
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeveloping degraders: principles and perspectives on design and chemical space†
Hannah J.
Maple
 *,
Nat
Clayden
,
Anne
Baron
,
Callum
Stacey
and
Robert
Felix
*,
Nat
Clayden
,
Anne
Baron
,
Callum
Stacey
and
Robert
Felix
Bio-Techne (Tocris), The Watkins Building, Atlantic Road, Avonmouth, Bristol, BS11 9QD, UK. E-mail: hannah.maple@bio-techne.com
First published on 12th August 2019
Abstract
Degraders (e.g. PROTACs, SNIPERs, degronimers etc.) are a new modality offering increasing potential both as tools for basic research and therapeutic development. They occupy chemical space that lies outside the classical Lipinski ‘Rule of 5’, which poses fresh challenges for achieving cell permeability and oral bioavailability. This study presents a comprehensive database of degrader structures from the peer reviewed literature, including both optimized degraders and first generation compounds, in order to provide a thorough assessment of the chemical space associated with this modality and identify common trends used during the ‘hit to lead’ process. The results provide insights into this new area of chemical space as well as pointers for degrader design, which we anticipate will be useful for researchers entering this field.
Introduction
The use of heterobifunctional small molecule degraders (e.g. PROTACs, SNIPERs, degronimers etc.) to elicit targeted protein degradation (TPD) is an area of increasing interest in chemical biology. The approach employs hybrid molecules with dual functionalities, one targeting a protein of interest, the other capable of recruiting an E3 ligase that draws in the target protein for ubiquitination and destruction by the proteasome. This modality effectively repurposes small molecule ligands to selectively degrade, rather than simply inhibit, target proteins of interest. A key feature is their catalytic mode of action; degraders can repeatedly engage and direct the ubiquitination of target molecules.1 They can therefore elicit a continued, strong response even at very low, sub-stoichiometric concentrations. Additionally, degraders can be used to knock-down proteins that although bound, are not effectively inhibited, by small molecules.2As tools for basic research, heterobifunctional degraders offer an attractive approach for inducing selective protein knockdown in a reversible and tuneable manner, without the requirement for genetic modification to cells. This has widely accepted therapeutic potential as an approach to target the ‘undruggable’ proteome and overcome common resistance mechanisms to current therapies. For example, in some cases antagonizing a target protein results in its upregulation, an acquired response that is difficult to overcome with standard inhibitors since the dose cannot be increased indefinitely. A degrader, on the other hand, might surmount this issue through the catalytic, sustained knock-down of a target protein.
The design of degraders is not, however, without its challenges; in particular conjugating two drug-like small molecules plus a linker will result in a compound with physicochemical properties that fall into chemical space beyond Lipinski's rule of 5 (Ro5),3,4 which provides guidelines for achieving molecules with oral bioavailability. Despite this, there are now multiple examples of degraders routinely achieving passive cellular permeability, oral bioavailability5 and even reports of blood–brain-barrier penetration (Arvinas, unpublished).
Strict application of the Lipinski ‘rules’ during medicinal chemistry campaigns is not universally considered beneficial. It has been argued that routinely and strictly applying rules based on data available over twenty years ago may not always be appropriate, particularly when considering more ‘challenging’ targets such as protein–protein interactions.6,7 Indeed, the recent increase in drug approvals for compounds outside the Ro58 has prompted several recent analyses of the chemical space beyond these boundaries to define chemical properties that can be altered to improve permeability and oral bioavailability of high MW compound classes.7,9,10
We have compiled a database of published degrader structures and classified them according to the constituent ligands, linker type, linker length, degradation effectiveness and physicochemical properties. The aim was to provide a review of the literature from a chemical space perspective and gain an understanding of the general principles that have been applied to the development of degraders. We have also profiled the typical linker types and lengths used during an early stage degrader project. It is worth noting that given the nascency of this field from a clinical perspective, the dataset of published degraders compiled here provides a valuable starting point for assessing the properties required to develop effective, cell permeable chemical tools for TPD. Degraders possessing good PK/PD properties and even oral bioavailability may differ significantly from the structures considered in this work, indeed, only 3% of the compounds surveyed here have associated, reported in vivo data.
The optimization process for degraders is complex and multi-faceted. Aside from the traditional measures of potency for small molecule ligands, several elegant studies have outlined how degrader optimization can require a holistic understanding of additional parameters, such as ternary complex formation, cooperativity and kinetics.11–13 At a fundamental level, the cellular permeability of degraders can be a significant factor contributing to the observed efficacy. Chessum et al.14 report this as a likely reason for poor efficacy of first-generation compounds that were subsequently used to guide design of effective second-generation degraders. As an additional consideration, transporter-mediated efflux is expected to be increasingly significant for higher MW compounds.9 Efflux via the ATP-binding cassette subfamily B member 1 (ABCB1) drug transporter was demonstrated for a heterobifunctional ALK degrader by Powell et al.,15 while a separate study reported the beneficial effects of pre-saturating drug transporters such as P-glycoprotein (PGP) with a high dose of cyclosporine A to improve activity of PTK degraders.16 Overall, it is expected that as a relatively new class of ‘Beyond Rule of 5’ (bRo5) compounds, attaining sufficient intracellular free concentration may be a challenge and that the literature to date may reflect some general trends in terms of key physicochemical properties that will provide a useful guide for research in this field.
Experimental
Physicochemical properties were calculated using ChemDraw® version 15.0.0 with the following exceptions: clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, calculated using the consensus value from SwissADME10 (http://www.swissadme.ch); number of aromatic rings, calculated using OSIRIS Datawarrior Version 5.0.0; and clogD calculated using FAFDrugs4.
P, calculated using the consensus value from SwissADME10 (http://www.swissadme.ch); number of aromatic rings, calculated using OSIRIS Datawarrior Version 5.0.0; and clogD calculated using FAFDrugs4.
Degrader score (Deg_S)
We define Deg_S as a single overall measure of degrader efficacy. The following parameters were taken into account: DC50; Dmax; observed degradation (in the absence of a DC50 value, the degradation profile for degraders was assessed by a subjective assessment of the available data, typically western blot); degrader concentration; incubation time. Observed degradation was reported together with degrader concentration and incubation time. Where multiple tests were published with the same degrader, the data taken was the lowest concentration/time at which the maximum response was observed. All scores were summed and then normalized against the total number of parameters included for each degrader (nu), to generate the final Deg_S value.The following scoring system was used (scores given in parenthesis).
DC50 (xd, nM): 0 < xd ≤ 30 (7); 30 < xd ≤ 50 (5); 50 < xd ≤ 100 (3); 100 < xd ≤ 500 (2); 500 < xd (1).
D max (xm, %): 95 < xm ≤ 100 (7); 85 < xm ≤ 95 (5); 65 < xm ≤ 85 (3); 35 < xm ≤ 65 (2); 0 < xm ≤ 35 (1).
Observed degradation (xo, %): 90 < xo ≤ 100 (7); 70 < xo ≤ 90 (5); 30 < xo ≤ 70 (3); 10 < xo ≤ 30 (2); 0 < xo ≤ 10 (1).
Degrader concentration (xc, μM): 0 < xc ≤ 0.1 (5); 0.1 < xc ≤ 1 (3); 1 < xc ≤ 3 (2); 3 < xc (1).
Incubation time (xt, hr): 0 < xt ≤ 4 (5); 4 < xt ≤ 12 (3); 12 < xt ≤ 24 (2); 24 < xt (1).
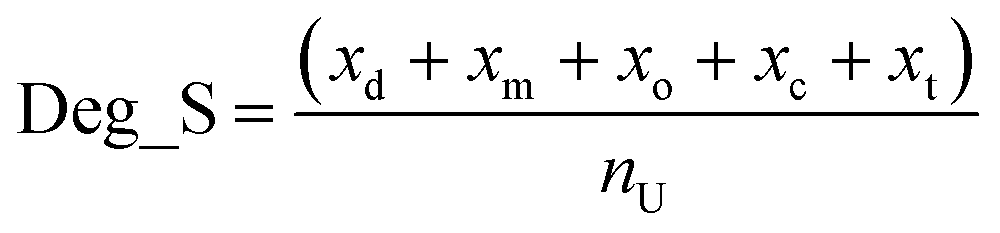
Data was normalized for principal component analysis by z-scoring. The software used for principal component analysis was Past version 3.20.17
Results and discussion
This study reports the analysis of 422 degraders from 73 articles published between 2014 and 2019. To the best of our knowledge these form a comprehensive set of studies reporting the discovery and development of novel degraders up until the time of writing and filtered to consider only those comprising small molecule components for both the E3 ligase ligand and target ligand. The dataset includes 70 different target ligands, for 39 different target proteins. The E3 ligase enzymes harnessed by the degraders in the dataset are: cereblon (CRBN); DDB1 and CUL4-associated factor 15 (DCAF15); inhibitor of apoptosis (IAP) proteins; murine double minute 2 (MDM2) and Von Hippel–Lindau (VHL), with the majority of degraders using either a CRBN or a VHL-targeting E3 ligase ligand (155 and 218 respectively). By including both the ‘final’ optimized compounds as well as all available published structures (including ESI† data) of degraders tested during development, the aim is to begin to capture any common strategies used during the degrader ‘hit-to-lead’ process.The degrader dataset compiled in this study comprised a range of physicochemical parameters: MW = 614–1413, clogP = −2.7–9, HBD = 1–10, HBA = 8–23, NRotB (number of rotatable bonds) = 6–49, NAr (number of aromatic rings) = 1–7, TPSA = 124–389. Experimental logP values were not determined as part of this work and we note that there are known limitations on the reliability of calculated logP. A comparison with published experimental values for a small subset of degraders reveals that calculated logP values are a reasonable reflection of the experimental data (see Table S1†). All compounds in the dataset were assigned a degrader score (Deg_S), weighted on parameters relating to the overall efficacy (for details see Experimental). It is challenging to objectively compare published degraders, since many studies do not report experimentally derived values relating to the degradation potency. Crews et al. originally defined the concept of the DC50 value (concentration at which the target protein is degraded by 50%) and the Dmax value (maximum percentage of target protein degraded).1 These values are important ways to ensure that work in the TPD field is inter-study comparable but are not always reported. Another variable factored into this study was the duration of degrader application before measurement of effects. There are published reports of degraders demonstrating delayed effects only after long (e.g. 48 hour) incubation with cells, which has in some cases been attributed to poor overall cell permeability.14 It is not possible, however, to directly correlate delay time to cell permeability as there are many other contributing factors, such as the kinetics of ternary complex formation, protein degradation rate and protein re-synthesis rate.18 Overall it is likely that degraders eliciting strong effects within a few hours of application are likely to be more effective compounds than those whose onset of effect is delayed by >8 hours.
A summary of the degrader dataset is given in Table 1. No clear correlations are apparent between Deg_S and an individual physicochemical property, which is unsurprising since the dataset incorporates a wide range of ligand classes and target proteins and, as previously discussed, overall efficacy will be dependent on multiple additional parameters. The exception to this is clogP, which does appear to increase with increasing Deg_S. This trend was also recently observed in a study by Steinebach et al. of CRBN–VHL hetero-PROTACs.19 It is noticeable that, despite the high molecular weight, the mean number of hydrogen bond donors is maintained within the Lipinski HBD ≤ 5 rule. The calculated AB-MPS metric is a predictor of oral absorption originally defined by DeGoey et al., according to the formula AB-MPS = Abs(clogD − 3) + NAr + NRotB.8 The lower the AB-MPS score, the greater the probability of absorption, with values ≤14 associated with a likelihood of oral absorption. No clear correlation is observed between Deg_S and AB-MPS suggesting that, for degraders, this metric is not in itself strongly predictive.
P, number of hydrogen bond donors (HBD) and acceptors (HBA), number of rotatable bonds (NRotB), number of aromatic rings (NAr), topological polar surface area (TPSA) and the AB-MPS metric8
| Deg_S | N | MW (Da) | clogP |
HBD | HBA | NRotB | NAr | TPSA (Å2) | AB-MPS |
|---|---|---|---|---|---|---|---|---|---|
| ≥0–<2 | 77 | 972(666–1277) | 4.6(0–9) | 4.1(1–7) | 13.1(8–19) | 24.0(8–40) | 4.4(3–6) | 213(133–293) | 31(14–47) |
| ≥2–<3 | 46 | 958(671–1245) | 4.7(−1–10) | 4.1(1–7) | 13.6(9–19) | 24.0(10–38) | 4.2(3–6) | 214(117–311) | 31(14–48) |
| ≥3–<4 | 93 | 1021(799–1243) | 4.5(0–9) | 4.5(1–8) | 14.5(10–19) | 27.0(15–39) | 4.2(2–7) | 230(143–316) | 34(22–47) |
| ≥4–<5 | 84 | 993(756–1231) | 5.1(2–9) | 4.2(2–6) | 13.5(8–19) | 24.8(10–40) | 4.5(2–7) | 214(153–274) | 31(16–47) |
| ≥5 | 122 | 977(752–1201) | 5.7(2–10) | 4.2(2–6) | 12.4(8–17) | 24.0(12–36) | 4.7(3–7) | 199(147–251) | 40(17–45) |
The TPSA values for the majority of degraders occupy a space below Whitty's observations that good solubility requires a TPSA of ≥0.23 × MW and (as expected) significantly above the expectation that good passive membrane permeability requires TPSA ≤ 140.20,21 A separate study considering high MW clinical candidate molecules showed that 92% compounds with MW > 700 Da have a TPSA/MW ratio of 0.15–0.3 (values that differed little for <700 Da compounds evaluated).22 Degraders scoring ≥4 in our dataset have a TPSA that scales with 0.11 × MW (R2 = 0.2). This is summarised in Fig. 1, which illustrates that, despite the majority of degraders not falling into Whitty's ‘aqueous soluble’ chemical space, they do fall broadly within the upper and lower bounds of a set of clinical candidate molecules with molecular weights >700 Da. The range of degrader MW and TPSA values together with their relative lipophilicity, is likely to result in poor aqueous solubility.14 In theory, the catalytic mode of degrader action allows sub-stoichiometric dosing, which effectively reduces the solubility requirement for a sufficiently potent degrader. Nonetheless, the propensity for poor aqueous solubility should be taken into account during degrader development.
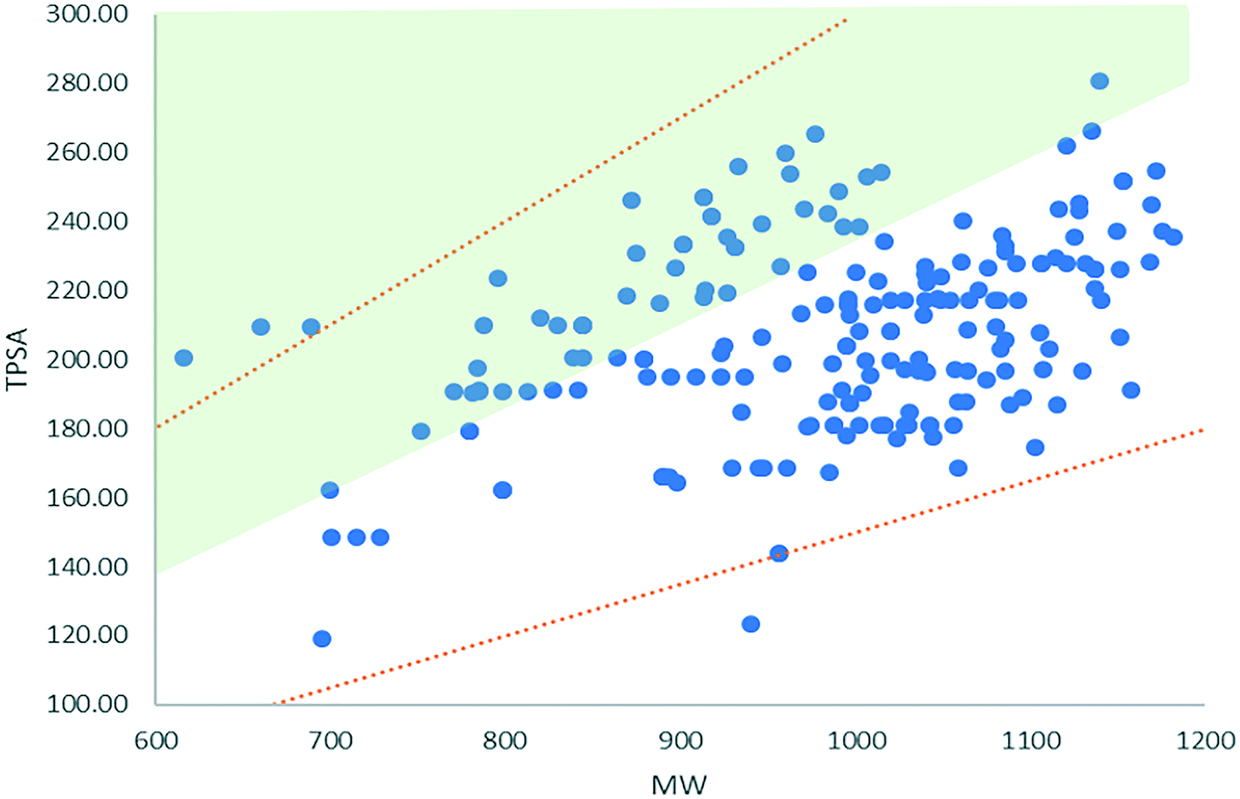 | ||
| Fig. 1 MW versus TPSA for degraders with scores ≥4 (blue). The green shaded area illustrates the space that complies with the observation that good solubility requires TPSA ≥ 0.23 × MW.19 The orange trendlines show the upper and lower bounds observed for 92% of clinical candidates >700 Da included in a study by Doak et al.22 | ||
We have compared physicochemical parameters for the degraders (taking the average values from compounds in the 4th quartile when ordered by increasing Deg_S) with other bRo5 analyses that were recently reviewed by Poongavanam et al.9 The studies included are as follows: orally absorbed drugs and clinical candidates with MW > 500Da;22 orally available preclinical compounds breaking >1 of Lipinski's rules in Abbvie's preclinical DMPK database;8 and orally available macrocyclic drugs.23 The results are summarised in the radar diagram shown in Fig. 2, which suggests that degraders occupy a differentiated physicochemical space from classical, orally available small molecule drugs (Ro5 and Veber), bRo5 oral preclinical and candidate small molecules (DeGoey and Kihlberg), and orally available macrocyclic drugs (Whitty).
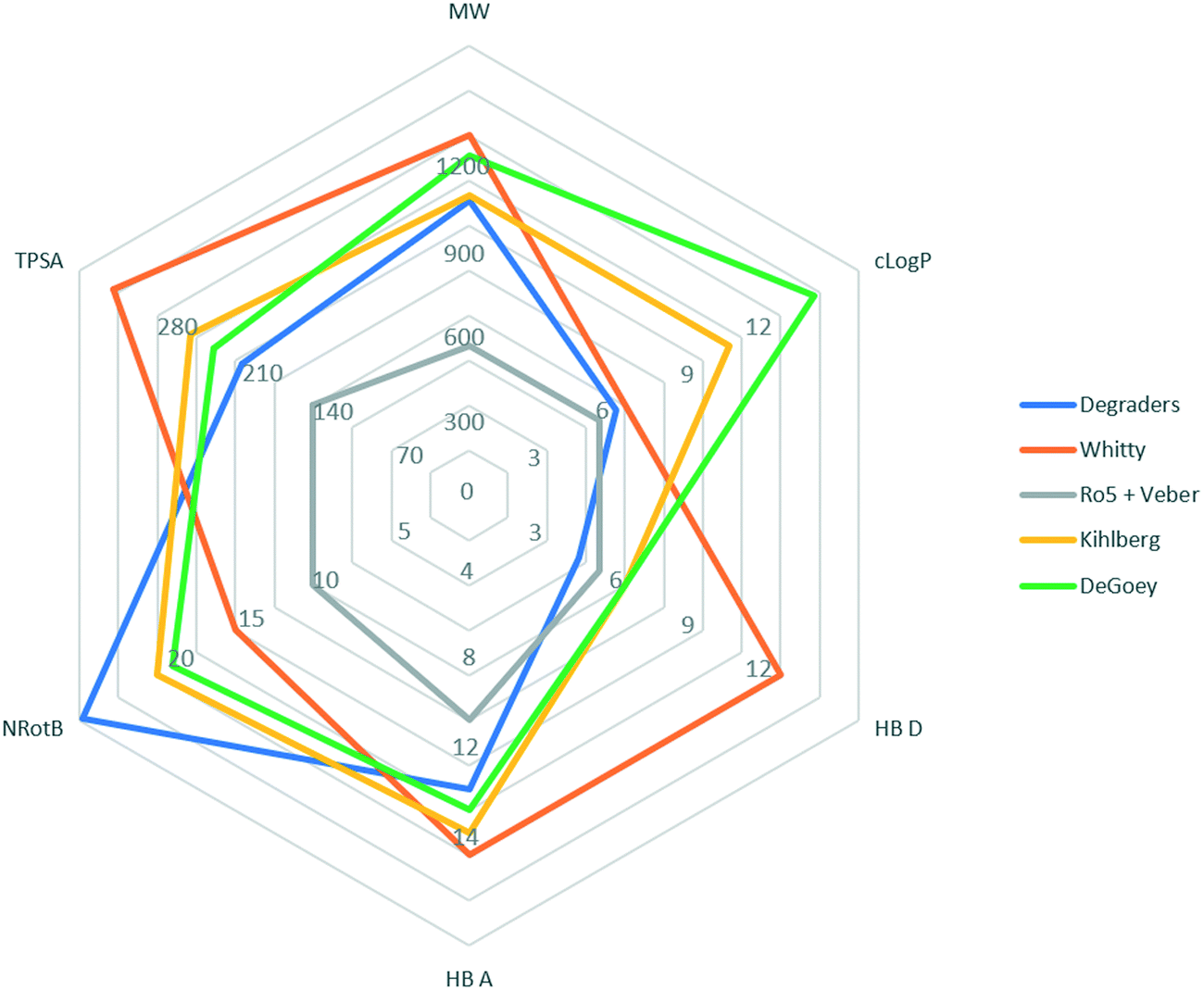 | ||
| Fig. 2 Radar diagram illustrating outer limits of chemical space occupied by compound classes from different published analyses: “Ro5 and Veber”;1,26 orally absorbed drugs and clinical candidates with MW > 500 Da, “Kihlberg”;20 orally available preclinical compounds breaking >1 of Lipinski's rules in Abbvie's preclinical DMPK database, “DeGoey”;6 orally available macrocyclic drugs, “Whitty”;21 plotted with the average values from compounds in the 4th quartile (by Deg_S) from the degrader dataset. | ||
Despite the broadly similar MW range for the bRo5 classes considered here, there are significant differences in the other parameters. Degraders fall within the original Lipinski rule for HBD count and are similar to oral macrocycles in terms of their clogP values. The degrader TPSA values are lower than all other bRo5 classes considered, suggesting that this parameter is actively controlled during degrader development. Notably the number of rotatable bonds is higher than all classes considered, which is expected given the prevalence of highly flexible PEG and alkyl linker typically groups employed in these molecules. The flexibility added by increased numbers of rotatable bonds is potentially significant in conferring cell permeability; a recent analysis suggests that dynamically exposed polarity is key for conferring permeability and solubility for bRo5 compound classes.24,25 Further studies are warranted to probe degrader low energy conformations and associated three dimensional properties such as the molecular PSA (MPSA). Conversely, during more advanced degrader optimization, there is evidence to suggest that introducing rigidity into the linker group can be beneficial.26–28 Increased rigidity will additionally reduce the entropic cost of binding and may be beneficial for DMPK properties.
Although not explicitly considered here, the prevalence of secondary amide bonds in degrader molecules is noteworthy. Due to their modular design, degraders are often synthesized through sequential coupling of E3 ligase ligand, linker and target ligand. Amide coupling reactions are frequently a convenient way to assemble these components. This can result in the incorporation of up to two amide bonds, in addition to any utilised in the target ligand/E3 ligase ligand. Amide groups contribute significantly to the polar surface area29 and desolvation of complexed water is considered a primary barrier to passive transport of peptides into cells.29,30 This is in addition to the hydrogen bond donor that amides contribute,14 and conformational restraints imposed by their partial double bond character, which may undesirably restrict flexibility in these molecules, particularly in the linker region. To address this, secondary amides have often been either removed,14,28 masked (ortho-substitution of an adjacent phenyl group with fluorine),14 or N-methylated.31
The small molecule ligands typically used to recruit different E3 ligases are structurally very different and as such confer distinct sets of properties to the final degrader molecule (Fig. 3). This may in turn impose different limitations/considerations during degrader development. The prototypical recruiter for VHL is the ligand, ‘VH 032’, developed by the Ciulli group,32 which is built around the hydroxyproline group found in the native HIF-1α substrate peptide. For CRBN, the series of immunomodulatory drugs (IMiDs): thalidomide, pomalidomide and lenalidomide are frequently employed as the E3 ligase recruiters. Another class of degrader, called ‘SNIPERs’, specifically recruit IAP E3 ligases. Although early results using bestatin analogs demonstrated proof of concept,33 second generation IAP recruiters such as those highlighted in Fig. 3 are now preferred as they enable generation of degraders with more potent activity and in vivo efficacy.34,35 Suitable vectors for attaching linker groups are generally well established, with alternative, underexplored linkage sites for VH 032 (from the phenyl ring,36,37 or from an (S)-methyl substituent28) offering further options for degrader design.
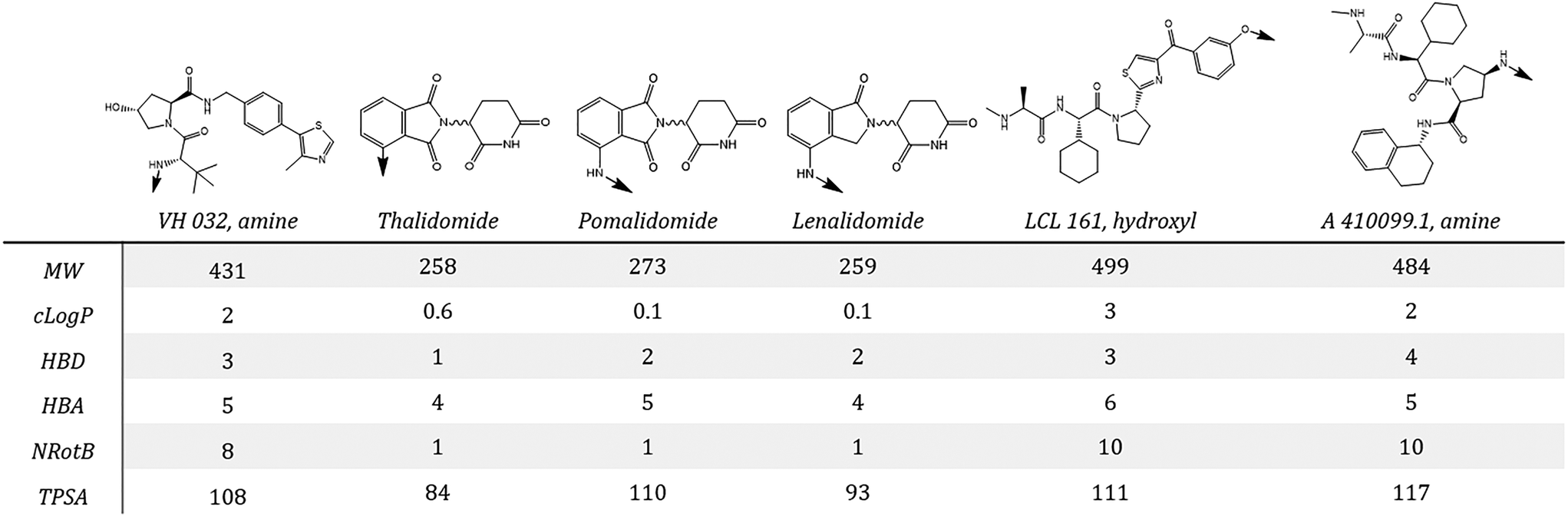 | ||
| Fig. 3 Chemical structures of commonly used VHL, CRBN and IAP E3 ligase ligands: VH 032, amine; thalidomide; pomalidomide; lenalidomide; LCL 161, hydroxyl and A 410099.1, amine (left to right) with corresponding physicochemical properties. Arrows indicate exit vectors for attachment to a linker group. | ||
An overall summary of the physicochemical parameters of degraders based on CRBN, VHL and IAP is given in Fig. 4. VH 032 and IAP ligands have a higher MW than the glutarimides, which is reflected in the final degrader MW being higher on average for these compounds. VH 032- and IAP-based degraders also have a higher average clogP and number of rotatable bonds compared with CRBN, which is a direct reflection of the properties of the E3 ligands themselves. The HBA count and TPSA for VHL- and IAP-based degraders are similar, with CRBN having a slightly higher average TPSA than the other two classes. The linker lengths used for each class of degrader are also included in Fig. 4, which show overall little difference between the E3 ligase recruiter types, except a narrowing of range for IAP-based degraders. The higher lipophilicity of VH 032- and IAP-compared to CRBN-ligands could potentially be usefully harnessed when developing a degrader that incorporates a very polar target ligand. For degraders based on target ligands that have a relatively high HBD count, the CRBN E3 ligase ligands may be beneficial in order to reduce the number of additional hydrogen bond donors introduced during the degrader development process. There are, however, well-documented reasons why the traditional CRBN ligands may be challenging to work with, such as poor stability38 and recruitment of ‘neosubstrates’39–41 that may confound resulting biological data.
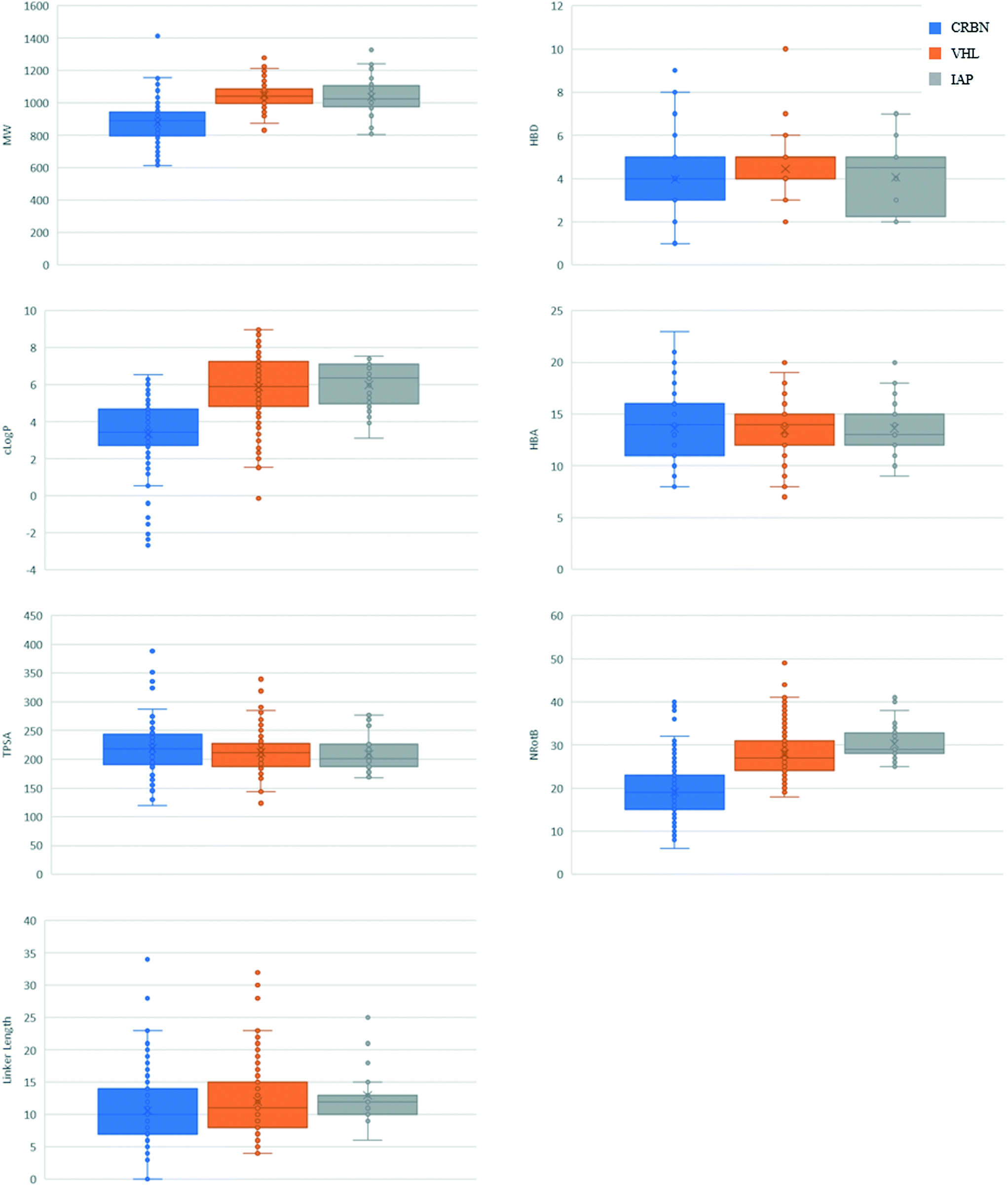 | ||
| Fig. 4 Box and whisker plots summarising properties of CRBN-, VHL- and IAP-based degraders. | ||
Development of further E3 ligase ligands that can be harnessed for degrader development is an area of current focus for the targeted protein degradation field. Expanding the toolbox of small molecule E3 ligase ligands will provide additional options and flexibility for degrader design. Recently published work in this area includes the successful recruitment of RNF11442 and DCAF1643 for novel degraders. Another E3 ligase, MDM2, has recently received renewed interest for degrader development. MDM2 was harnessed using the small molecule ligand, nutlin 3, for the first reported ‘all small molecule’ degrader.44 While this provided proof of concept, further work to fully explore the potential of MDM2 for degrader development has only recently been published, demonstrating not only that the MDM2 ligand, idasanutlin, can be used to generate nanomolar-potency degraders against BRD4, but that the resulting upregulation of the tumor suppressor p53, provided a synergistic antiproliferative effect.45
To investigate any trends used during degrader optimization in more depth, the two principal groups in the dataset, CRBN- and VHL-recruiting degraders, were selected for further study. These two groups were chosen for further investigation since they represent the largest proportion (88%) of the dataset. To probe any possible correlation between Deg_S and physicochemical properties for these degraders, data from the two groups was normalized (z-scoring) and subject to principle components analysis (PCA). The correlation loadings for PC1 versus PC2 are shown in Fig. 5. The Deg_S values for CRBN-based degraders correlate with increasing clogP, linker length, number of rotatable bonds, MW and HBA count. There is a strong negative correlation with HBD count. TPSA correlates negatively with Deg_S and there is a weak negative correlation with the number of amide bonds. For VHL-based degraders, the overall profile is similar but less pronounced. Increasing Deg_S positively correlates with increased clogP, MW, number of rotatable bonds and linker length. There are weak negative correlations with TPSA, HBD count and number of amide bonds. The more pronounced results observed in this study for CRBN-based versus VHL-based degraders may at least in part be explained by the properties of the respective ligands, as illustrated in Fig. 3. There is significantly less scope to adjust certain key physicochemical parameters with VH 032-based degraders, since this ligand in itself already contributes more strongly to the overall MW, clogP, HBD and NRotB of the final degrader molecule.
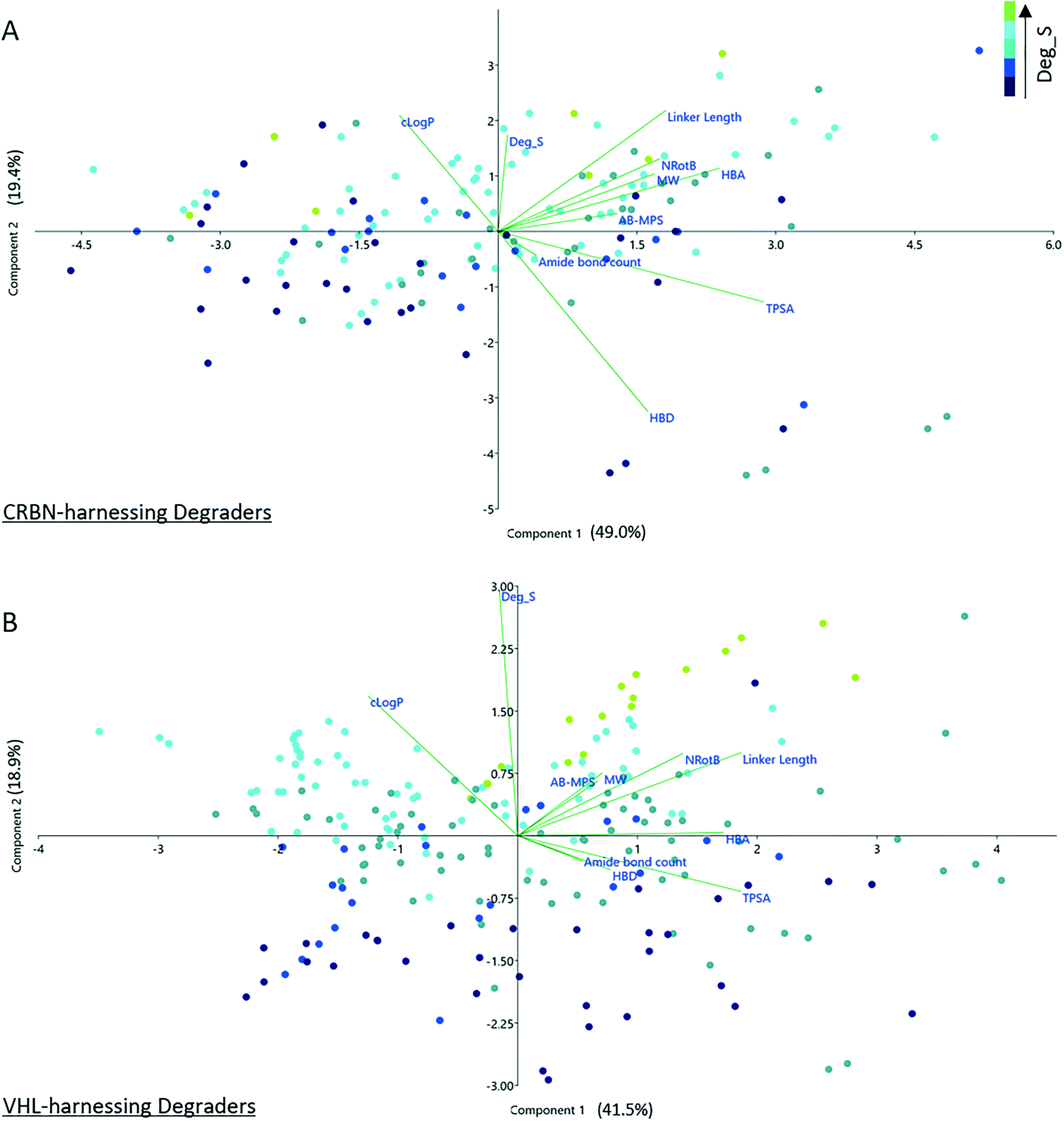 | ||
| Fig. 5 PCA analysis of CRBN-based degraders (a) and VHL-based degraders (b). The percentage variance explained by each principal component is given in brackets next to each axis label. Each variable is shown as a vector. Angles between vectors indicate their degree of correlation. Positively correlated variables are grouped together (≈0° angle). Negatively correlated ones are positioned on opposite sides of the plot origin (≈180° degrees). A 90° angle between two variables indicate that they are uncorrelated. | ||
The optimisation process for degraders may involve alterations to the E3 ligase ligand, linker, ligand for the target protein and both exit vectors.11 Currently, a priori prediction of structure–activity relationships with degrader molecules is challenging and empirical effort is required, although there are examples emerging of design guided by structural biology46 and computational approaches.47,48 The structure and length of the linker component is critical since this drives/impacts several determinants of the final degrader activity and mechanism of action, factors including: degrader conformation and binding orientation, ternary complex formation,46 selectivity47 and also physicochemical properties. The Ciulli group have pioneered the use of X-ray co-crystal structures of degrader-induced ternary complexes to perform structure-based optimization of degraders and predict optimal ligand exit vectors and linker lengths. This can be used to favour the formation of highly cooperative ternary complexes, which can improve degrader selectivity and efficacy.46 Computational approaches are also showing promise as a complementary tool for generating ternary complex poses in the absence of X-ray data to support structure-based design. Although at an earlier stage, they show potential for aiding rational design of degraders and potentially revealing alternative ensembles that reflect the plasticity of the overall ternary complex.47,48
The possibilities for chemical linker design are endless, which is a daunting prospect for new researchers entering this field. In practise, however, 64% of published degrader molecules use either alkyl or ethylene glycol repeating units, which alters little (63%) when only high (Deg_S = ≥4) scoring degraders are considered. These groups provide a way to explore optimal linker lengths, with the choice of alkyl or PEG groups allowing influence over the resulting physicochemical properties, TPSA and clogP in particular. Elaborating on the basic polyethylene glycol core to incorporate different alkyl chain lengths has been used in several studies19,36 and this is the next most prevalent class of linker design (16% of all degraders in this dataset). Incorporation of rigidifying groups (ring systems, alkyne groups) has also been used in several studies.26,35 From the dataset considered here, the percentages of degraders incorporating solely alkyl or ethylene glycol repeating units are 23% and 40% respectively, which alters to 28% and 35% respectively for high (Deg_S = ≥4) scoring degraders. The slight shift towards alkyl linkers for high scoring degraders reflects the correlation observed in Fig. 5 between increased Deg_S with increased clogP and decreased TPSA.
Conclusions
Heterobifunctional degraders represent an exciting new chemical modality, both from a basic research and a pharmaceutical perspective. We have reviewed the published literature to generate a ‘Degrader Database’ of molecules that have been synthesized at all stages of the pre-clinical development process. We have compared the physicochemical properties exhibited by efficacious degraders with other published sets of bRo5 compounds and find that degraders occupy a differentiated physicochemical space, results that are in agreement with an analysis published during preparation of this manuscript.49 Properties such as hydrogen bond donor count and TPSA are noticeably reduced compared to other bRo5 compounds with similar MW and degraders typically do comply with the original Lipinski HBD ≤ 5 rule. Conversely, the number of rotatable bonds in degrader molecules is significantly higher than all other classes considered. This property is conferred by the predominant use of flexible linker groups, an aspect that may alter for degraders progressing into clinical development. These general trends are reflected in principal component analyses that include the measure of degrader effectiveness ‘Deg_S’ defined and used in this study.Some general principles can be drawn from this study in terms of degrader design. For both predominant classes of degraders (CRBN- and VHL-recruiting), increased degrader score is correlated with increasing clogP and decreasing TPSA and HBD count. Based on this study, we suggest that the HBD count is kept ≤5 and we observe that for the highest scoring (Deg_S) degraders, the TPSA does not exceed 250 Å2. We note that increased lipophilicity of degraders positively correlates with Deg_S and that the highest scoring degraders have an average clogP of 6. Despite the vast possibilities for linker design, the majority of Degraders published to date employ simple alkyl or ethylene glycol repeating units. In most cases, these linker types appear to be sufficient for the generation of potent degraders, which may be useful as tool compounds or as candidates for further development. We anticipate that this study will provide a useful summary of the field from a chemical perspective and offer some broad guidelines for degrader development.
Conflicts of interest
There are no conflicts to declare.References
- D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611 CrossRef CAS PubMed.
- L. N. Gechijian, D. L. Buckley, M. A. Lawlor, J. M. Reyes, J. Paulk, C. J. Ott, G. E. Winter, M. A. Erb, T. G. Scott, M. Xu, H.-S. Seo, S. Dhe-Paganon, N. P. Kwiatkowski, J. A. Perry, J. Qi, N. S. Gray and J. E. Bradner, Nat. Chem. Biol., 2018, 14, 405–412 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS.
- T. Neklesa, L. B. Snyder, R. R. Willard, N. Vitale, J. Pizzano, D. A. Gordon, M. Bookbinder, J. Macaluso, H. Dong, C. Ferraro, G. Wang, J. Wang, C. M. Crews, J. Houston, A. P. Crew and I. Taylor, J. Clin. Oncol., 2019, 37, 259 Search PubMed.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- M. D. Shultz, J. Med. Chem., 2019, 62, 1701–1714 CrossRef CAS PubMed.
- D. A. DeGoey, H.-J. Chen, P. B. Cox and M. D. Wendt, J. Med. Chem., 2018, 61, 2636–2651 CrossRef CAS PubMed.
- V. Poongavanam, B. C. Doak and J. Kihlberg, Curr. Opin. Chem. Biol., 2018, 44, 23–29 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- S. L. Fisher and A. J. Phillips, Curr. Opin. Chem. Biol., 2018, 44, 47–55 CrossRef CAS PubMed.
- K. M. Riching, S. Mahan, C. R. Corona, M. McDougall, J. D. Vasta, M. B. Robers, M. Urh and D. L. Daniels, ACS Chem. Biol., 2018, 13, 2758–2770 CrossRef CAS PubMed.
- M. J. Roy, S. Winkler, S. J. Hughes, C. Whitworth, M. Galant, W. Farnaby, K. Rumpel and A. Ciulli, ACS Chem. Biol., 2019, 14, 361–368 CrossRef CAS.
- N. E. A. Chessum, S. Y. Sharp, J. J. Caldwell, A. E. Pasqua, B. Wilding, G. Colombano, I. Collins, B. Ozer, M. Richards, M. Rowlands, M. Stubbs, R. Burke, P. C. McAndrew, P. A. Clarke, P. Workman, M. D. Cheeseman and K. Jones, J. Med. Chem., 2018, 61, 918–933 CrossRef CAS PubMed.
- C. E. Powell, Y. Gao, L. Tan, K. A. Donovan, R. P. Nowak, A. Loehr, M. Bahcall, E. S. Fischer, P. A. Jänne, R. E. George and N. S. Gray, J. Med. Chem., 2018, 61, 4249–4255 CrossRef CAS PubMed.
- J. Popow, H. Arnhof, G. Bader, H. Berger, A. Ciulli, D. Covini, C. Dank, T. Gmaschitz, P. Greb, J. Karolyi-Özguer, M. Koegl, D. B. McConnell, M. Pearson, M. Rieger, J. Rinnenthal, V. Roessler, A. Schrenk, M. Spina, S. Steurer, N. Trainor, E. Traxler, C. Wieshofer, A. Zoephel and P. Ettmayer, J. Med. Chem., 2019, 62, 2508–2520 CrossRef CAS.
- O. Hammer, D. Harper and P. Ryan, PAST: Paleontological Statistics Software Package for Education and Data Analysis, 2001, vol. 4 Search PubMed.
- Y. Zhang, C. Loh, J. Chen and N. Mainolfi, Drug Discovery Today: Technol., 2019, 31, 53–60 CrossRef.
- C. Steinebach, H. Kehm, S. Lindner, L. P. Vu, S. Köpff, Á. López Mármol, C. Weiler, K. G. Wagner, M. Reichenzeller, J. Krönke and M. Gütschow, Chem. Commun., 2019, 55, 1821–1824 RSC.
- A. Whitty, M. Zhong, L. Viarengo, D. Beglov, D. R. Hall and S. Vajda, Drug Discovery Today, 2016, 21, 712–717 CrossRef CAS PubMed.
- A. Whitty, L. A. Viarengo and M. Zhong, Org. Biomol. Chem., 2017, 15, 7729–7735 RSC.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- E. A. Villar, D. Beglov, S. Chennamadhavuni, J. A. Porco Jr, D. Kozakov, S. Vajda and A. Whitty, Nat. Chem. Biol., 2014, 10, 723 CrossRef CAS PubMed.
- M. Rossi Sebastiano, B. C. Doak, M. Backlund, V. Poongavanam, B. Over, G. Ermondi, G. Caron, P. Matsson and J. Kihlberg, J. Med. Chem., 2018, 61, 4189–4202 CrossRef CAS PubMed.
- B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601–2611 CrossRef CAS PubMed.
- Y. Li, J. Yang, A. Aguilar, D. McEachern, S. Przybranowski, L. Liu, C.-Y. Yang, M. Wang, X. Han and S. Wang, J. Med. Chem., 2019, 62, 448–466 CrossRef CAS PubMed.
- C. Qin, Y. Hu, B. Zhou, E. Fernandez-Salas, C.-Y. Yang, L. Liu, D. McEachern, S. Przybranowski, M. Wang, J. Stuckey, J. Meagher, L. Bai, Z. Chen, M. Lin, J. Yang, D. N. Ziazadeh, F. Xu, J. Hu, W. Xiang, L. Huang, S. Li, B. Wen, D. Sun and S. Wang, J. Med. Chem., 2018, 61, 6685–6704 CrossRef CAS PubMed.
- X. Han, C. Wang, C. Qin, W. Xiang, E. Fernandez-Salas, C.-Y. Yang, M. Wang, L. Zhao, T. Xu, K. Chinnaswamy, J. Delproposto, J. Stuckey and S. Wang, J. Med. Chem., 2019, 62, 941–964 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS.
- J. E. Bock, J. Gavenonis and J. A. Kritzer, ACS Chem. Biol., 2013, 8, 488–499 CrossRef CAS PubMed.
- Z. I. Bassi, M. C. Fillmore, A. H. Miah, T. D. Chapman, C. Maller, E. J. Roberts, L. C. Davis, D. E. Lewis, N. W. Galwey, K. E. Waddington, V. Parravicini, A. L. Macmillan-Jones, C. Gongora, P. G. Humphreys, I. Churcher, R. K. Prinjha and D. F. Tough, ACS Chem. Biol., 2018, 13, 2862–2867 CrossRef CAS PubMed.
- C. Galdeano, M. S. Gadd, P. Soares, S. Scaffidi, I. Van Molle, I. Birced, S. Hewitt, D. M. Dias and A. Ciulli, J. Med. Chem., 2014, 57, 8657–8663 CrossRef CAS PubMed.
- Y. Itoh, R. Kitaguchi, M. Ishikawa, M. Naito and Y. Hashimoto, Bioorg. Med. Chem., 2011, 19, 6768–6778 CrossRef CAS PubMed.
- N. Ohoka, K. Okuhira, M. Ito, K. Nagai, N. Shibata, T. Hattori, O. Ujikawa, K. Shimokawa, O. Sano, R. Koyama, H. Fujita, M. Teratani, H. Matsumoto, Y. Imaeda, H. Nara, N. Cho and M. Naito, J. Biol. Chem., 2017, 292, 4556–4570 CrossRef CAS PubMed.
- C. P. Tinworth, H. Lithgow, L. Dittus, Z. I. Bassi, S. E. Hughes, M. Muelbaier, H. Dai, I. E. D. Smith, W. J. Kerr, G. A. Burley, M. Bantscheff and J. D. Harling, ACS Chem. Biol., 2019, 14, 342–347 CrossRef CAS PubMed.
- B. E. Smith, S. L. Wang, S. Jaime-Figueroa, A. Harbin, J. Wang, B. D. Hamman and C. M. Crews, Nat. Commun., 2019, 10, 131 CrossRef PubMed.
- C. Maniaci, S. J. Hughes, A. Testa, W. Chen, D. J. Lamont, S. Rocha, D. R. Alessi, R. Romeo and A. Ciulli, Nat. Commun., 2017, 8, 830 CrossRef PubMed.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- P. P. Chamberlain, A. Lopez-Girona, K. Miller, G. Carmel, B. Pagarigan, B. Chie-Leon, E. Rychak, L. G. Corral, Y. J. Ren, M. Wang, M. Riley, S. L. Delker, T. Ito, H. Ando, T. Mori, Y. Hirano, H. Handa, T. Hakoshima, T. O. Daniel and B. E. Cathers, Nat. Struct. Mol. Biol., 2014, 21, 803 CrossRef CAS PubMed.
- J. Krönke, N. D. Udeshi, A. Narla, P. Grauman, S. N. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer, X. Li, C. Ciarlo, E. Hartman, N. Munshi, M. Schenone, S. L. Schreiber, S. A. Carr and B. L. Ebert, Science, 2014, 343, 301–305 CrossRef PubMed.
- G. Lu, R. E. Middleton, H. Sun, M. Naniong, C. J. Ott, C. S. Mitsiades, K.-K. Wong, J. E. Bradner and W. G. Kaelin, Science, 2014, 343, 305–309 CrossRef CAS PubMed.
- J. N. Spradlin, X. Hu, C. C. Ward, S. M. Brittain, M. D. Jones, L. Ou, M. To, A. Proudfoot, E. Ornelas, M. Woldegiorgis, J. A. Olzmann, D. E. Bussiere, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Nat. Chem. Biol., 2019, 15, 747–755 CrossRef CAS PubMed.
- X. Zhang, V. M. Crowley, T. G. Wucherpfennig, M. M. Dix and B. F. Cravatt, Nat. Chem. Biol., 2019, 15, 737–746 CrossRef CAS PubMed.
- A. R. Schneekloth, M. Pucheault, H. S. Tae and C. M. Crews, Bioorg. Med. Chem. Lett., 2008, 18, 5904–5908 CrossRef CAS PubMed.
- J. Hines, S. Lartigue, H. Dong, Y. Qian and C. M. Crews, Cancer Res., 2019, 79, 251–262 CrossRef CAS PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K.-H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514 CrossRef CAS PubMed.
- R. P. Nowak, S. L. DeAngelo, D. Buckley, Z. He, K. A. Donovan, J. An, N. Safaee, M. P. Jedrychowski, C. M. Ponthier, M. Ishoey, T. Zhang, J. D. Mancias, N. S. Gray, J. E. Bradner and E. S. Fischer, Nat. Chem. Biol., 2018, 14, 706–714 CrossRef CAS PubMed.
- M. L. Drummond and C. I. Williams, J. Chem. Inf. Model., 2019, 59, 1634–1644 CrossRef CAS PubMed.
- S. D. Edmondson, B. Yang and C. Fallan, Bioorg. Med. Chem. Lett., 2019, 29, 1555–1564 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Dataset of degraders analysed in this study. See DOI: 10.1039/c9md00272c |
| This journal is © The Royal Society of Chemistry 2019 |