
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycans in drug discovery
Pablo
Valverde
a,
Ana
Ardá
a,
Niels-Christian
Reichardt
b,
Jesús
Jiménez-Barbero
 *acd and
Ana
Gimeno
*a
*acd and
Ana
Gimeno
*a
aCIC bioGUNE, Bizkaia Technology Park, Building 800, 48162 Derio, Bizkaia, Spain. E-mail: jjbarbero@cicbiogune.es
bCIC biomaGUNE, Paseo Miramon 182, 20014 Donostia, Gipuzkoa, Spain
cIkerbasque, Basque Foundation for Science, 48013 Bilbao, Bizkaia, Spain
dDepartment of Organic Chemistry II, University of the Basque Country, UPV/EHU, 48940 Leioa, Bizkaia, Spain
First published on 26th July 2019
Abstract
Glycans are key players in many biological processes. They are essential for protein folding and stability and act as recognition elements in cell–cell and cell–matrix interactions. Thus, being at the heart of medically relevant biological processes, glycans have come onto the scene and are considered hot spots for biomedical intervention. The progress in biophysical techniques allowing access to an increasing molecular and structural understanding of these processes has led to the development of effective therapeutics. Indeed, strategies aimed at designing glycomimetics able to block specific lectin–carbohydrate interactions, carbohydrate-based vaccines mimicking self- and non-self-antigens as well as the exploitation of the therapeutic potential of glycosylated antibodies are being pursued. In this mini-review the most prominent contributions concerning recurrent diseases are highlighted, including bacterial and viral infections, cancer or immune-related pathologies, which certainly show the great promise of carbohydrates in drug discovery.
Introduction
Glycans are essential biomolecules in nature. They can be found as simple monosaccharides or forming more complex structures such as naturally occurring glycoconjugates, where the sugar moieties are attached to proteins or lipids. Being the most abundant molecules in living organisms, carbohydrates play many important biological roles, including their function as an energy source through glucose metabolism or as a structural component of cell walls.1 It is noteworthy that the biological roles of glycans go further beyond.2 Complex glycans are required for the proper folding of newly synthesized polypeptides and are involved in the stability, solubility and trafficking of the final glycoproteins. Moreover, being at the surface of all types of cells, carbohydrates drive critical biological functions that rely on the specific recognition of glycan structures by other biomolecules (Fig. 1). These interactions are fundamental and trigger important processes including cell signalling, proliferation and differentiation or tissue development. Indeed, the molecular recognition of glycans is at the heart of pathological processes such as bacterial adhesion, viral infection, inflammation or immune system activation.3 Consequently, either involved in self- or non-self-recognition, glycans are attractive targets for a wide range of medical applications. In fact, carbohydrates are more and more recognized as hot spots for biomedical intervention in drug discovery programs. However, in balance with their biological roles, the exploitation of sugars as biomedical targets has been poorly developed. Certainly, the molecular basis governing glycan functions is still not fully understood, probably as a consequence of the huge structural complexity and heterogeneity of glycans.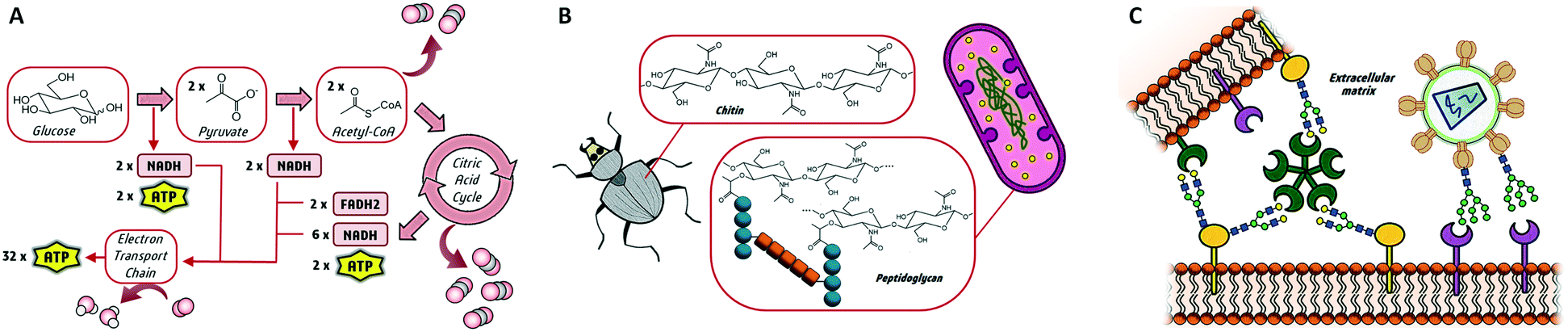 | ||
| Fig. 1 Three main biological roles of glycans as (A) energy source, (B) structural elements and (C) molecular recognition elements in cell–cell and cell-pathogen interactions. | ||
The wide variety of monosaccharides which can be differently linked, together with the non-template driven glycan biosynthesis, increases the number of glycan structures present in nature. In fact, glycosylation in life is a complex process which varies among different cells and tissues and is affected by cell microenvironment.4,5 These factors have hindered the methodological evaluation of the glycome and have slowed down the progress in glycosciences.6 Nonetheless, advances in glycan synthesis and the development of highly sensitive and informative analytical tools are providing increasing knowledge in the complex correlation between glycan structure and function. The progress in MS instrumentation allows mapping of glycan attachment sites, profiling glycan structural variations, and determining detailed fine structures of carbohydrates.7 In addition, new synthetic strategies have allowed access to pure and high amounts of defined complex glycan structures.8 The combination of these novel synthetic approaches with high-throughput screening methods, such as those based on microarrays,9 has become a potent tool for the discovery of glycan receptors and the study of structure–activity relationships. Moreover, the progress in high-resolution techniques including NMR, X-ray or EM has revealed fine structural details controlling glycan recognition. In parallel, computational tools are being developed for the refinement of X-ray crystal structures containing glycans10 and the revolution in electron imaging methods approaching atomic resolution has provided access to glycoconjugate images.11 Similarly, technical advances in sensitivity and resolution and the use of labelling strategies have allowed addressing NMR studies of large saccharides that provide a much more accurate 3D view of the glycan structure and conformation.12 Moreover, specific NMR experiments including STD-NMR, trNOESY or those focused on protein-based methods have been successfully designed to characterize glycan interactions in solution.13 Major endeavours are being applied to study these interactions under mimicked physiological conditions14,15 or even using living cells.16 The growing understanding of structure–activity relationships is triggering a boost in the application of glycans in medicinal chemistry. We and others have reviewed the most prominent contributions to the design of glycomimetics, glycan-based vaccines and therapeutics and highlighted the great promise of carbohydrates in drug discovery.17 This non-exhaustive review also aims to show the recent advances in the field over the past years, which definitely show the biomedical relevance of carbohydrates. Small molecule glycomimetics, glycopeptides and glycoproteins as well as a brief description of carbohydrate-based vaccines are included.
Small molecule glycomimetics
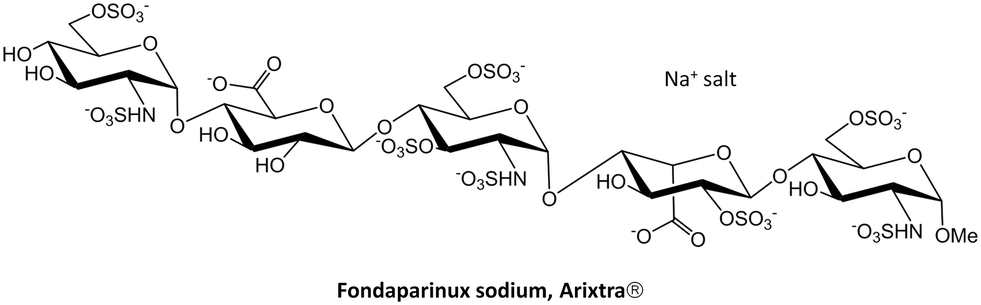 | ||
| Scheme 1 Chemical structure of fondaparinux sodium salt, commercially known as Arixtra. | ||
Fondaparinux is a particular case of a structurally well-defined heparin,24 but as mentioned above, heparins are generally polydisperse drugs prepared from animal tissues.25 Research is still pursuing the development of efficient synthetic methodologies to produce monodisperse heparin oligosaccharides in large scale, both natural fragments and modified scaffolds. In fact, notable advances are enabling the synthesis and evaluation of new heparin-like fragments as potential therapeutics. The incorporation of one-pot-based strategies26 and iterative synthetic schemes has permitted an easier and faster access to very large heparin-like oligomers.27 Moreover, the programmed inclusion of different protective groups at specific positions allows further modifications of the original macromolecules.28 Some of these modifications also include heparin labelling to facilitate the purification steps29 or tagging to monitor the tissue distribution of heparin-based therapeutic candidates in biological studies.30 Additionally, enzymatically driven protocols have allowed the synthesis of large libraries of heparin oligosaccharides ranging from 6 to 9 sugar units and with different sulfation patterns.31 Also, monodisperse heparin sulfate with anticoagulant activity has been directly prepared in human cells by recombinant expression of human serglycin.32 All these methodologies have permitted a thorough analysis of those structural factors affecting heparin binding to relevant biomedical targets, especially the position/distribution or amount of sulfate groups in the heparin scaffold. Precise attention has been paid to the interaction of heparin fragments with tau, β-amyloid and α-synuclein aggregates,33 the pro-angiogenic cytokines VEGF165 and FGF-2,34 the chemokines CXCL8 and CXCL12 (ref. 35) or the Robo1 receptor.36
 | ||
| Fig. 2 C-glycoside inhibitors of LecB bearing sulfonamide moieties. X-ray crystal structures of the interaction with LecB (pdb 5MB1 and 5MAY). | ||
While LecB displays strong affinity for Fuc-containing oligosaccharides, LecA is a galactose-specific lectin. The systematic analysis of monofluorinated galactopyranosides where the hydroxyl groups of the natural sugar were replaced one by one allowed establishing the fundamental sugar interacting features and provided the basis for designing potent and stable glycomimetics for LecA.44 Interestingly, Titz and coworkers have reported a covalent inhibitor bearing an electrophilic epoxide, which exploits the reactivity towards a specific cysteine residue on the carbohydrate binding domain of the protein. This irreversible covalent bond with LecA permanently blocks the protein activity (Fig. 3).45
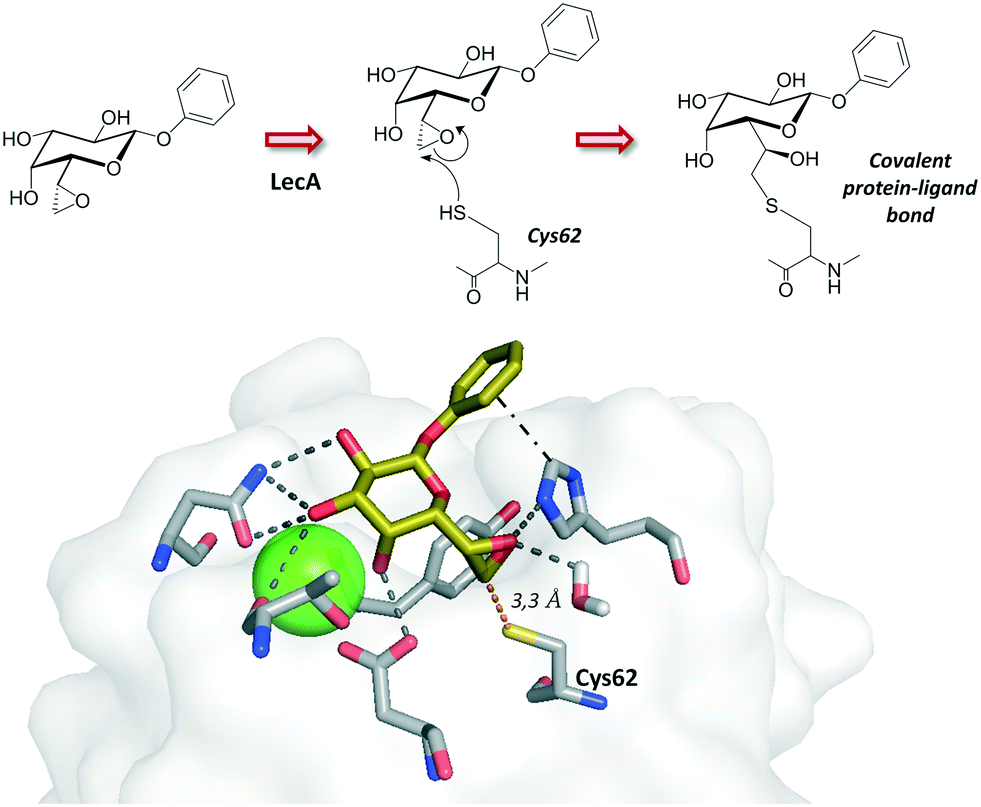 | ||
| Fig. 3 Covalent inhibitor described for LecA. The crystal structure for the non-covalent mode interaction is depicted (pdb 5MIM). | ||
Among bacterial diseases, one of the most widely occurring infections results from the interaction between FimH, a lectin located at the tip of bacterial type 1 pili, and mannosylated glycoproteins on the urothelial mucosa. The first crystal structure of FimH bound to butyl mannose was described in 2005 and established the basis for sugar binding.46 The mannose is located in a deep pocket surrounded by hydrophobic residues (also known as tyrosine gate), whose architecture has been exploited to design potent inhibitors. In fact, compounds bearing aliphatic or aromatic aglycones attached to α-D-Man have been synthesized and evaluated as potential drugs.47 The use of fluorinated aglycones, which maximize π–π stacking with the tyrosine gate, provided low nM antagonists with the best affinities reported to date (Fig. 4).48 In addition, prodrugs with improved oral bioavailability were also obtained by phosphorylation of FimH antagonists.49
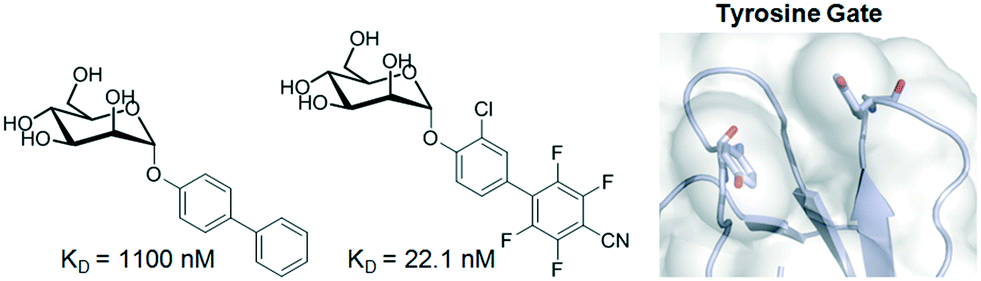 | ||
| Fig. 4 Mannoside inhibitors of FimH displaying aromatic aglycones. | ||
Traditionally, common strategies for the design of potent glycomimetics have been focused on enhancing the contribution of the binding enthalpy by increasing the number and strength of protein–ligand interactions, thus providing higher affinities. However, this conception is changing in medicinal chemistry and it is moving towards the further analysis of the binding entropy contribution. In the case of FimH antagonists, septanose glycomimetics provided much the same H-bond network as that of their parent hexoses but suffered from entropic penalties due to the rigidification of the molecule upon binding.50 Furthermore, tools such as KinITC have recently emerged for evaluating the binding kinetics, parameters which have been usually neglected.51
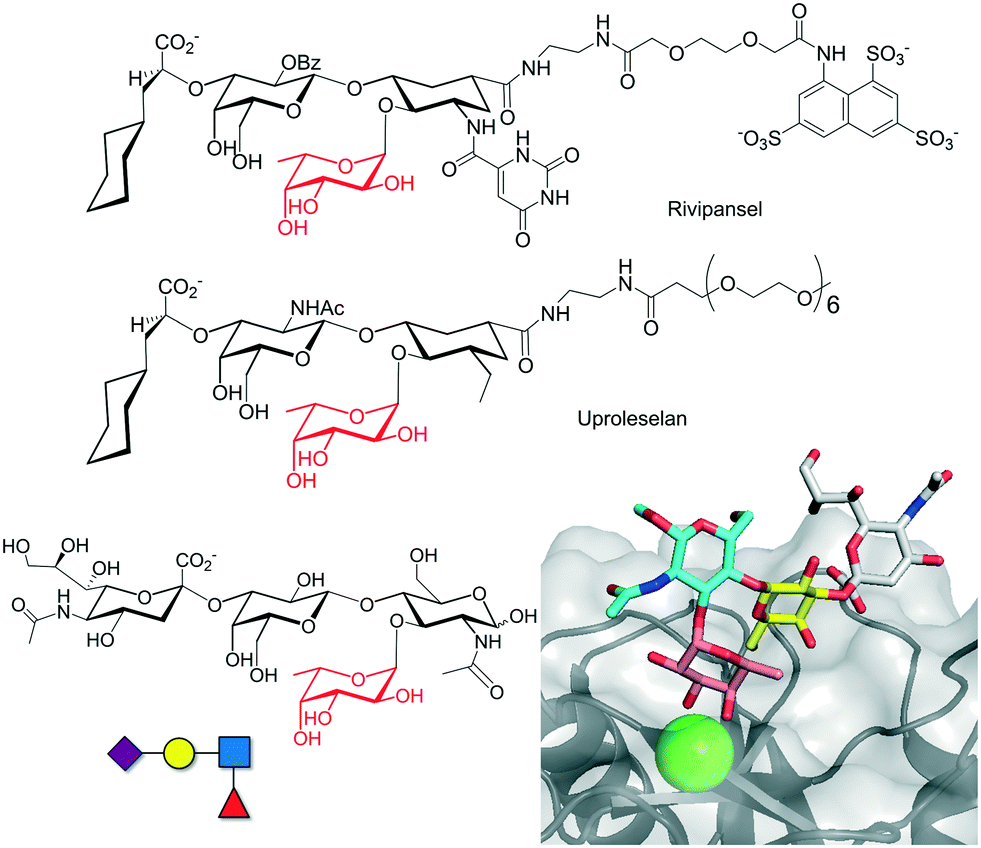 | ||
| Fig. 5 Chemical structure of selectin inhibitors rivipansel and uproleselan. The chemical structure of sLex antigen and the crystal structure of the interaction with selectin are depicted at the bottom (pdb 1G1T). | ||
The discovery of new potent inhibitors is an active area of research. Ernst and co-workers used an NMR-guided fragment screening for designing nM affinity E-selectin inhibitors based on the attachment of the sLex scaffold, which binds to the primary binding site, to a second site ligand.52 Additional strategies based on the use of glycopolymers with a multivalent presentation of sLex or the single sugars Fuc, Gal, or Neu5Ac have also provided very promising results for inhibiting E-, P- and L-selectins.53 Other lectins such as DC-SIGN, Siglecs and galectins, with important functions in inflammation or immune system activation, have also been considered as important targets. Sialic acid sugars, working as ligands of the immunosuppressive sialic acid binding immunoglobulin-like (Siglec) lectins, are emerging as important regulators of the immune system. Aberrant sialic acid–Siglec interactions have been associated with multiple diseases including autoimmunity, infection, inflammation, aging or cancer. Therefore, glycomimetics targeting Siglecs have been pursued, especially for Siglec-2 (also called CD22).54 Sialoside mimetics carrying modifications on C-2, C-4, C-5 and C-9 of the sialic acid molecule were firstly envisaged, but later multiple site modifications led to more potent inhibitors.55 However, most of these glycomimetics do not have enough avidity to compete with natural glycoprotein ligands,56 and thus, multivalent presentation on polymers or nanoparticles provided more successful results.57 Interestingly, Paulson and co-workers have designed Siglec ligands based on di- and trivalent natural N-glycan scaffolds which showed low nM/high pM avidity and were capable of being endocytosed by CD22 on B lymphoma cells (Fig. 6).58 Feasibly, the individual branches of the chemically modified N-glycans interact simultaneously with multiple CD22 receptors, increasing the binding affinity.
 | ||
| Fig. 6 Trivalent modified N-glycans described as pM inhibitors of Siglec-2. | ||
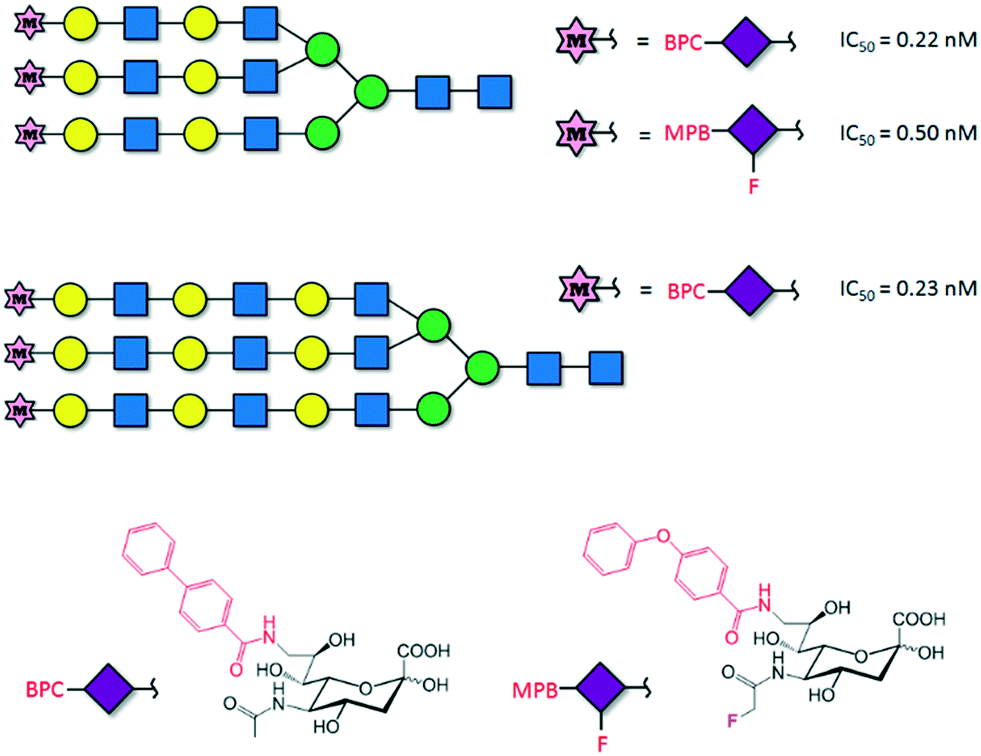
Single protein–carbohydrate interactions are usually weak and therefore chemical strategies must be applied to enhance the efficacy of glycomimetics. This is the case of the interaction between DC-SIGN and its natural ligands. DC-SIGN is a human C-type lectin receptor (CLR) located on the surface of dendritic cells that recognizes Lewis-type and high-mannose antigens. DC-SIGN introduces tolerance against self-antigens but also recognizes antigens from numerous pathogens (HIV, Ebola), a process that initiates an immune response or results in propagation and escape from the immune system. The structural features of the binding between natural sugars, including the blood type antigens and DC-SIGN, have been explored59 and revealed that the development of high-affinity ligands towards DC-SIGN has to overcome several drawbacks: the low affinity of the monovalent binding events, usually in the mM range, and the cross-reactivity among CLRs. Hence, considerable efforts have been made to develop high-affinity and selective glycomimetics. Indeed, approaches based on targeting extended binding sites have been investigated recently so as to improve the binding affinity. Although the strategy has not been applied yet to the design of potent DC-SIGN inhibitors, a library of 986 fragments has been screened using SPR, and five unknown secondary binding sites were validated using 1H-15N HSQC NMR spectroscopy.60 Interestingly, the understanding of key binding structural features was successfully exploited for designing highly specific glycomimetics for DC-SIGN.61 The rational modification of first-generation glycomimetics based on dimannosides provided μM affinity inhibitors that specifically recognize DC-SIGN vs. langerin, a similar CLR. Structural studies indicated that the introduction of a positive group on position C6 of the interacting sugar would reduce the affinity for langerin, since the stabilizing contacts with K313 on the binding site of the protein would be abolished. In fact, the introduction of a positively charged amino group in this position enhanced the affinity and selectivity towards DC-SIGN (Fig. 7A).
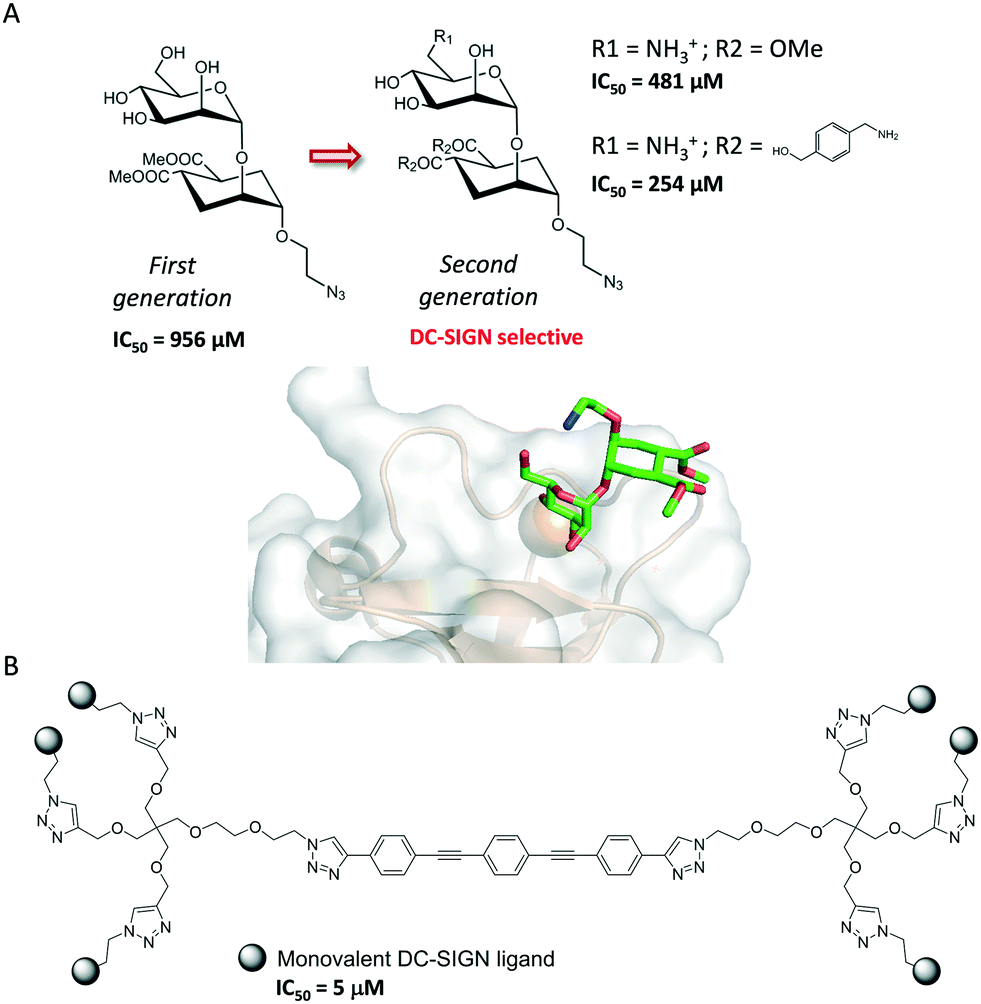 | ||
| Fig. 7 (A) First- and second-generation DC-SIGN glycomimetics. The X-ray crystal structure of DC-SIGN bound to a second-generation glycomimetic is also represented (pdb 2XR5). (B) DC-SIGN multivalent ligand. | ||
Notably, the structure of this compound is still far from that of an ideal drug-like molecule. Indeed, the affinity should be much more increased. The structurally open and water-exposed binding domain of DC-SIGN has hampered the development of potent glycomimetics and in fact, multivalency has been resorted to in order to increase the binding affinity (Fig. 7B).62 Recently, quantum dots surrounded by a dense array of mono- and disaccharides have been used as probes to gain insights into the binding mode of these multivalent interactions.63 Galectins, which comprise a large family of β-D-galactoside-binding lectins, are also involved in many biological functions with important implications in cancer progression, inflammation, immune responses, fibrosis and heart diseases.64 In fact, the blockage of galectins by high-potency glycomimetics is being pursued as a therapeutic strategy65 and several related clinical trials are currently ongoing.66,67 A huge effort has been made on the synthesis of mono- and multivalent glycomimetics68 allowing access to promising galectin ligands.69 In addition, the structural binding features of galectins have been deeply investigated. We have recently demonstrated how ligand flexibility modulates the thermodynamics and kinetics of the protein–sugar binding process.70 Particularly, we found that histo blood group antigens, which are quite rigid, bind to human galectin-3 with improved affinity due to their more favourable entropy of binding. Concerning this issue, a TF-mimetic with rather restricted conformational flexibility that efficiently binds to galectin-3 with a significant gain on the binding entropy has been recently reported.71 Moreover, recent neutron crystallography studies of galectin-3 provided a detailed view of the H-bonding network in the binding site of the protein.72 In particular, for galectin-3, the introduction of aromatic groups on the galactose C3 position of lactose, which established additional favourable cation–π interactions with the protein, provided potent compounds.73 Moreover, studies with aromatic thiodigalactoside derivatives, with improved hydrolytic stability, yielded a low nanomolar inhibitor,74 TD139,75 which has been approved by the US Food and Drug Administration for the treatment of idiopathic pulmonary fibrosis. Subsequent structurally driven tuning of these molecules provided selective and potent nM inhibitors.76–78 The introduction of highly fluorinated aryltriazole groups on C3 favoured the interactions with the protein, reaching KD values under 1–2 nM. In fact, this strategy has recently led to the development of aminopyrimidine–galactose hybrids with enhanced selectivity (>300-fold) towards galectin-3 over galectin-1.79 Similarly, the use of asymmetrical thiodigalactosides with C3-aryltriazolyl and O3-coumaryl groups has also led to high-affinity and selective galectin-3 inhibitors (Scheme 2).80
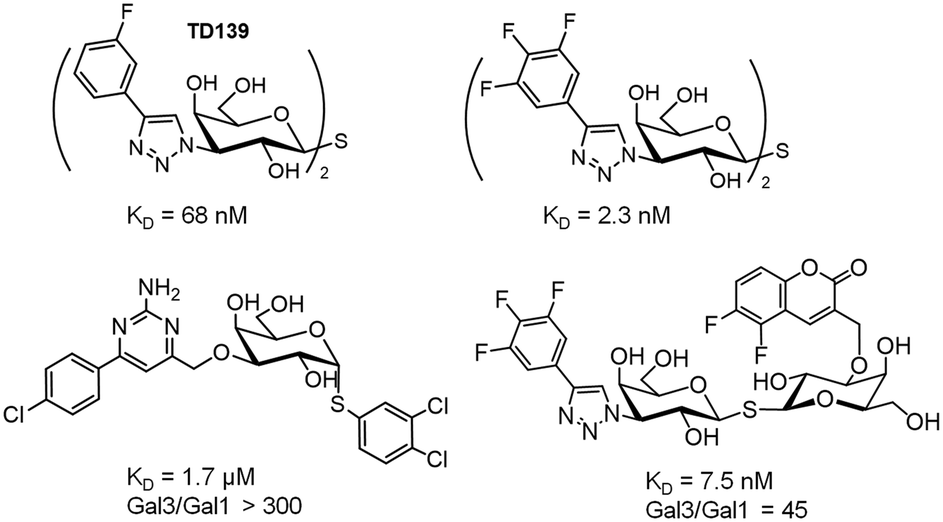 | ||
| Scheme 2 High-affinity and selective thiodigalactoside glycomimetics of galectin-3. | ||
Therapeutic glycopeptides
Glycopeptide antibiotics (GPAs) such as vancomycin, teicoplanin, bleomycin and ristocetin were discovered a long time ago. However, more than 50 years after it was first introduced, vancomycin still constitutes a standard therapy against serious Gram-positive infections. In fact, the development of new antibiotic glycopeptides is still a current area of research.81 The general mechanism of action of glycopeptides such as vancomycin entails the binding to lipid II inhibiting the synthesis of peptidoglycans, a vital structural component of the bacterial cell wall. Although the sugar moiety does not participate in the direct contacts between vancomycin and lipid II, it plays a key role in the back-to-back dimerization of vancomycin that increases its lipid II binding affinity.82 Moreover, it has been suggested that the sugar residue provides steric hindrance and limits the conformational flexibility of the vancomycin molecule, driving it toward lipid II binding.83 Indeed, peptide or protein glycosylation exponentially expands the structure and biological functions of the corresponding glycoconjugate. Factors such as the shape of the interacting glycopeptide or glycoprotein, the glycan density or additional carbohydrate-independent interactions modulate their solubility, stability and molecular recognition.84 However, the complexity and heterogeneity of the glycosylation process in natively expressed glycoproteins has hampered the complete understanding of those effects modulating their biological functions. Consequently, the synthetic preparation of homogeneous glycopeptides and glycoproteins has been pursued and allows extension of the overview beyond the “elemental” sugar–protein recognition phenomenon. In fact, synthetic glycopeptides also known as neoglycopeptides, capable of imitating the multivalent display of carbohydrates at the cell surface, have become an important objective as therapeutic compounds, and several reports appeared in the past years about targeting medically relevant lectins.85 Glycopeptide dendrimers,86 glycopeptide nanoparticles87 or glycopolyproline scaffolds88 have been successfully designed to enhance lectin avidity.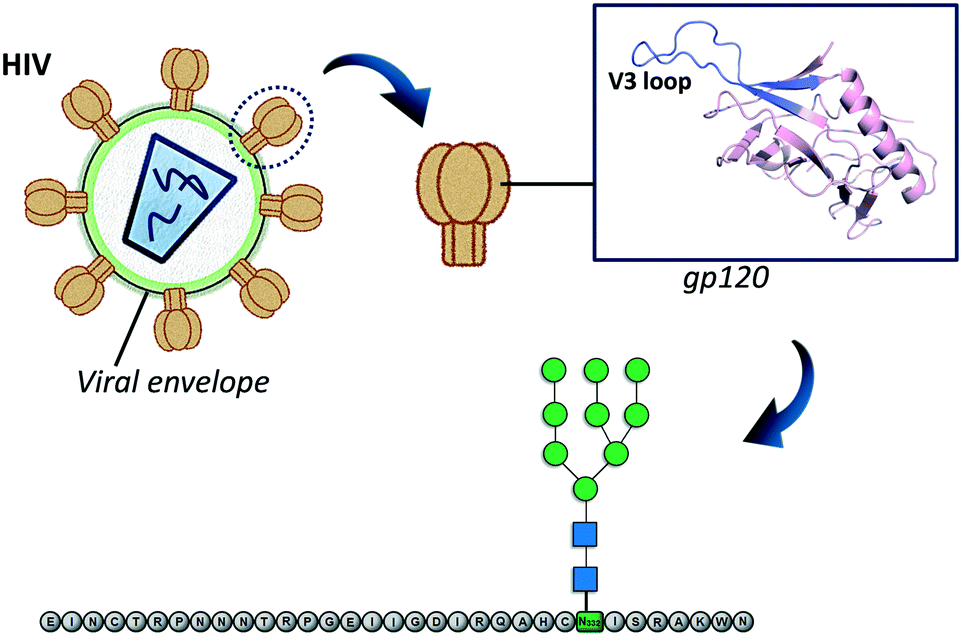 | ||
| Fig. 8 Schematic representation of the employed approach to design anti-HIV immunogenic glycopeptides. | ||
 | ||
| Fig. 9 (A) Model of normal densely glycosylated MUC1 and tumor-associated (TA) MUC1 with truncated glycosylation. (B) MUC1 synthetic glycopeptide bearing modifications at the peptide backbone and at the glycosidic linkage. | ||
The use of unnatural glycopeptides not only improves resistance against enzymatic degradation, but also overcomes cancer immune escape mechanisms. Indeed, when the S-glycoside mimetic carrying a fluoroproline was conjugated to gold nanoparticles and administered to mice, it provided a significant immune response, eliciting antibodies against cancer-related natural MUC1 antigens. The combination with immunostimulants is required for the activation of the T-cell-dependent pathways and the production of high-affinity IgG antibodies. Similarly, conjugation to carrier proteins, such as tetanus toxoid, bovine serum albumin100 or bacteriophages,101 delivers potent MUC1 glycopeptide vaccines that selectively target tumour-associated MUC1 on tumour cells and not those on normal epithelial cells.
Carbohydrate-based vaccines
Carbohydrates with differentiating glycosylation patterns of pathogens or malignant host cells could be used as antigens for vaccine development.102 However, glycans in general are poorly immunogenic and unable to elicit a T-cell memory response. As a unique exception, polysaccharides with alternating positive and negative charges present at the surface of some Gram-positive bacteria are able to activate T cells.103 Apart from this, vaccine development is usually achieved by conjugation of glycan antigens to appropriate carriers such as proteins, peptides or nanoparticles. In fact, chemical ligation has an impact on the vaccine efficiency and different methods have been developed.104 As described in the previous section, tumor-associated carbohydrate antigens represent specific targets for cancer immunotherapy, and vaccine design based on the synthetic modification of natural TACAs has provided promising results showing potent responses with reduced immunotolerance.105 Conjugates carrying bacterial glycans have also been extensively employed for vaccine preparation and the topic has been recently reviewed.106 Initially, the polysaccharide coats and major virulence factors of bacteria were isolated from natural sources, and then chemically conjugated to carrier proteins. Although these vaccination agents were capable of eliciting a T-cell-dependent immune response, the heterogeneity of the samples complicated the manufacturing and introduced undesirable side effects. More recently, the progress in carbohydrate chemistry and methods to determine the immunogenic glycan epitopes107 has permitted the development of rationally designed antibacterial and antifungal vaccines. However, bacterial pathogens such as Haemophilus influenza type b (Hib), Neisseria meningitidis, and Streptococcus pneumoniae continue to cause deadly diseases and the design of safe and highly efficacious vaccines is still an active area of research. For instance, Prevnar13 and Synflorix are currently employed pneumococcal vaccines, though they only cover 13 and 10, respectively, of more than 90 existing serotypes and consequently fail to offer a wide coverage. In this regard, Seeberger and co-workers have recently shown that glycoconjugates containing oligosaccharides of different S. pneumoniae serotypes can be co-formulated or used in combination with marketed vaccines to generate a multivalent vaccine which induces a strong antibody response.108 Other novel approaches such as the use of outer membrane vesicles109 or TL4 ligands as carriers110 are being developed. Generally, such factors including the glycan epitope, the carrier protein, the chemical ligation of the sugar part or the activation mechanism of the immune system make it difficult to rationalize the response to vaccines, and thus, vaccine design results in a very challenging task. Interestingly, it has been described that the polysaccharide structure influences the antigen processing and in turn the mechanism underlying the adaptive immune response.111Glycoproteins and therapeutic applications
The application of proteins as therapeutics in the pharmaceutical field has been incredibly expanded over the past years, proteins being one of the actual best-selling products for the treatment of a wide range of diseases and pathologies. In this regard, a great percentage of these proteins habitually display post-translational modifications (PTMs), and glycan attachment is one of the most prominent structural changes. More than half of the commercially available therapeutic proteins are glycoproteins, which are obtained through molecular biology protocols using living cell systems. Since the glycan profile is essentially heterogeneous the recent research still pursues the development and application of robust protocols to analyse the glycan profile of both commercial and potentially therapeutic glycoproteins,112–114 trying to better understand how those different glycoforms affect the product quality in terms of effectiveness, stability, pharmacokinetics and toxicity.115,116 Usually, the carbohydrate content of intact glycoproteins is directly assessed by chromatographic separation coupled to mass spectrometry detection. Recently, a novel workflow has been designed which uses relatively simple LC or LC/MS methods to characterize complex glycosylated proteins and antibodies at different levels (intact glycoprotein, fragments cleaved by different proteases, released glycans).117 Even so, minor changes in the N-glycosylation pattern are difficult to detect in the extracted ion chromatograms. In this regard, Planinc et al. have proposed an interesting approach oriented to combine the sensitivity of MS detection devices with subsequent principal component analysis (PCA) and classification through soft independent modelling by class analogy (SIMCA) that can be applied to the evaluation of previously released glycans via enzymatic cleavage.118 Lately, a new and robust capillary electrophoresis-based method coupled to ESI-MS has permitted the determination of the glycan content of diverse known mAbs, highlighting the reproducibility and the accuracy of the results by comparison with a HILIC-based reference method.119 Additionally, the applicability of lectin microarrays120 to screen the glycans of therapeutic glycoproteins, especially monoclonal antibodies, has also been highlighted.121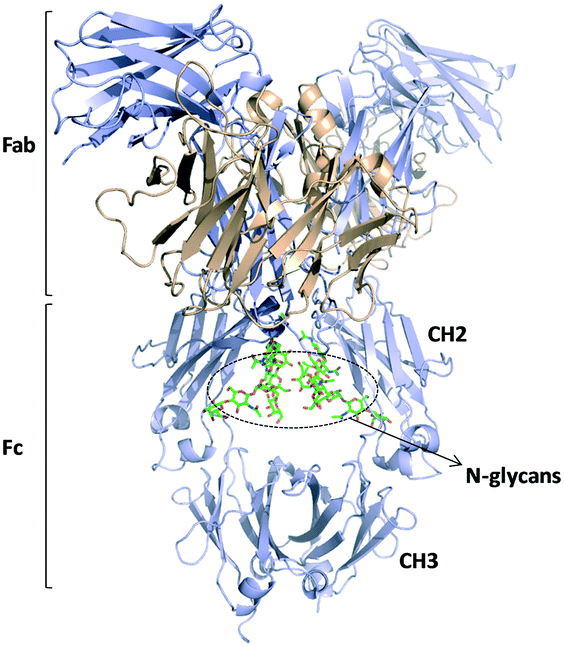 | ||
| Fig. 10 Schematic representation of an IgG1 antibody structure (pdb 1HZH).124 Fab and Fc regions are indicated and the glycans present in the CH2 domains are highlighted. | ||
For instance, reported evidence already exists about the effects of non-reducing galactose and fucose terminal residues on the immune effector functions of antibodies.125 Recently, an antibody constituted by a non-glycosylated F8-based diabody has been engineered, in which each F8 unit is fused via the C-terminus to a glycosylated interleukin-9 (IL9).126 With this system, Venetz and co-workers demonstrated that variations in the N-linked glycans deeply changed the efficiency of the F8 fragment to target the tumor cells. By quantitative distribution analyses, they interestingly found a positive correlation between the level of sialylation in IL9 N-glycans and the capability of the F8 moiety to specifically bind the spliced extradomain A (EDA) of fibronectin. Similarly, the importance of controlling microheterogeneity in the production of mAbs with therapeutic purposes has been underlined. Indeed, it has been proposed that discrepancies in efficiency and safety among mAbs could be explained by differences in the surface charge of each glycoform as a consequence of the existing heterogeneous glycan environments.127
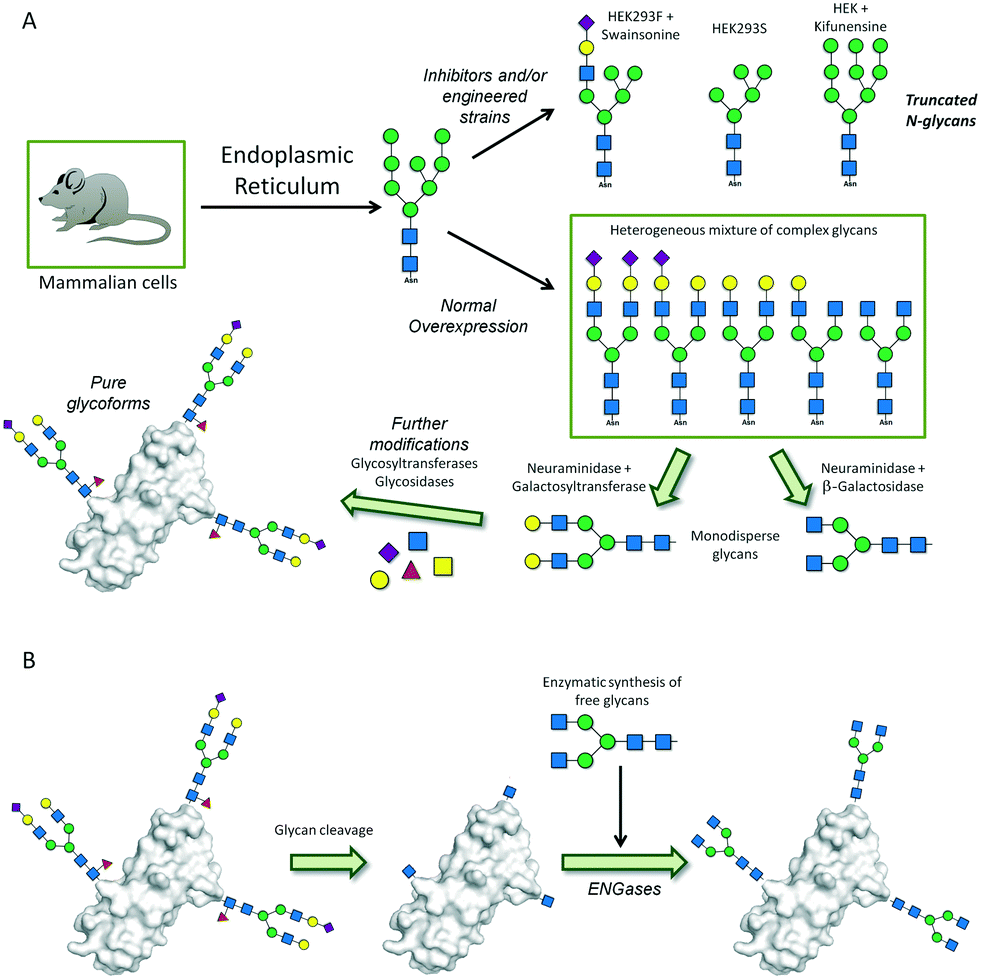 | ||
| Fig. 11 Biosynthetic approaches for the preparation of homogeneous glycoproteins. (A) Previous and post enzyme-based remodeling of the overexpressed glycoforms. (B) Cleavage and subsequent incorporation of the desired oligosaccharide chain. | ||
Besides enzyme-based remodeling, special engineered yeast strains with altered biosynthetic routes are likewise used to afford mammalian-like glycans.135 A modified yeast strain has been prepared by disparity mutagenesis, which is glycosylation-deficient and exclusively produces high-mannose-type N-glycans (Man9).136 An alternative strategy for glycoprotein preparation is total chemical synthesis or semi-synthesis. Native chemical ligation (NCL) techniques allow the chemoselective linkage of peptidic fragments through coupling of a C-terminal thioester to an N-terminal cysteine residue.137 This approach still has some important limitations, especially for quite long proteins that would require multiple ligations. The chemical lability of thioesters, also subjected to epimerization equilibria, and the low percentage of cysteine residues in protein sequences constitute the major drawbacks. Consequently, and given the complexity and variability of peptide fragments that may be needed, this area of research remains under study and optimization. The use of β-mercapto amino acid analogues instead of natural cysteines, and the utilization of peptide hydrazides, which are more stable, as substitutes for peptide thioesters are some of the alternatives that have been described so far.138
Conclusions
In the past years, many contributions have demonstrated that the exploitation of carbohydrates in medicinal chemistry opens a broad spectrum of therapeutic possibilities. Despite the technical challenges associated with the intrinsic complexity of glycans, tailor-made small glycomimetics have been designed for blocking specific carbohydrate–lectin interactions. The success of rationally driven approaches for the development of new drugs is still to be widely demonstrated, although it is clearly improving. In fact, several glycomimetics are already in clinical trials. Nevertheless, more specific targets for glycomimetics still have to be uncovered. The enormous progress in synthetic strategies, analytical tools and high-resolution biophysical techniques has allowed advances in the study of complex glycosylated structures, including glycoproteins. Dendrimers, nanoparticles and other polyvalent structures have been introduced as a strategy to increase avidity, not to mention the therapeutic potential of specifically designed glycopeptides. The development of glycopeptide-based vaccines aimed to fight diseases such as cancer or HIV is an active area of research. Similarly, potent anti-bacterial vaccines have been developed. In this regard, more attention should be paid to the mechanisms underlying the immune response, where the role of glycan epitopes remains not fully understood. However, the awareness of the key role of glycans also on the antibody effectiveness, stability, pharmacokinetics and toxicity has broken the limits of their therapeutic applications. Definitely, progress in glycoprotein expression and their structural analysis is beginning to overcome the challenge derived from the complexity of large glycoconjugates. Furthermore, aspects directed to the study of the cell-surface microenvironment effects are also being addressed. Surely, the increasing understanding of all aspects of glycoscience will allow the development of novel and more effective therapies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council for financial support (ERC-2017-AdG, project number 788143-RECGLYCANMR). We also thank Instituto de Salud Carlos III of Spain, ISCIII (grant PRB3 IPT17/0019 to A. G.) and Agencia Estatal Investigación of Spain, AEI (grants CTQ2015-64597-C2-1-P and RTI2018-094751-B-C21 and the Severo Ochoa Excellence Accreditation SEV-2016-0644).Notes and references
- R. D. Cummings and M. E. Etzler, Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press, New York, 2nd edn, 2009, ch. 6 Search PubMed.
- A. Varki, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Nat. Rev. Nephrol., 2019, 15, 346–366 CrossRef.
- H. J. Joshi, Y. Narimatsu, K. T. Schjoldager, H. L. P. Tytgat, M. Aebi, H. Clausen and A. Halim, Cell, 2018, 172, 632–632.e2 CrossRef CAS PubMed.
- F. Schwarz and M. Aebi, Curr. Opin. Struct. Biol., 2011, 21, 576–582 CrossRef CAS.
- J. Nilsson, A. Halim, A. Grahn and G. Larson, Glycoconjugate J., 2013, 30, 119–136 CrossRef CAS PubMed.
- (a) A. V. Everest-Dass, E. S. X. Moh, C. Ashwood, A. M. M. Shathili and N. H. Packer, Expert Rev. Proteomics, 2018, 15, 165 CrossRef CAS PubMed; (b) M. J. Kailemia, G. Xu, M. Wong, Q. Li, E. Goonatilleke, F. Leon and C. B. Lebrilla, Anal. Chem., 2018, 90, 208 CrossRef CAS PubMed.
- L. Krasnova and C.-H. Wong, J. Am. Chem. Soc., 2019, 1411, 3735–3754 CrossRef PubMed.
- T. M. Puvirajesinghe and J. E. Turnbull, Microarrays, 2016, 5, 3 CrossRef PubMed.
- J. Agirre, G. J. Davies, K. S. Wilson and K. D. Cowtan, Curr. Opin. Struct. Biol., 2017, 44, 39–47 CrossRef CAS PubMed.
- (a) D. Sirohi, Z. Chen, L. Sun, T. Klose, T. C. Pierson, M. G. Rossmann and R. J. Kuhn, Science, 2016, 352, 467–470 CrossRef CAS; (b) H. B. Gristick, H. Wang and P. J. Bjorkman, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 822–828 CrossRef CAS.
- A. Ardá, H. Coelho, B. Fernández de Toro, S. Galante, A. Gimeno, A. Poveda, J. Sastre, L. Unione, P. Valverde, F. Javier Cañada and J. Jiménez-Barbero, Special Periodic Reports. Carbohydrate Chemistry, ed. A. P. Rauter, T. Lindhorst and Y. Queneau, RSC, 2017, ch. 2, vol. 42 Search PubMed.
- A. Ardá and J. Jiménez-Barbero, Chem. Commun., 2018, 54, 4761–4769 RSC.
- A. Diniz, J. S. Dias, J. Jiménez-Barbero, F. Marcelo and E. J. Cabrita, Chem. – Eur. J., 2017, 23, 13213–13220 CrossRef CAS PubMed.
- S. C. Purcell and K. Godula, Interface Focus, 2019, 9, 20180080 CrossRef PubMed.
- E. Luchinat and L. Banci, IUCrJ, 2017, 4, 108–118 CrossRef CAS PubMed.
- (a) A. Fernández-Tejada, F. J. Cañada and J. Jiménez-Barbero, ChemMedChem, 2015, 10, 1291–1295 CrossRef; (b) A. Fernández-Tejada, F. J. Cañada and J. Jiménez-Barbero, Chem. – Eur. J., 2015, 21, 10616–10628 CrossRef PubMed; (c) J. E. Hudak and C. R. Bertozzi, Chem. Biol., 2014, 21, 16–37 CrossRef CAS PubMed; (d) S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed; (e) M. Dalziel, M. Crispin, C. N. Scanlan, N. Zitzmann and R. A. Dwek, Science, 2014, 343, 1235681 CrossRef PubMed; (f) R. Hevey, Pharmaceuticals, 2019, 12, 55 CrossRef.
- R. J. Linhardt, J. Med. Chem., 2003, 46, 2551–2564 CrossRef CAS PubMed.
- (a) M. Guerrini, D. Beccati, Z. Shriver, A. Naggi, K. Viswanathan, A. Bisio, I. Capila, J. C. Lansing, S. Guglieri, B. Fraser, A. Al-Hakim, N. S. Gunay, Z. Zhang, L. Robinson, L. Buhse, M. Nasr, J. Woodcock, R. Langer, G. Venkataraman, R. J. Linhardt, B. Casu, G. Torri and R. Sasisekharan, Nat. Biotechnol., 2008, 26, 669–675 CrossRef CAS PubMed; (b) D. Keire, B. Mulloy, C. Chase, A. Al-Hakim, D. Cairatti, E. Gray, J. Hogwood, T. Morris, P. Mourao, M. D. Soares and A. Szajek, BioPharm Int., 2015, 28, 36–42 Search PubMed.
- S. Mohamed and D. R. Coombe, Pharmaceuticals, 2017, 10, 78 CrossRef PubMed.
- J. M. Herbert, M. Petitou, J. C. Lormeau, R. Cariou, J. Necciari, H. N. Magnani, P. Zandberg, R. G. M. van Amsterdam, C. A. A. van Boeckel and D. G. Meuleman, Cardiovasc. Drug Rev., 1997, 15, 1–26 CrossRef CAS.
- (a) D. Fisher, B. Xing, J. Dill, H. Li, H. H. Hoang, Z. Z. Zhao, X. L. Yang, R. Bachoo, S. Cannon, F. M. Longo, M. Sheng, J. Silver and S. X. Li, J. Neurosci., 2011, 31, 14051–14066 CrossRef CAS PubMed; (b) Y. J. Shen, A. P. Tenney, S. A. Busch, K. P. Horn, F. X. Cuascut, K. Liu, Z. G. He, J. Silver and J. G. Flanagan, Science, 2009, 326, 592–596 CrossRef CAS PubMed; (c) C. H. Coles, Y. Shen, A. P. Tenney, C. Siebold, G. C. Sutton, W. Lu, J. T. Gallagher, E. Y. Jones, J. G. Flanagan and A. R. Aricescu, Science, 2011, 332, 484–488 CrossRef CAS PubMed.
- (a) Y. Ohtake and S. X. Li, Brain Res., 2015, 1619, 22–35 CrossRef CAS PubMed; (b) A. W. Stoker, Semin. Cell Dev. Biol., 2015, 37, 90–97 CrossRef CAS PubMed.
- X. Dai, W. Liu, Q. Zhou, C. Cheng, C. Yang, S. Wang, M. Zhang, P. Tang, H. Song, D. Zhang and Y. Qin, J. Org. Chem., 2016, 81(1), 162–184 CrossRef CAS PubMed.
- U. Bhaskar, E. Sterner, A. M. Hickey, A. Onishi, F. Zhang, J. S. Dordick and R. J. Linhardt, Appl. Microbiol. Biotechnol., 2012, 93, 1–16 CrossRef PubMed.
- S. Dey, H. Lo and C. H. Wong, J. Am. Chem. Soc., 2019, 141, 10309–10314 CrossRef CAS PubMed.
- (a) S. U. Hansen, G. J. Miller, G. C. Jayson and J. M. Gardiner, Org. Lett., 2013, 15(1), 88–91 CrossRef CAS PubMed; (b) S. U. Hansen, G. J. Miller, M. J. Cliff, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2015, 6(11), 6158–6164 RSC.
- (a) M. Baráth, S. U. Hansen, C. E. Dalton, G. C. Jayson, G. J. Miller and J. M. Gardiner, Molecules, 2015, 20(4), 6167–6180 CrossRef PubMed; (b) S. Dey and C. H. Wong, Chem. Sci., 2018, 9(32), 6685–6691 RSC.
- C. Zong, A. Venot, O. Dhamale and G. J. Boons, Org. Lett., 2013, 15(2), 342–345 CrossRef CAS PubMed.
- S. U. Hansen, G. J. Miller, C. Cole, G. Rushton, E. Avizienyte, G. C. Jayson and J. M. Gardiner, Nat. Commun., 2013, 4, 2016 CrossRef PubMed.
- X. Zhang, V. Pagadala, H. M. Jester, A. M. Lim, T. Q. Pham, A. P. Goulas, J. Liu and R. J. Linhardt, Chem. Sci., 2017, 8(12), 7932–7940 RSC.
- M. S. Lord, B. Cheng, F. Tang, J. G. Lyons, J. Rnjak-Kovacina and J. M. Whitelock, Metab. Eng., 2016, 38, 105–114 CrossRef CAS PubMed.
- B. E. Stopschinski, B. B. Holmes, G. M. Miller, V. A. Manon, J. Vaquer-Alicea, W. L. Prueitt, L. C. Hsieh-Wilson and M. I. Diamond, J. Biol. Chem., 2018, 293(27), 10826–10840 CrossRef CAS.
- (a) E. Avizienyte, C. L. Cole, G. Rushton, G. J. Miller, A. Bugatti, M. Presta, J. M. Gardiner and G. C. Jayson, PLoS One, 2016, 11(8), e0159739 CrossRef; (b) G. J. Miller, S. U. Hansen, E. Avizienyte, G. Rushton, C. L. Cole, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2013, 4, 3218–3222 RSC.
- G. C. Jayson, S. U. Hansen, G. J. Miller, C. L. Cole, G. Rushton, E. Avizienyte and J. M. Gardiner, Chem. Commun., 2015, 51(72), 13846–13849 RSC.
- C. Zong, R. Huang, E. Condac, Y. Chiu, W. Xiao, X. Li, W. Lu, M. Ishahara, S. Wang, A. Ramiah, M. Stickney, P. Azadi, I. J. Amster, K. W. Moremen, L. Wang, J. S. Sharp and G. J. Boons, J. Am. Chem. Soc., 2016, 138(39), 13059–13067 CrossRef CAS PubMed.
- L. Unione, A. Gimeno, P. Valverde, I. Calloni, H. Coelho, S. Mirabella, A. Poveda, A. Ardá and J. Jimenez-Barbero, Curr. Med. Chem., 2017, 24, 4057–4080 CrossRef CAS PubMed.
- R. Sommer, S. Wagner, A. Varrot, C. M. Nycholat, A. Khaledi, S. Häussler, J. C. Paulson, A. Imberty and A. Titz, Chem. Sci., 2016, 7, 4990–5001 RSC.
- S. Wang, L. Dupin, M. Noel, C. J. Carroux, L. Renaud, T. Gehin, A. Meyer, E. Souteyrand, J. J. Vasseur, G. Vergoten, Y. Chevolot, F. Morvana and S. Vidal, Chem. – Eur. J., 2016, 22, 11785–11794 CrossRef CAS PubMed.
- K. Buffet, I. Nierengarten, N. Galanos, E. Gillon, M. Holler, A. Imberty, S. E. Matthews, S. Vidal, S. P. Vincenta and J. F. Nierengarten, Chem. – Eur. J., 2016, 22, 2955–2963 CrossRef CAS.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- R. Sommer, D. Hauck, A. Varrot, S. Wagner, A. Audfray, A. Prestel, H. M. Mçller, A. Imberty and A. Titz, ChemistryOpen, 2015, 4, 756–767 CrossRef CAS.
- R. Sommer, S. Wagner, K. Rox, A. Varrot, D. Hauck, E.-C. Wamhoff, J. Schreiber, T. Ryckmans, T. Brunner, C. Rademacher, R. W. Hartmann, M. Brönstrup, A. Imberty and A. Titz, J. Am. Chem. Soc., 2018, 140, 2537–2545 CrossRef CAS PubMed.
- V. Denavit, D. Lainé, C. Bouzriba, E. Shanina, É. Gillon, S. Fortin, C. Rademacher, A. Imberty and D. Giguère, Chem. – Eur. J., 2019, 25, 4478–4490 CrossRef CAS PubMed.
- S. Wagner, D. Hauck, M. Hoffmann, R. Sommer, I. Joachim, R. Müller, A. Imberty, A. Varrota and A. Titz, Angew. Chem., Int. Ed., 2017, 56, 16559–16564 CrossRef CAS.
- J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C. S. Hung, J. Pinkner, R. Slattegard, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2005, 55, 441–455 CrossRef CAS.
- (a) S. Kleeb, L. Pang, K. Mayer, D. Eris, A. Sigl, R. C. Preston, P. Zihlmann, T. Sharpe, R. P. Jakob, D. Abgottspon, A. S. Hutter, M. Scharenberg, X. Jiang, G. Navarra, S. Rabbani, M. Smiesko, N. Ludin, J. Bezencon, O. Schwardt, T. Maiera and B. Ernst, J. Med. Chem., 2015, 58, 2221–2239 CrossRef CAS PubMed; (b) C. Jarvis, Z. Han, V. Kalas, R. Klein, J. S. Pinkner, B. Ford, J. Binkley, C. K. Cusumano, Z. Cusumano, L. Mydock-McGrane, S. J. Hultgren and J. W. Janetka, ChemMedChem, 2016, 11, 367–373 CrossRef CAS PubMed.
- W. Schönemann, J. Cramer, T. Mühlethaler, B. Fiege, M. Silbermann, S. Rabbani, P. Dätwyler, P. Zihlmann, R. P. Jakob, C. P. Sager, M. Smieško, O. Schwardt, T. Maier and B. Ernst, ChemMedChem, 2019, 14, 749–757 CrossRef PubMed.
- S. Kleeb, X. Jiang, P. Frei, A. Sigl, J. Bezencon, K. Bamberger, O. Schwardt and B. Ernst, J. Med. Chem., 2016, 59(7), 3163–3182 CrossRef CAS.
- C. P. Sager, B. Fiege, P. Zihlmann, R. Vannam, S. Rabbani, R. P. Jakob, R. C. Preston, A. Zalewski, T. Maier, M. W. Peczuh and B. Ernst, Chem. Sci., 2018, 9, 646–654 RSC.
- P. Zihlmann, M. Silbermann, T. Sharpe, X. Jiang, T. Mühlethaler, R. P. Jakob, S. Rabbani, C. P. Sager, P. Frei, T. Maier and B. Ernst, Chem. – Eur. J., 2018, 24, 13049–13057 CrossRef CAS.
- J. Egger, C. Weckerle, B. Cutting, O. Schwardt, S. Rabbani, K. Lemme and B. Ernst, J. Am. Chem. Soc., 2013, 135, 9820–9828 CrossRef CAS PubMed.
- K. E. Moog, M. Barz, M. Bartneck, F. Beceren-Braun, N. Mohr, Z. Wu, L. Braun, J. Dernedde, E. A. Liehn, F. Tacke, T. Lammers, H. Kunz and R. Zentel, Angew. Chem., Int. Ed., 2016, 55, 1416–1421 Search PubMed.
- T. Angata, C. M. Nycholat and M. S. Macauley, Trends Pharmacol. Sci., 2015, 36, 645–660 CrossRef CAS.
- C. Büll, T. Heise, G. J. Adema and T. J. Boltje, Trends Biochem. Sci., 2016, 41, 519–531 CrossRef PubMed.
- J. Ereño-Orbea, T. Sicard, H. Cui, M. T. Mazhab-Jafari, S. Benlekbir, A. Guarné, J. L. Rubinstein and J.-P. Julien, Nat. Commun., 2017, 8, 764 CrossRef.
- H. Prescher, A. Schweizer, E. Kuhfeldt, L. Nitschke and R. Brossmer, ChemBioChem, 2017, 18, 1216–1225 CrossRef CAS.
- W. Peng and J. C. Paulson, J. Am. Chem. Soc., 2017, 139, 12450–12458 CrossRef CAS PubMed.
- (a) H. Feinberg, R. Castelli, K. Drickamer, P. H. Seeberger and W. I. Weis, J. Biol. Chem., 2007, 282, 4202–4209 CrossRef CAS PubMed; (b) A. Holla and A. Skerra, Protein Eng., Des. Sel., 2011, 24, 659–669 CrossRef CAS PubMed; (c) K. Brzezicka, B. Echeverria, S. Serna, A. Van Diepen, C. H. Hooke and N.-C. Reichardt, ACS Chem. Biol., 2015, 10, 1290–1302 CrossRef CAS PubMed; (d) K. Pederson, D. A. Mitchell and J. H. Prestegard, Biochemistry, 2014, 53, 5700–5709 CrossRef CAS PubMed; (e) J. D. Martínez, P. Valverde, S. Delgado, C. Romanò, B. Linclau, N. C. Reichardt, S. Oscarson, A. Ardá, J. Jiménez-Barbero and F. J. Cañada, Molecules, 2019, 24, E2337 CrossRef PubMed; (f) P. Valverde, S. Delgado, J. D. Martínez, J.-B. Vendeville, B. Linclau, J. Malassis, N. C. Reichardt, F. J. Cañada, J. Jiménez-Barbero and A. Ardá, ACS Chem. Biol., 2019, 14, 1660–1671 CrossRef CAS PubMed.
- J. Aretz, H. Baukmann, E. Shanina, J. Hanske, R. Wawrzinek, V. A. Zapolśkii, P. H. Seeberger, D. E. Kaufmann and C. Rademacher, Angew. Chem., Int. Ed., 2017, 56, 7292–7296 CrossRef CAS PubMed.
- V. Porkolab, E. Chabrol, N. Varga, S. Ordanini, I. Sutkevičiūtė, M. Thepau, M. J. Garcia-Jimenez, E. Girard, P. M. Nieto, A. Bernardi and F. Fieschi, ACS Chem. Biol., 2018, 13, 600 CrossRef CAS PubMed.
- (a) B. Bertolotti, I. Sutkeviciute, M. Ambrosini, R. Ribeiro-Viana, J. Rojo, F. Fieschi, H. Dvořáková, M. Kašáková, K. Parkan, M. Hlaváčková, K. Nováková and J. Moravcová, Org. Biomol. Chem., 2017, 15, 3995 RSC; (b) S. Ordanini, N. Varga, V. Porkolab, M. Thépaut, L. Belvisi, A. Bertaglia, A. Palmioli, A. Berzi, D. Trabattoni, M. Clerici, F. Fieschi and A. Bernardi, Chem. Commun., 2015, 51, 3816 RSC; (c) A. Berzi, S. Ordanini, B. Joosten, D. Trabattoni, A. Cambi, A. Bernardi and M. Clerici, Sci. Rep., 2016, 6, 35373 CrossRef CAS PubMed.
- Y. Guo, I. Nehlmeier, E. Poole, C. Sakonsinsiri, N. Hondow, A. Brown, Q. Li, S. Li, J. Whitworth, Z. Li, A. Yu, R. Brydson, W. B. Turnbull, S. Pöhlmann and D. Zhou, J. Am. Chem. Soc., 2017, 139, 11833–11844 CrossRef CAS PubMed.
- L. Johannes, R. Jacob and H. Leffler, J. Cell Sci., 2018, 131, jcs208884 CrossRef PubMed.
- R. P. M. Dings, M. C. Miller, R. J. Griffin and K. H. Mayo, Int. J. Mol. Sci., 2018, 19, 905 CrossRef PubMed.
- A. Girard and J. L. Magnani, Trends Glycosci. Glycotechnol., 2018, 30, SE211–SE220 CrossRef.
- H. Blanchard, X. Yu, P. M. Collins and K. Bum-Erdene, Expert Opin. Ther. Pat., 2014, 24, 1053–1065 CrossRef CAS PubMed.
- V. L. Campo, M. F. Marchiori, L. C. Rodrigues and M. Dias-Baruffi, Glycoconjugate J., 2016, 3, 853–876 CrossRef PubMed.
- D. Laaf, P. Bojarová, L. Elling and V. Kren, Trends Biotechnol., 2019, 37, 402–415 CrossRef CAS PubMed.
- A. Gimeno, S. Delgado, P. Valverde, S. Bertuzzi, M. A. Berbis, J. Echavarren, A. Lacetera, S. Martín-Santamaria, A. Surolia, F. J. Cañada, J. Jimenez-Barbero and A. Ardá, Angew. Chem., Int. Ed., 2019, 58, 7268–7272 CrossRef CAS.
- S. Santarsia, A. S. Grosso, F. Trovão, J. Jiménez-Barbero, A. L. Carvalho, C. Nativi and F. Marcelo, ChemMedChem, 2018, 13, 2030–2036 CrossRef CAS PubMed.
- F. Manzoni, J. Wallerstein, T. E. Schrader, A. Ostermann, L. Coates, M. Akke, M. P. Blakeley, E. Oksanen and D. T. Logan, J. Med. Chem., 2018, 61, 4412–4420 CrossRef CAS PubMed.
- R. Tellez-Sanz, L. García-Fuentes and A. Vargas-Berenguel, Curr. Med. Chem., 2013, 20, 2979–2990 CrossRef CAS.
- I. Cumpstey, A. Sundin, H. Leffler and U. J. Nilsson, Angew. Chem., Int. Ed., 2005, 44, 5110–5112 CrossRef CAS PubMed.
- T. Delaine, P. Collins, A. MacKinnon, G. Sharma, J. Stegmayr, V. K. Rajput, S. Mandal, I. Cumpstey, A. Larumbe, B. A. Salameh, B. Kahl-Knutsson, H. van Hattum, M. V. Scherpenzeel, R. J. Pieters, T. Sethi, H. Schambye, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, ChemBioChem, 2016, 17, 1759–1770 CrossRef CAS PubMed.
- T.-J. Hsieh, H.-Y. Lin, Z. Tu, T.-C. Lin, S.-C. Wu, Y.-Y. Tseng, F.-T. Liu, S.-T. D. Hsu and C.-H. Lin, Sci. Rep., 2016, 6, 29457 CrossRef CAS PubMed.
- V. K. Rajput, A. MacKinnon, S. Mandal, P. Collins, H. Blanchard, H. Leffler, T. Sethi, H. Schambye, B. Mukhopadhyay and U. J. Nilsson, J. Med. Chem., 2016, 59, 8141–8147 CrossRef CAS PubMed.
- F. R. Zetterberg, K. Peterson, R. E. Johnsson, T. Brimert, M. Håkansson, D. T. Logan, H. Leffler and U. J. Nilsson, ChemMedChem, 2018, 13, 133–137 CrossRef CAS PubMed.
- A. Dahlqvist, F. R. Zetterberg, H. Leffler and U. J. Nilsson, Med. Chem. Commun., 2019, 10, 913–925 RSC.
- K. Peterson, R. Kumar, O. Stenström, P. Verma, P. R. Verma, M. Hakansson, B. Kahl-Knutsson, F. Zetterberg, H. Leffler, M. Akke, D. T. Logan and U. J. Nilsson, J. Med. Chem., 2018, 61, 1164–1175 CrossRef CAS.
- M. A. T. Blaskovich, K. A. Hansford, M. S. Butler, Z. Jia, A. E. Mark and M. A. Cooper, ACS Infect. Dis., 2018, 4, 715–735 CrossRef CAS.
- V. Kren and T. Rezanka, FEMS Microbiol. Rev., 2008, 32, 858–889 CrossRef CAS PubMed.
- J. Kaplan, B. D. Korty, P. H. Axelsen and P. J. Loll, J. Med. Chem., 2001, 44, 1837–1840 CrossRef CAS PubMed.
- S. Ng, E. Lin, P. I. Kitov, K. F. Tjhung, O. O. Gerlits, L. Deng, B. Kasper, A. Sood, B. M. Paschal, P. Zhang, C.-C. Ling, J. S. Klassen, C. J. Noren, L. K. Mahal, R. J. Woods, L. Coates and R. Derda, J. Am. Chem. Soc., 2015, 137, 5248–5251 CrossRef CAS PubMed.
- S. Behren and U. Westerlind, Molecules, 2019, 24, 100 CrossRef PubMed.
- (a) M. Bergmann, G. Michaud, R. Visini, X. Jin, E. Gillon, A. Stocker, A. Imberty, T. Darbre and J.-L. Reymond, Org. Biomol. Chem., 2016, 14, 138–148 RSC; (b) G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J.-L. Reymond, Chem. Sci., 2016, 7, 166–182 RSC.
- C. Bonduelle, H. Oliveira, C. Gauche, J. Huang, A. Heise and S. Lecommandoux, Chem. Commun., 2016, 52, 11251–11254 RSC.
- S.-F. Huang, C.-H. Lin, Y.-T. Lai, C.-L. Tsai, T.-J. R. Cheng and S.-K. Wang, Chem. – Asian J., 2018, 13, 686–700 CrossRef CAS PubMed.
- H. Cai, J. Orwenyo, J. P. Giddens, Q. Yang, R. Zhang, C. C. LaBranche, D. C. Montefiori and L.-X. Wang, Cell Chem. Biol., 2017, 21, 1513–1522 CrossRef PubMed.
- H. Cai, R. Zhang, J. Orwenyo, J. P. Giddens, Q. Yang, C. C. LaBranche, D. Montefiori and L.-X. Wang, J. Med. Chem., 2018, 61, 10116–10125 CrossRef CAS PubMed.
- A. Escolano, H. B. Gristick, M. E. Abernathy, J. Merkenschlager, R. Gautam, T. Y. Oliveira, J. Pai, A. P. West Jr, C. O. Barnes, A. A. Cohen, H. Wang, J. Golijanin, D. Yost, J. R. Keeffe, Z. Wang, P. Zhao, K. Yao, J. Bauer, L. Nogueira, H. Gao, A. V. Voll, D. C. Montefiori, M. S. Seaman, A. Gazumyan, M. Silva, A. T. McGuire, L. Stamatatos, D. J. Irvine, L. Wells, M. A. Martin, P. J. Bjorkman and M. C. Nussenzweig, Nature, 2019, 570, 468–473 CrossRef CAS PubMed.
- N. Martínez-Sáez, J. M. Peregrina and F. Corzana, Chem. Soc. Rev., 2017, 46, 7154–7175 RSC.
- H. Coelho, T. Matsushita, G. Artigas, H. Hinou, F. J. Cañada, R. Lo-Man, C. Leclerc, E. J. Cabrita, J. Jiménez-Barbero, S.-I. Nishimura, F. Garcia-Martín and F. Marcelo, J. Am. Chem. Soc., 2015, 137, 12438–12441 CrossRef CAS PubMed.
- N. Martínez-Sáez, J. Castro-López, J. Valero-González, D. Madariaga, I. Compañón, V. J. Somovilla, M. Salvadó, J. L. Asensio, J. Jiménez-Barbero, A. Avenoza, J. H. Busto, G. J. L. Bernardes, J. M. Peregrina, R. Hurtado-Guerrero and F. Corzana, Angew. Chem., 2015, 127, 9968–9972 CrossRef.
- I. A. Bermejo, I. Usabiaga, I. Compañón, J. Castro-López, A. Insausti, J. A. Fernandez, A. Avenoza, J. H. Busto, J. Jiménez-Barbero, J. L. Asensio, J. M. Peregrina, G. Jiménez-Osés, R. Hurtado-Guerrero, E. J. Cocinero and F. Corzana, J. Am. Chem. Soc., 2018, 140, 9952–9960 CrossRef CAS PubMed.
- N. Martínez-Sáez, N. T. Supekar, M. A. Wolfert, I. A. Bermejo, R. Hurtado-Guerrero, J. L. Asensio, J. Jiménez-Barbero, J. H. Busto, A. Avenoza, G.-J. Boons, J. M. Peregrina and F. Corzana, Chem. Sci., 2016, 7, 2294–2301 RSC.
- D. Straßburger, M. Glaffig, N. Stergiou, S. Bialas, P. Besenius, E. Schmitt and H. Kunz, ChemBioChem, 2018, 19, 1142–1146 CrossRef PubMed.
- I. Compañón, A. Guerreiro, V. Mangini, J. Castro-López, M. Escudero-Casao, A. Avenoza, J. H. Busto, S. Castillón, J. Jiménez-Barbero, J. L. Asensio, G. Jiménez-Osés, O. Boutureira, J. M. Peregrina, R. Hurtado-Guerrero, R. Fiammengo, G. J. L. Bernardes and F. Corzana, J. Am. Chem. Soc., 2019, 141, 4063–4072 CrossRef PubMed.
- V. J. Somovilla, I. A. Bermejo, I. S. Albuquerque, N. Martínez-Sáez, J. Castro-López, F. Garcia Martin, I. Compañón, H. I. Hinou, S.-I. Nishimura, J. Jiménez-Barbero, J. L. Asensio, A. Avenoza, J. H. Busto, R. Hurtado-Guerrero, J. M. Peregrina, G. J. L. Bernardes and F. Corzana, J. Am. Chem. Soc., 2017, 139, 18255–18261 CrossRef CAS PubMed.
- B. Palitzsch, N. Gaidzik, N. Stergiou, S. Stahn, S. Hartmann, B. Gerlitzki, N. Teusch, P. Flemming, E. Schmitt and H. Kunz, Angew. Chem., Int. Ed., 2016, 55, 2894–2898 CrossRef CAS PubMed.
- X. Wu, Z. Yin, C. McKay, C. Pett, J. Yu, M. Schorlemer, T. Gohl, S. Sungsuwan, S. Ramadan, C. Baniel, A. Allmon, R. Das, U. Westerlind, M. G. Finn and X. Huang, J. Am. Chem. Soc., 2018, 140, 16596–16609 CrossRef CAS PubMed.
- M. A. Wolfert and G.-J. Boons, Nat. Chem. Biol., 2013, 9, 776–784 CrossRef CAS PubMed.
- L. Sun, D. R. Middleton, P. L. Wantuch, A. Ozdilek and F. Y. Avic, Glycobiology, 2016, 26, 1029–1040 CrossRef CAS PubMed.
- F. Berti and R. Adamo, Chem. Soc. Rev., 2018, 47, 9015–9025 RSC.
- (a) T.-C. Chang, Y. Manabe, Y. I. Fujimoto, S. Ohshima, Y. Kametani, K. Kabayama, Y. Nimura, C.-C. Lin and K. I. Fukase, Angew. Chem., Int. Ed., 2018, 57, 8219–8224 CrossRef CAS PubMed; (b) M.-M. Wei, Y. S. Wang and X. S. Ye, Med. Res. Rev., 2018, 38, 1003–1026 CrossRef PubMed.
- (a) C. Colombo, O. Pitirollo and L. Lay, Molecules, 2018, 23, 1712 CrossRef PubMed; (b) S. A. Krumm and K. J. Doores, Current Topics in Microbiology and Immunology, Springer, Berlin, Heidelberg, 2018 Search PubMed; (c) J. Hütter and B. Lepenies, Methods Mol. Biol., 2015, 1331, 1–10 CrossRef PubMed.
- C. L. Anish, B. Schumann, C. L. Pereira and P. H. Seeberger, Chem. Biol., 2014, 21, 38–50 CrossRef CAS PubMed.
- P. Kaplonek, N. Khan, K. Reppe, B. Schumann, M. Emmadi, M. P. Lisboa, F.-F. Xu, A. D. J. Calow, S. G. Parameswarappa, M. Witzenrath, C. L. Pereira and P. H. Seeberger, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 13353–13358 CrossRef CAS PubMed.
- (a) T. C. Stevenson, C. Cywes-Bentley, T. D. Moeller, K. B. Weyant, D. Putnam, Y.-F. Chang, B. D. Jones, G. B. Pier and M. P. DeLisa, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3106–E3115 CrossRef CAS PubMed; (b) F. Micoli, S. Rondini, R. Alfini, L. Lanzilao, F. Necchi, A. Negrea, O. Rossi, C. Brandt, S. Clare, P. Mastroeni, R. Rappuoli, A. Saul and C. A. MacLennan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10428–10433 CrossRef CAS PubMed.
- Q. Li and Z. Guo, Molecules, 2018, 23, 1583 CrossRef PubMed.
- X. Sun, G. Stefanetti, F. Berti and D. L. Kasper, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 193–198 CrossRef CAS PubMed.
- S. Carillo, S. Mittermayr, A. Farrell, S. Albrecht and J. Bones, Methods Mol. Biol., 2017, 1603, 227–241 CrossRef CAS PubMed.
- B. Bobaly, V. D'Atri, A. Goyon, O. Colas, A. Beck, S. Fekete and D. Guillarme, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1060, 325–335 CrossRef CAS PubMed.
- A. Resemann, W. Jabs, A. Wiechmann, E. Wagner, O. Colas, W. Evers, E. Belau, L. Vorwerg, C. Evans, A. Beck and D. Suckau, mAbs, 2016, 8, 318–330 CrossRef CAS PubMed.
- D. Reusch and M. L. Tejada, Glycobiology, 2015, 25, 1325–1334 CrossRef CAS.
- L. Liu, J. Pharm. Sci., 2015, 104, 1866–1884 CrossRef CAS.
- E. Largy, F. Cantais, G. Van Vyncht, A. Beck and A. Delobel, J. Chromatogr. A, 2017, 1498, 128–146 CrossRef CAS PubMed.
- A. Planinc, B. Dejaegher, Y. V. Heyden, J. Viaene, S. Van Praet, F. Rappez, P. Van Antwerpen and C. Delporte, Anal. Bioanal. Chem., 2017, 409, 477–485 CrossRef CAS.
- J. Giorgetti, V. D'Atri, J. Canonge, A. Lechner, D. Guillarme, O. Colas, E. Wagner-Rousset, A. Beck, E. Leize-Wagner and Y. François, Talanta, 2018, 178, 530–537 CrossRef CAS.
- L. Zhang, S. Luo and B. Zhang, mAbs, 2016, 8, 205–215 CrossRef CAS PubMed.
- L. Zhang, S. Luo and B. Zhang, mAbs, 2016, 8, 524–535 CrossRef CAS PubMed.
- F. Cymera, H. Beck, A. Rohde and D. Reusch, Biologicals, 2018, 52, 1–11 CrossRef PubMed.
- F. Higel, A. Seidl, F. Sörgel and W. Friess, Eur. J. Pharm. Biopharm., 2016, 100, 94–100 CrossRef CAS PubMed.
- E. O. Saphire, P. W. H. I. Parren, R. Pantophlet, M. B. Zwick, G. M. Morris, P. M. Rudd, R. A. Dwek, R. L. Stanfield, D. R. Burton and L. A. Wilson, Science, 2001, 293, 1155–1159 CrossRef CAS PubMed.
- W. Li, Z. Zhu, W. Chen, Y. Feng and D. S. Dimitrov, Front. Immunol., 2017, 8, 1554 CrossRef PubMed.
- D. Venetz, C. Hess, C. Lin, M. Aebi and D. Neri, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(7), 2000–2005 CrossRef CAS PubMed.
- B. Hintersteiner, N. Lingg, E. Janzek, O. Mutschlechner, H. Loibner and A. Jungbauer, Biotechnol. J., 2016, 11(12), 1617–1627 CrossRef CAS.
- A. W. Barb, D. J. Falconer and G. P. Subedi, Methods Enzymol., 2019, 614, 239–261 Search PubMed.
- S. Yanaka, H. Yagi, R. Yogo, M. Y. Utsumi and K. Kato, J. Biomol. NMR, 2018, 71, 193–202 CrossRef CAS.
- M. Dicker and R. Strasser, Expert Opin. Biol. Ther., 2015, 15(10), 1501–1516 CrossRef.
- Q. Yang and L. Wang, Methods Enzymol., 2017, 597, 265–281 CAS.
- Y. Mimura, T. Kato, R. Saldova, R. O'Flaherty, T. Izumi, Y. Mimura-Kimura, T. Utsunomiya, Y. Mizukami, K. Yamamoto, T. Matsumoto and P. M. Rudd, Protein Cell, 2018, 9, 47–62 CrossRef CAS PubMed.
- A. J. Fairbanks, Chem. Soc. Rev., 2017, 46, 5128–5146 RSC.
- T. B. Parsons, W. B. Struwe, J. Gault, K. Yamamoto, T. A. Taylor, R. Raj, K. Wals, S. Mohammed, C. V. Robinson, J. L. P. Benesch and B. G. Davis, Angew. Chem., Int. Ed., 2016, 55, 2361–2367 CrossRef CAS PubMed.
- T. Nagasu, Y. Shimma, Y. Nakanishi, J. Kuromitsu, K. Iwama, K. Nakayama, K. Suzuki and Y. Jigami, Yeast, 1992, 8, 535–547 CrossRef CAS.
- H. Abe, Y. Takaoka, Y. Chiba, N. Sato, S. Ohgiya, A. Itadani, M. Hirashima, C. Shimoda, Y. Jigami and K. Nakayama, Glycobiology, 2009, 19, 428–436 CrossRef CAS PubMed.
- C. Unverzagt and Y. Kajihara, Curr. Opin. Chem. Biol., 2018, 46, 130–137 CrossRef.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645–7649 CrossRef CAS.
- R. Chang, A. Al Maghribi, V. Vanderpoel, A. Ing, V. Vasilevko, D. H. Cribbs, R. Boado, W. M. Pardridge and R. Sumbria, Alzheimer's Dementia, 2018, 14, 1086–1087 CrossRef.
- W. Liu, L. Wen, T. Xie, H. Wang, J. Gong and X. Yang, J. Neurosurg., 2016, 127, 1–8 Search PubMed.
- A. Tankiewicz-Kwedlo, J. M. Hermanowicz, T. Domaniewski, K. Pawlak, M. Rusak, A. Pryczynicz, A. Surazynski, T. Kaminski, A. Kazberuk and D. Pawlak, Br. J. Pharmacol., 2018, 175, 743–762 CrossRef CAS PubMed.
- S. Rasaneh and M. R. Dadras, Biomed. Tech., 2015, 60, 485–490 CAS.
- A. Kesavan, P. Ilaiyaraja, W. S. Beaula, V. V. Kumari, J. S. Lal, C. Arunkumar, G. Anjana, S. Srinivas, A. Ramesh, S. K. Rayala, D. Ponraju and G. Venkatraman, Eur. J. Pharm. Biopharm., 2015, 96, 255–263 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |