
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh affinity rigidified AT2 receptor ligands with indane scaffolds†
Charlotta
Wallinder
a,
Christian
Sköld
a,
Sara
Sundholm
a,
Marie-Odile
Guimond
c,
Samir
Yahiaoui
a,
Gunnar
Lindeberg
a,
Nicole
Gallo-Payet
c,
Mathias
Hallberg
 *b and
Mathias
Alterman
a
*b and
Mathias
Alterman
a
aDepartment of Medicinal Chemistry, BMC, Uppsala University, P.O. Box 574, SE-751 23 Uppsala, Sweden
bThe Beijer Laboratory, Department of Pharmaceutical Biosciences, Division of Biological Research on Drug Dependence, BMC, Uppsala University, P.O. Box 591, SE-751 24 Uppsala, Sweden. E-mail: Mathias.Hallberg@farmbio.uu.se
cService of Endocrinology, Faculty of Medicine and Health Sciences, University of Sherbrooke, Sherbrooke, J1H 5N4 Quebec, Canada
First published on 18th November 2019
Abstract
Rigidification of the isobutyl side chain of drug-like AT2 receptor agonists and antagonists that are structurally related to the first reported selective AT2 receptor agonist 1 (C21) delivered bioactive indane derivatives. Four enantiomer pairs were synthesized and the enantiomers were isolated in an optical purity >99%. The enantiomers 7a, 7b, 8a, 8b, 9a, 9b, 10a and 10b bind to the AT2 receptor with moderate (Ki = 54–223 nM) to high affinity (Ki = 2.2–7.0 nM). The enantiomer with positive optical rotation (+) exhibited the highest affinity at the receptor. The indane derivatives 7b and 10a are among the most potent AT2 receptor antagonists reported so far. As illustrated by the enantiomer pairs 7a/b and 10a/b, an alteration at the stereogenic center has a pronounced impact on the activation process of the AT2 receptor, and can convert agonists to antagonists and vice versa.
Introduction
The hypertensive octapeptide angiotensin II (Ang II) activates mainly two receptor subtypes; the AT1 receptor and the AT2 receptor. Selective AT1 receptor antagonists have been used as antihypertensives (the Sartans)1,2 for more than 25 years, while neither AT2 receptor agonists nor antagonists have yet been launched as pharmaceuticals. An AT2 receptor antagonist, EMA401 (olodanrigan) was in clinical trials and aimed for management of neuropathic pain.3–5 Moreover, in recent years also AT2 receptor agonists have emerged as a potencial new class of pharmaceutical agents5,6 and activation of AT2 receptor results in a variety of physiological responses.6,7 The expression of the AT2 receptor is often low in healthy adults and is predominately observed in renal, cardiovascular, and brain tissues. However, during certain pathological conditions, such as myocardial infarction, vascular injury, brain ischemia, renal failure, and cutaneous wounds, the AT2 receptor expression is up-regulated.6,7 Notably, receptor activation stimulates neurite outgrowth in neuronal cells, one of the first steps in neuronal differentiation.8Peptides and pseudopeptides that act as selective AT2 receptor agonists have been known for a long time and used for comprehensive studies of physiological effects of AT2 receptor activation.9–17
In 2004, our group disclosed the first selective non-peptide and drug-like receptor AT2 receptor agonist C21/M024 (1)18 (Fig. 1). Subsequently a series of structurally related agonists were reported.19–23 Compound 1 has been evaluated in several experimental disease models and is entering phase II clinical trials as a potential treatment for idiopathic pulmonary fibrosis. For recent reviews and on the discovery of C21 (1) and brief surveys of its pharmacology, see references.24–26
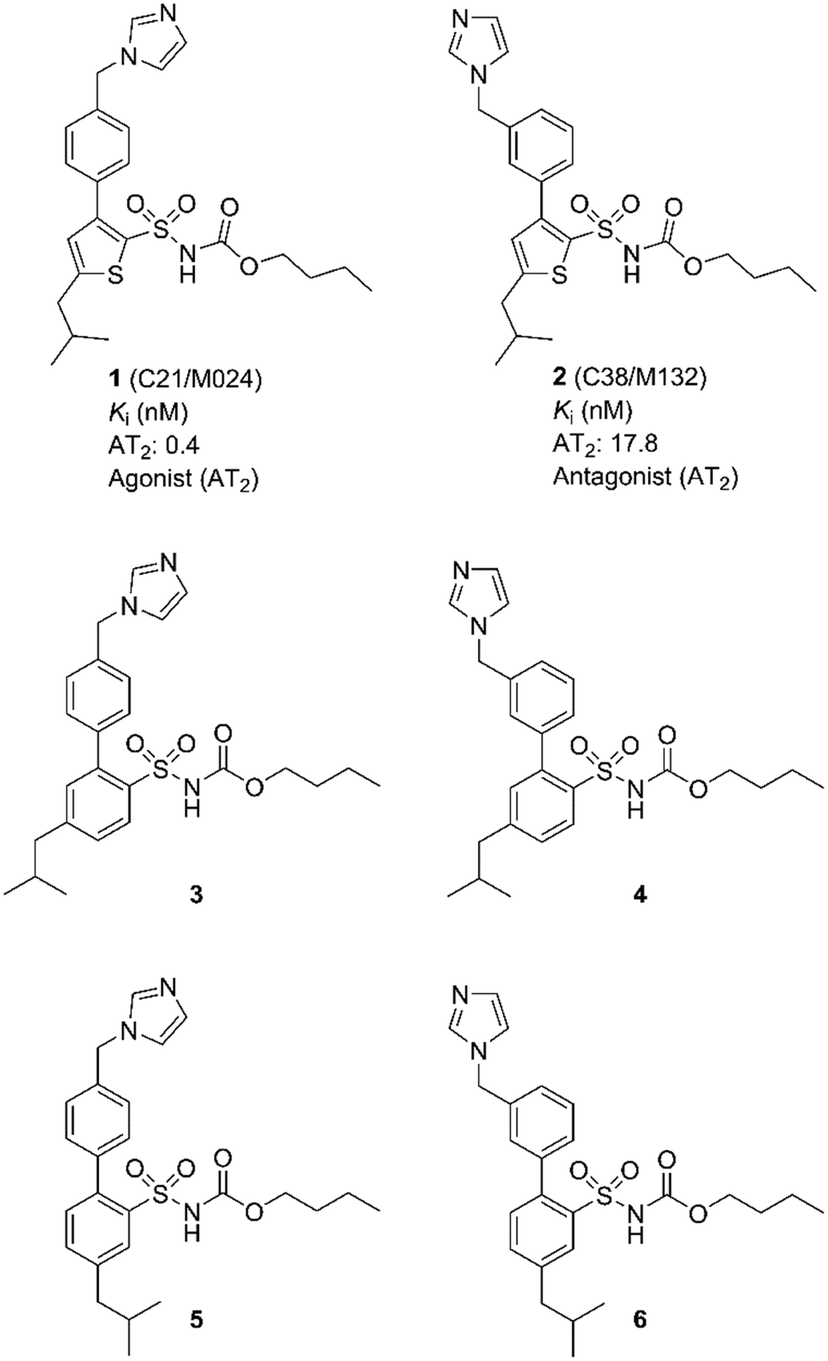 | ||
| Fig. 1 Structures of compounds 1–6. | ||
We observed that a migration of the methylene imidazole substituent from the para position of the agonist 1 to the meta position resulted in a compound C38/M132 (2) that acted as an AT2 receptor antagonist.27,28 Analogously, the biphenyl derivative 3 that is a potent AT2 receptor agonist was transformed to the antagonist 4 through the same structural manipulation (Fig. 1).29
Furthermore, we reported that both compound 5 and 6 with the methylene imidazole group in the para position and in the meta position, respectively, exhibited agonism although their binding affinities to the AT2 receptor were 10-fold lower. Thus, the relative positions of the methylene imidazole and the isobutyl substituent seemed of importance in determining the functional activity of this class of selective AT2 receptor biaryl derivatives (Fig. 1).29 It can be hypothesized that the imidazole rings of 1–6i.e. both agonists and antagonists, could bind to the same interaction point of the AT2 receptor, while the isobutyl groups of agonists and antagonists are forced to be positioned differently and cause the difference in functionality observed. Hence, the topography and location of the lipophilic side chains of 1–6 might be the primary determinants of functionality.
These data and the proposed hypothesis encouraged us to assess the impact and importance of the orientation of the isobutyl side chain in space for a) binding affinity to the AT2 receptor and b) functional response (agonism or antagonism). It is known that a structurally related non selective AT1 receptor agonist (L-162782) encompassing the same isobutyl group as in 1–6 was converted to a non selective AT1 receptor antagonist (L-162389) after deletion of one single methyl group from the side chain.30 Notably, regarding affinity to the AT1 receptor, there are no examples known of any AT2 receptor ligand encompassing the imidazole ring that is found in 1–6 that demonstrate any affinity at the AT1 receptor.
Eight enantiomers, with conformationally constrained isobutyl groups, 7a, 7b, 8a, 8b, 9a, 9b, 10a, 10b, were prepared and their ability to bind to the AT2 receptor and their capacity to activate or block the AT2 receptor were examined (Fig. 2). In essence, there are two classes of ligands: a class with a 1-isopropyl, 4-aryl, 5-sulfonyl arrangement (7 and 8) and a class with a 3-isopropyl, 6-aryl, 5-sulfonyl arrangement (9 and 10) with the aryl group substituted in either the 3′-position or the 4′-position. The isopropyl substituent in each forms a chiral centre.
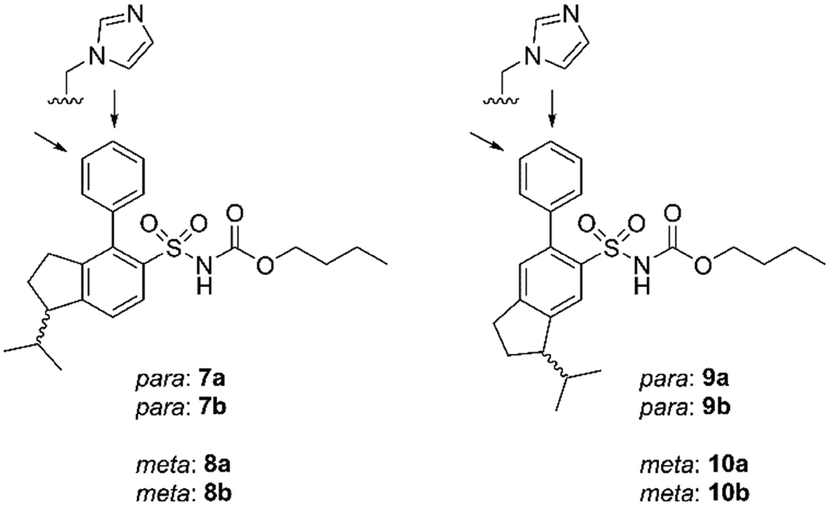 | ||
| Fig. 2 Structures of compounds 7a/b, 8a/b, 9a/b, 10a/b. | ||
Results
Chemistry
The synthesis of the rigidified analogues (7a/b, 8a/b, 9a/b and 10a/b) started with the synthesis of 1-isopropylindane (12), which was prepared essentially as described by Phillips et al.31 Briefly, 1-indanone was alkylated in a Grignard reaction with freshly prepared 2-propylmagnesium bromide. Spontaneous elimination of the alcohol group upon acidic work-up rendered the unsaturated compound 11 which was hydrogenated directly using PtO2 as the catalyst. The saturated compound 12 was isolated in a total yield of 52%. Compound 12 was subsequently treated with chlorosulfonic acid to yield the positional isomers 13 and 14 in 43%. These two compounds were separated by column chromatography and isolated in 16% and 14% yield, respectively. The isolated sulfonyl chlorides 13 and 14 were directly protected with tert-butylamine to provide the sulfonamides 15 and 16, in excellent yields. The enantiomers of 15 and 16 were separated using chiral preparative HPLC (Scheme 1). | ||
| Scheme 1 Reagents: (a) 2-propylmagnesium bromide, diethyl ether, toluene, HCl (aq); (b) H2 (40 psi), PtO2, ethanol; (c) chlorosulfonic acid, DCM; (d) tert-butylamine, DCM. | ||
Chiral GC-MS was used to determine the enantiomeric purity of the isolated (+) (15a and 16a) and (−) (15b and 16b) forms of 15 and 16, which in both cases proved to be >99%. The pure enantiomers were transformed into the corresponding boronic acids by selective ortho-lithiation and subsequent boronation and were used in the next step without further purification.31 The crude boronic acids were coupled with compounds 17 and 18, which were prepared by the N-alkylation of imidazole by 3- and 4-bromobenzylbromide, respectively. The subsequent Suzuki coupling reactions performed by microwave irradiation (150 °C, 5 min)32 afforded compounds 21a/b, 22a/b in Scheme 2 and compounds 24a/b, 25a/b in Scheme 3. Compounds 21a/b and 22a/b were isolated by HPLC as the TFA salts in moderate yields. We were unfortunately not able to isolate the Suzuki products from boronic acids 20a/b. Compounds 16a and 16b gave rise to only one boronic acid each (compounds 23a and 23b), probably due to steric hindrance induced by the isopropyl group (Scheme 3).
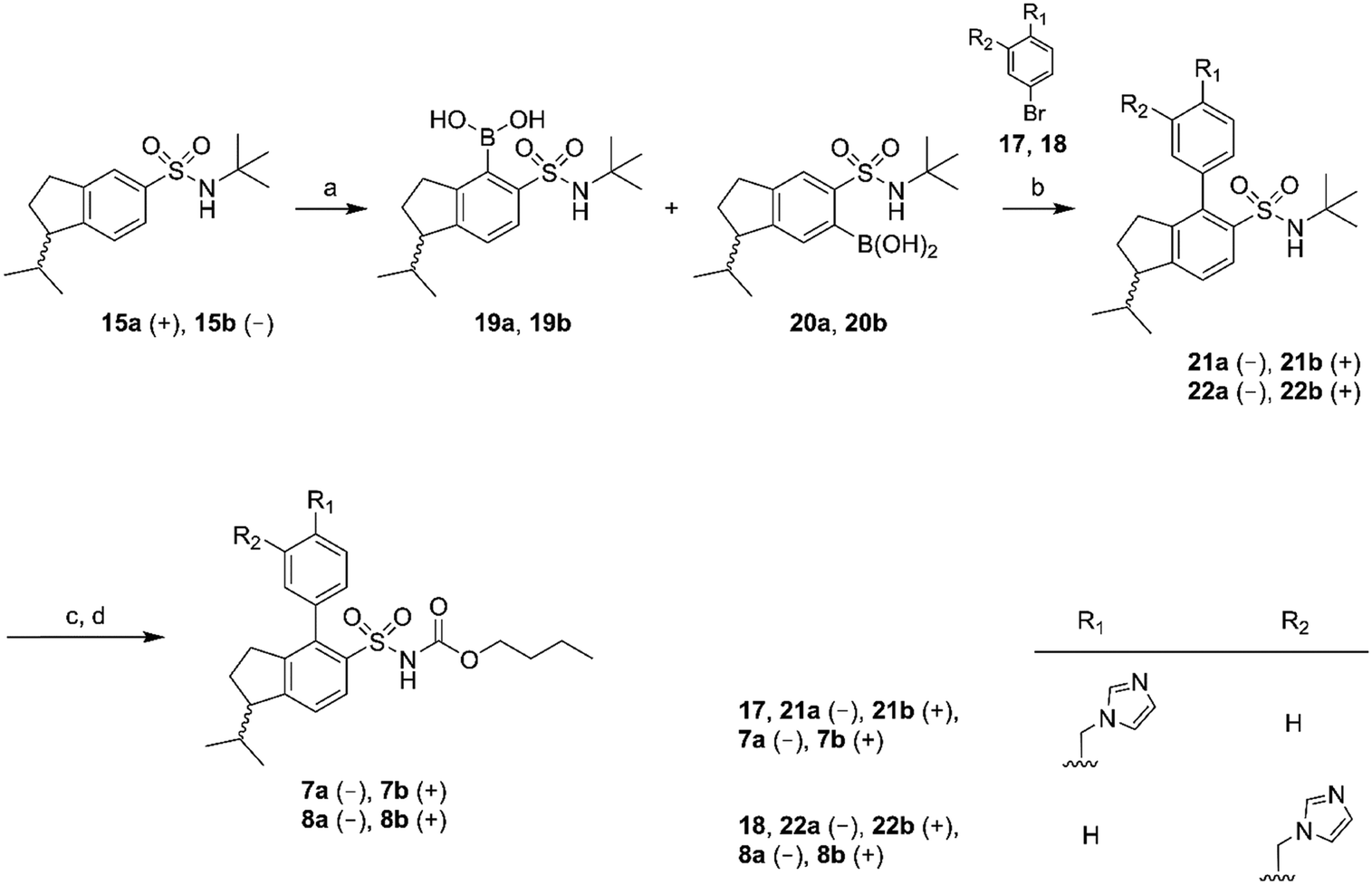 | ||
| Scheme 2 Reagents: (a) n-BuLi, THF, triisopropyl borate, HCl (aq); (b) Pd(PPh3)4, Na2CO3, toluene, ethanol, H2O; (c) BCl3, DCM; (d) n-butyl chloroformate, Na2CO3, DCM, H2O. | ||
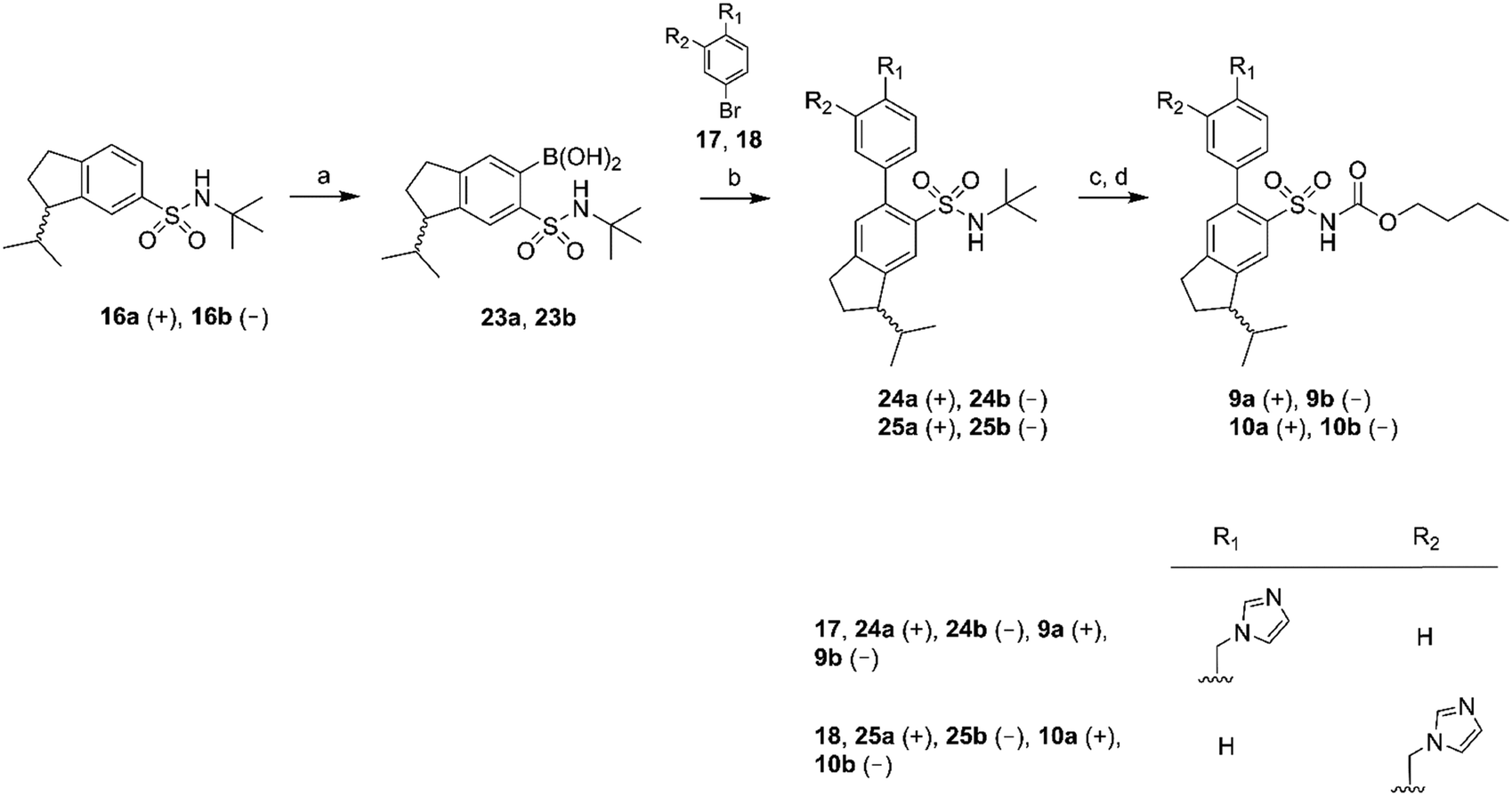 | ||
| Scheme 3 Reagents: (a) n-BuLi, THF, triisopropyl borate, HCl (aq); (b) Pd(PPh3)4, Na2CO3, toluene, ethanol, H2O; (c) BCl3, DCM; (d) n-butyl chloroformate, Na2CO3, DCM, H2O. | ||
All isolated Suzuki products (21a/b, 22a/b, 24a/b and 25a/b) were deprotected with BCl3 (ref. 33) and subsequently acylated with n-butyl chloroformate in dichloromethane, water and Na2CO3 to yield the final products 7a/b, 8a/b, 9a/b and 10a/b in 48–61% yield, after purification by preparative HPLC (Schemes 2 and 3).
Binding assay
The indane analogues, compounds 7a/b, 8a/b, 9a/b and 10a/b, were evaluated for AT2 receptor affinity in radio-ligand assays by displacement of [125I]Ang II from AT2 receptors in pig uterus membrane as previously described.34 The natural substrate Ang II and the selective AT1 receptor antagonist losartan were used as reference substances.35 The evaluation of the rigidified enantiomers was focused on AT2 receptor affinity, since we previously observed that no affinity to the AT1 receptor is retained when modifying the lower part of this structure class.19 The results of the binding studies are presented in Table 1. In summary, the 1-isopropyl, 4-aryl, 5-sulfonyl arrangement is less suited to binding than the 3-isopropyl, 6-aryl, 5-sulfonyl arrangement, which all retain potent affinity. C21 (1) exhibited a Ki value of 0.4 nM in the same assay, as previously reported.18| Compound | Optical rotation | K i ± SD (nM) | Function (10 nM) |
|---|---|---|---|
| 7a | (−) | 127.4 ± 4.6 | Agonist |
| 7b | (+) | 2.8 ± 0.2 | Antagonist |
| 8a | (−) | 223.1 ± 5.8 | Antagonist |
| 8b | (+) | 53.5 ± 1.0 | Antagonist |
| 9a | (+) | 2.2 ± 0.2 | Agonist |
| 9b | (−) | 5.9 ± 0.1 | Agonist |
| 10a | (+) | 3.2 ± 0.1 | Antagonist |
| 10b | (−) | 7.0 ± 0.2 | Agonist |
Functional assay
All rigidified compounds were evaluated for functionality in a neurite outgrowth assay with NG108-15 cells. We have previously shown that NG108-15 cells express mainly the AT2 receptor and that a three-that treatment with Ang II or the selective peptidic AT2 receptor agonist CGP-42112 (Nα-nicotinoyl-Tyr-(Nα-Cbz-Arg)-Lys-His-Pro-Ile)36,37 induces neurite outgrowth.8,28The signaling pathways involve a sustained increase in Rap1/B-Raf/p42/p44mapk activity and activation of the nitric oxide/guanylyl cyclase/cGMP pathway.38,39 Cells were treated in the absence or presence of Ang II, 7a/b, 8a/b, 9a/b and 10a/b. Single concentration determinations were applied. After three days of treatment, cells were examined under a phase-contrast microscope and micrographs were taken. To establish an adequate test concentration, the compounds were evaluated at various concentrations ranging from 0.1 nM to 100 nM. It was only at the highest concentration of compounds 7a/b that any evidence of cell death was observed, and that observation was most probably due to a higher concentration of DMSO (verified by a control test, data not shown). All compounds were tested at a concentration of 10 nM, the antagonists (all but 7b) were also tested at 100 nM to verify that the lack of neurite outgrowth was not attributed to too low concentration and the higher concentration did not change the results (data not shown). Co-incubation with the selective AT2 receptor antagonist PD 123319 reduced the induced neurite outgrowth, verifying that the effect was mediated through the AT2 receptor. Treatment with PD 123319 alone did not alter the morphology compared to untreated cells (data not shown), as previously reported.28 The antagonistic effect was verified through co-incubation with Ang II resulting in reduced neurite outgrowth. The functionalities are summarised in Table 1 and an overview is presented in Fig. 6. A brief comparison of the functionalities of the compounds examined is available in the Discussion, vide infra. The neurite outgrowth results are shown in Fig. 3 and 4.
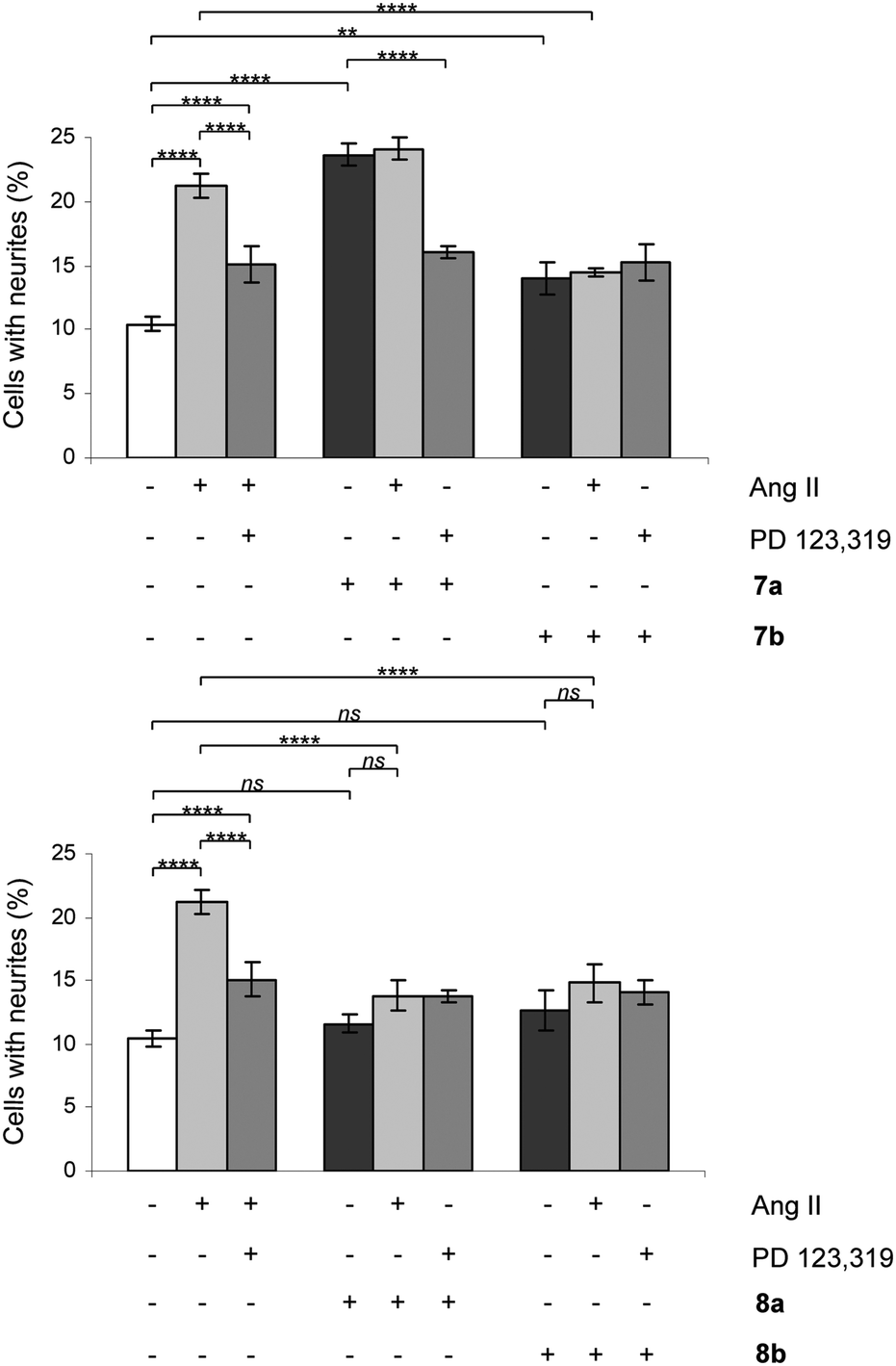 | ||
| Fig. 3 Effect of compounds 7a/b and 8a/b on neurite outgrowth in NG108-15 cells. The cells were plated at a cell density of 3.6 × 104 cells/Petri dishes (35 mm) and were cultured for three days in the absence or presence of 100 nM Ang II, 10 nM 7a, 10 nM 7b, 10 nM 8a or 10 nM 8b alone or in combination with 10 μM PD 123319 or 100 nM Ang II. Cells with at least one neurite longer than a cell body were counted as positive for neurite outgrowth. The number of cells with neurites was expressed as the percentage of the total number in the micrographs (at least 400 cells according to the experiment). The results are significant according to two-way ANOVA: ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; ns = not significant. | ||
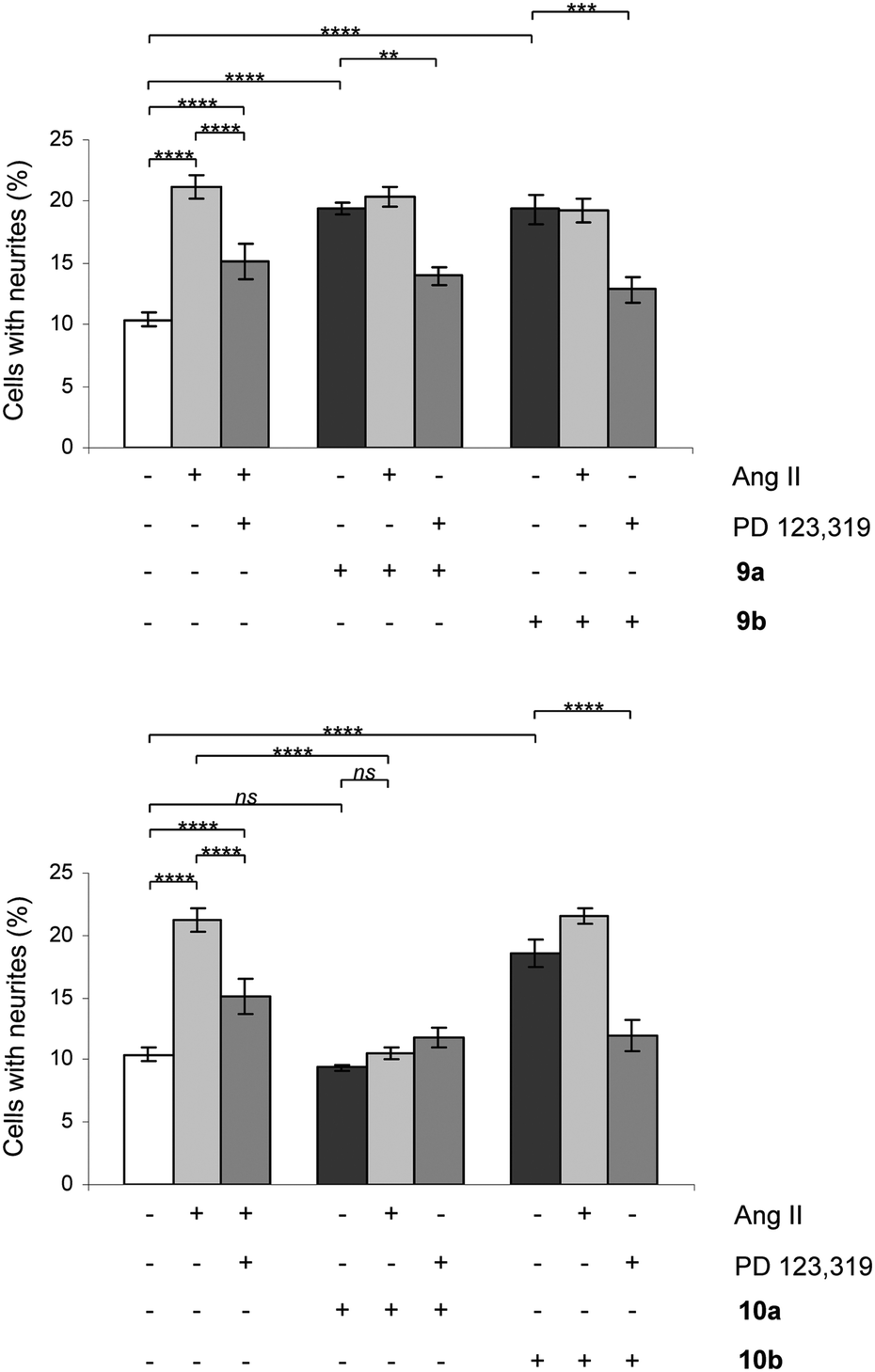 | ||
| Fig. 4 Effect of compounds 9a/b and 10a/b on neurite outgrowth in NG108-15 cells. The cells were plated at a cell density of 3.6 × 104 cells/Petri dishes (35 mm) and were cultured for three days in the absence or presence of 100 nM Ang II, 10 nM 9a, 10 nM 9b, 10 nM 10a or 10 nM 10b alone or in combination with 10 μM PD 123319 or 100 nM Ang II. Cells with at least one neurite longer than a cell body were counted as positive for neurite outgrowth. The number of cells with neurites was expressed as the percentage of the total number in the micrographs (at least 400 cells according to the experiment). The results are significant according to two-way ANOVA: ****, p < 0.0001; ***, p < 0.001; **, p < 0.01; ns = not significant. | ||
Discussion
The two rigidified enantiomers 7a and 7b displayed large differences in the biological properties as compared to the non rigidified analogue 3. Compound 7a (−) lost almost 200 times in affinity, but retained a strong agonistic effect, while the other enantiomer 7b (+) retained high affinity (Ki = 2.8 nM) but the functionality was interconverted to antagonism (Table 1 and Fig. 3). The enantiomer pair 8a/b, resulting from the rigidification of 4, both lost affinity compared to 4, where compound 8a (−) showed the highest loss. Notably, both compounds retained the antagonistic function (Table 1 and Fig. 3).Upon rigidification of the biphenyl analogues with the isobutyl chain in the meta position in relation to the sulfonylcarbamate group all compounds showed a slightly higher affinity (Table 1). The enantiomers 9a and 9b, resulting from the rigidification of 5, both retained the agonistic properties. The enantiomer pair from the rigidification of 6, 10a/b, on the other hand demonstrated opposite effects, the (−) enantiomer 10b retained the agonistic effect while the (+) enantiomer 10a functioned as an antagonist (Table 1 and Fig. 4). Ang II stimulation after pretreatment with 10a gave the same percentage of neurite outgrowth as untreated control cells, while pretreatment with PD 123319 resulted in a significantly increased number of stimulated cells (****, p < 0.0001). This could be an indication that, as previously shown for compound 4,29 this class of AT2 receptor antagonists is more efficient in inhibiting Ang II induced neurite outgrowth compared to PD 123319.
From a comparison of the agonist 1 and the antagonist 2 it seems evident that the position of the methyleneimidazole side chain might affect the functional response of this class of compounds.
Thus, the reason for the observed difference in functionality could be attributed to different positions and molecular interactions of the imidazole ring with the AT2 receptor, when having all other structural features superimposed.
From molecular modeling an alternative hypothesis emerges and suggests that the difference in functionality could instead be due to the positioning of the isobutyl side chain. When the imidazole rings and acidic nitrogen in the sulfonylcarbamate group are superimposed the isobutyl side chains of 1 and 2 are located differently according to modeling. We decided to study this structural feature of AT2 receptor ligands in some more detail in an attempt to elucidate structural requirements for agonistic and antagonistic response. We reported that independent of the methylene imidazole position, both an antagonist (4) and an agonist (6) could be obtained by moving the isobutyl side chain.29 This suggests that the structural feature responsible for functionality is the spatial relationship between the imidazole ring and the isobutyl side chain.
To further delineate the structural features important for the functional response, analogues with rigidified alkyl groups mimicking the isobutyl side chain were synthesized and evaluated. The resulting compounds were enantiomeric pairs with different directions of the isobutyl mimicking group, see Table 1 and Fig. 5. In the case of compounds 5 and 6, with the isobutyl group in the meta position to the sulfonylcarbamate group, the rigid analogues 9a/b and 10a/b respectively, showed an improved affinity to the receptor, possibly due to an improved fit to the receptor. However, in the case of the para substituted isobutyl compounds 3 and 4, the affinity decreased for the rigidified analogues, 7a/b and 8a/b respectively. The agonist 7a lost nearly 200 times in affinity even though it induced the highest degree of neurite outgrowth among this set of compounds (Fig. 5). A rationale for this observation is not obvious. The antagonist 7b on the other hand turned out to be the antagonist with the highest affinity found in this compound class. All selective nonpeptide AT2 receptor antagonists we have reported so far have exhibited lower affinities than the corresponding agonists. The antagonists 7b and 10a are indeed the first compounds to show Ki values in the single digit nM range.27,29 For comparison the AT2R antagonists 2 (C38/M132) and 4 display Ki values of 17.8 nM and 12.3 nM, respectively and PD123319 that is structurally very different from the ligands discussed herein exhibits an IC50 of 34 nM.18,40
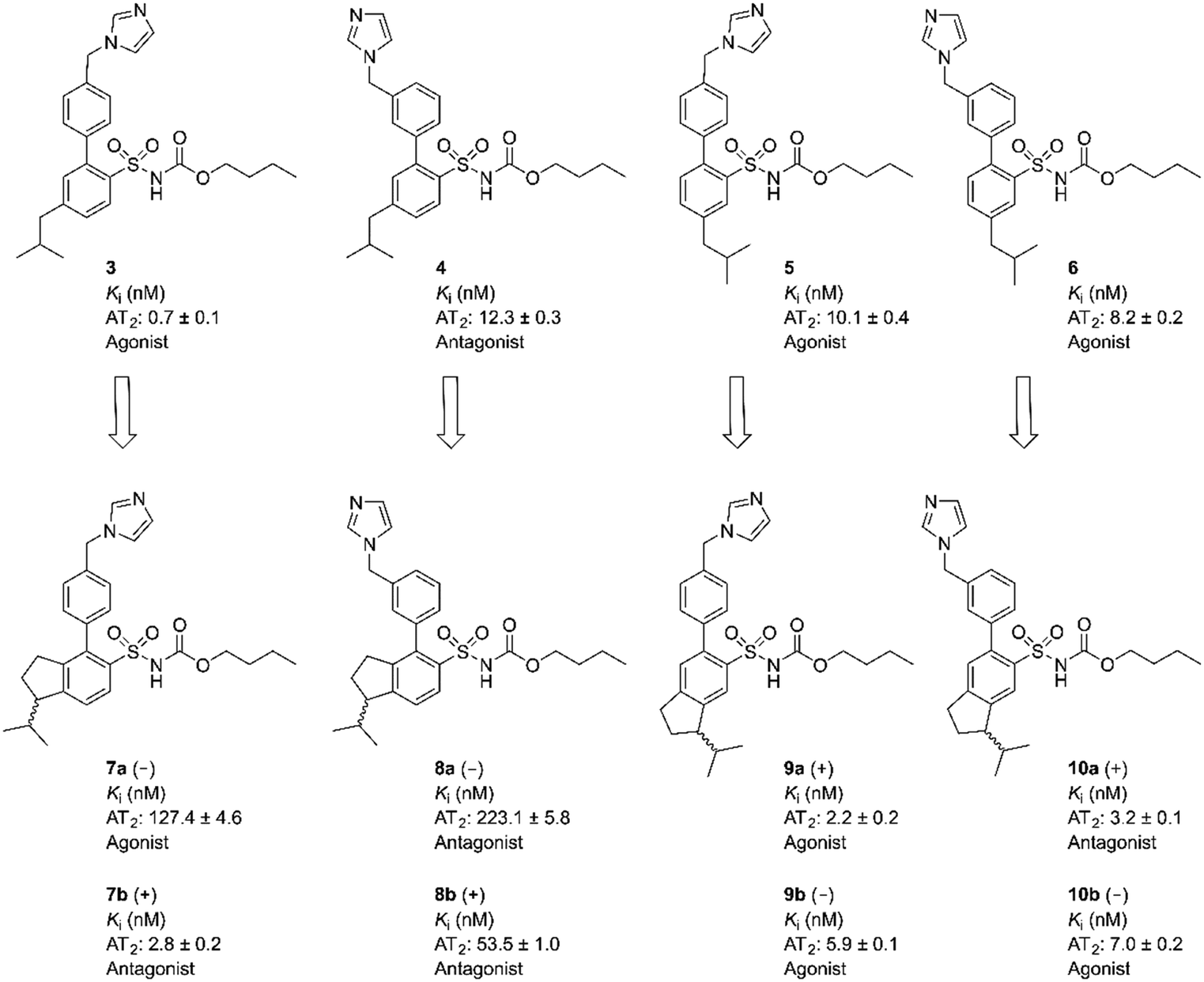 | ||
| Fig. 5 Overview over the investigated structures and their biological activities at the AT2 receptor. | ||
We have previously observed potent inhibition of Ang II-induced neurite outgrowth from this class of AT2 receptor antagonists at low concentrations, as compared to the corresponding Ki-values.27–29 One hypothesis for this effect may be that a partial inhibition of the signaling pathway is sufficient to completely inhibit neurite outgrowth, the final effect of AT2 receptor stimulation in NG108-15 cells. This hypothesis is in agreement with the observation that the TrkA inhibitor α-cyano-(3,5-di-t-butyl-4-hydroxy)thiocinnamide (AG879)41 only partially inhibits the p42/p44mapk pathway, but that its effect on neurite outgrowth is total.42 When studying the effect of the antagonist 2 on AT2 receptor signaling pathway a higher level of inhibition was observed at 100 nM compared to 10 nM (inhibition of p42/p44mapk and Rap1), while the difference between the two concentrations was less pronounced in the inhibition of neurite outgrowth induced by Ang II or compound 1 (C21/M024).28 Thus a partial occupation of AT2 receptor binding sites by these new antagonists could induce partial inhibition of the signaling pathway and, therefore, inhibit Ang II-induced neurite outgrowth. Nevertheless, since the functional experiments were performed on intact cells, the possibility of interactions with other growth stimulating systems cannot be excluded. Thus, not only AT2R but also interactions with other endogenous receptors might account, at least partly for the observed functional response exerted by this series of compounds.
The present study revealed that the functional response was sensitive to the direction of the isobutyl side chain. In two cases agonists with an isobutyl chain in the para and meta positions in relation to the sulfonyl carbonate group (3 and 6) were rigidified to analogues, which act as antagonists, compound 7b (+) and compound 10a (+), respectively. The reason for this is presumably that the bioactive conformation responsible for stabilizing the active conformation of the AT2 receptor cannot be adopted by 7b and 10a.
However, the structural difference between the enantiomers 7a (agonist) and 7b (antagonist) and the enantiomers 10a (antagonist) and 10b (agonist), respectively, are small. To visualize and exemplify the small structural differences, low energy conformations of model compounds of these enantiomer pairs are shown superimposed in Fig. 6 (see ESI† for details). All-atom superposition resulted in RMSD = 2.64 Å and 3.02 Å for 7a/b and 10a/b model structure conformations, respectively. However, the main structural differences clearly reside in the area of the isopropyl substituent, showed by RMSD = 0.62 Å and 0.63 Å, respectively, when excluding the aliphatic part of the indane moiety and the isopropyl substituent in the RMSD calculations. In two cases the functional response was unaffected by rigidification of the isobutyl side chain (4 to 8a/b and 5 to 9a/b, Fig. 6). Thus, the spatial relationship between the structural features in 3 and 6 are sensitive to the direction of the isopropyl group regarding functional response while the spatial arrangement in 4 and 5 render no difference in functionality attributed to the direction of the isopropyl side chain (Fig. 5). This suggests that it is not only the position of the methyleneimidazole, but the spatial relationship between the imidazole ring and the isobutyl side chain that determine the functional activity. Thus, a minor structural alteration can convert these AT2 receptor ligands from agonists to antagonists. Observations that subtle structural modification is sufficient to convert functionalities of nonpeptide ligands to peptide-activated GPCRs have previously been reported.30,43,44
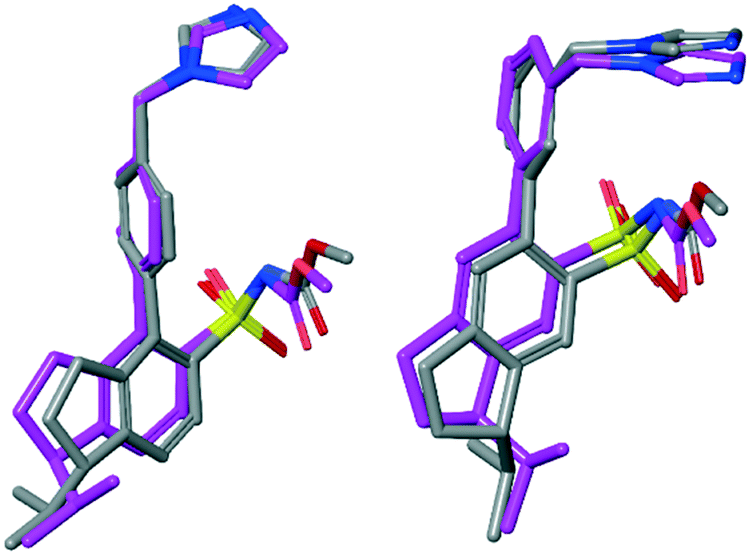 | ||
| Fig. 6 Low energy conformations of enantiomer pairs superimposed visualizing the small structural difference between an agonist and an antagonist. Left: Model compounds of 7a (−) (agonist) and 7b (+) (antagonist) superimposed. Right: Model compounds of 10a (+) (antagonist) and 10b (−) (agonist) superimposed. | ||
Concerning peptide-activated GPCRs, and the corresponding endogenous peptide ligands, although the latter are often large, the actual activating or effector domain of the peptide is frequently relatively small and other domains of the full length peptide are required to reach high affinity and selectivity.45 The fact that [Ile8]AngII acts as an antagonist (or partial agonist) at the AT1 receptor while the native [Phe8]AngII acts an agonist, constitute one example from the renin–angiotensin system (RAS) that small changes in the effector domain of peptides can have a large impact on functionality.46 Notable, the classic antihypertensive peptides and AT1 receptor antagonists saralasin and sarile with lipophilic side chains in the C-terminal, possibly interacting with the same binding site as the isopropyl group of the nonpeptides 7–10 act as agonists at the AT2 receptor.15
Conclusion
In summary, it is demonstrated herein that rigidification of the isobutyl side chain, common in AT2 receptor ligands, by incorporation of an indane scaffold delivers ligands that bind to the AT2 receptor with moderate to high affinity. In all four cases of enantiomer pairs synthesized and studied (7a/b, 8a/b, 9a/b and 10a/b), the enantiomer with positive optical rotation (+) exhibited the highest affinity at the receptor. To the best of our knowledge, 7b and 10a are the most potent nonpeptide AT2 receptor antagonists reported. As illustrated by the enantiomer pairs 7a/b and 10a/b, a minor alteration at the stereogenic center can make a great impact on the receptor activation process of the AT2 receptor, that analogously previously was demonstrated in the case of two AT1 receptor ligands. Although, we were never able to determine the absolute configuration of the isolated pure enantiomers, the results presented herein illustrate that small alterations in the topography of the ligands exert a pronounced impact on both affinity and functionality.Experimental part
Chemistry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Na2CO3 was added and the reaction mixture was cooled to 0 °C and n-butyl chloroformate was added and the reaction was left to reach room temperature over night. The reaction mixture was diluted with CHCl3 organic and the phases were separated. The organic phase was washed with water and brine, dried over MgSO4 and evaporated. The residue was purified on preparative RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give the pure products 7a, 7b, 8a, 8b, 9a, 9b, 10a and 10b.
:EtOAc 10:1) to provide compound 13 in 16% yield (1.09 g, mmol) and compound 14 in 14% yield (0.98 g, mmol). The total yield of the product mixture reached 43% (2.94 g, 11.4 mmol, 13:mix:14 = 1:0.8:0.9).
:1 mixture of isohexane and isopropanol and separated on chiral, preparative HPLC (isohexane/isopropanol 95:5 for compound 15 and 93:7 for compound 16) to give the pure enantiomers 15a, 15b, 16a and 16b. The enantiomeric purity of all isolated enantiomers were >99% according to chiral GC-MS (see ESI† for chromatograms).
:mix:15b, 1:1:1). 15a: [α]22D = +11.7° (c, 1.025, CHCl3); 15b: [α]22D = −12.2° (c, 1.080, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.70–7.67 (m, 2H), 7.27–7.24 (m, 1H), 4.53 (brs, 1H), 3.17–3.12 (m, 1H), 2.98–2.81 (m, 2H), 2.18–2.08 (m, 2H), 1.96–1.87 (m, 1H), 1.23 (s, 9H), 1.00 (d, J = 7.0 Hz, 3H), 0.76 (d, J = 7.0 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 151.6, 145.8, 141.4, 125.2, 124.6, 123.0, 54.7, 51.2, 31.7, 30.9, 30.3, 26.6, 21.2, 17.7. Anal. calcd. for C16H25NO2S: C, 65.05; H, 8.53; N, 4.47. Found (15a): C, 64.87; H, 8.52; N, 4.61. Found (15b): C, 65.14; H, 8.66; N, 4.56.
:mix:16b, 1:0.5:0.8). 16a: [α]23D = +33.4° (c, 0.800, CHCl3); 16b: [α]23D = −32.7° (c, 1.045, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.69–7.65 (m, 2H), 7.29–7.25 (m, 1H), 4.52 (brs, 1H), 3.18–3.13 (m, 1H), 2.98–2.81 (m, 2H), 2.20–2.08 (m, 2H), 1.95–1.86 (m, 1H), 1.21 (s, 9H), 1.00 (d, J = 6.8 Hz, 3H), 0.76 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.8, 147.6, 141.2, 125.6, 124.8, 123.1, 54.6, 51.0, 31.9, 30.9, 30.3, 26.7, 21.1, 17.7. Anal. calcd. for C16H25NO2S: C, 65.05; H, 8.53; N, 4.47. Found (16a): C, 64.89; H, 8.43; N, 4.43. Found (16b): C, 64.83; H, 8.57; N, 4.54.
:MeOH, 20:1) and about half of the product mixture was subsequently separated by RP-HPLC (H2O/MeCN with 0.1% TFA) to yield the pure compound 21a as the TFA salt in >13% (53 mg, 0.094 mmol). An analytical sample was desalted by washing with 2 M Na2CO3 for determination of optical rotation and NMR analysis. [α]21D = −23.6° (c, 0.315, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.98 (d, J = 8.1 Hz, 1H), 7.59–7.58 (m, 1H), 7.39–7.37 (m, 1H), 7.34–7.32 (m, 1H), 7.26 (dd, J = 8.1, 1.0 Hz, 1H), 7.24–7.19 (m, 2H), 7.13–7.12 (m, 1H), 6.95–6.94 (m, 1H), 5.18 (s, 2H), 3.43 (s, 1H), 3.22–3.17 (m, 1H), 2.53–2.40 (m, 2H), 2.22–2.11 (m, 1H), 2.09–2.00 (m, 1H), 1.85–1.76 (m, 1H), 1.07 (s, 9H), 1.01 (d, J = 6.8 Hz, 3H), 0.78 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 151.5, 146.0, 140.1, 138.1, 137.6, 136.1, 135.2, 130.7, 130.6, 130.2, 127.0, 126.95, 126.90, 123.6, 119.5, 54.6, 51.6, 50.6, 31.6, 30.8, 30.1, 26.3, 21.2, 17.7. HRMS (M + H+): 452.2366 C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and about half of the product mixture was subsequently separated by RP-HPLC (H2O/MeCN with 0.1% TFA) to yield the pure compound 21b as the TFA salt in >14% (58 mg, 0.103 mmol). An analytical sample was desalted by washing with 2 M Na2CO3 for determination of optical rotation and NMR analysis. [α]22D = +18.2° (c, 0.550, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.97 (d, J = 8.1 Hz, 1H), 7.59–7.58 (m, 1H), 7.39–7.36 (m, 1H), 7.34–7.31 (m, 1H), 7.26 (dd, J = 8.1, 0.9 Hz, 1H), 7.24–7.18 (m, 2H), 7.13–7.11 (m, 1H), 6.96–6.94 (m, 1H), 5.2 (s, 2H), 3.4 (s, 1H), 3.22–3.16 (m, 1H), 2.53–2.40 (m, 2H), 2.22–2.10 (m, 1H), 2.09–1.99 (m, 1H), 1.85–1.75 (m, 1H), 1.07 (s, 9H), 1.01 (d, J = 6.9 Hz, 3H), 0.78 (d, J = 6.9 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 151.5, 146.0, 140.1, 138.1, 137.6, 136.1, 135.2, 130.7, 130.5, 130.1, 127.0, 126.9, 126.9, 123.6, 119.5, 54.6, 51.6, 50.6, 31.5, 30.8, 30.1, 26.3, 21.2, 17.7. HRMS (M + H+): 452.2377 C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and about half of the product mixture was subsequently separated by RP-HPLC (H2O/MeCN with 0.1% TFA) to yield the pure compound 22a as the TFA salt in >19% (74 mg, 0.131 mmol). An analytical sample was desalted by washing with 2 M Na2CO3 for determination of optical rotation and NMR analysis. Compound 22a was observed as two rotamers at 24 °C in NMR in the ratio 1:1. Increasing the temperature to 70 °C (CD3CN) resulted in one set of peaks, a part from one of the methylgroups which had not fully merged. The spectra recorded at 24 °C are reported here. [α]22D = −19.8° (c, 0.430, CHCl3). 1H NMR (CDCl3, 24 °C), δ, ppm: 7.99–7.95 (m, 1H), 7.57–7.54 (m, 1H), 7.45–7.39 (m, 1H), 7.34–7.24 (m, 3H), 7.20–7.13 (m, 1H), 7.09–7.07 (m, 1H), 6.97–6.93 (m, 1H), 5.15 (s, 2H), 3.44–3.40 (m, 1H), 3.22–3.16 (m, 1H), 2.54–2.12 (m, 2H), 2.21–2.12 (m, 1H), 2.09–1.98 (m, 1H), 1.85–1.75 (m, 1H), 1.07–1.04 (m, 9H), 1.03–0.99 (m, 3H), 0.80–0.78 (m, 3H). 13C NMR (CDCl3, 24 °C), δ, ppm: 151.5, 145.9, 140.03, 140.00, 138.81, 138.74, 137.5, 136.4, 136.3, 135.25, 135.21, 130.1, 129.7, 129.6, 129.5, 129.3, 128.8, 128.7, 127.02, 126, 99, 126.97, 126.94, 123.64, 123.60, 119.4, 54.58, 54.56, 51.63, 51.56, 50.83, 50.81, 31.6, 31.5, 30.9, 30.6, 30.10, 30.08, 26.4, 26.3, 21.22, 21.15, 17.72, 17.68. HRMS (M + H+): 452.2371 C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and about half of the product mixture was subsequently separated by RP-HPLC (H2O/MeCN with 0.1% TFA) to yield the pure compound 22b as the TFA salt in >22% (91 mg, 0.161 mmol). An analytical sample was desalted by washing with 2 M Na2CO3 for determination of optical rotation and NMR analysis. Compound 22b was observed as two rotamers at 24 °C in NMR in the ratio 1:1. Increasing the temperature to 70 °C (CD3CN) resulted in one set of peaks a part from one of the methylgroups which had not fully merged. The spectra recorded at 24 °C are reported here. [α]22D = +18.0° (c, 0.615, CHCl3). 1H NMR (CDCl3, 24 °C), δ, ppm: 7.99–7.97 (m, 1H), 7.68–7.55 (m, 1H), 7.45–7.40 (m, 1H), 7.34–7.25 (m, 3H), 7.21–7.14 (m, 1H), 7.12–7.08 (m, 1H), 6.99–6.94 (m, 1H), 5.16 (s, 2H), 3.40–3.38 (m, 1H), 3.22–3.17 (m, 1H), 2.54–2.33 (m, 2H), 2.22–2.12 (m, 1H), 2.09–1.99 (m, 1H), 1.85–1.76 (m, 1H), 1.06–1.05 (m, 9H), 1.08–1.00 (m, 3H), 0.82–0.77 (m, 3H). 13C NMR (CDCl3, 24 °C), δ, ppm: 151.5, 145.9, 140.03, 139.99, 138.8, 138.7, 137.5, 136.34, 136.26, 135.25, 135.20, 130.1, 129.7, 129.6, 129.5, 129.3, 128.82, 128.76, 127.02, 127.00, 126.97, 126.95, 123.64, 123.60, 119.5, 54.58, 54.55, 51.62, 51.55, 50.84, 50.83, 31.56, 31.49, 30.9, 30.6, 30.10, 30.08, 26.4, 26.3, 21.22, 21.15, 17.71, 17.67. HRMS (M + H+): 452.2375 C26H34N3O2S requires 452.2372.
:MeOH 10:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give compound 7a as a white solid in 61% yield (16.1 mg, 0.0325 mmol). The carbon signals from the imidazole were unfortunately not detected in 13C NMR. [α]23D = −31.7° (c, 0.400, CHCl3). 1H NMR (CDCl3), δ, ppm: 8.12 (d, J = 8.1 Hz, 1H), 7.74–7.50 (m, 1H), 7.33 (dd, J = 8.1, 1.0 Hz, 1H), 7.29–7.26 (m, 1H), 7.22–7.19 (m, 1H), 7.17–7.13 (m, 2H), 7.04–6.76 (m, 2H), 5.13 (s, 2H), 4.04 (t, J = 6.6 Hz, 2H), 3.22–3.17 (m, 1H), 2.49–2.36 (m, 2H), 2.22–2.14 (m, 1H), 2.08–1.99 (m, 1H), 1.85–1.76 (m, 1H), 1.55–1.47 (m, 2H), 1.30–1.22 (m, 2H), 1.02 (d, J = 6.8 Hz, 3H), 0.87 (t, J = 7.3 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 153.0, 151.3, 146.3, 138.0, 136.3, 135.4, 135.2, 130.0, 129.6, 129.4, 127.32, 127.26, 123.6, 66.4, 51.8, 51.1, 31.4, 30.77 (CH2), 30.76 (CH), 26.3, 21.2, 19.0, 17.8, 13.8. HRMS (M + H+): 496.2277 C27H34N3O4S requires 496.2270.
:MeOH 10:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give compound 7b as a white solid in 61% yield (16.5 mg, 0.0333 mmol). All carbon signals from the imidazole were unfortunately not detected in 13C NMR. [α]23D = +30.2° (c, 0.365, CHCl3).1H NMR (CDCl3), δ, ppm: 8.12 (d, J = 8.1 Hz, 1H), 7.70–7.46 (m, 1H), 7.35–32 (m, 1H), 7.29–7.26 (m, 1H), 7.22–7.19 (m, 1H), 7.18–7.13 (m, 2H), 7.04–6.76 (m, 2H), 5.11 (s, 1H), 4.04 (t, J = 6.6 Hz, 2H), 3.22–3.17 (m, 1H), 2.49–2.36 (m, 2H), 2.22–2.14 (m, 1H), 2.08–1.99 (m, 1H), 1.85–1.76 (m, 1H), 1.54–1.47 (m, 2H), 1.30–1.20 (m, 2H), 1.02 (d, J = 6.8 Hz, 3H), 0.88 (t, J = 7.4 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 153.0, 151.4, 146.3, 137.9, 136.3, 135.5, 135.3, 130.0, 129.6, 129.4, 128.6, 127.3, 127.2, 123.6, 119.4, 66.4, 51.8, 51.0, 31.4, 30.70 (CH2), 30.68 (CH), 26.3, 21.2, 19.0, 17.8, 13.8. HRMS (M + H+): 496.2260 C27H34N3O4S requires 496.2270.
:MeOH 10:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give compound 8a as a white solid in 59% yield (20.8 mg, 0.0420 mmol). Compound 8a was observed as two rotamers at 24 °C in NMR. Due to high temperature sensitivity no high temperature experiments were performed since this was performed on the protected compound (22a) and the spectra recorded at 24 °C are reported here. [α]23D = −23.2° (c, 0.590, CHCl3). 1H NMR (CDCl3), δ, ppm: 8.20–8.15 (m, 1H), 7.36–7.12 (m, 5H), 7.05–6.84 (m, 2H), 6.78–6.73 (m, 1H), 5.05–4.75 (m, 2H), 4.11–4.00 (m, 2H), 3.25–3.16 (m, 1H), 2.53–2.32 (m, 2H), 2.24–2.14 (m, 1H), 2.09–2.00 (m, 1H), 1.87–1.76 (m, 1H), 1.57–1.50 (m, 2H), 1.32–1.23 (m, 2H), 1.03 (d, J = 6.8 Hz, 3H), 0.91–0.86 (m, 3H), 0.83–0.80 (m, 3H). 13C NMR (CDCl3), δ, ppm: 152.5, 152.41, 152.37, 145.93, 145.92, 138.7, 138.6, 137.0, 136.4, 136.3, 136.0, 135.99, 135.54, 135.50, 129.8, 129.7, 129.2, 129.1, 128.9, 128.7, 128.61, 128.58, 127.8, 125.54, 125.51, 123.5, 119.4, 65.94, 65.90, 51.80, 51.76, 51.00, 31.3, 30.84, 30.80, 30.62, 26.34, 26.30, 21.2, 19.0, 17.9, 17.8, 13.8. HRMS (M + H+): 496.2252 C27H34N3O4S requires 496.2270.
:MeOH 10:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give compound 8b as a white solid in 48% yield (17.2 mg, 0.0347 mmol). Compound 8b was observed as two rotamers at 24 °C in NMR. Due to high temperature sensitivity no high temperature experiments were performed since this was performed on the protected compound (22b) and the spectra recorded at 24 °C are reported here. [α]23D = +22.8° (c, 0.555, CHCl3). 1NMR (CDCl3), δ, ppm: 8.20–8.15 (m, 1H), 7.36–7.12 (m, 5H), 7.05–6.84 (m, 2H), 6.79–6.74 (m, 1H), 5.04–4.76 (m, 2H), 4.12–4.00 (m, 2H), 3.25–3.16 (m, 1H), 2.53–2.32 (m, 2H), 2.24–2.15 (m, 1H), 2.09–2.00 (m, 1H), 1.87–1.76 (m, 1H), 1.57–1.50 (m, 2H), 1.33–1.22 (m, 2H), 1.03 (d, J = 6.8 Hz, 3H), 0.91–0.86 (m, 3H), 0.83–0.80 (m, 3H). 13C NMR (CDCl3), δ, ppm: 152.6, 152.30, 152.25, 146.0, 145.9, 138.63, 138.60, 137.0, 136.4, 136.3, 136.0, 135.9, 135.63, 135.60, 129.8, 129.7, 129.2, 129.1, 128.9, 128.7, 128.63, 128.60, 128.1, 125.58, 125.56, 123.5, 119.4, 66.0, 51.81, 51.77, 50.9, 31.37, 31.35, 30.83, 30.80, 30.63, 26.35, 26.30, 21.3, 19.04, 19.03, 17.9, 17.8, 13.8. HRMS (M + H+): 496.2264 C27H34N3O4S requires 496.2270.
:MeOH, 20:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to yield the pure compound 24a as a white solid in >22% (31.2 mg, 0.069 mmol). [α]23D = +13.5° (c, 0.455, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.94 (d, J = 1.1 Hz, 1H), 7.72–7.69 (m, 1H), 7.51–7.48 (m, 2H), 7.21–7.24 (m, 2H), 7.12–7.13 (m, 1H), 7.06–7.07 (m, 1H), 6.94–6.95 (m, 1H), 5.19 (s, 2H), 3.53 (s, 1H), 3.23–3.18 (m, 1H), 2.98–2.82 (m, 2H), 2.23–2.12 (m, 2H), 1.98–1.89 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.99 (s, 9H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.3, 146.8, 140.5, 140.0, 137.6, 137.4, 135.9, 130.8, 129.4, 128.2, 127.0, 124.3, 119.5, 54.4, 51.0, 50.8, 31.8, 30.8, 29.9, 26.6, 21.1, 17.7. HRMS (M + H+): 452.2363, C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to yield the pure compound 24b as a white solid in >19% (23.4 mg, 0.052 mmol). [α]23D = −13.9° (c, 0.445, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.94 (d, J = 1.1 Hz, 1H), 7.69 (m, 1H), 7.51–7.48 (m, 2H), 7.24–7.20 (m, 2H), 7.12–7.13 (m, 1H), 7.07–7.06 (m, 1H), 6.94–6.95 (m, 1H), 5.19 (s, 2H), 3.52 (s, 1H), 3.23–3.18 (m, 1H), 2.98–2.82 (m, 2H), 2.23–2.12 (m, 2H), 1.98–1.89 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.99 (s, 9H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.3, 146.8, 140.5, 140.0, 137.6, 137.4, 135.9, 130.8, 129.5, 128.2, 127.0, 124.3, 119.5, 54.4, 51.0, 50.8, 31.8, 30.8, 29.9, 26.6, 21.1, 17.7. HRMS (M + H+): 452.2375, C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to yield the pure compound 25a as a white solid in >21% (29.9 mg, 0.066 mmol). [α]23D = +13.1° (c, 0.470, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.94 (d, J = 1.1 Hz, 1H), 7.72–7.67 (m, 1H), 7.45–7.39 (m, 3H), 7.21–7.18 (m, 1H), 7.12–7.08 (m, 1H), 7.07–7.06 (m, 1H), 7.02–6.97 (m, 1H), 5.17 (s, 2H), 3.48 (s, 1H), 3.23–3.18 (m, 1H), 2.98–2.82 (m, 2H), 2.23–2.12 (m, 2H), 1.98–1.89 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.97 (s, 9H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.3, 146.8, 141.0, 140.0, 137.6, 137.3, 135.9, 129.9, 129.7, 129.4, 128.7, 128.2, 127.0, 124.4, 119.6, 54.4, 51.12, 51.11, 31.8, 30.8, 29.9, 26.6, 21.1, 17.7. HRMS (M + H+): 452.2379, C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to yield the pure compound 25b as a white solid in >8.9% (11.3 mg, 0.025 mmol). [α]23D = −14.0° (c, 0.400, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.95 (d, J = 1.1 Hz, 1H), 7.80–7.79 (m, 1H), 7.44–7.39 (m, 3H), 7.23–7.20 (m, 1H), 7.12–7.11 (m, 1H), 7.07–7.06 (m, 1H), 7.01–7.00 (m, 1H), 5.19 (s, 2H), 3.47 (s, 1H), 3.24–3.19 (m, 1H), 2.98–2.83 (m, 2H), 2.24–2.12 (m, 2H), 1.98–1.90 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.98 (s, 9H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.4, 146.9, 141.1, 140.0, 137.6, 137.2, 135.7, 130.0, 129.8, 129.0, 128.8, 128.2, 127.1, 124.5, 119.6, 54.5, 51.2, 51.0, 31.8, 30.9, 30.0, 26.6, 21.1, 17.7. HRMS (M + H+): 452.2368, C26H34N3O2S requires 452.2372.
:1). Na2CO3 was added and the reaction mixture was cooled to 0 °C and n-butyl chloroformate was added and the reaction was left to reach room temperature over night. The reaction mixture was diluted with CHCl3 organic and the phases were separated. The organic phase was washed with water and brine, dried over MgSO4 and evaporated. The residue was purified on preparative RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give the pure products 7a, 7b, 8a, 8b, 9a, 9b, 10a and 10b.
:EtOAc 10:1) to provide compound 13 in 16% yield (1.09 g, mmol) and compound 14 in 14% yield (0.98 g, mmol). The total yield of the product mixture reached 43% (2.94 g, 11.4 mmol, 13:mix:14 = 1:0.8:0.9).
:1 mixture of isohexane and isopropanol and separated on chiral, preparative HPLC (isohexane/isopropanol 95:5 for compound 15 and 93:7 for compound 16) to give the pure enantiomers 15a, 15b, 16a and 16b. The enantiomeric purity of all isolated enantiomers were >99% according to chiral GC-MS (see ESI† for chromatograms).
:mix:15b, 1:1:1). 15a: [α]22D = +11.7° (c, 1.025, CHCl3); 15b: [α]22D = −12.2° (c, 1.080, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.70–7.67 (m, 2H), 7.27–7.24 (m, 1H), 4.53 (brs, 1H), 3.17–3.12 (m, 1H), 2.98–2.81 (m, 2H), 2.18–2.08 (m, 2H), 1.96–1.87 (m, 1H), 1.23 (s, 9H), 1.00 (d, J = 7.0 Hz, 3H), 0.76 (d, J = 7.0 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 151.6, 145.8, 141.4, 125.2, 124.6, 123.0, 54.7, 51.2, 31.7, 30.9, 30.3, 26.6, 21.2, 17.7. Anal. calcd. for C16H25NO2S: C, 65.05; H, 8.53; N, 4.47. Found (15a): C, 64.87; H, 8.52; N, 4.61. Found (15b): C, 65.14; H, 8.66; N, 4.56.
:mix:16b, 1:0.5:0.8). 16a: [α]23D = +33.4° (c, 0.800, CHCl3); 16b: [α]23D = −32.7° (c, 1.045, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.69–7.65 (m, 2H), 7.29–7.25 (m, 1H), 4.52 (brs, 1H), 3.18–3.13 (m, 1H), 2.98–2.81 (m, 2H), 2.20–2.08 (m, 2H), 1.95–1.86 (m, 1H), 1.21 (s, 9H), 1.00 (d, J = 6.8 Hz, 3H), 0.76 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.8, 147.6, 141.2, 125.6, 124.8, 123.1, 54.6, 51.0, 31.9, 30.9, 30.3, 26.7, 21.1, 17.7. Anal. calcd. for C16H25NO2S: C, 65.05; H, 8.53; N, 4.47. Found (16a): C, 64.89; H, 8.43; N, 4.43. Found (16b): C, 64.83; H, 8.57; N, 4.54.
:MeOH, 20:1) and about half of the product mixture was subsequently separated by RP-HPLC (H2O/MeCN with 0.1% TFA) to yield the pure compound 21a as the TFA salt in >13% (53 mg, 0.094 mmol). An analytical sample was desalted by washing with 2 M Na2CO3 for determination of optical rotation and NMR analysis. [α]21D = −23.6° (c, 0.315, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.98 (d, J = 8.1 Hz, 1H), 7.59–7.58 (m, 1H), 7.39–7.37 (m, 1H), 7.34–7.32 (m, 1H), 7.26 (dd, J = 8.1, 1.0 Hz, 1H), 7.24–7.19 (m, 2H), 7.13–7.12 (m, 1H), 6.95–6.94 (m, 1H), 5.18 (s, 2H), 3.43 (s, 1H), 3.22–3.17 (m, 1H), 2.53–2.40 (m, 2H), 2.22–2.11 (m, 1H), 2.09–2.00 (m, 1H), 1.85–1.76 (m, 1H), 1.07 (s, 9H), 1.01 (d, J = 6.8 Hz, 3H), 0.78 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 151.5, 146.0, 140.1, 138.1, 137.6, 136.1, 135.2, 130.7, 130.6, 130.2, 127.0, 126.95, 126.90, 123.6, 119.5, 54.6, 51.6, 50.6, 31.6, 30.8, 30.1, 26.3, 21.2, 17.7. HRMS (M + H+): 452.2366 C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and about half of the product mixture was subsequently separated by RP-HPLC (H2O/MeCN with 0.1% TFA) to yield the pure compound 21b as the TFA salt in >14% (58 mg, 0.103 mmol). An analytical sample was desalted by washing with 2 M Na2CO3 for determination of optical rotation and NMR analysis. [α]22D = +18.2° (c, 0.550, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.97 (d, J = 8.1 Hz, 1H), 7.59–7.58 (m, 1H), 7.39–7.36 (m, 1H), 7.34–7.31 (m, 1H), 7.26 (dd, J = 8.1, 0.9 Hz, 1H), 7.24–7.18 (m, 2H), 7.13–7.11 (m, 1H), 6.96–6.94 (m, 1H), 5.2 (s, 2H), 3.4 (s, 1H), 3.22–3.16 (m, 1H), 2.53–2.40 (m, 2H), 2.22–2.10 (m, 1H), 2.09–1.99 (m, 1H), 1.85–1.75 (m, 1H), 1.07 (s, 9H), 1.01 (d, J = 6.9 Hz, 3H), 0.78 (d, J = 6.9 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 151.5, 146.0, 140.1, 138.1, 137.6, 136.1, 135.2, 130.7, 130.5, 130.1, 127.0, 126.9, 126.9, 123.6, 119.5, 54.6, 51.6, 50.6, 31.5, 30.8, 30.1, 26.3, 21.2, 17.7. HRMS (M + H+): 452.2377 C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and about half of the product mixture was subsequently separated by RP-HPLC (H2O/MeCN with 0.1% TFA) to yield the pure compound 22a as the TFA salt in >19% (74 mg, 0.131 mmol). An analytical sample was desalted by washing with 2 M Na2CO3 for determination of optical rotation and NMR analysis. Compound 22a was observed as two rotamers at 24 °C in NMR in the ratio 1:1. Increasing the temperature to 70 °C (CD3CN) resulted in one set of peaks, a part from one of the methylgroups which had not fully merged. The spectra recorded at 24 °C are reported here. [α]22D = −19.8° (c, 0.430, CHCl3). 1H NMR (CDCl3, 24 °C), δ, ppm: 7.99–7.95 (m, 1H), 7.57–7.54 (m, 1H), 7.45–7.39 (m, 1H), 7.34–7.24 (m, 3H), 7.20–7.13 (m, 1H), 7.09–7.07 (m, 1H), 6.97–6.93 (m, 1H), 5.15 (s, 2H), 3.44–3.40 (m, 1H), 3.22–3.16 (m, 1H), 2.54–2.12 (m, 2H), 2.21–2.12 (m, 1H), 2.09–1.98 (m, 1H), 1.85–1.75 (m, 1H), 1.07–1.04 (m, 9H), 1.03–0.99 (m, 3H), 0.80–0.78 (m, 3H). 13C NMR (CDCl3, 24 °C), δ, ppm: 151.5, 145.9, 140.03, 140.00, 138.81, 138.74, 137.5, 136.4, 136.3, 135.25, 135.21, 130.1, 129.7, 129.6, 129.5, 129.3, 128.8, 128.7, 127.02, 126, 99, 126.97, 126.94, 123.64, 123.60, 119.4, 54.58, 54.56, 51.63, 51.56, 50.83, 50.81, 31.6, 31.5, 30.9, 30.6, 30.10, 30.08, 26.4, 26.3, 21.22, 21.15, 17.72, 17.68. HRMS (M + H+): 452.2371 C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and about half of the product mixture was subsequently separated by RP-HPLC (H2O/MeCN with 0.1% TFA) to yield the pure compound 22b as the TFA salt in >22% (91 mg, 0.161 mmol). An analytical sample was desalted by washing with 2 M Na2CO3 for determination of optical rotation and NMR analysis. Compound 22b was observed as two rotamers at 24 °C in NMR in the ratio 1:1. Increasing the temperature to 70 °C (CD3CN) resulted in one set of peaks a part from one of the methylgroups which had not fully merged. The spectra recorded at 24 °C are reported here. [α]22D = +18.0° (c, 0.615, CHCl3). 1H NMR (CDCl3, 24 °C), δ, ppm: 7.99–7.97 (m, 1H), 7.68–7.55 (m, 1H), 7.45–7.40 (m, 1H), 7.34–7.25 (m, 3H), 7.21–7.14 (m, 1H), 7.12–7.08 (m, 1H), 6.99–6.94 (m, 1H), 5.16 (s, 2H), 3.40–3.38 (m, 1H), 3.22–3.17 (m, 1H), 2.54–2.33 (m, 2H), 2.22–2.12 (m, 1H), 2.09–1.99 (m, 1H), 1.85–1.76 (m, 1H), 1.06–1.05 (m, 9H), 1.08–1.00 (m, 3H), 0.82–0.77 (m, 3H). 13C NMR (CDCl3, 24 °C), δ, ppm: 151.5, 145.9, 140.03, 139.99, 138.8, 138.7, 137.5, 136.34, 136.26, 135.25, 135.20, 130.1, 129.7, 129.6, 129.5, 129.3, 128.82, 128.76, 127.02, 127.00, 126.97, 126.95, 123.64, 123.60, 119.5, 54.58, 54.55, 51.62, 51.55, 50.84, 50.83, 31.56, 31.49, 30.9, 30.6, 30.10, 30.08, 26.4, 26.3, 21.22, 21.15, 17.71, 17.67. HRMS (M + H+): 452.2375 C26H34N3O2S requires 452.2372.
:MeOH 10:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give compound 7a as a white solid in 61% yield (16.1 mg, 0.0325 mmol). The carbon signals from the imidazole were unfortunately not detected in 13C NMR. [α]23D = −31.7° (c, 0.400, CHCl3). 1H NMR (CDCl3), δ, ppm: 8.12 (d, J = 8.1 Hz, 1H), 7.74–7.50 (m, 1H), 7.33 (dd, J = 8.1, 1.0 Hz, 1H), 7.29–7.26 (m, 1H), 7.22–7.19 (m, 1H), 7.17–7.13 (m, 2H), 7.04–6.76 (m, 2H), 5.13 (s, 2H), 4.04 (t, J = 6.6 Hz, 2H), 3.22–3.17 (m, 1H), 2.49–2.36 (m, 2H), 2.22–2.14 (m, 1H), 2.08–1.99 (m, 1H), 1.85–1.76 (m, 1H), 1.55–1.47 (m, 2H), 1.30–1.22 (m, 2H), 1.02 (d, J = 6.8 Hz, 3H), 0.87 (t, J = 7.3 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 153.0, 151.3, 146.3, 138.0, 136.3, 135.4, 135.2, 130.0, 129.6, 129.4, 127.32, 127.26, 123.6, 66.4, 51.8, 51.1, 31.4, 30.77 (CH2), 30.76 (CH), 26.3, 21.2, 19.0, 17.8, 13.8. HRMS (M + H+): 496.2277 C27H34N3O4S requires 496.2270.
:MeOH 10:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give compound 7b as a white solid in 61% yield (16.5 mg, 0.0333 mmol). All carbon signals from the imidazole were unfortunately not detected in 13C NMR. [α]23D = +30.2° (c, 0.365, CHCl3).1H NMR (CDCl3), δ, ppm: 8.12 (d, J = 8.1 Hz, 1H), 7.70–7.46 (m, 1H), 7.35–32 (m, 1H), 7.29–7.26 (m, 1H), 7.22–7.19 (m, 1H), 7.18–7.13 (m, 2H), 7.04–6.76 (m, 2H), 5.11 (s, 1H), 4.04 (t, J = 6.6 Hz, 2H), 3.22–3.17 (m, 1H), 2.49–2.36 (m, 2H), 2.22–2.14 (m, 1H), 2.08–1.99 (m, 1H), 1.85–1.76 (m, 1H), 1.54–1.47 (m, 2H), 1.30–1.20 (m, 2H), 1.02 (d, J = 6.8 Hz, 3H), 0.88 (t, J = 7.4 Hz, 3H), 0.80 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 153.0, 151.4, 146.3, 137.9, 136.3, 135.5, 135.3, 130.0, 129.6, 129.4, 128.6, 127.3, 127.2, 123.6, 119.4, 66.4, 51.8, 51.0, 31.4, 30.70 (CH2), 30.68 (CH), 26.3, 21.2, 19.0, 17.8, 13.8. HRMS (M + H+): 496.2260 C27H34N3O4S requires 496.2270.
:MeOH 10:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give compound 8a as a white solid in 59% yield (20.8 mg, 0.0420 mmol). Compound 8a was observed as two rotamers at 24 °C in NMR. Due to high temperature sensitivity no high temperature experiments were performed since this was performed on the protected compound (22a) and the spectra recorded at 24 °C are reported here. [α]23D = −23.2° (c, 0.590, CHCl3). 1H NMR (CDCl3), δ, ppm: 8.20–8.15 (m, 1H), 7.36–7.12 (m, 5H), 7.05–6.84 (m, 2H), 6.78–6.73 (m, 1H), 5.05–4.75 (m, 2H), 4.11–4.00 (m, 2H), 3.25–3.16 (m, 1H), 2.53–2.32 (m, 2H), 2.24–2.14 (m, 1H), 2.09–2.00 (m, 1H), 1.87–1.76 (m, 1H), 1.57–1.50 (m, 2H), 1.32–1.23 (m, 2H), 1.03 (d, J = 6.8 Hz, 3H), 0.91–0.86 (m, 3H), 0.83–0.80 (m, 3H). 13C NMR (CDCl3), δ, ppm: 152.5, 152.41, 152.37, 145.93, 145.92, 138.7, 138.6, 137.0, 136.4, 136.3, 136.0, 135.99, 135.54, 135.50, 129.8, 129.7, 129.2, 129.1, 128.9, 128.7, 128.61, 128.58, 127.8, 125.54, 125.51, 123.5, 119.4, 65.94, 65.90, 51.80, 51.76, 51.00, 31.3, 30.84, 30.80, 30.62, 26.34, 26.30, 21.2, 19.0, 17.9, 17.8, 13.8. HRMS (M + H+): 496.2252 C27H34N3O4S requires 496.2270.
:MeOH 10:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to give compound 8b as a white solid in 48% yield (17.2 mg, 0.0347 mmol). Compound 8b was observed as two rotamers at 24 °C in NMR. Due to high temperature sensitivity no high temperature experiments were performed since this was performed on the protected compound (22b) and the spectra recorded at 24 °C are reported here. [α]23D = +22.8° (c, 0.555, CHCl3). 1NMR (CDCl3), δ, ppm: 8.20–8.15 (m, 1H), 7.36–7.12 (m, 5H), 7.05–6.84 (m, 2H), 6.79–6.74 (m, 1H), 5.04–4.76 (m, 2H), 4.12–4.00 (m, 2H), 3.25–3.16 (m, 1H), 2.53–2.32 (m, 2H), 2.24–2.15 (m, 1H), 2.09–2.00 (m, 1H), 1.87–1.76 (m, 1H), 1.57–1.50 (m, 2H), 1.33–1.22 (m, 2H), 1.03 (d, J = 6.8 Hz, 3H), 0.91–0.86 (m, 3H), 0.83–0.80 (m, 3H). 13C NMR (CDCl3), δ, ppm: 152.6, 152.30, 152.25, 146.0, 145.9, 138.63, 138.60, 137.0, 136.4, 136.3, 136.0, 135.9, 135.63, 135.60, 129.8, 129.7, 129.2, 129.1, 128.9, 128.7, 128.63, 128.60, 128.1, 125.58, 125.56, 123.5, 119.4, 66.0, 51.81, 51.77, 50.9, 31.37, 31.35, 30.83, 30.80, 30.63, 26.35, 26.30, 21.3, 19.04, 19.03, 17.9, 17.8, 13.8. HRMS (M + H+): 496.2264 C27H34N3O4S requires 496.2270.
:MeOH, 20:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to yield the pure compound 24a as a white solid in >22% (31.2 mg, 0.069 mmol). [α]23D = +13.5° (c, 0.455, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.94 (d, J = 1.1 Hz, 1H), 7.72–7.69 (m, 1H), 7.51–7.48 (m, 2H), 7.21–7.24 (m, 2H), 7.12–7.13 (m, 1H), 7.06–7.07 (m, 1H), 6.94–6.95 (m, 1H), 5.19 (s, 2H), 3.53 (s, 1H), 3.23–3.18 (m, 1H), 2.98–2.82 (m, 2H), 2.23–2.12 (m, 2H), 1.98–1.89 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.99 (s, 9H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.3, 146.8, 140.5, 140.0, 137.6, 137.4, 135.9, 130.8, 129.4, 128.2, 127.0, 124.3, 119.5, 54.4, 51.0, 50.8, 31.8, 30.8, 29.9, 26.6, 21.1, 17.7. HRMS (M + H+): 452.2363, C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to yield the pure compound 24b as a white solid in >19% (23.4 mg, 0.052 mmol). [α]23D = −13.9° (c, 0.445, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.94 (d, J = 1.1 Hz, 1H), 7.69 (m, 1H), 7.51–7.48 (m, 2H), 7.24–7.20 (m, 2H), 7.12–7.13 (m, 1H), 7.07–7.06 (m, 1H), 6.94–6.95 (m, 1H), 5.19 (s, 2H), 3.52 (s, 1H), 3.23–3.18 (m, 1H), 2.98–2.82 (m, 2H), 2.23–2.12 (m, 2H), 1.98–1.89 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.99 (s, 9H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.3, 146.8, 140.5, 140.0, 137.6, 137.4, 135.9, 130.8, 129.5, 128.2, 127.0, 124.3, 119.5, 54.4, 51.0, 50.8, 31.8, 30.8, 29.9, 26.6, 21.1, 17.7. HRMS (M + H+): 452.2375, C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to yield the pure compound 25a as a white solid in >21% (29.9 mg, 0.066 mmol). [α]23D = +13.1° (c, 0.470, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.94 (d, J = 1.1 Hz, 1H), 7.72–7.67 (m, 1H), 7.45–7.39 (m, 3H), 7.21–7.18 (m, 1H), 7.12–7.08 (m, 1H), 7.07–7.06 (m, 1H), 7.02–6.97 (m, 1H), 5.17 (s, 2H), 3.48 (s, 1H), 3.23–3.18 (m, 1H), 2.98–2.82 (m, 2H), 2.23–2.12 (m, 2H), 1.98–1.89 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.97 (s, 9H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.3, 146.8, 141.0, 140.0, 137.6, 137.3, 135.9, 129.9, 129.7, 129.4, 128.7, 128.2, 127.0, 124.4, 119.6, 54.4, 51.12, 51.11, 31.8, 30.8, 29.9, 26.6, 21.1, 17.7. HRMS (M + H+): 452.2379, C26H34N3O2S requires 452.2372.
:MeOH, 20:1) and subsequently by RP-HPLC (H2O/MeCN with 0.05% HCOOH) to yield the pure compound 25b as a white solid in >8.9% (11.3 mg, 0.025 mmol). [α]23D = −14.0° (c, 0.400, CHCl3). 1H NMR (CDCl3), δ, ppm: 7.95 (d, J = 1.1 Hz, 1H), 7.80–7.79 (m, 1H), 7.44–7.39 (m, 3H), 7.23–7.20 (m, 1H), 7.12–7.11 (m, 1H), 7.07–7.06 (m, 1H), 7.01–7.00 (m, 1H), 5.19 (s, 2H), 3.47 (s, 1H), 3.24–3.19 (m, 1H), 2.98–2.83 (m, 2H), 2.24–2.12 (m, 2H), 1.98–1.90 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.98 (s, 9H), 0.79 (d, J = 6.8 Hz, 3H). 13C NMR (CDCl3), δ, ppm: 149.4, 146.9, 141.1, 140.0, 137.6, 137.2, 135.7, 130.0, 129.8, 129.0, 128.8, 128.2, 127.1, 124.5, 119.6, 54.5, 51.2, 51.0, 31.8, 30.9, 30.0, 26.6, 21.1, 17.7. HRMS (M + H+): 452.2368, C26H34N3O2S requires 452.2372.
Biology
000–85000 cpm, 0.03 nM) and variable concentrations of test substance (compounds 7a, 7b, 8a, 8b, 9a, 9b, 10a and 10b). Samples were incubated at 25 °C for 1.5 h, and binding was terminated by filtration through Whatman GF/B glass-fiber filter sheets, which had been presoaked overnight with 0.3% polyethylamine, using a Brandel cell harvester. The filters were washed with 3 × 3 mL of Tris-HCl (pH 7.4) and transferred to tubes. The radioactivity was measured in a γ-counter. The characteristics of the Ang II binding AT2 receptor was determined by using six different concentrations (0.03–5 nmol L−1) of the labeled [125I]-AngII. Nonspecific binding was determined in the presence of 1 μM Ang II. The specific binding was determined by subtracting the nonspecific binding from the total bound [125I]AngII. The apparent dissociation constant Ki values were calculated from IC50 values using the Cheng–Prusoff equation (Kd = 0.73 ± 0.06 nM, [L] = 0.057 nM). The binding data were best fitted with a one-site fit. All determinations were performed in triplicate.
Cells were treated during three days, once a day (first treatment 24 hours after plating), and micrographs were taken the fourth day. For all experiments, cells were plated at the same initial density of 3.6 × 104 cells/35 mm Petri dish. To determine a good test concentration, all compounds were tested at various concentrations ranging from 0.1 nM to 100 nM. It was only at the highest concentration of compounds 7a and 7b that any evidence of cell death was observed, and that was most probably due to a higher concentration of DMSO (due to low solubility). Cells were treated without (control cells), or with [Val5]-angiotensin II (100 nM) or with compound 7a (10 nM), 7b (10 nM), 8a (10 and 100 nM), 8b (10 and 100 nM), 9a (10 nM), 9b (10 nM), 10a (10 and 100 nM) or 10b (10 nM) in the absence or in the presence of PD 123319 (10 μM), an AT2 receptor antagonist. The antagonist was introduced daily 30 min prior to Ang II, compound 7a, 7b, 8a, 8b, 9a, 9b, 10a or 10b. Compound 7a (10 nM), 7b (10 nM), 8a (10 and 100 nM), 8b (10 and 100 nM), 9a (10 nM), 9b (10 nM), 10a (10 and 100 nM) or 10b (10 nM) were also tested in the presence of Ang II (100 nM) where the compounds were introduced daily 30 min prior to Ang II, to evaluate antagonistic properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge support from the Swedish Research Council, from the Swedish Foundation for Strategic Research, from Kjell and Märta Beijer Foundation, Knut and Alice Wallenberg Foundation, from the Canadian Institutes of Health Research to N. G. P. and M. D. Payet (MOP 27912), from the Alzheimer's Society of Canada to N. G. P. and by the program of Canada Research Chair to N. G. P., and Vicore Pharma AB. N. G. P. is a holder of a Canadian Research Chair in Endocrinology of the Adrenal Gland. We also thank Dr. Milad Botros for technical assistance and Dr. Luke R. Odell for linguistic advice.References
- W. H. Birkenhäger and P. W. de Leeuw, J. Hypertens., 1999, 17, 873–881 CrossRef PubMed.
- M. Burnier, Circulation, 2001, 103, 904–912 CrossRef CAS PubMed.
- A. S. C. Rice, R. H. Dworkin, T. D. McCarthy, P. Anand, C. Bountra, P. I. McCloud, J. Hill, G. Cutter, G. Kitson, N. Desem and M. Raff, Lancet, 2014, 383, 1637–1647 CrossRef CAS.
- M. T. Smith, B. D. Wyse and S. R. Edwards, Pain Med., 2013, 14, 692–705 CrossRef PubMed.
- J. M. Keppel Hesselink and M. E. Schatman, J. Pain Res., 2017, 10, 439–443 CrossRef PubMed.
- U. M. Steckelings, E. Kaschina and T. Unger, Peptides, 2005, 26, 1401–1409 CrossRef CAS PubMed.
- S. H. Padia and R. M. Carey, Pflugers Arch., 2013, 465, 99–110 CrossRef CAS PubMed.
- L. Laflamme, M. Gasparo, J. M. Gallo, M. D. Payet and N. Gallo-Payet, J. Biol. Chem., 1996, 271, 22729–22735 CrossRef CAS PubMed.
- S. Whitebread, M. Mele, B. Kamber and M. de Gasparo, Biochem. Biophys. Res. Commun., 1989, 163, 284–291 CrossRef CAS PubMed.
- J. Georgsson, U. Rosenstrom, C. Wallinder, H. Beaudry, B. Plouffe, G. Lindeberg, M. Botros, F. Nyberg, A. Karlen, N. Gallo-Payet and A. Hallberg, Bioorg. Med. Chem., 2006, 14, 5963–5972 CrossRef CAS PubMed.
- R. C. Speth and K. H. Kim, Biochem. Biophys. Res. Commun., 1990, 169, 997–1006 CrossRef CAS PubMed.
- U. Rosenström, C. Sköld, B. Plouffe, G. Lindeberg, M. Botros, F. Nyberg, G. Wolf, A. Karlén, N. Gallo-Payet and A. Hallberg, J. Med. Chem., 2005, 48, 4009–4024 CrossRef PubMed.
- F. Magnani, C. G. Pappas, T. Crook, V. Magafa, P. Cordopatis, S. Ishiguro, N. Ohta, J. Selent, S. Bosnyak, E. S. Jones, I. P. Gerothanassis, M. Tamura, R. E. Widdop and A. G. Tzakos, ACS Chem. Biol., 2014, 9, 1420–1425 CrossRef CAS PubMed.
- E. S. Jones, M. P. Del Borgo, J. F. Kirsch, D. Clayton, S. Bosnyak, I. Welungoda, N. Hausler, S. Unabia, P. Perlmutter, W. G. Thomas, M. I. Aguilar and R. E. Widdop, Hypertension, 2011, 57, 570–576 CrossRef CAS PubMed.
- M. O. Guimond, M. Hallberg, N. Gallo-Payet and C. Wallinder, ACS Med. Chem. Lett., 2014, 5, 1129–1132 CrossRef CAS PubMed.
- M. Hallberg, J. Savmarker and A. Hallberg, Curr. Protein Pept. Sci., 2017, 18, 809–818 CAS.
- G. T. Wagenaar, H. Laghmani el, M. Fidder, R. M. Sengers, Y. P. de Visser, L. de Vries, R. Rink, A. J. Roks, G. Folkerts and F. J. Walther, Am. J. Physiol., 2013, 305, L341–L351 CAS.
- Y. Wan, C. Wallinder, B. Plouffe, H. Beaudry, A. K. Mahalingam, X. Wu, B. Johansson, M. Holm, M. Botoros, A. Karlen, A. Pettersson, F. Nyberg, L. Fandriks, N. Gallo-Payet, A. Hallberg and M. Alterman, J. Med. Chem., 2004, 47, 5995–6008 CrossRef CAS PubMed.
- X. Wu, Y. Wan, A. K. Mahalingam, A. M. S. Murugaiah, B. Plouffe, M. Botros, A. Karlén, M. Hallberg, N. Gallo-Payet and M. Alterman, J. Med. Chem., 2006, 49, 7160–7168 CrossRef CAS PubMed.
- A. M. S. Murugaiah, C. Wallinder, A. K. Mahalingam, X. Wu, Y. Wan, B. Plouffe, M. Botros, A. Karlén, M. Hallberg, N. Gallo-Payet and M. Alterman, Bioorg. Med. Chem., 2007, 15, 7166–7183 CrossRef CAS PubMed.
- C. Wallinder, M. Botros, U. Rosenström, M.-O. Guimond, H. Beaudry, F. Nyberg, N. Gallo-Payet, A. Hallberg and M. Alterman, Bioorg. Med. Chem., 2008, 16, 6841–6849 CrossRef CAS PubMed.
- A. K. Mahalingam, Y. Wan, A. M. S. Murugaiah, C. Wallinder, X. Wu, B. Plouffe, M. Botros, F. Nyberg, A. Hallberg, N. Gallo-Payet and M. Alterman, Bioorg. Med. Chem., 2010, 18, 4570–4590 CrossRef CAS PubMed.
- J. Liu, Q. Liu, X. Yang, S. Xu, H. Zhang, R. Bai, H. Yao, J. Jiang, M. Shen, X. Wu and J. Xu, Bioorg. Med. Chem., 2013, 21, 7742–7751 CrossRef CAS PubMed.
- M. Hallberg, C. Sumners, U. M. Steckelings and A. Hallberg, Med. Res. Rev., 2018, 38, 602–624 CrossRef CAS PubMed.
- M. Larhed, M. Hallberg and A. Hallberg, Med. Chem. Rev., 2018, 51, 69–82 Search PubMed.
- Y. Wang, M. Del Borgo, H. W. Lee, D. Baraldi, B. Hirmiz, T. A. Gaspari, K. M. Denton, M. I. Aguilar, C. S. Samuel and R. E. Widdop, Front. Pharmacol., 2017, 8, 564 CrossRef PubMed.
- A. M. S. Murugaiah, X. Wu, C. Wallinder, A. K. Mahalingam, Y. Wan, C. Sköld, M. Botros, M.-O. Guimond, A. Joshi, F. Nyberg, N. Gallo-Payet, A. Hallberg and M. Alterman, J. Med. Chem., 2012, 55, 2265–2278 CrossRef CAS PubMed.
- M.-O. Guimond, C. Wallinder, M. Alterman, A. Hallberg and N. Gallo-Payet, Eur. J. Pharmacol., 2013, 699, 160–171 CrossRef CAS PubMed.
- C. Wallinder, C. Sköld, M. Botros, M.-O. Guimond, M. Hallberg, N. Gallo-Payet, A. Karlén and M. Alterman, ACS Med. Chem. Lett., 2015, 6, 178–182 CrossRef CAS PubMed.
- S. Perlman, C. M. Costa-Neto, A. A. Miyakawa, H. T. Schambye, S. A. Hjorth, A. C. Paiva, R. A. Rivero, W. J. Greenlee and T. W. Schwartz, Mol. Pharmacol., 1997, 51, 301–311 CrossRef CAS PubMed.
- D. D. Phillips, J. A. Cimildoro, P. Scheiner and W. A. Johnson, J. Org. Chem., 1958, 23, 786–788 CrossRef CAS.
- M. Larhed and A. Hallberg, J. Org. Chem., 1996, 61, 9582–9584 CrossRef CAS.
- Y. Wan, X. Wu, A. K. Mahalingam and M. Alterman, Tetrahedron Lett., 2003, 44, 4523–4525 CrossRef CAS.
- A. H. Nielsen, K. Schauser, H. Winther, V. Dantzer and K. Poulsen, Clin. Exp. Pharmacol. Physiol., 1997, 24, 309–314 CrossRef CAS PubMed.
- D. J. Carini, J. V. Duncia, P. E. Aldrich, A. T. Chiu, A. L. Johnson, M. E. Pierce, W. A. Price, J. B. I. Santella, G. J. Wells, R. R. Wexler, P. C. Wong, S.-E. Yoo and P. B. M. W. M. Timmermans, J. Med. Chem., 1991, 34, 2525–2547 CrossRef CAS PubMed.
- B. Buisson, S. P. Bottari, M. de Gasparo, N. Gallo-Payet and M. D. Payet, FEBS Lett., 1992, 309, 161–164 CrossRef CAS PubMed.
- V. Brechler, P. W. Jones, N. R. Levens, M. de Gasparo and S. P. Bottari, Regul. Pept., 1993, 44, 207–213 CrossRef CAS PubMed.
- L. Gendron, L. Laflamme, N. Rivard, C. Asselin, M. D. Payet and N. Gallo-Payet, Mol. Endocrinol., 1999, 13, 1615–1626 CrossRef CAS PubMed.
- L. Gendron, F. Cote, M. D. Payet and N. Gallo-Payet, Neuroendocrinology, 2002, 75, 70–81 CrossRef CAS PubMed.
- A. M. Murugaiah, X. Wu, C. Wallinder, A. K. Mahalingam, Y. Wan, C. Skold, M. Botros, M. O. Guimond, A. Joshi, F. Nyberg, N. Gallo-Payet, A. Hallberg and M. Alterman, J. Med. Chem., 2012, 55, 2265–2278 CrossRef CAS PubMed.
- A. Levitzki and A. Gazit, Science, 1995, 267, 1782–1788 CrossRef CAS PubMed.
- B. Plouffe, M. O. Guimond, H. Beaudry and N. Gallo-Payet, Endocrinology, 2006, 147, 4646–4654 CrossRef CAS PubMed.
- E. E. Sugg, Annual Reports in Medicinal Chemistry, 1997, vol. 32, pp. 277–283 Search PubMed.
- M. Wijtmans, D. J. Scholten, L. Roumen, M. Canals, H. Custers, M. Glas, M. C. A. Vreeker, F. J. J. de Kanter, C. de Graaf, M. J. Smit, I. J. P. de Esch and R. Leurs, J. Med. Chem., 2012, 55, 10572–10583 CrossRef CAS PubMed.
- J. S. Blakeney, R. C. Reid, G. T. Le and D. P. Fairlie, Chem. Rev., 2007, 107, 2960–3041 CrossRef CAS PubMed.
- M. C. Khosla, R. A. Leese, W. L. Maloy, A. T. Ferreira, R. R. Smeby and F. M. Bumpus, J. Med. Chem., 1972, 15, 792–795 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chiral GC-MS chromatograms of compound 15 and 16, HPLC purity of the test compounds and computational details. See DOI: 10.1039/c9md00402e |
| This journal is © The Royal Society of Chemistry 2019 |