), 156.4, 140.8 (d, JCF = 8.9 Hz), 139.3, 139.1 (d, JCF = 11.5 Hz), 137.5, 135.4, 134.2, 132.9, 128.8, 126.0, 125.8, 124.6, 124.5, 116.1 (d, JCF = 2.6 Hz), 111.9 (d, JCF = 21.2 Hz), 104.7 (d, JCF = 26.5 Hz), 65.5, 61.3, 30.2, 29.4, 21.5 (d, JCF = 2.0 Hz), 20.9, 15.4. 19F NMR (376 MHz, CDCl3) δ −113.29. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C25H27FN3O3S, 468.1751; found, 468.1751.
:EtOAc 83:17 ≥ 0:100) to give BOC-protected amine 10 as a yellow foam (358 mg, 60%). 1H-NMR (400 MHz, CDCl3) δ 7.87–7.78 (m, 2H), 7.77 (d, J = 7.9 Hz, 1H), 7.48–7.43 (m, 2H), 7.40 (dd, J = 8.4, 6.9 Hz, 1H), 7.30 (dd, J = 6.9, 0.9 Hz, 1H), 6.47 (td, J = 6.8, 0.9 Hz, 1H), 5.67 (s, 1H), 5.56 (d, J = 7.2 Hz, 1H), 4.23 (s, 2H), 3.71 (s, 3H), 3.63 (dd, J = 11.9, 6.5 Hz, 1H), 3.39 (dd, J = 11.9, 1.7 Hz, 1H), 1.42 (s, 9H); 13C-NMR (100 MHz, CDCl3) δ 159.6, 154.1, 151.6, 138.3, 136.4, 134.0, 133.4, 131.8, 128.8, 127.9, 127.8, 126.4, 125.8, 125.5, 123.8, 115.6, 80.8, 69.1, 60.7, 36.0, 32.7, 28.3; HRMS (ESI+) (m/z): [M + H+]+ calcd. for C24H27N2O4S+, 439.1686; found, 439.1691.
:MeOH 99:1 ≥ 92:8) to give amine 11 as a yellow foam (85 mg, 32%). 1H-NMR (400 MHz, CDCl3) δ 7.89–7.80 (m, 2H), 7.78 (d, J = 8.0 Hz, 1H), 7.51–7.43 (m, 2H), 7.41 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 6.8 Hz, 1H), 5.88 (dd, J = 7.3, 1.7 Hz, 1H), 5.69 (s, 1H), 4.26 (s, 2H), 3.73 (s, 3H), 3.66 (dd, J = 12.0, 7.1 Hz, 1H), 3.16 (dd, J = 12.0, 1.6 Hz, 1H), 2.48 (s, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.6, 151.3, 137.3, 136.4, 134.0, 133.5, 131.9, 128.9, 127.9, 127.9, 126.4, 125.8, 125.5, 123.9, 115.3, 72.9, 60.8, 35.7, 32.7; HRMS (ESI+) (m/z): [M + H+]+ calcd. for C19H19N2O2S+, 339.1162; found, 339.1167.
:EtOAc 90:10 ≥ 20:80, Rf = 0.3), then dissolved in DMF (0.5 ml) and phenyl acetylene (4.2 μl, 0.038 mmol), sodium ascorbate (1.5 mg, 0.0077 mmol), CuSO4 (0.61 mg, 0.0038 mmol) and H2O (50 μl) were added. The mixture was stirred for 4 hours and then diluted with EtOAc and washed with brine. The organic layer was dried (Na2SO4), filtered and concentrated under reduced pressure. Purified using preparative HPLC (H2O:MeCN 90:10 ≥ 0:100 with 0.75% formic acid over 40 minutes) to give 1,2,3-triazole 12 as a yellow solid (4 mg, 45%). 1H-NMR (400 MHz, CDCl3) δ 8.20 (s, 1H), 7.89–7.84 (m, 1H), 7.82–7.77 (m, 4H), 7.49–7.45 (m, 2H), 7.44–7.37 (m, 3H), 7.35–7.28 (m, 3H), 5.68 (t, J = 1.1 Hz, 1H), 4.35 (d, J = 12.7 Hz, 1H), 4.33, 4.22 (ABq, JAB = 17.2 Hz, 2H), 4.05 (dd, J = 12.5, 7.1 Hz, 1H), 3.79 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 160.0, 153.4, 148.1, 138.6, 137.2, 134.1, 133.0, 131.9, 130.2, 129.1, 129.0, 128.9, 128.5, 128.2, 128.1, 126.6, 126.0, 125.7, 123.9, 121.0, 115.4, 73.0, 61.0, 33.9, 33.0; HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H23N4O2S+, 467.1536; found, 467.1540.
:MeCN to give isopropyl amine 26 as a white solid (32 mg, 43%). 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 7.99–7.93 (m, 1H), 7.90–7.82 (m, 2H), 7.56–7.48 (m, 5H), 7.43 (d, J = 7.0 Hz, 1H), 7.30 (apparent t, J = 7.7 Hz, 2H), 7.05 (apparent t, J = 7.4 Hz, 1H), 5.48 (d, J = 8.9 Hz, 1H), 5.16 (s, 1H), 4.41, 4.29 (ABq, JAB = 17.1 Hz, 2H), 3.87 (dd, J = 11.9, 9.1 Hz, 1H), 3.62 (br d, J = 6.1 Hz, 1H), 3.50 (d, J = 12.0 Hz, 1H), 3.32–3.25 (m, 1H), 1.13 (d, J = 6.2 Hz, 3H), 1.11 (d, J = 6.2 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 166.0, 159.3, 154.5, 146.0, 138.7, 134.7, 133.5, 131.5, 128.8, 128.6, 127.9, 127.3, 126.2, 125.8, 125.7, 124.2, 123.5, 120.8, 119.0, 112.8, 64.7, 48.9, 33.4, 31.5, 23.45, 23.42. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C28H28N3O2S, 470.1897; found, 470.1895.
:MeCN to give dimethylamine 27 as a white solid (39 mg, 55%). 1H NMR (600 MHz, DMSO-d6) δ 10.39 (s, 1H), 7.96 (d, J = 7.7 Hz, 1H), 7.91 (d, J = 7.9 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.59–7.45 (m, 5H), 7.40 (d, J = 7.0 Hz, 1H), 7.30 (apparent t, J = 7.8 Hz, 2H), 7.06 (apparent t, J = 7.4 Hz, 1H), 5.45 (d, J = 9.1 Hz, 1H), 5.31 (s, 1H), 4.35, 4.30 (ABq, JAB = 16.7 Hz, 2H), 3.92 (dd, J = 12.0, 9.2 Hz, 1H), 3.55 (d, J = 11.9 Hz, 1H), 2.71 (s, 6H). 13C NMR (151 MHz, DMSO) δ 166.0, 159.5, 156.1, 147.3, 138.7, 134.8, 133.5, 131.5, 128.8, 128.6, 127.6, 127.2, 126.3, 125.8, 125.6, 125.1, 124.0, 123.6, 119.0, 113.0, 63.8, 42.6, 41.8, 33.8, 31.9. HRMS (ESI+) (m/z): [M + Na+]+ calcd. for C27H25N3O2SNa+, 478.1565; found, 478.1557.
:MeCN to give carbamate 28 as a white solid (29 mg, 38%). 1H NMR (600 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.89* (s, 0.8H), 8.62* (s, 0.2H), 8.01–7.93 (m, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.87–7.80 (m, 1H), 7.59–7.47 (m, 5H), 7.41 (d, J = 7.0 Hz, 1H), 7.30 (apparent t, J = 7.7 Hz, 2H), 7.06 (t, J = 7.4 Hz, 1H), 5.52 (dd, J = 9.1, 1.6 Hz, 1H), 5.22* (s, 0.2H), 5.15* (s, 0.8H), 4.17, 4.12 (ABq, JAB = 17.1 Hz, 2H), 3.96 (apparent t, J = 10.6 Hz, 1H), 3.65 (s, 3H), 3.56 (d, J = 11.9 Hz, 1H). 13C NMR (151 MHz, DMSO) δ 165.7, 159.6, 155.7, 154.8, 149.8, 138.6, 133.5, 131.5, 128.8, 128.6, 128.1, 127.5, 126.3, 125.8, 125.7, 124.1, 123.6, 119.0, 112.4, 110.9, 65.0, 52.1, 33.6, 31.9. HRMS (ESI+) (m/z): [M + Na+]+ calcd. for C27H23N3O4SNa+, 508.1301; found, 508.1298.*Note: rotameric signals.
:MeCN to give amide 29 as a white solid (37 mg, 50%). 1H NMR (600 MHz, DMSO-d6) δ 10.40 (s, 1H), 9.41 (s, 1H), 7.98–7.94 (m, 1H), 7.88 (d, J = 8.3 Hz, 1H), 7.87–7.83 (m, 1H), 7.59–7.45 (m, 5H), 7.41 (d, J = 7.0 Hz, 1H), 7.30 (apparent t, J = 7.7 Hz, 2H), 7.06 (apparent t, J = 7.4 Hz, 1H), 5.50 (d, J = 9.1 Hz, 1H), 5.14 (s, 1H), 4.14, 4.08 (ABq, JAB = 17.3 Hz, 2H), 3.94 (dd, J = 12.0, 9.1 Hz, 1H), 3.56 (d, J = 12.0 Hz, 1H), 2.02 (s, 3H). 13C NMR (151 MHz, DMSO) δ 169.5, 165.8, 159.6, 154.5, 148.7, 138.6, 133.6, 133.5, 131.5, 128.8, 128.6, 128.1, 127.5, 126.3, 125.8, 125.7, 124.2, 123.6, 119.0, 112.3, 111.3, 64.9, 33.8, 31.8, 22.4. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H24N3O3S+, 470.1533; found, 470.1536.
:MeCN to give methyl sulfonamide 30 as a white solid (41 mg, 52%). 1H NMR (600 MHz, DMSO-d6) δ 10.40 (s, 1H), 9.31 (s, 1H), 7.97 (d, J = 7.8 Hz, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.58–7.47 (m, 5H), 7.43 (d, J = 7.0 Hz, 1H), 7.30 (apparent t, J = 7.8 Hz, 2H), 7.06 (apparent t, J = 7.4 Hz, 1H), 5.50 (d, J = 9.0 Hz, 1H), 5.04 (s, 1H), 4.47, 4.31 (ABq, JAB = 17.7 Hz, 2H), 4.04–3.99 (m, 1H), 3.60 (d, J = 11.9 Hz, 1H), 3.22 (s, 3H). 13C NMR (151 MHz, DMSO) δ 165.6, 159.4, 156.3, 151.6, 138.6, 133.8, 133.5, 131.5, 128.8, 128.6, 128.3, 127.6, 126.3, 125.9, 125.7, 124.2, 123.6, 119.0, 112.7, 109.9, 65.0, 43.3, 34.4, 32.1. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C26H24N3O4S2+, 506.1203; found, 506.1200.
:MeCN (5:1) afforded the amide 32 as an orange powder (43 mg, 49%). 1H-NMR (600 MHz, DMSO-d6) δ 10.55 (s, 1H), 9.32 (s, 1H), 7.98–7.95 (m, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.88–7.85 (m, 1H), 7.54–7.49 (m, 3H), 7.43 (d, J = 6.9 Hz, 1H), 7.30–7.26 (m, 1H), 7.10 (s, 1H), 6.76–6.72 (m, 1H), 5.48 (dd, J = 9.3, 2.0 Hz, 1H), 5.04 (s, 1H), 4.46, 4.32 (ABq, JAB = 17.7 Hz, 2H), 4.01 (dd, J = 11.9, 9.3 Hz, 1H), 3.59 (dd, J = 11.9, 2.0 Hz, 1H), 3.21 (s, 3H), 2.27 (s, 3H). 13C-NMR (151 MHz, DMSO-d6) δ 165.9, 162.0 (d, JCF = 141.0 Hz), 159.4, 156.3, 151.6, 140.6 (d, JCF = 9.6 Hz), 139.8 (d, JCF = 12.1 Hz), 133.8, 133.5, 131.5, 128.6, 128.3, 127.6, 126.3, 125.9, 125.7, 124.2, 115.4 (CF broad peak), 112.7, 110.8 (d, JCF = 21.2 Hz), 109.9, 103.2 (d, JCF = 26.4 Hz), 65.0, 43.3, 34.4, 21.0 (d, JCF = 2.0 Hz). HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H25FN3O4S2, 538.1265; found, 538.1273.
:MeCN (5:1) afforded the amide 33 as beige powder (66 mg, 78%). 1H-NMR (600 MHz, DMSO-d6) δ 10.32 (s, 1H), 9.32 (s, 1H), 7.99–7.95 (m, 1H), 7.92–7.85 (m, 2H), 7.55–7.49 (m, 3H), 7.44–7.42 (m, 1H), 7.41–7.39 (m, 1H), 7.31–7.27 (m, 1H), 7.18 (t, J = 7.8 Hz, 1H), 6.90–6.86 (m, 1H), 5.49 (dd, J = 9.2, 2.0 Hz, 1H), 5.04 (s, 1H), 4.46, 4.32 (ABq, JAB = 17.7 Hz, 2H), 4.01 (dd, J = 11.8, 9.2 Hz, 1H), 3.58 (dd, J = 11.8, 2.0 Hz, 1H), 3.21 (s, 3H), 2.25 (s, 3H). 13C-NMR (151 MHz, DMSO-d6) δ 165.6, 159.5, 156.2, 151.6, 138.5, 138.1, 133.9, 133.5, 131.5, 128.7, 128.6, 128.3, 127.6, 126.3, 125.9, 125.7, 124.33, 124.25, 119.6, 116.2, 112.7, 109.9, 65.0, 43.3, 34.4, 32.1, 21.1. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H26N3O4S2, 520.1359; found, 520.1362.
:EtOAc 60:40 ≥ 0:100) to give methyl ester 36 as a yellow foam (8.12 g, 85%). 1H-NMR (400 MHz, CDCl3) δ 7.90–7.73 (m, 3H), 7.52–7.41 (m, 2H), 7.41–7.28 (m, 6H), 7.24–7.18 (m, 1H), 5.93 (s, 1H), 5.80–5.65 (m, 1H), 5.54 (dd, J = 8.4, 2.4 Hz, 1H), 5.20–5.10 (m, 2H), 4.27–4.05 (m, 2H), 3.76 (s, 3H), 3.73–3.63 (m, 1H), 3.55–3.45 (m, 1H); 13C-NMR (100 MHz, CDCl3) δ 168.3, 161.0, 154.9, 154.8, 148.4, 135.9, 134.1, 132.9, 132.0, 128.9, 128.7, 128.5, 128.3, 128.1, 127.7, 126.5, 126.0, 125.6, 124.0, 115.5, 111.5, 67.8, 64.0, 53.5, 34.9, 32.2; HRMS (ESI+) (m/z): [M + H+]+ calcd. for C28H25N2O5S+, 501.1479; found, 501.1476.
:EtOAc 25:75 ≥ 0:100) followed by preperative HPLC (H2O:MeCN 90:10 ≥ 0:100 with 0.75% formic acid over 40 minutes) to give 1,2,4-triazole 37 as a yellow solid (135 mg, 39%). 1H-NMR (400 MHz, CDCl3) δ 7.87–7.85 (m, 4H), 7.72 (d, J = 8.4 Hz, 1H), 7.46–7.36 (m, 2H), 7.35–7.21 (m, 9H), 7.11 (d, J = 7.1 Hz, 1H), 6.48 (s, 1H), 6.20 (d, J = 7.3 Hz, 1H), 5.68 (s, 1H), 5.13 (s, 2H), 4.17, 3.95 (ABq, JAB = 17.6 Hz, 2H), 3.78–3.66 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 161.7, 158.3, 157.6, 155.7, 155.1, 157.7, 136.0, 134.0, 132.9, 131.9, 130.0, 128.8, 128.8, 128.7, 128.7, 128.4, 128.2, 128.0, 127.8, 126.6, 126.5, 125.9, 125.6, 124.1, 115.0, 112.4, 67.7, 60.8, 34.7, 33.7; LRMS (m/z): [M + H+]+ calcd. for C34H28N5O3S, 586.2+; found, 586.3.
:MeCN 90:10 ≥ 0:100 with 0.75% formic acid over 40 minutes) to give methyl sulfonamide 38 as a yellow powder (8 mg, 53%). 1H-NMR (400 MHz, DMSO-d6) δ 14.27 (br s, 1H), 9.34 (br s, 1H), 8.01–7.95 (m, 1H), 7.94–7.86 (m, 4H), 7.57–7.42 (m, 7H), 6.17 (dd, J = 8.0, 1.1 Hz, 1H), 5.09 (s, 1H), 4.48, 4.34 (ABq, JAB = 17.2 Hz, 2H), 4.10 (dd, J = 11.5, 8.2 Hz, 1H), 3.55 (dd, J = 11.5, 1.1 Hz, 1H), 3.22 (s, 3H); 13C-NMR (150 MHz, DMSO-d6) δ 161.6, 158.4, 157.5, 150.6, 134.1, 133.0, 132.0, 130.2, 128.9 (2C), 128.8, 128.1, 128.1, 126.6, 126.6, 126.0, 125.6, 124.4, 115.7, 111.8, 60.8, 43.7, 35.5, 33.5;. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H24N5O3S2+, 530.1315; found, 530.1314.
:EtOAc 88:12 ≥ 0:100) to give 40 as a colorless oil (1.39 g, 64%) [α]D 2° (c 0.0035, DCM). 1H-NMR (400 MHz, CDCl3) δ 7.86–7.80 (m, 2H), 7.71–7.66 (m, 2H), 5.10–5.03 (m, 1H), 4.66 (t, J = 1.8 Hz, 2H), 3.72 (s, 3H), 3.58 (dd, J = 11.3, 8.6 Hz, 1H), 3.48 (dd, J = 11.3, 9.6 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 170.8, 170.0, 167.4, 134.4, 132.1, 123.9, 78.1, 53.0, 39.8, 35.9. HRMS (ESI+) (m/z): [M + Na+]+ calcd. for C14H12N2O4SNa+, 327.0410; found, 327.0411.
:EtOAc 88:12 ≥ 0:100) to give methyl ester 41 as a brown foam (742 mg, 62%). [α]D −4° (c 0.0025, DCM); 1H-NMR (400 MHz, CDCl3) δ 7.98–7.91 (m, 2H), 7.76–7.69 (m, 4H), 7.67 (d, J = 8.3 Hz, 1H), 7.40–7.32 (m, 2H), 7.29 (t, J = 7.4 Hz, 1H), 7.22 (d, J = 6.9 Hz, 1H), 5.79 (s, 1H) 5.54 (dd, J = 8.5, 2.6 Hz, 1H), 4.07, 3.99 (ABq, JAB = 17.1 Hz, 2H), 3.78 (s, 3H), 3.73 (dd, J = 11.9, 8.4 Hz, 1H), 3.53 (dd, J = 11.9, 2.7 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ 167.9, 166.5, 166.4, 160.8, 154.2, 149.4, 134.7, 134.6, 133.8, 132.0, 131.6, 131.4, 131.3, 128.6, 128.1, 128.0, 126.4, 125.7, 125.4, 123.9, 123.9, 123.8, 115.5, 106.7, 63.9, 53.4, 34.7, 32.3. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C28H21N2O5S+, 497.1166; found, 497.1166.
:MeOH 99:1 ≥ 88:12 with 1% AcOH) to give carboxylic acid 42 as a brown solid (955 mg, 78%). 1H-NMR (400 MHz, DMSO-d6) δ 13.60 (br s, 1H), 7.98–7.86 (m, 5H), 7.83–7.73 (m, 2H), 7.48–7.34 (m, 3H), 7.33–7.28 (m, 1H), 5.50 (s, 1H), 5.46 (dd, J = 9.0, 2.4 Hz, 1H), 4.13, 4.07 (ABq, JAB = 17.4 Hz, 2H), 3.95 (dd, J = 11.9, 9.2 Hz, 1H), 3.59 (dd, J = 11.9, 2.1 Hz, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 169.0, 166.4, 166.2, 159.6, 154.0, 150.8, 135.2, 135.2, 133.4, 132.7, 131.2, 130.9, 128.6, 128.0, 127.6, 126.2, 125.7, 125.4, 123.9, 123.8, 123.7, 113.4, 105.3, 63.7, 33.8, 32.0. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H19N2O5S+, 483.1009; found, 483.1007.
:EtOAc 80:20 ≥ 0:100, the column was prewashed with heptane:EtOAc:TEA 80:20:2 to neutralize the silica gel) to give BOC-protected amine 43 as a pale yellow foam (610 mg, 57%). 1H-NMR (600 MHz, CDCl3) δ 7.82–7.76 (m, 2H), 7.71–7.67 (m, 4H), 7.63 (d, J = 8.3 Hz, 1H), 7.34–7.29 (m, 2H), 7.24 (t, J = 7.9 Hz, 1H), 7.17 (d, J = 7.0 Hz, 1H), 6.60 (t, J = 7.4 Hz, 1H), 5.77 (s, 1H), 5.73 (d, J = 7.8 Hz, 1H), 3.97 (s, 2H), 3.66 (dd, J = 11.8, 6.6 Hz, 1H), 3.28 (d, J = 11.8 Hz, 1H), 1.41 (s, 9H). 13C-NMR (151 MHz, CDCl3) δ 166.7, 166.4, 160.0, 153.9, 153.9, 149.0, 134.6, 133.7, 131.9, 131.5, 131.3, 131.2, 128.6, 128.0, 127.9, 126.3, 125.7, 125.3, 123.9, 123.9, 123.7, 115.8, 106.2, 80.8, 69.7, 36.2, 34.6, 28.2. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C31H28N3O5S+, 554.1744; found, 554.1742.
:MeOH 99:1 ≥ 90:10) to give amine 44 as a pale yellow foam (201 mg, 88%). 1H-NMR (600 MHz, CDCl3) δ 7.85–7.79 (m, 2H), 7.74–7.68 (m, 4H), 7.68 (d, J = 8.4 Hz, 1H), 7.36–7.30 (m, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.22 (d, J = 7.0 Hz, 1H), 5.93 (dd, J = 7.3, 1.9 Hz, 1H), 5.81 (s, 1H), 4.01 (s, 2H), 3.67 (dd, J = 11.9, 7.5 Hz, 1H), 3.12 (d, J = 11.9, 1.9 Hz, 1H), 2.59 (s, 2H). 13C-NMR (151 MHz, CDCl3) δ 166.7, 166.5, 161.3, 154.1, 148.1, 134.6, 133.8, 132.1, 131.6, 131.4, 131.3, 128.6, 128.1, 127.9, 126.4, 125.7, 125.4, 123.9, 123.8, 115.5, 106.2, 74.2, 35.5, 34.6. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C26H20N3O3S+, 454.1220; found, 454.1222.
:EtOAc 90:10 ≥ 20:80) to give a yellow oil containing the azide. Redissolved in a mixture of DMF (1.86 ml) and H2O (0.186 ml) and added sodium ascorbate (61.5 mg, 0.310 mmol), CuSO4 (24.8 mg, 0.155 mmol) and phenyl acetylene (28.4 μl, 0.257 mmol). The reaction was stirred at room temperature for 3.5 hours. Diluted with EtOAc and washed with brine. EDTA (50 eq.) was added and the water phase was then acidified to pH 5 and left to stir overnight. Extracted with EtOAc. The combined organic layers were dried (Na2SO4), filtered and concentrated. Purified using column chromatography on silica gel (heptane:EtOAc 88:12 ≥ 0:100) to give 1,2,3-triazole 45 as a colorless solid (9 mg, 1.4%). 1H-NMR (400 MHz, DMSO-d6) δ 8.70 (s, 1H), 8.01–7.89 (m, 4H), 7.89–7.82 (m, 3H), 7.81–7.73 (m, 2H), 7.52–7.38 (m, 5H), 7.36 (t, J = 7.4 Hz, 2H), 7.29 (d, J = 7.4 Hz, 1H), 5.52 (s, 1H), 4.32 (dd, J = 13.1, 7.6 Hz, 1H), 4.17, 4.10 (ABq, JAB = 17.4 Hz, 2H), 3.87 (d, J = 12.8 Hz, 1H). 13C-NMR (100 MHz, DMSO-d6) δ 166.3, 166.2, 159.1, 155.3, 151.4, 145.9, 135.3, 135.2, 133.4, 132.4, 131.1, 130.9, 130.1, 129.0, 128.6, 128.2, 128.1, 127.6, 126.3, 125.8, 125.5, 125.3, 124.0, 123.9, 123.6, 121.8, 113.6, 105.8, 74.1, 35.0, 33.9. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C34H24N5O3S+, 582.1594; found, 582.1593.
:MeCN 95:5 ≥ 0:100 with 0.75% formic acid over 40 minutes) and then freeze dried to give amine 46 as a colorless solid (7 mg, 99%). 1H-NMR (600 MHz, CDCl3) δ 8.20 (s, 1H), 7.87–7.75 (m, 5H), 7.48–7.35 (m, 5H), 7.34–7.30 (m, 2H), 7.24 (d, J = 7.1 Hz, 1H), 6.10 (s, 1H), 5.66 (s, 1H), 4.50, 4.33 (ABq, JAB = 17.7 Hz, 2H), 4.27 (d, J = 12.5 Hz, 1H), 4.06 (dd, J = 12.6, 7.3 Hz, 1H), 3.23 (s, 3H); 13C-NMR (151 MHz, CDCl3) δ 160.3, 157.9, 149.8, 148.1, 134.1, 132.6, 132.0, 129.9, 129.0, 128.9, 128.7, 128.4, 128.1, 126.7, 126.1, 126.0, 125.7, 124.2, 121.3, 116.1, 111.3, 73.8, 44.0, 35.7, 33.6. HRMS (ESI+) (m/z): [M + H+]+ calcd. for C27H24N5O3S2+, 530.1315; found, 530.1313.
Author contributions
S. B., Å. G., F. A. initiated the project. M. K., A. G. C., J. A. D. G., and A. E. G. L. developed the synthetic routes. M. K., A. G. C., J. A. D. G., A. E. G. L., and P. S. performed chemical synthesis and structural analysis of compounds. C. N.-O. tested the compounds in reinfect and cytotoxicity experiments. J. S. and W. B. did reinfect preliminary testing of first compounds. Å. G., J. S. and E. W. performed animal experiments. R. S. measured and calculated pharmacokinetic data. M. K., C. N.-O., S. B., Å. G., A. G. C., R. S., K. V. and F. A. analyzed data and wrote the manuscript.Abbreviations
| AUC | Area under curve |
| C. trachomatis | Chlamydia trachomatis |
| CL | Clearance |
| DIPEA | N,N-Diisopropylethylamine |
| DMEM | Dulbecco's modified eagle medium |
| EB | Elementary body |
| FBS | Fetal bovine serum |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| HBSS | Hanks balanced salt solution |
| IFU | Inclusion forming unit |
| MOI | Multiplicity of infection |
| MWI | Microwave irradiation |
| RB | Reticulate body |
| rt | Room temperature |
| TBTU | O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate |
| IV | Intravenously |
| PO | Per oss (oral) |
Conflicts of interest
Sven Bergström and Fredrik Almqvist are cofounders of Quretech Bio AB.Acknowledgements
We acknowledge funding from the Swedish Research Council (F. A. and S. B.), the Knut and Alice Wallenberg Foundation (F. A. and S. B.), the Göran Gustafsson Foundation (F. A.), the Kempe Foundation (F. A.), and the Swedish Foundation for Strategic Research (F. A.). We thank Ingela Nilsson and Berit Sondén for technical assistance and Fritiof Pontén for valuable comments on the manuscript.References
- World Health Organization, Global incidence and prevalence of selected curable sexually transmitted infections – 2008, 978 92 4 150383 9, World Health Organization, Geneva, Switzerland, 2012.
- J. A. Whittum-Hudson and A. P. Hudson, Human chlamydial infections: persistence, prevalence, and outlook for the future, Nat. Sci. Soc., 2005, 13, 371–382 CrossRef
.
- A. Dautry-Varsat, A. Subtil and T. Hackstadt, Recent insights into the mechanisms of Chlamydia entry, Cell. Microbiol., 2005, 7, 1714–1722 CAS
- A. Omsland, B. S. Sixt, M. Horn and T. Hackstadt, Chlamydial metabolism revisited: interspecies metabolic variability and developmental stage-specific physiologic activities, FEMS Microbiol. Rev., 2014, 38, 779–801 CrossRef CAS PubMed
- K. Hybiske and R. S. Stephens, Mechanisms of host cell exit by the intracellular bacterium Chlamydia, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11430–11435 CrossRef CAS PubMed
- W. M. Geisler, Diagnosis and Management of Uncomplicated Chlamydia trachomatis Infections in Adolescents and Adults: Summary of Evidence Reviewed for the 2015 Centers for Disease Control and Prevention Sexually Transmitted Diseases Treatment Guidelines, Clin. Infect. Dis., 2015, 61, S774–S784 CrossRef PubMed
- M. R. Hammerschlag and S. A. Kohlhoff, Treatment of chlamydial infections, Expert Opin. Pharmacother., 2012, 13, 545–552 CrossRef CAS PubMed
- C. Jernberg, S. Löfmark, C. Edlund and J. K. Jansson, Long-term impacts of antibiotic exposure on the human intestinal microbiota, Microbiology, 2010, 156, 3216–3223 CrossRef CAS PubMed
- P. Engström, K. S. Krishnan, B. D. Ngyuen, E. Chorell, J. Normark, J. Silver, R. J. Bastidas, M. D. Welch, S. J. Hultgren, H. Wolf-Watz, R. H. Valdivia, F. Almqvist and S. Bergström, A 2-pyridone-amide inhibitor targets the glucose metabolism pathway of Chlamydia trachomatis, mBio, 2014, 6, e02304–e02314 CrossRef PubMed
- J. A. Good, J. Silver, C. Nunez-Otero, W. Bahnan, K. S. Krishnan, O. Salin, P. Engstrom, R. Svensson, P. Artursson, A. Gylfe, S. Bergstrom and F. Almqvist, Thiazolino 2-pyridone amide inhibitors of Chlamydia trachomatis infectivity, J. Med. Chem., 2016, 59, 2094–2108 CrossRef CAS PubMed
- J. A. D. Good, M. Kulén, J. Silver, K. S. Krishnan, W. Bahnan, C. Núñez-Otero, I. Nilsson, E. Wede, E. de Groot, Å. Gylfe, S. Bergström and F. Almqvist, Thiazolino 2-pyridone amide isosteres as inhibitors of Chlamydia trachomatis infectivity, J. Med. Chem., 2017, 60, 9393–9399 CrossRef CAS PubMed
- B. N. Ames, J. McCann and E. Yamasaki, Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagenicity test, Mutat. Res., 1975, 31, 347–364 CAS
- M. Kulén, M. Lindgren, S. Hansen, A. G. Cairns, C. Grundström, A. Begum, I. van der Lingen, K. Brännström, M. Hall, U. H. Sauer, J. Johansson, A. E. Sauer-Eriksson and F. Almqvist, Structure-based design of inhibitors targeting PrfA, the master virulence regulator of Listeria monocytogenes, J. Med. Chem., 2018, 61, 4165–4175 CrossRef PubMed
- J. R. Dunetz, Y. Xiang, A. Baldwin and J. Ringling, General and Scalable Amide Bond Formation with Epimerization-Prone Substrates Using T3P and Pyridine, Org. Lett., 2011, 13, 5048–5051 CrossRef CAS PubMed
- V. Åberg, M. Sellstedt, M. Hedenström, J. S. Pinkner, S. J. Hultgren and F. Almqvist, Design, synthesis and evaluation of peptidomimetics based on substituted bicyclic 2-pyridones-targeting virulence of uropathogenic E. coli, Bioorg. Med. Chem., 2006, 14, 7563–7581 CrossRef PubMed
- M. D. Evans, J. Ring, A. Schoen, A. Bell, P. Edwards, D. Berthelot, R. Nicewonger and C. M. Baldino, The accelerated development of an optimized synthesis of 1,2,4-oxadiazoles: application of microwave irradiation and statistical design of experiments, Tetrahedron Lett., 2003, 44, 9337–9341 CrossRef CAS
- Code:logP(o/w). Molecular Operating Environment (MOE), version 2016.08, Chemical Computing Group, Quebec, Canada
- G. M. Castanedo, P. S. Seng, N. Blaquiere, S. Trapp and S. T. Staben, Rapid synthesis of 1,3,5-substituted 1,2,4-triazoles from carboxylic acids, amidines, and hydrazines, J. Org. Chem., 2011, 76, 1177–1179 CrossRef CAS PubMed
- J. Yang, M. Jamei, K. R. Yeo, A. Rostami-Hodjegan and G. T. Tucker, Misuse of the well-stirred model of hepatic drug clearance, Drug Metab. Dispos., 2007, 35, 501–502 CrossRef CAS PubMed
- H. D. Caldwell, J. Kromhout and J. Schachter, Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis, Infect. Immun., 1981, 31, 1161–1176 CAS
- S. Marwaha, H. Uvell, O. Salin, A. E. Lindgren, J. Silver, M. Elofsson and Å. Gylfe, N-acylated derivatives of sulfamethoxazole and sulfafurazole inhibit intracellular growth of Chlamydia trachomatis, Antimicrob. Agents Chemother., 2014, 58, 2968–2971 CrossRef PubMed
- I. Hubatsch, E. G. E. Ragnarsson and P. Artursson, Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers, Nat. Protoc., 2007, 2, 2111 CrossRef CAS PubMed
- K. Riccardi, S. Cawley, P. D. Yates, C. Chang, C. Funk, M. Niosi, J. Lin and L. Di, Plasma protein binding of challenging compounds, J. Pharm. Sci., 2015, 104, 2627–2636 CrossRef CAS PubMed
- H. Emtenäs, L. Alderin and F. Almqvist, An enantioselective ketene−imine cycloaddition method for synthesis of substituted ring-fused 2-pyridinones, J. Org. Chem., 2001, 66, 6756–6761 CrossRef PubMed
- P. Baranczewski, A. Stanczak, K. Sundberg, R. Svensson, Å. Wallin, J. Jansson, P. Garberg and H. Postlind, Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development, Pharmacol. Rep., 2006, 58(4), 453–472 CAS
Footnotes |
| † These authors contributed equally. |
| ‡ Division of Infection Medicine, BMC, Lund University, Sweden. |
| § ReViral Ltd, NETpark Plexus, Thomas Wright Way, Sedgefield, TS21 3FD, United Kingdom. |
| This journal is © The Royal Society of Chemistry 2019 |