
Synthetic studies on the reverse antibiotic natural products, the nybomycins†‡
Oliver A.
Bardell-Cox
 ab,
Andrew J. P.
White
a,
Luis
Aragón
*b and
Matthew J.
Fuchter
*a
ab,
Andrew J. P.
White
a,
Luis
Aragón
*b and
Matthew J.
Fuchter
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, London, W12 OBZ, UK. E-mail: m.fuchter@imperial.ac.uk
bCell Cycle Group, MRC London Institute of Medical Sciences, Du Cane Road, London W12 0NN, UK. E-mail: luis.aragon@lms.mrc.ac.uk
First published on 24th May 2019
Abstract
Antimicrobial resistance (AMR) is a serious issue that could have severe consequences if steps are not taken. The nybomycin natural products have the potential to extend the clinical efficacy of the marketed fluoroquinolone class of antibiotics through a ‘reverse antibiotic’ approach. However, only very limited structure–activity relationships are known for these fascinating compounds, in part due to challenges with their synthesis. Here we report a new scalable and robust synthetic route to the nybomycin natural products to aid in the development of this series. Through this synthesis, we report the antibiotic activity of novel analogues of this family confirming the selectivity for fluoroquinolone resistant bacteria and potential future opportunities for further optimisation.
Antibiotic resistance is a worldwide issue. Globally, AMR causes around 700
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year at a low estimate.1 This figure is likely to be an underestimate as incidences are hard to attribute and sometimes not reported. However, with the rise of AMR and the lack of new clinical options, this figure is projected to rise to around 10 million deaths in 2050.1 That equates to one death every 3 seconds.
000 deaths per year at a low estimate.1 This figure is likely to be an underestimate as incidences are hard to attribute and sometimes not reported. However, with the rise of AMR and the lack of new clinical options, this figure is projected to rise to around 10 million deaths in 2050.1 That equates to one death every 3 seconds.
The quinolone class of antibiotics, which include ciprofloxacin and levofloxacin, are used to treat a variety of bacterial infections including from both Gram-positive and Gram-negative organisms.2 They are one of the most prescribed classes of antibiotics.3 The targets for these antibiotics are the homologous type II topoisomerase enzymes, DNA gyrase and topoisomerase IV.4,5 Both of these enzymes relieve the strain (or amount of supercoiling) generated in DNA as a consequence of its unwinding during DNA replication.6–8 Fluoroquinolones function by stabilising the cleaved complex of DNA gyrase (or topoisomerase IV) and DNA, leading to fragmentation of the DNA and death of the bacterial cells.8–10 The main cause of resistance to this class of antibiotics occurs when there is a mutation in one of the key residues in the quinolone-resistance determining region (QRDR) of the enzyme.9,11 These mutations disrupt a water-mediated magnesium bridge linking the quinolone to the protein via the non-catalytic Mg2+ ion, resulting in a significant loss of affinity of the fluoroquinolone to the enzyme.6,9,12,13
Resistance to fluoroquinolones was noted rapidly upon initial clinical usage and the resistance rate continues to increase in most bacteria species.14 Given the success of these antibiotics, new approaches are needed to re-establish their clinical effectiveness. One emerging approach is the use of a ‘reverse antibiotic’,15,16 a concept experimentally verified in vitro with the fluoroquinolone ciprofloxacin and the natural product deoxynybomycin.16 In this approach (Fig. 1), deoxynybomycin was identified to be active against fluoroquinolone-resistant bacteria through binding to QRDR mutant type II topoisomerase enzymes, S84L gyrA and E84K parC. Importantly however, resistance to deoxynybomycin occured by a back-mutation to the wild-type type II topoisomerase enzymes, thus re-sensitising the bacteria to fluoroquinolones. In theory, the combined usage of fluoroquinolones and nybomycin natural products could trap bacteria in an ‘evolutionary loop’ where they will always be sensitive to one of the antibiotics. This approach breaks the cycle of needing to constantly develop new antibiotics for resistant bacteria and may be a powerful way to tackle AMR, especially once further developed. Furthermore, development of the nybomycin series may provide alternative antibiotics against this validated bacterial target class, given some of the side effects observed for the fluoroquinolones.17,18
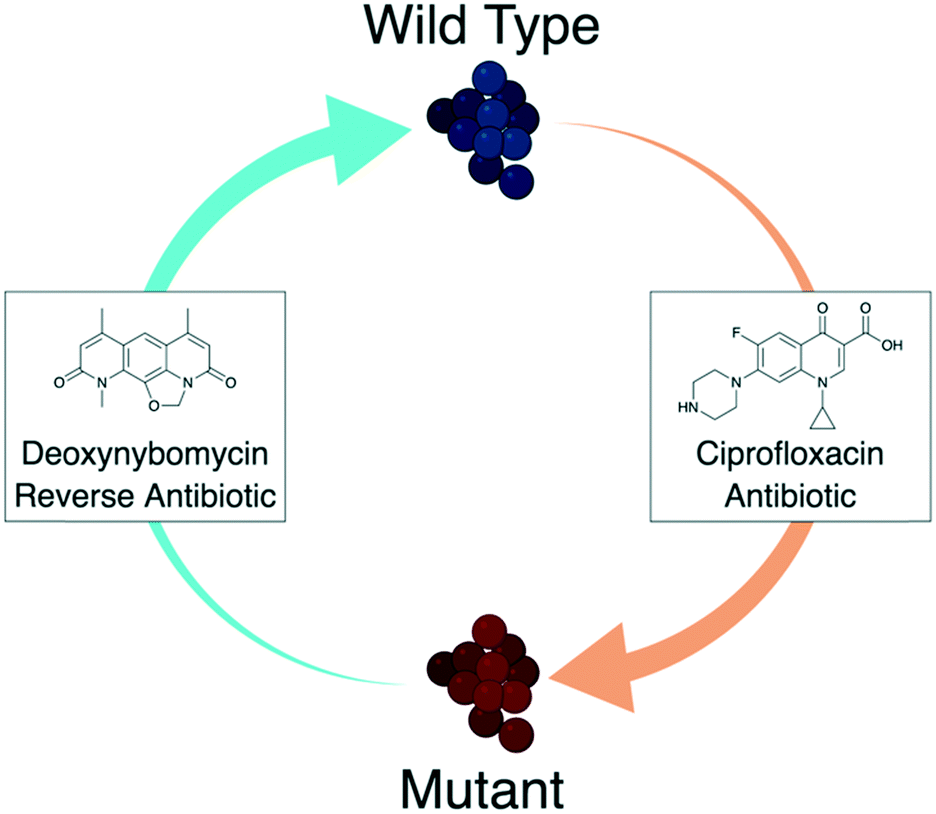 | ||
| Fig. 1 Diagrammatic representation of the ‘reverse antibiotic’ concept. Ciprofloxacin (a fluoroquinolone antibiotic) is an effective treatment of wild type S. aureus shown in blue. Target mutations lead to a mutant strain, shown in red, that is resistant to ciprofloxacin. Deoxynybomycin (a reverse antibiotic) can then be used to treat the mutant S. aureus. Importantly, the mutation required to gain resistance to deoxynybomycin reverts back to the wild-type target, which re-sensitises the S. aureus to fluoroquinolone antibiotics. This could be an important approach in breaking the constant need to discover new antibiotics with novel modes of action.15,16 Image created with http://BioRender.com. | ||
Deoxynybomycin belongs to the nybomycin series of natural products. Both nybomycin and deoxynybomycin are reported to be highly active against the fluoroquinolone resistant bacteria strains (FQR) but inactive against the fluoroquinolone sensitive strains (FQS) and are, thus far, the only example of a ‘reverse antibiotic’.15,16 Whilst the nybomycins show a lack of activity against Gram-negative bacteria,19 it has been shown that structural modification of the scaffold can lead to broad spectrum activity.19 Despite the key importance this places upon this natural product series, very limited medicinal chemistry exploration of the nybomycin scaffold has been reported.16,19 Such medicinal chemistry studies are clearly needed: both nybomycin and deoxynybomycin have poor aqueous solubility and selectivity issues over the human topoisomerase isoforms.20 Optimisation will be needed to alleviate these liabilities, in order for this class of antibiotics to progress into clinical exploration. Here we report our synthetic studies on the nybomycins, aiming to develop a reliable and reproducible synthesis that is amenable to analogue synthesis. Through this route, we report some initial structure activity relationships (SAR) of the nybomycin series', identifying novel structural determinants for their antibiotic activity.
Although nybomycin and deoxynybomycin are natural products that were first reported in 1955,21 their isolation is far from straight-forward22 and the fermentation process is inefficient (1000 L is reported to give 350–200 mg of nybomycin).23 The previously reported total syntheses are also relatively low yielding (13%,24 11%,16 0.84%25,26 overall yield). An overview of the prior synthetic approaches is shown in Fig. 2. Route 1, developed by Rinehart and Forbis,25,26 disregards the symmetry of the molecule and constructs the central framework sequentially. Given this, in addition to some low yielding steps, an overall yield of 0.84% is obtained for the synthesis of deoxynybomycin, which is clearly not ideal for analogue synthesis. Routes 2 and 3, developed by Nussbaum and co-workers24 and Hergenrother and co-workers16,27,28 respectively, better exploit the symmetrical nature of the molecule, reducing the overall number of linear steps.
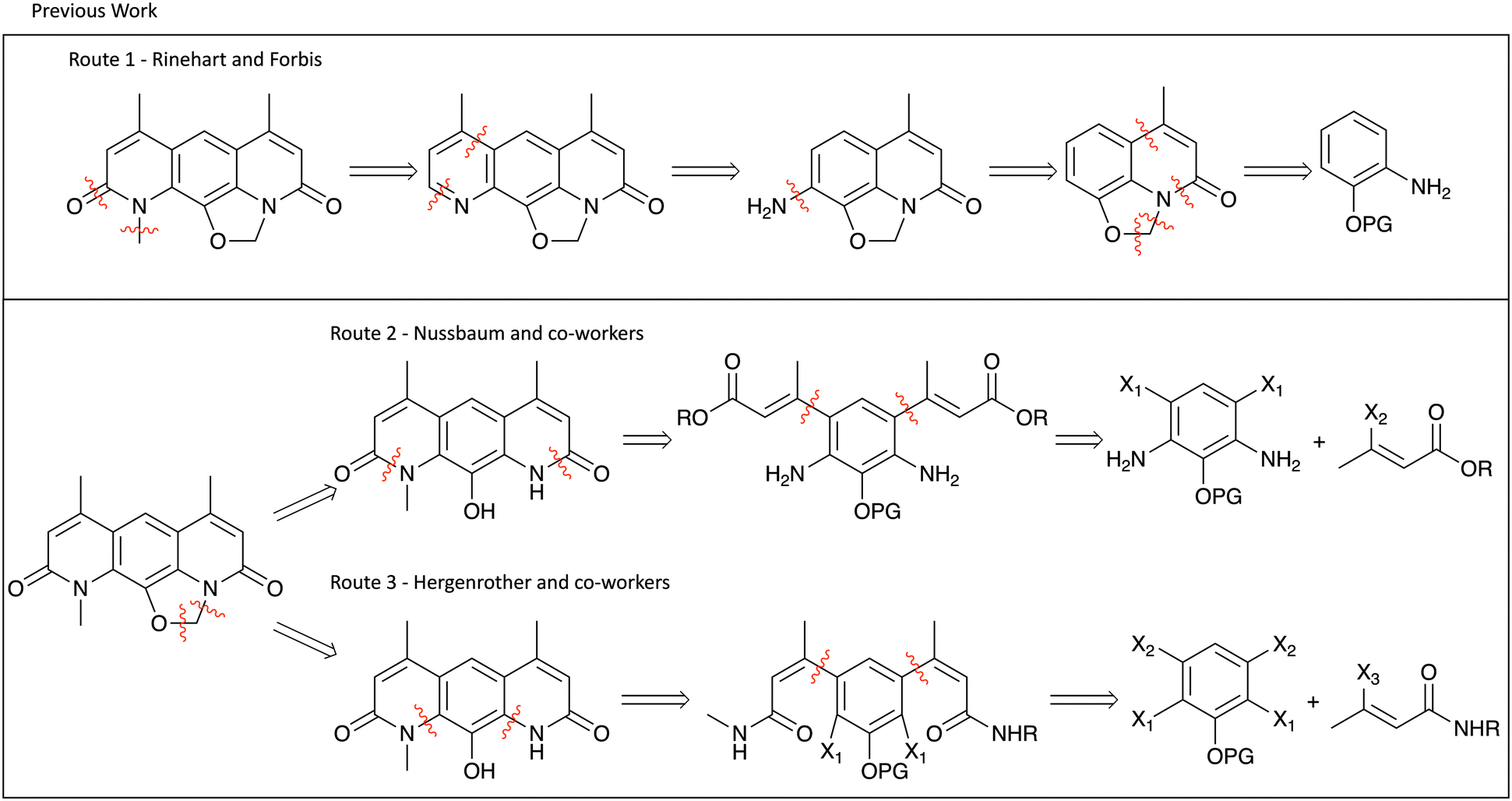 | ||
| Fig. 2 Previously reported routes to deoxynybomycin. | ||
Route 3 was identified as the preferred route for further medicinal chemistry as it had the best overall yield and offered the most reliable synthesis of deoxynybomycin. The cis-iodo-alkene 1 and borylated core 2 intermediates, shown in Scheme 1, were synthesised in accordance with the literature protocols.27–29 However, the reported Suzuki-Miyaura cross-coupling failed to yield any of the desired compound 3 in our hands. A screen for alternative reaction conditions was therefore carried out; altering the catalyst, ligand, base and solvent. Regrettably, all conditions surveyed failed to yield any of the desired cross-coupled product. Due to the relatively unactivated nature of aryl boronic ester 2 and the base instability of the iodo-alkene 1, we proposed that swapping the coupling partners would improve the robustness of this capricious cross-coupling reaction.
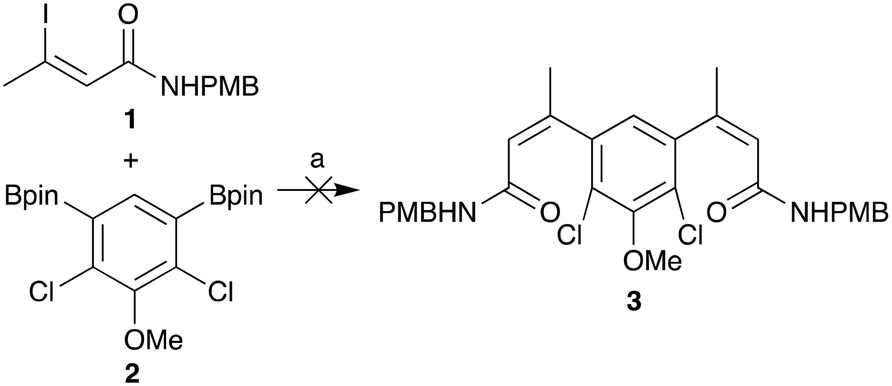 | ||
| Scheme 1 Unsuccessful Suzuki-Miyaura cross-coupling of 1 and 2 to form 3 using the previously reported conditions.27–29 (a) Pd(dppf)Cl2·CH2Cl2, K2CO3, DME, H2O, 85 °C. | ||
Borylated-alkene 4 was synthesised as shown in Scheme 2. This was achieved in a high yield in accordance with the work of Aggarwal and co-workers from ethyl but-2-ynoate using bis(pinacolato)diboron, sodium tert-butoxide and copper(II) chloride.30 Determination of the stereoisomer formed in this reaction was challenging, but mechanistically it was more likely to be the Z-isomer following syn-addition: incorrect stereochemistry for the latter cyclisation. This material was progressed in the hope that in situ isomerisation would occur, either heat or transition metal mediated, during palladium catalysed amination. Pleasingly, cross-coupling of the borylated alkene 4 and the iodo core 5 progressed in an excellent yield (97%). This validated the approach to exchange to coupling partners in order to further optimise this chemistry. However, prior to cyclisation, the ethyl ester 6 needed to be converted to the amide 8, via the carboxylic acid 7. Unfortunately, base-mediated hydrolysis of the di-ester 6 proved problematic and the desired product could not be obtained due to degradation.
 | ||
| Scheme 2 Attempted formation of amide 8. (a) B2pin2, NaOtBu, Cu(II)Cl2, xantphos, MeOH, THF, rt, 77%, (b) Pd(dppf)Cl2·CH2Cl2, K2CO3, DME, H2O, 85 °C, 97%. (c) NaOH, H2O, THF. | ||
In order to avoid such degradation, the chemistry was repeated using N-(4-methoxybenzyl)but-2-ynamide (9). The formation of the alkyne amide 9 was achieved from but-2-ynoic acid using propylphosphonic anhydride (T3P) in good yields (Scheme 3). Alkyne amide 9 then underwent hydroboration as before, to yield the (presumably) syn-hydroborated alkene amide 10.30 Product 10 was then successfully coupled with the iodo core 5 using the previous conditions, again in an excellent yield (86%). The next step was to cyclise the compound to form the core of deoxynybomycin 11. Using the Buchwald-Hartwig conditions reported by Hergenrother and co-workers,28,29 no evidence of cyclised material was observed and only the starting amide remained. A screen of alternative reaction conditions was carried out varying the catalyst, base and solvent, as well as alternative pathways such as nucleophilic aromatic substitution (SNAr), but none of these conditions successfully delivered the product. We attribute the failure of these reactions to the incorrect stereochemistry of the alkene and lack of in situ isomerisation. We therefore instead explored alternative methods to access the correct stereochemistry to facilitate cyclisation.
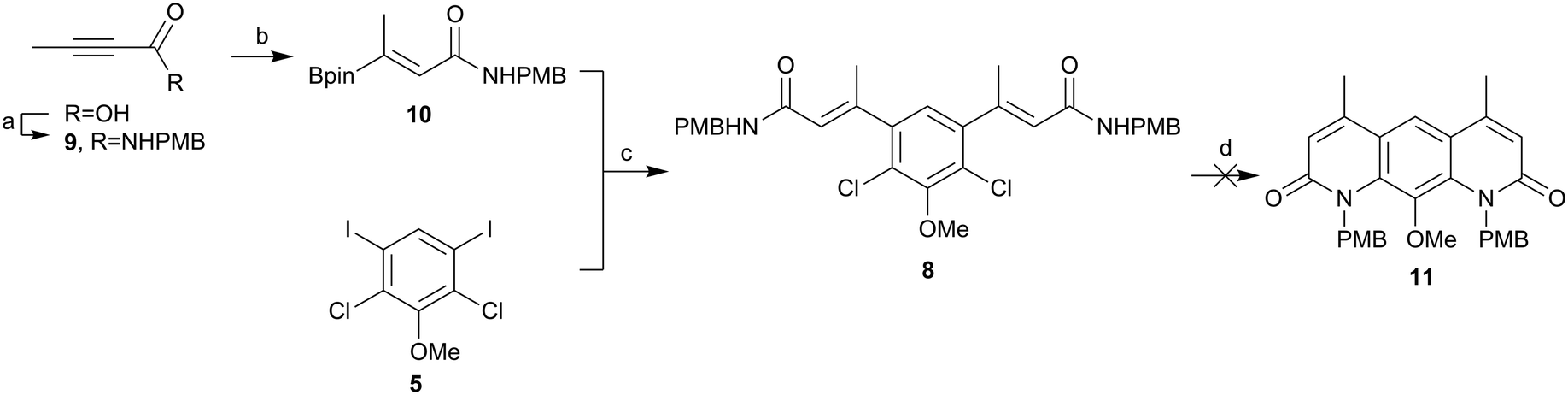 | ||
| Scheme 3 Attempted synthesis of 11. (a) NH2PMB, T3P, NEt3, THF, rt, 84%, (b) B2pin2, NatOBu, Cu(II)Cl2, xantphos, MeOH, THF, rt, 97%, (c) Pd(dppf)Cl2·CH2Cl2, K2CO3, DME, H2O, 85 °C, 86%, (d) Pd G2 Xphos, Xphos, K2CO3, i-PrOH, 90 °C. | ||
Hydroboration of an alkyne typically occurs to provide the anti-Markovnikov adduct with syn-selectivity via a four-membered transition state. Although such regiochemistry is required for our coupling intermediate, the stereochemistry is incorrect. Given our failed attempt above, E-stereochemistry of the hydroboration product would be required in order for the resultant coupled product to be in the correct stereochemistry to cyclise. Sawamura and co-workers have previously reported an anti-hydroboration of alkynyl esters to give hydroborated alkenes with the desired regio- and stereochemistry.31 Given the previous challenges with ester hydrolysis once attached to the core, synthesis of the amide coupling partner 12 was attempted (Scheme 4). Unfortunately the anti-hydroboration chemistry developed by Sawamura and co-workers did not work for substrate 9. Instead of hydroboration, reduction was the preferred pathway and the major isolated product was (E)-N-(4-methoxybenzyl)but-2-enamide (13).
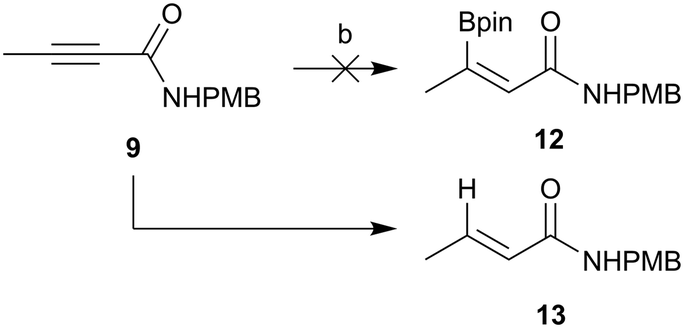 | ||
| Scheme 4 Attempted synthesis of amide coupling partner 12. (a) HBpin, P(Bu)3, 60 °C, MeCN, 47%. | ||
Therefore, we returned to an ester coupling partner, shown in Scheme 5. Pleasingly, ethyl but-2-ynoate was hydroborated in a reasonable yield (60%) and ∼9:1 anti:syn. Evidence for the alternative stereochemistry of this reaction came from the 1H NMR data. There was a downfield shift of 0.43 ppm in the alkene proton and 0.22 ppm in the alkene methyl group when compared to the previously synthesised addition product, assumed to be syn. Coupling of this material 14 to iodo core 5 progressed in excellent yield (98%). However, base-mediated hydrolysis once again proved be problematic. Conditions using different hydroxide sources, reaction times, temperatures, solvents and solvent ratios were screened, shown in the ESI.‡ The best conditions – using NaOH (3M) in MeOH – yielded small amounts of the di-acid 16, but purifying this compound proved problematic and telescoping the crude material into the amide formation using T3P as the coupling agent or via the formation of the acyl chloride did not yield any of the desired di-amide 3. Direct amination of ester 15 using trimethylaluminum also failed.
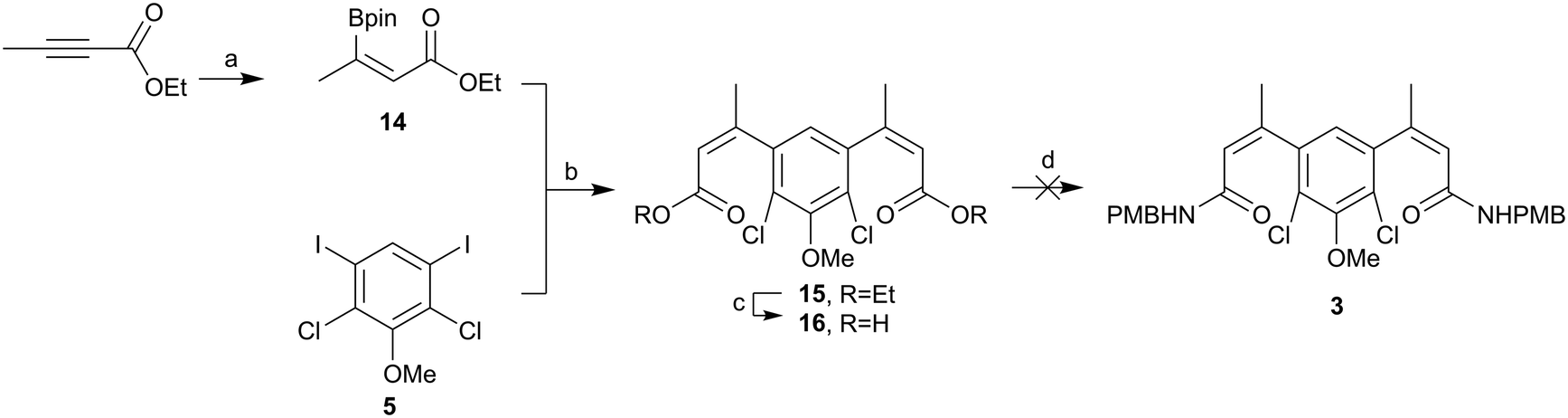 | ||
| Scheme 5 Attempted synthesis of amide 3. (a) HBpin, P(Me)3, THF, rt, 60%, (b) Pd(dppf)Cl2·CH2Cl2, K2CO3, DME, H2O, 85 °C, 98%, (c) NaOH, H2O, MeOH, rt, (d) (i) (COCl)2, rt, (ii) NH2PMB, NEt3, DCM, rt. | ||
Given the apparent base lability of the intermediate 15, we changed to the use of a tert-butyl ester, which would allow formation of carboxylic acid 16 through acid-mediated tert-butyl cleavage (Scheme 6). Alkyne 17 was synthesised as reported previously in the literature,32via an elimination and deprotonation of a mixture of cis/trans 1-bromopropene with nBuLi, then trapping the lithiated alkyne anion formed with di-tert-butyl dicarbonate. Pleasingly, the hydroboration developed by Sawamura and co-workers31 worked well on alkyne 17 to give alkene 18 in an acceptable yield (52%). This material was purified by distillation and crystals were grown from a mixture of pentane and diethyl ether to allow for determination of the geometry across the double bond by a single-crystal X-ray diffraction. As evident from the X-ray structure of alkene 18 (Fig. 3), the desired anti-addition product was indeed obtained. This information compared to the 1H NMR data let us unequivocally assign the stereochemistry of alkene 4 as Z.
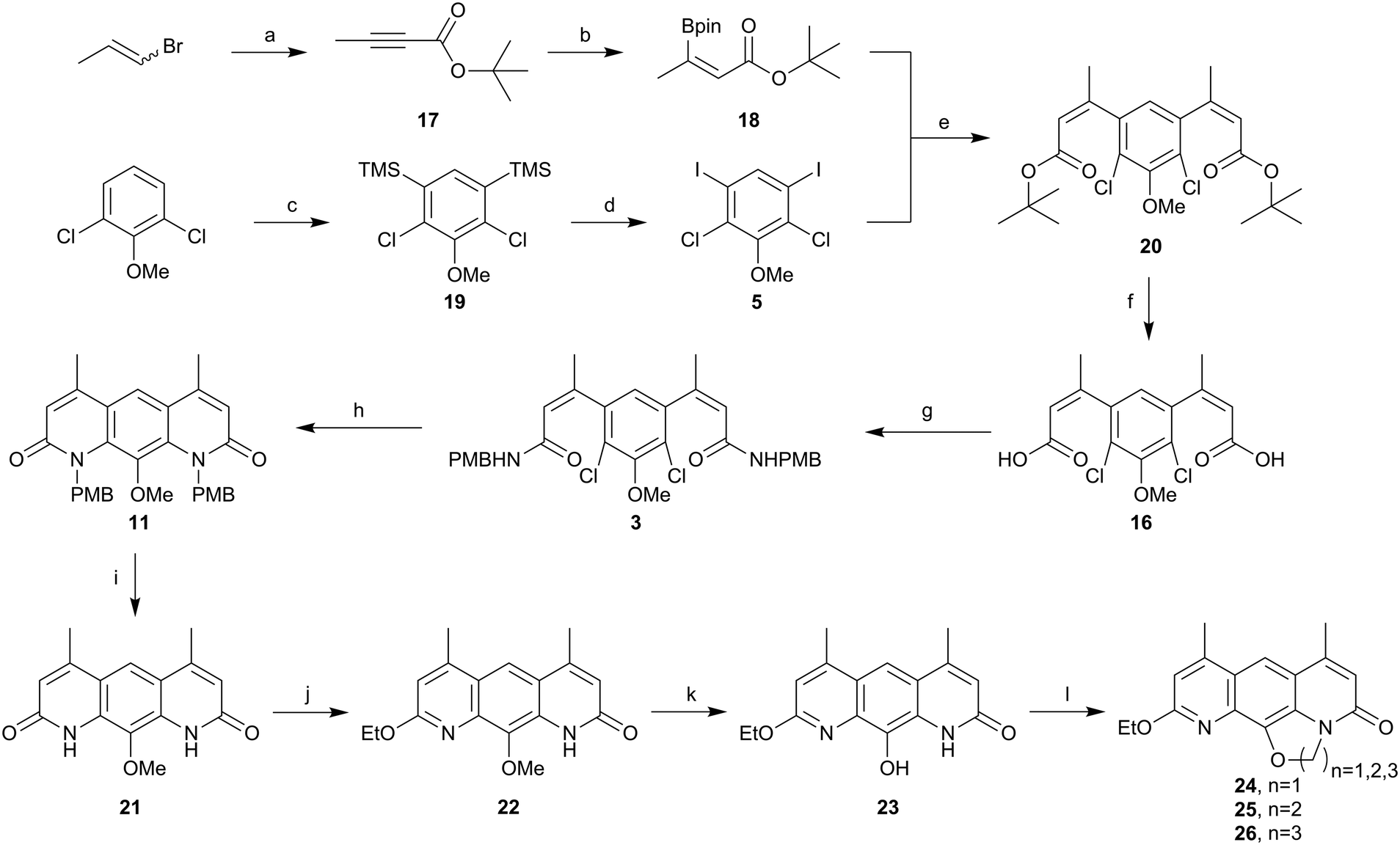 | ||
| Scheme 6 Synthesis of ethoxy-pyridine analogues of 24, 25 and 26. (a) n-BuLi, Boc2O, THF, −78 °C, 98%, (b) HBpin, P(Me)3, THF, rt, 52%, (c) LDA, TMSCl, THF, −78 °C, quant, (d) ICl, DCM, rt, 86%, (e) Pd(dppf)Cl2·CH2Cl2, K2CO3, DME, H2O, 85 °C, 75%, (f) TFA, DCM, rt, 95%, (g) (i) (COCl)2, rt, (ii) NH2PMB, NEt3, DCM, rt, 96%, (h) Pd G2 Xphos, Xphos, K2CO3, i-PrOH, 90 °C, 99%, (i) TFA, rt, 98%, (j) EtI, K2CO3, DMSO, 70 °C, 59%, (k) BBr3, DCM, rt, 41%, (l) CH2Br2, K2CO3, DMSO, 85 °C, 14% for 24, BrCH2CH2Br, K2CO3, DMSO, 85 °C, 13% for 25, BrCH2CH3CH2Br, K2CO3, DMF, 110 °C, 18% for 26. | ||
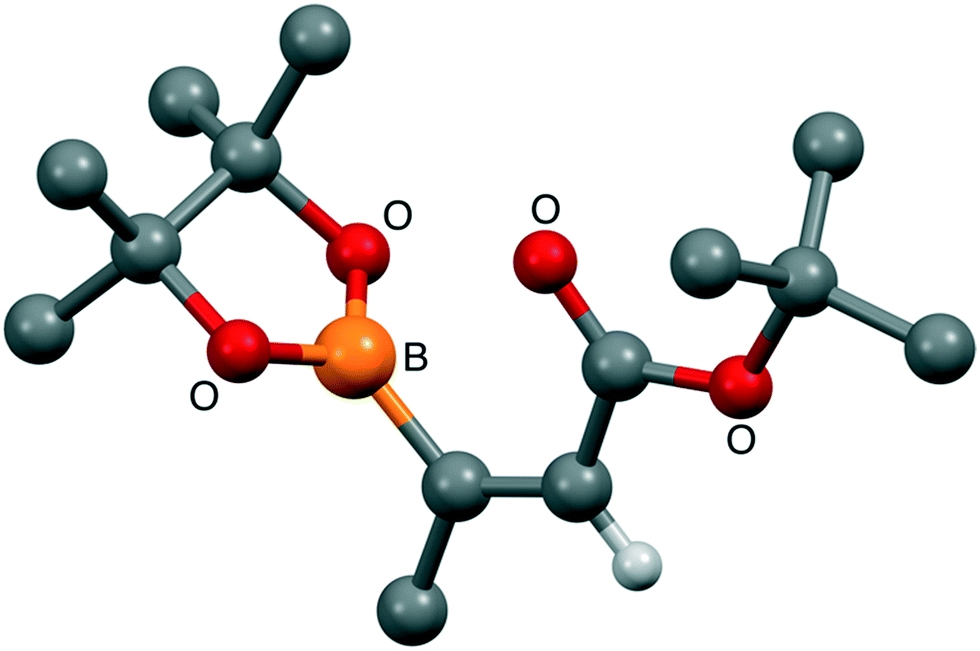 | ||
| Fig. 3 Structural conformation of 18. X-ray structure of 18, confirming anti-hydroboration had occurred and the coupling partner had the desired conformation (E). | ||
The key coupling step proceeded cleanly and efficiently to yield the di-ester 20 in a good yield (75%) and proved to be robust and suitable to gram scale synthesis (Scheme 6). Treatment of 20 with trifluoroacetic acid afforded the desired di-acid 16 in an excellent yield (95%), which was cleanly converted to the di para-methoxybenzyl (PMB) amide 3 by first formation of the di-acyl chloride and then treatment with excess PMB amine. Amide 3 was cyclised using the Buchwald–Hartwig amination conditions used by Hergenrother and co-workers27,28 to yield 11. Deprotection of the PMB protecting groups was achieved using trifluoroacetic acid, resulting in 21. The solubility of compound 21 was very poor in a range of organic solvents, but ethylation using iodoethane in DMSO at 85 °C with potassium carbonate as the base gave a more soluble product. After analysis of the product by 13C NMR and comparison with similar compounds in the literature,16 this reaction was found to have afforded the O-alkylated ethoxy-pyridine 22 as the major reaction product. The chemoselectivity of this alkylation was contradictory to the literature, which suggested N-alkylation to be preferred.33–35 However, we took this as an opportunity to explore the antibacterial effect of these novel nybomycin-like analogues and probe novel structure–activity relationships. Chemoselective demethylation of anisole 22 was achieved using BBr3 and the product 23 could be isolated via precipitation with water and filtration. The resulting off-white solid 23 was cyclised using 1,1-dibromomethane for 24, 1,2-dibromoethane for 25 and 1,3-dibromopropane for 26, to access three novel nybomycin-like analogues for anti-bacterial testing.
Attention then returned to the synthesis of 30. To avoid O-ethylation the N-ethyl N-PMB amide 27 was prepared, prior to cyclisation, as shown in Scheme 7. Di-acid 16 underwent an unsymmetrical amide coupling with a 1:1 mixture of ethylamine and PMB amine to give the statistical mixture of the amide products. These products could be separated by column chromatography affording the desired amine 27 in a 42% yield and the undesired di-PMB amide 3 which could be recycled for the synthesis of the ethoxy-pyridines above. The desired amide 27 was cyclised under Buchwald–Hartwig amination conditions using Xphos Pd G2.36 This gave good conversion (85%) to the cyclic product 28 which was then globally deprotected using HBr at reflux. Treatment of the reaction mixture with water gave an off-white precipitate which was isolated via filtration to give 29. This material could then be cyclised using 1,1-dibromomethane for 30, 1,2-dibromoethane for 31 and 1,3-dibromopropane for 32, to two novel nybomycin analogues for anti-bacterial testing and comparison with the previously described compound 30.16
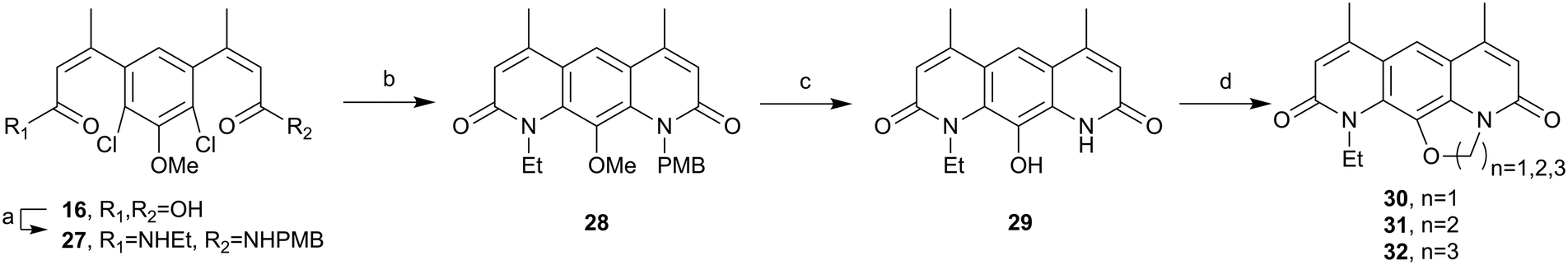 | ||
| Scheme 7 Synthesis of deoxynybomycin analogues 30, 31 and 32. (a) (i) (COCl)2, rt, (ii) NH2Et, NH2PMB, NEt3, DCM, rt, 42%, (b) Pd G2 Xphos, Xphos, K2CO3, i-PrOH, 85 °C, 85%, (c) HBr, 110 °C, 91%, (d) CH2Br2, K2CO3, DMSO, 85 °C, 33% for 30, BrCH2CH2Br, K2CO3, DMSO, 110 °C, 28% for 31, BrCH2CH3CH2Br, K2CO3, DMF, 110 °C, 44% for 32. | ||
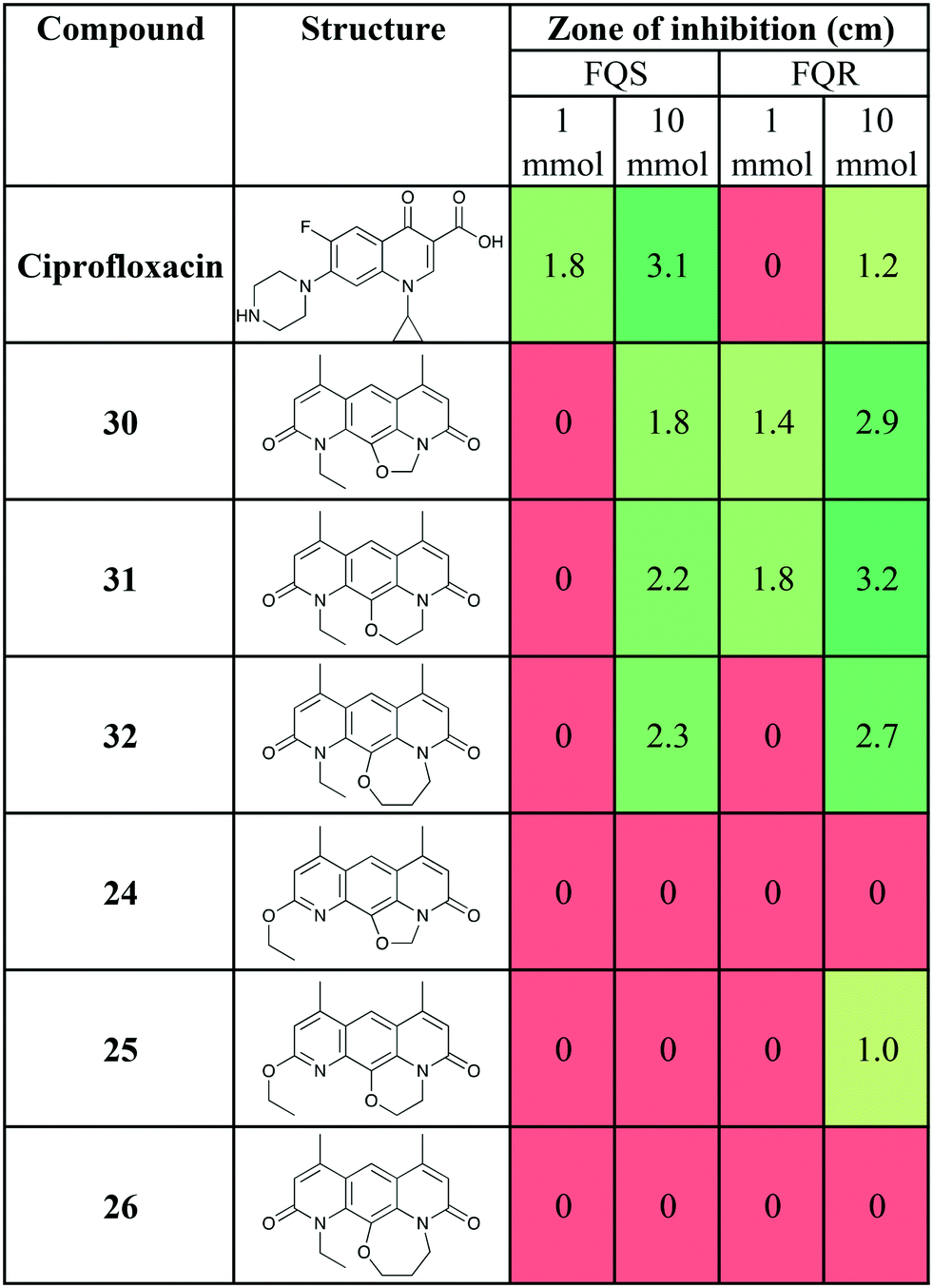
The antibiotic activity of 30 and novel analogues (24, 25, 26, 31 and 32) were determined using the Kirby–Bauer disc diffusion assay37 using the protocol described by Sridhar and co-workers,38 with the fluoroquinolone sensitive (FQS) S. aureus strain SH1000 and the fluoroquinolone resistant (FQR) S. aureus strain USA300 JE2 (Table 1). S. aureus USA300 JE2 has a mutated DNA gyrase (S84L) and topoisomerase IV (S80Y) which causes resistance to ciprofloxacin as well as additional mutations that confer resistance to other antibiotic classes.39
|
As previously reported,15,16 compound 30 showed the desired mutant selectivity, resulting in effective killing of USA300 JE2. Ring expanded analogues 31 and 32 gave comparable activity, with the 6 membered ring (31) tentatively the most active. This initial result will have to be confirmed using an additional susceptibility testing method.
Interestingly, compounds 24, 25, and 26, where the oxygen of the pyridone was alkylated rather than the nitrogen, showed greatly reduced activity compared to 30, 31, and 32. 24 was inactive at a concentration of 32 mmol in both the FQR strain and FQS strain (data not shown). However, compound 25 with the 6-membered ring, showed some activity against the FQR strain at 10 mmol. This novel SAR is intriguing and suggests that further investigation may be warranted. Such work is ongoing and will be reported in due course.
Conclusions
In summary we have developed a robust and scalable route to the nybomycin scaffold which can facilitate the synthesis diverse array of analogues. The key differentiating step of our approach was to switch the coupling partners of the capricious Suzuki-Miyaura cross-coupling, resulting in scalable coupling yields up to 98%. Our route only involves the use of column chromatography three times which further enables scalability. We believe this route should allow for much needed medicinal chemistry exploration of the nybomycin ‘reverse antibiotics’. Indeed, we have initially surveyed five novel analogues which indicate key structure determinants for antibiotic activity and opportunities for further optimisation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MJF would like to thank the EPSRC for an Established Career Fellowship (EP/R00188X/1). The Aragon laboratory is supported by the UK Medical Research Council. OBC was supported by a Cross Faculty Collaborative studentship from the London Institute of Medical Research (LMS), which receives its core funding from the UK Medical Research Council. The authors would like to give thanks Dr Andrew Edwards for providing the SH1000 and USA300 JE2 bacterial strains, Peter Haycock for NMR support, Adam Jarmuz for microbiology support and Dr. Lisa Haigh for mass spectrometry.Notes and references
- J. O'Neill, Rev. Antimicrob. Resist., 2015, 1–42 Search PubMed.
- M. I. Andersson and A. P. MacGowan, J. Antimicrob. Chemother., 2003, 51, 1–11 CrossRef CAS PubMed.
- K. J. Aldred, R. J. Kerns and N. Osheroff, Biochemistry, 2014, 53, 1565–1574 CrossRef CAS PubMed.
- H. Fukuda and K. Hiramatsu, Antimicrob. Agents Chemother., 1999, 43, 410–412 CrossRef CAS PubMed.
- D. C. Hooper, Drugs, 1999, 58, 6–10 CrossRef CAS PubMed.
- K. Drlica, M. Malik, R. J. Kerns and X. Zhao, Antimicrob. Agents Chemother., 2008, 52, 385–392 CrossRef CAS PubMed.
- K. Drlica and X. Zhao, Microbiol. Mol. Biol. Rev., 1997, 61, 377–392 CAS.
- C. Levine, H. Hiasa and K. J. Marians, Biochim. Biophys. Acta, Gene Struct. Expression, 1998, 1400, 29–43 CrossRef CAS.
- L. S. Redgrave, S. B. Sutton, M. A. Webber and L. J. V. Piddock, Trends Microbiol., 2014, 22, 438–445 CrossRef CAS PubMed.
- K. Drlica, Curr. Opin. Microbiol., 1999, 2, 504–508 CrossRef CAS PubMed.
- H. Yoshida, M. Bogaki, M. Nakamura and S. Nakamura, Antimicrob. Agents Chemother., 1991, 34, 1271–1272 CrossRef PubMed.
- C. Sissi, B. Cheng, V. Lombardo, Y.-C. Tse-Dinh and M. Palumbo, Gene, 2013, 524, 253–260 CrossRef CAS PubMed.
- K. J. Aldred, S. A. McPherson, C. L. Turnbough, R. J. Kerns and N. Osheroff, Nucleic Acids Res., 2013, 41, 4628–4639 CrossRef CAS PubMed.
- A. Dalhoff, Interdiscip. Perspect. Infect. Dis., 2012, 2012, 1–37 CrossRef PubMed.
- K. Hiramatsu, M. Igarashi, Y. Morimoto, T. Baba, M. Umekita and Y. Akamatsu, Int. J. Antimicrob. Agents, 2012, 39, 478–485 CrossRef CAS PubMed.
- E. I. Parkinson, J. S. Bair, B. A. Nakamura, H. Y. Lee, H. I. Kuttab, E. H. Southgate, S. Lezmi, G. W. Lau and P. J. Hergenrother, Nat. Commun., 2015, 6, 1–9 Search PubMed.
- N. L. Millar, S. Siebert and I. B. McInnes, Nature, 2019, 566, 326 CrossRef CAS PubMed.
- J. Marchant, Nature, 2018, 555, 431–433 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- P. J. Hergenrother and A. P. Ley, WO2018/237140A1, 2018.
- F. Strelitz, H. Flon and I. N. Asheshov, Proc. Natl. Acad. Sci. U. S. A., 1955, 41, 620–624 CrossRef CAS PubMed.
- S. Adelmann, T. Baldhoff, B. Koepcke and G. Schembecker, J. Chromatogr. A, 2013, 1274, 54–64 CrossRef CAS PubMed.
- J. A. Pope, R. A. Nelson, C. P. Schaffner, R. T. Rosen and R. C. Pandey, J. Ind. Microbiol., 1990, 6, 61–69 CrossRef CAS PubMed.
- F. Nussbaum, A. Ebbinghaus, A. Mayer-Bartschmid, W. Zitzmann, W. Wiese, M. Stadler and S. Anlauf, EP2130831A1, 2009.
- K. L. Rinehart, G. Leadbetter, R. A. Larson and R. M. Forbis, J. Am. Chem. Soc., 1970, 92, 6995–6996 CrossRef CAS.
- R. M. Forbis and K. L. Rinehart, J. Am. Chem. Soc., 1973, 95, 5003–5013 CrossRef CAS PubMed.
- E. I. Parkinson, J. S. Bair, M. Cismesia and P. J. Hergenrother, ACS Chem. Biol., 2013, 8, 2173–2183 CrossRef CAS PubMed.
- J. S. Bair, R. Palchaudhuri and P. J. Hergenrother, J. Am. Chem. Soc., 2010, 132, 5469–5478 CrossRef CAS PubMed.
- E. I. Parkinson and P. J. Hergenrother, Acc. Chem. Res., 2015, 48, 2715–2723 CrossRef CAS PubMed.
- A. P. Pulis, P. Fackler and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 4382–4385 CrossRef CAS PubMed.
- K. Nagao, A. Yamazaki, H. Ohmiya and M. Sawamura, Org. Lett., 2018, 20, 1861–1865 CrossRef CAS PubMed.
- A. Otaka, E. Mitsuyama, T. Kinoshita, H. Tamamura and N. Fujii, J. Org. Chem., 2000, 65, 4888–4899 CrossRef CAS PubMed.
- K. K. Park and J. J. Lee, Tetrahedron, 2004, 60, 2993–2999 CrossRef CAS.
- V. V. Kravtsova, E. Y. Kovalenko, I. V. Ukrainets, A. A. Tkach and V. I. Mamchur, Chem. Heterocycl. Compd., 2010, 46, 850–855 CrossRef.
- G. A. Kraus and S. Kesavan, Tetrahedron Lett., 2005, 46, 1111–1113 CrossRef CAS.
- M. R. Biscoe, B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2008, 6686–6687 CrossRef CAS PubMed.
- A. W. Bauer, J. C. Sherris, M. Turck and W. M. M. Kirby, Am. J. Clin. Pathol., 1966, 45, 493–496 CrossRef CAS PubMed.
- N. Vineetha, R. A. Vignesh and D. Sridhar, Int. J. Appl. Res., 2015, 1, 624–631 Search PubMed.
- M. Vestergaard, B. Leng, J. Haaber, M. S. Bojer, C. S. Vegge and H. Ingmer, Front. Microbiol., 2016, 7, 1–10 Search PubMed.
Footnotes |
| † Raw data files are openly available DOI: 10.14469/hpc/5498 |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, characterisation data, NMR spectra, further X-ray data. CCDC 1908213. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9md00207c |
| This journal is © The Royal Society of Chemistry 2019 |