
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Scaffold hybridization strategy towards potent hydroxamate-based inhibitors of Flaviviridae viruses and Trypanosoma species†
Erofili
Giannakopoulou
a,
Vasiliki
Pardali
a,
Efseveia
Frakolaki
b,
Vasileios
Siozos
b,
Vassilios
Myrianthopoulos
a,
Emmanuel
Mikros
a,
Martin C.
Taylor
c,
John M.
Kelly‡
c,
Niki
Vassilaki§
b and
Grigoris
Zoidis¶
 *a
*a
aSchool of Health Sciences, Faculty of Pharmacy, Department of Pharmaceutical Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis-Zografou, GR-15771 Athens, Greece. E-mail: zoidis@pharm.uoa.gr
bMolecular Virology Laboratory, Hellenic Pasteur Institute, Vas. Sofias Avenue, GR-11521, Athens, Greece
cDepartment of Pathogen Molecular Biology, London School of Hygiene and Tropical Medicine, Keppel Street, London WC1E 7HT, UK
First published on 16th May 2019
Abstract
Infections with Flaviviridae viruses, such as hepatitis C virus (HCV) and dengue virus (DENV) pose global health threats. Infected individuals are at risk of developing chronic liver failure or haemorrhagic fever respectively, often with a fatal outcome if left untreated. Diseases caused by tropical parasites of the Trypanosoma species, T. brucei and T. cruzi, constitute significant socioeconomic burden in sub-Saharan Africa and continental Latin America, yet drug development is under-funded. Anti-HCV chemotherapy is associated with severe side effects and high cost, while dengue has no clinically approved therapy and antiparasitic drugs are outdated and difficult to administer. Moreover, drug resistance is an emerging concern. Consequently, the need for new revolutionary chemotherapies is urgent. By utilizing a molecular framework combination approach, we combined two distinct chemical entities with proven antiviral and trypanocidal activity into a novel hybrid scaffold attached by an acetohydroxamic acid group (CH2CONHOH), aiming at derivatives with dual activity. The novel spiro-carbocyclic substituted hydantoin analogues were rationally designed, synthesized and evaluated for their potency against three HCV genotypes (1b, 3a, 4a), DENV and two Trypanosoma species (T. brucei, T. cruzi). They exhibited significant EC50 values and remarkable selectivity indices. Several modifications were undertaken to further explore the structure activity relationships (SARs) and confirm the pivotal role of the acetohydroxamic acid metal binding group.
Introduction
Hepatitis C virus (HCV) is a global health burden with ∼71 million people estimated to be infected.1 It is a main cause of chronic liver disease and can lead to liver cirrhosis and hepatocellular carcinoma. HCV is a member of the Flaviviridae family and belongs to the Hepacivirus genus.2 The viral genome is a 9600 nucleotide-long single-stranded RNA of positive polarity and encodes a single polyprotein precursor, which is processed into structural proteins (core, E1 and E2), p7 required for assembly and release of virus particles and six non-structural (NS) proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B).3,4 The NS proteins form a membrane-associated replicase complex (RC) in association with host cell factors. HCV is classified into 8 major genotypes and at least 86 sub/genotypes (GTs),5 with genotype 1 being the most predominant type worldwide.6Until recently, the standard of care for HCV was ribavirin and pegylated–interferon alpha (PEG–IFN), which had severe side effects and low sustained virologic response (SVR) rates.7 Since 2011, direct-acting antivirals (DAAs) targeting the viral NS3/4A protease, the NS5B RNA-dependent RNA polymerase (RdRp) and the NS5A phosphoprotein were introduced (Fig. 1)8–11 and allowed the implementation of interferon-free treatment schedules,12,13 which are able to attain SVR in more than 90% of patients.14 DAAs efficacy differs between genotypes, with genotype 3 elimination rate being lower than the others.15,16
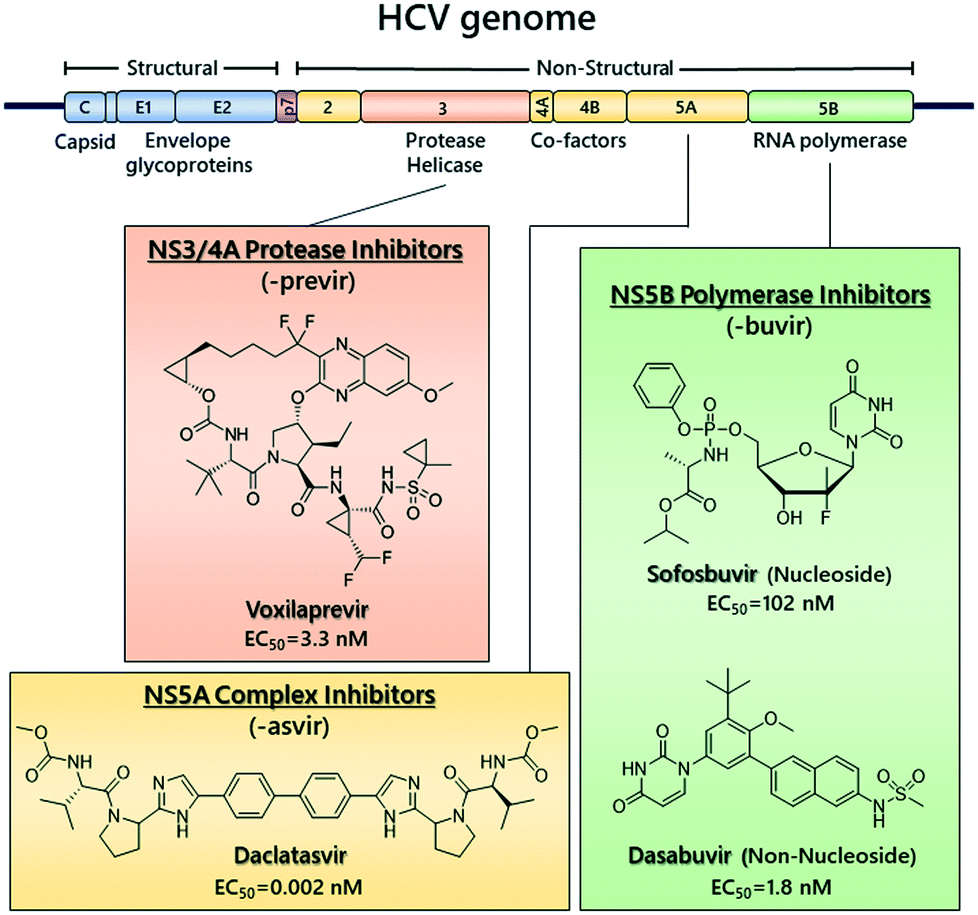 | ||
| Fig. 1 Representative chemical structures of clinically approved DAAs and their respective EC50s against HCV genotype 1b. | ||
However, there is a risk for the development of antiviral drug resistance, depending on the regimen and the genotype.17 Moreover, available DAAs are costly, limiting their widespread clinical use, and, as a result, therapy remains low on a global scale.18 Thus, there is still a need for novel effective drugs of lower cost.
The mosquito-borne dengue virus (DENV) is another member of the Flaviviridae family, belonging to the Flavivirus genus. It causes widely distributed and endemic, visceral and central nervous system diseases.19 Symptoms of infection range from mild (dengue fever) to the more severe dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS).20 The recently approved dengue vaccine has only limited overall efficacy21 and there is no approved antiviral therapy.22 DENV genome consists of a positive single-stranded RNA of ∼11 kb in length, encoding for the viral polyprotein, which is processed into structural proteins (C, prM, E) and non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5).23,24
Bivalent metal ions are important for the stability and activity of various HCV and DENV proteins, including NS3 helicase/protease, RdRp polymerase and HCV NS5A protein.25–30
Human African trypanosomiasis (HAT), also known as sleeping sickness, is a vector-borne parasitic disease with 65 million people being vulnerable to infection.31 HAT occurs in 36 countries of sub-Saharan Africa and it is caused by infection with protozoan parasites belonging to the Trypanosoma brucei sp. T. b. gambiense is the causative agent of a chronic form of the disease (gambiense HAT, gHAT). It is prevalent in central and western Africa, and can take years or decades to produce a fatal outcome. T. b. rhodesiense is responsible for an acute form disease, causing death within weeks or months of infection. Rhodesiense HAT (rHAT) is found in eastern and southern Africa.32,33 Clinical progression of both forms of HAT is divided in two stages. The early hemolymphatic stage and the late meningoencephalitic stage when the parasites enter the central nervous system causing neurological damage and sleep disturbances.34
Treatment approaches of HAT consist of five drugs: pentamidine, suramin, melarsoprol, eflornithine, and nifurtimox–eflornithine combination therapy (NECT) (Fig. 2). Nevertheless, their increasing levels of resistance, their limited efficacy, toxicity and adverse side effects, make HAT treatment a daunting challenge.35 Fexinidazole and acoziborole (Fig. 2) are two potential drug candidates, currently being in advanced clinical trials.36,37
 | ||
| Fig. 2 Currently available marketed drugs for treatment of HAT and Chagas disease. Highlighted regions are reported to play a major role in the mechanism of action. Chemical structures of potential drug candidates fexinidazole and acoziborole. | ||
American trypanosomiasis, or Chagas disease, is a potentially life-threatening infection caused by the protozoan parasite Trypanosoma cruzi. It is endemic in continental Latin America, with a devastating effect upon 6–7 million people,38 and it is characterized by two distinct clinical phases: an acute mostly asymptomatic phase, and a chronic phase characterized mainly by cardiac disorders.39 The two drugs available, nifurtimox and benznidazole (Fig. 2), are only partially effective and toxic side effects limit their use.40
Unfortunately, there is no efficient disease management for both parasitic infections and the World Health Organization has listed them among the neglected tropical diseases (NTDs). Therefore, in absence of vaccines, development of novel effective therapeutics is an imperative for their treatment and control.33
Several groups of trypanosomatid metalloenzymes have been identified as playing an important role in the metabolism and viability of parasites. Amongst the most widely explored are histone deacetylases (HDACs), trypanosome alternative oxidase (TAO) and carbonic anhydrase (CA).41–48
Metalloenzymes' inhibition appears attractive as a chemotherapeutic approach against viruses and parasites.47–53 It has been proved that the vast majority of metalloenzyme inhibitors are chelating agents employing metal binding groups to coordinate the active site metal ion.54,55 As a continuation of our research on metal chelating agents,56,57 we were interested in developing novel combined scaffolds demonstrating both antiviral and antiparasitic properties.
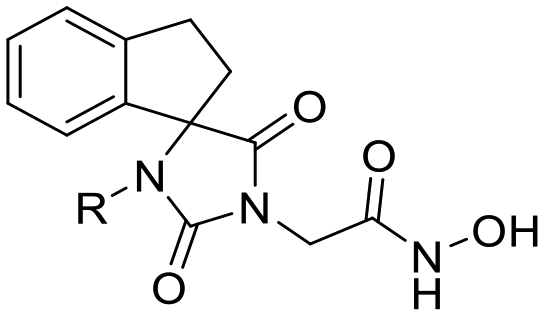
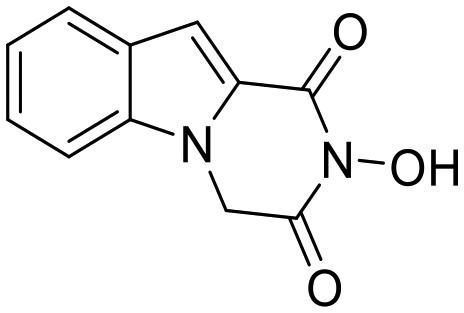
In this study, we report the rational design and synthesis of novel spiro-hydantoin analogues, attached by the acetohydroxamic acid moiety (CH2CONHOH) as the potential metal binding functional group (Fig. 3). Several bicyclic fused rings were used as spiro-substituents on the basic 2,4-diketoimidazolidine scaffold, while methyl- and benzyl-groups were incorporated at the amide nitrogen atom of hydantoin, to enhance binding affinity and improve drug-likeness of the compounds. The novel analogues were assessed as antiviral and trypanocidal agents and exhibited remarkable inhibitory activity with respect to their parent compounds I and II. More specifically, they were evaluated for their effect on RNA replication and cell viability on different HCV genotypes (1b, 3a, 4a) and DENV. Their antiparasitic activity was assessed based on their ability to inhibit proliferation of cultured bloodstream form T. brucei and T. cruzi epimastigotes in vitro.
 | ||
| Fig. 3 Indole–flutimide analogue (I) with antiviral activity, cycloheptane spiro-substituted 2,6-DKP analogue (II) with trypanocidal activity and novel synthesized analogues reported in this study. Metal binding groups are indicated in plum elliptical shape. | ||
Results and discussion
Rational design – theoretical calculations
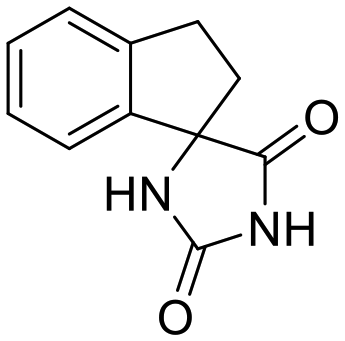
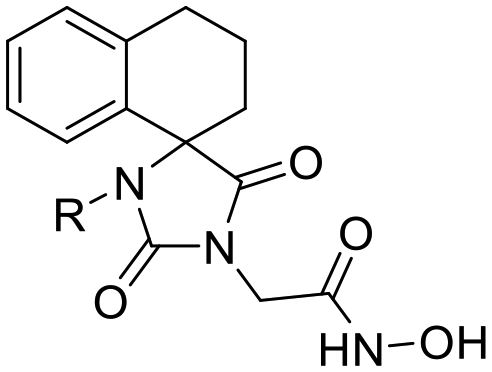
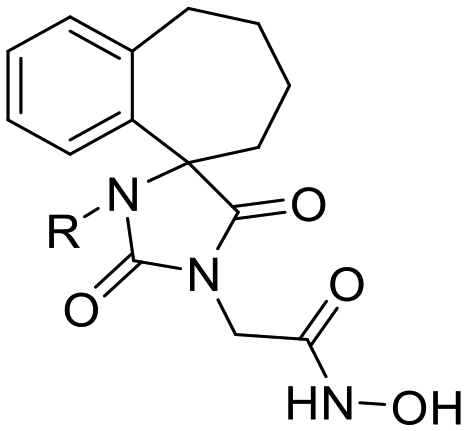
Previous studies by our group have resulted in compounds carrying metal chelating moieties that demonstrated a highly promising inhibition capacity against either pathogenic viruses, including influenza and HCV,56 or parasites of the Trypanosoma genus.57,58 The two above-mentioned series of analogues, although structurally diverse, were based on rational incorporation of metal chelating moieties on scaffolds with pronounced drug-likeness, such as the combination of flutimide with the indole system (I) or incorporation of the acetohydroxamic acid moiety on a 2,6-diketopiperazine (2,6-DKP) scaffold (II) (Fig. 3). In the present study, a conceptual approach to further exploit the bioactivity of molecules with potential metal chelating efficacy toward designing effectors with inhibitory activity against both disease-related viruses and Trypanosoma parasites is reported. The design was based on combination of the exocyclic acetohydroxamic acid pharmacophore, bearing well proven trypanocidal activity, with a lipophilic tail based on a bicyclic system-substituted hydantoin that could effectively mimic the antiviral flutimide scaffold yet with increased steric bulk, thus affording a novel synthetically tractable heterocyclic scaffold as presented in detail in Fig. 3.To counterbalance possible issues of water solubility arising from the tricyclic system of the antiviral lead I, we selected to enhance the sp3 character of the novel scaffold by introducing a spiro connection between the acetohydroxamic group-carrying hydantoin and the indane system. In this case, resembling the spiro trypanocidal lead II would contribute toward avoiding possible Lipinski's rule-of-five violations. Hence, the indole nitrogen atom was converted to a carbon, giving rise to a hydrophobic indane (compound 16). Moreover, with the aim of evaluating the contribution of the critical parameter of lipophilicity on biological activity, extensions of the indane system to tetralin or benzocycloheptane were considered (compounds 17 and 18), while the addition of alkyl substituents such as methyl- or benzyl-groups on N3 of hydantoin was also explored (compounds 16a, 17a, 18a and 16b, 17b, 18b respectively). To confirm the importance of the exocyclic acetohydroxamic acid moiety on antiviral and trypanocidal activity, the derivative 4, lacking metal chelating ability, was evaluated as a negative control (Tables 1 and 2). The derivative 16 carrying the smallest steric bulk, i.e. an indane system with no substitution on N3, was selected for probing the validity of our approach.
| Structure | Cmpd | R | HCV-1b | HCV-3a | HCV-4a | DENV-2 | CC50c (μM) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50a (μM) | SIb | EC50a (μM) | SIb | EC50a (μM) | SIb | EC50a (μM) | SIb | ||||
| a EC50 (median effective concentration) values represent the mean ± SD of three independent experiments, each carried out in triplicate. Calculated against HCV genotype 1b (Huh5-2, HCV 1b replicon cells), genotype 3a (Huh7.5-3a, HCV 3a replicon cells), genotype 4a (Huh7.5-4a, HCV 4a replicon cells). b Selectivity index: CC50/EC50. c CC50 (median cytotoxic concentration against Huh5-2, HCV replicon cells) values represent the mean ± SD of three independent experiments, each carried out in triplicate. | |||||||||||
|
16 | H | 2.92 ± 0.01 | >68 | 2.97 ± 0.21 | >67 | 0.58 ± 0.04 | >345 | 96.34 ± 1.00 | >2 | >200 |
| 16a | CH3 | 1.41 ± 0.01 | 19 | — | — | 0.21 ± 0.02 | 128 | — | — | 26.91 ± 0.67 | |
| 16b | CH2Ph | 0.22 ± 0.01 | 34 | — | — | 0.14 ± 0.01 | 54 | — | — | 7.53 ± 1.29 | |

|
17 | H | 1.41 ± 0.35 | >142 | 2.37 ± 0.17 | >84 | 0.38 ± 0.04 | >526 | 61.49 ± 3.86 | >3 | >200 |
| 17a | CH3 | 1.45 ± 0.07 | 99 | — | — | 0.21 ± 0.01 | 682 | — | — | 143.2 ± 0.37 | |
| 17b | CH2Ph | 0.32 ± 0.01 | 28 | — | — | 0.07 ± 0.02 | 127 | — | — | 8.90 ± 0.77 | |

|
18 | H | 0.25 ± 0.02 | 794 | 0.08 ± 0.01 | 2480 | 0.10 ± 0.00 | 1984 | 16.29 ± 2.17 | 12 | 198.4 ± 2.24 |
| 18a | CH3 | 1.18 ± 0.07 | 95 | — | — | 0.11 ± 0.01 | 1015 | — | — | 111.6 ± 12.43 | |
| 18b | CH2Ph | 1.00 ± 0.05 | 5 | — | — | 0.12 ± 0.01 | 45 | — | — | 5.45 ± 0.27 | |
|
4 | H | >10 | — | — | — | — | — | — | — | >10 |
|
I | — | 126.30 ± 14.20 | >1.6 | — | — | — | — | — | — | >200 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D and pKa values
D and pKa values
| Structure | Cmpd | R | T. brucei | T. cruzi | L6 cells CC50c (μM) | logDd (pH 7.4) |
pKae | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50a (μM) | EC90a (μM) | SIb | EC50a (μM) | EC90a (μM) | SIb | ||||||
|
a Concentrations required to inhibit growth of T. brucei and T. cruzi by 50% and 90%, respectively. EC50 and EC90 data are the mean of triplicate experiments ± SEM.
b Selectivity indices were calculated as the ratio of the CC50 for L6 cells to EC50 for T. brucei or T. cruzi respectively.
c Cytotoxicity was determined by establishing the concentration required to inhibit growth of cultured L6 cells by 50% (CC50). Data are the mean of triplicate experiments ± SEM.
d logD at pH 7.4 determined using Marvin.62
e pKa values for the oxime proton determined from MM preoptimized geometries at the DFT(b3lyp) level with the cc-pVTZ(+) basis set using the Jaguar pKa module of Maestro.63–66
|
|||||||||||

|
16 | H | 0.44 ± 0.08 | 0.80 ± 0.02 | 609 | >30 | >30 | — | 268 ± 9 | −0.24 | 9.1 |
| 16a | CH3 | 1.22 ± 0.39 | 1.93 ± 0.45 | 451 | >30 | — | — | 550 ± 24 | −0.01 | 9.0 | |
| 16b | CH2Ph | 0.101 ± 0.003 | 0.192 ± 0.003 | 62 | 1.70 ± 0.06 | 3.01 ± 0.36 | 4 | 6.27 ± 0.60 | 1.71 | 9.5 | |
|
17 | H | 0.71 ± 0.12 | 1.27 ± 0.02 | 590 | >30 | >30 | — | 419 ± 55 | 0.21 | 9.3 |
| 17a | CH3 | 1.91 ± 0.23 | 4.69 ± 0.10 | 15 | 27.6 ± 5.3 | 42.1 ± 2.4 | 1 | 28.2 ± 1.0 | 0.43 | 9.0 | |
| 17b | CH2Ph | 0.050 ± 0.003 | 0.077 ± 0.021 | 63 | 0.96 ± 0.03 | 1.71 ± 0.07 | 3 | 3.17 ± 0.48 | 2.16 | 9.2 | |
|
18 | H | 0.40 ± 0.04 | 0.52 ± 0.05 | 313 | >30 | — | — | 125 ± 12 | 0.65 | 9.3 |
| 18a | CH3 | 0.23 ± 0.01 | 0.30 ± 0.01 | 71 | 10.6 ± 0.6 | 15.3 ± 0.2 | 2 | 16.4 ± 1.1 | 0.88 | 9.2 | |
| 18b | CH2Ph | 0.013 ± 0.002 | 0.018 ± 0.001 | 604 | 0.495 ± 0.058 | 0.94 ± 0.033 | 16 | 7.85 ± 0.33 | 2.60 | 9.4 | |

|
4 | H | >50 | — | — | >500 | — | — | >500 | 0.89 | n.a. |

|
II | — | 1.87 ± 0.08 | 2.53 ± 0.29 | — | — | — | — | — | — | — |
Compound 16 was synthesized and subsequently tested against HCV (replicon 1b) and bloodstream form T. brucei in vitro, exhibiting significant inhibitory activity against both targets. More specifically, the EC50 value for HCV was in the low micromolar range (EC5016 = 2.92 μM, Table 1) and for T. brucei was in the nanomolar range (EC5016 = 0.44 μM, Table 2), identifying this particular acetohydroxamic acid derivative as a promising and original lead compound. All the aforementioned designed analogues were also synthesized and tested against two different genotypes of HCV (1b and 4a) and against two Trypanosoma species. Analogues 16, 17 and 18 were also tested against HCV genotype 3a and DENV. The resulting EC50 and cytotoxicity values are presented in Tables 1 and 2.
Chemistry
As depicted in Scheme 1, the commercially available ketones 1–3 (Scheme 1A) were subjected to Bucherer–Bergs reaction with ammonium carbonate and potassium/sodium cyanide in EtOH/H2O. Treatment of the resulting hydantoins 4–6 with potassium bis(trimethylsilyl)amide in dry THF afforded the corresponding potassium imidate salts. The latter intermediates were converted to the respective benzyl esters 7–9 upon SN2 reaction with benzyl bromoacetate in dry DMF, as described in our previously published protocols.59,60 Catalytic hydrogenolysis (Pd/C 10%) of the aforementioned benzyl esters led to the carboxylic acids 10–12, which were further converted to the corresponding O-benzyl hydroxamates 13–15 upon coupling with O-benzylhydroxylamine·HCl in the presence of CDI or EDCI·HCl, HOBt. Finally, removal of the protective group was accomplished with hydrogenolysis, yielding almost quantitatively the target acetohydroxamic acids 16–18. | ||
| Scheme 1 A) Starting ketones B) synthesis of the target acetohydroxamic acid analogues 16–18, 16a–18a and 16b–18b. Reagents and conditions: (a) method A: NaCN (2.27 eq.), (NH4)2CO3 (6 eq.), EtOH/H2O (1.1:1), reflux, 8 h–11 days; | method B: KCN (1.3 eq.), (NH4)2CO3 (5 eq.), EtOH/H2O (1:3), MW (100 W), 120 °C, 25 min–1 h; (b) (i) [(CH3)3Si]2NK (1.02 eq.), dry THF (0–5 °C) and then r.t., 20 min–1 h, argon; (ii) BrCH2COOCH2Ph (1.05 eq.), dry DMF, 35–38 °C, 48 h, argon; (c) (i) NaH (1.2 eq.), dry DMF, (0–5 °C) and then r.t., 15 min, argon; (ii) PhCH2Br (1.2 eq.), 60 °C, 7 days, argon; (d) H2, Pd/C 10%, EtOH/AcOEt 3:1, 50–55 psi, 40–45 °C, 3 h; (e) method A: EDCI·HCl (1.2 eq.), HOBt (1.2 eq.), PhCH2ONH2·HCl (1.2 eq.), TEA (5.8 eq.), dry CH2Cl2/dry DMF, 30–35 °C, 40 h, argon; | method B: (i) CDI (1.2 eq.), dry THF, 28–30 °C, 1 h, argon; (ii) PhCH2ONH2·HCl (1.2 eq.), TEA (1.82 eq.), 28–30 °C, 24 h, argon; (f) H2, Pd/C 10%, EtOH/AcOEt 3:1, 50–55 psi, 40–45 °C, 3 h. C) Methyl esters obtained because of unexpected transesterification reaction (pink frames). | ||
For the synthesis of the N-alkylated analogues, the precursors benzyl esters 7–9 were reacted with NaH and the corresponding alkyl halide in dry DMF. Surprisingly, in the case of N-methylation procedure, transesterification products (methyl esters) were also isolated (Scheme 1C), along with the expected N-methylated benzyl esters.59 Employment of the previously described methodology, catalytic hydrogenolysis of the benzyl esters 7a–9a and 7b–9b, followed by amidation and deprotection of the amides 13a–15a and 13b–15b, afforded the desired N-alkylated acetohydroxamic acids 16a–18a and 16b–18b.
Biology
Screening of compounds using an HCV genotype 1b replicon system. The effects of analogues 16–18, 16a–18a, 16b–18b and 4 on HCV RNA replication and cell viability were determined in the stable cell line Huh5-2 containing an HCV genotype 1b (strain Con1) replicon encoding the firefly luciferase gene.61 In this cell system, the level of expressed luciferase directly correlates with the level of HCV RNA replication. Using serial dilutions of the compounds, the half maximal effective concentration (EC50) and the median cytotoxic concentration (CC50) were obtained, by measuring HCV replication-driven luciferase activity and intracellular ATP levels, respectively (Table 1, Fig. 4A). Dose–response curve analysis is presented for the most potent analogue 18 (Fig. 4B). Selectivity indices (SI) = CC50/EC50 were calculated for all analogues synthesized.
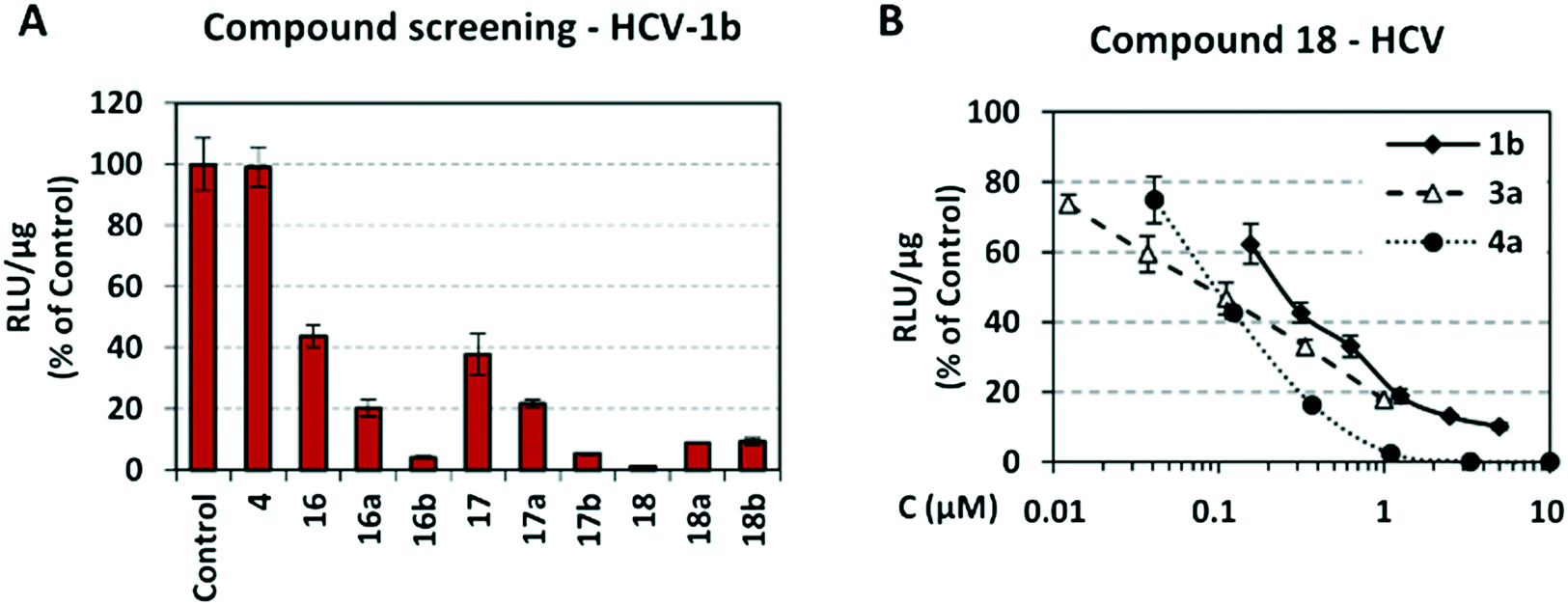 | ||
| Fig. 4 A) Screening of compounds against HCV genotype 1b replication. Huh5-2 cells, harboring HCV genotype 1b (Con1) subgenomic replicon, were seeded at 30% confluency and treated for 72 h with the compounds at 3 μM. B) Activity of compound 18, against HCV genotype 1b, 3a and 4a RNA replication, using dose–response curve analysis. Huh5-2, Huh7.5-3a and Huh7.5-4a cells, harboring subgenomic replicons of HCV genotype 1b (Con1), 3a (S52) or 4a (ED43), respectively, were seeded at 30% confluency and treated for 72 h with serial dilutions of 18. In both A and B, as a marker of viral RNA replication, firefly luciferase activity was determined and calculated as relative light units (RLU) per μg of total protein. Values obtained with compound-treated cells were expressed as percentage of those obtained with cells treated with the solvent DMSO (control). Bars represent mean values obtained from three separate experiments in triplicate. Error bars represent standard deviation (SD). | ||
As shown in Table 1, acetohydroxamic acid analogues were potent against HCV-1b replication, with EC50 values ranging from 0.22 to 2.92 μM. Compounds 16b, 17b and 18 exhibited the highest inhibitory activity, with EC50s in the submicromolar range (0.22 μM, 0.32 μM and 0.25 μM, respectively). We observed that the replacement of indane ring of 16 with the bulkier tetralin (17) and benzocycloheptane (18) rings progressively increased the activity of the compounds. It is noteworthy that the activity of 18 (EC50 = 0.25 μM) was 12-fold better than lead compound 16 (EC50 = 2.92 μM). A marked improvement was also observed on the EC50 value of 16 (43-fold lower), compared to the structurally related fused indole–flutimide analogue I (EC50 = 126.3 μM),56 validating the idea of incorporating a spiro connection between hydantoin and the indane ring. Introduction of a methyl substituent on the amide nitrogen atom of the 2,4-diketoimidazolidine scaffold of 16 led to a 2-fold increase in the activity (EC5016a = 1.41 μM). However, this methylation had almost no effect in the potency of 17 (EC5017 = 1.41 μM; EC5017a = 1.45 μM) and resulted in lower inhibitory potency for the N-methylated analogue 18a (EC5018a = 1.18 μM). The addition of the bulky lipophilic benzyl substituent to the same nitrogen atom of the hydantoin scaffold led to lower EC50 values for the resulting analogues 16b (EC50 = 0.22 μM) and 17b (EC50 = 0.32 μM), as compared to the addition of the methyl substituent. On the other hand, the benzyl group decreased the activity of compound 18b 4-fold with respect to the corresponding NH-analogue 18. In the case of the hydantoin 4, the lack of activity underlines the importance of the acetohydroxamic acid metal binding group for the antiviral potency of the compounds.
Concerning the cytotoxicity evaluation (Table 1), all the non-substituted analogues 16, 17 and 18 exhibited high CC50 values (≥200 μM), which resulted in remarkable selectivity indices. The most promising analogue appears to be compound 18, with SI value of 794. It is of great interest that the incorporation of an alkyl substituent onto the amide nitrogen atom progressively increased the cytotoxicity of the compounds, in agreement with the higher lipophilicity determined by calculated logD values (Table 2); the bulkier the lipophilic substituent, the lower the CC50 value.
Activity of the compounds against other HCV genotypes. As the compounds exhibited significant potency against HCV genotype 1b, they were selected for further testing against other HCV genotypes. Specifically, we determined viral replication-derived luciferase activity in Huh7.5-3a stable cell line for compounds 16, 17 and 18 and in Huh7.5-4a stable cell line for compounds 16–18, 16a–18a and 16b–18b, containing subgenomic replicons of HCV genotype 3a (isolate S52) and 4a (isolate ED43), respectively.
Similar to genotype 1b, the activity of 16, 17 and 18 against HCV genotype 3a ranged from low micromolar to nanomolar, and the increase in ring size conferred higher potency. Specifically, EC50 values were below 2.97 μM, with the most promising analogue being compound 18 (EC5018 = 0.08 μM; SI 18 = 2480), whose activity was 3-fold higher compared to the one exhibited in HCV-1b.
Biological evaluation of the hydroxamic acid derivatives on HCV-4a indicated enhanced activity as compared to genotypes 1b and 3a. All the novel synthesized analogues inhibited HCV-4a replication at submicromolar concentrations, ranging from 0.07 μM for 17b to 0.58 μM for the indane substituted analogue 16. Tetralin substitution increased the activity by 1.5 times (EC5017 = 0.38 μM) and benzocycloheptane by 6 times (EC5018 = 0.10 μM).
Moreover, N-methyl substitution on the hydantoin ring had a favorable effect on the antiviral activity, but only in the context of the indane- and tetralin-substituted analogues (EC5016a = 0.21 μM; EC5017a = 0.21 μM). The N-methylated analogue 18a exhibited potency similar (in the order of 0.1 μM) to the parent compound 18. The same trend was observed for the N-benzyl counterparts 16b and 17b, which were found to be 4 and 5.5 times more potent than their parent compounds 16 and 17, respectively, whereas compound 18b was equipotent (EC5018b = 0.12 μM) to the NH and N-methyl congeners (EC5018 = 0.10 μM and EC5018a = 0.11 μM, respectively). The increased activity of the compounds in HCV genotype 4a, as compared to the other genotypes, resulted in higher SI values, which ranged from >345 to 1984 for the NH analogues 16, 17 and 18. As for the N-methylated analogues, the most selective were 17a (SI 682) and 18a (SI 1015). Finally, out of all compounds, the most promising appeared to be 18, whose SI values were significantly high: 794, 2480 and 1984 for genotypes 1b, 3a and 4a, respectively.
Validation of compounds 17a and 18 activity with additional assays. The inhibition profile of the most potent and safe compounds from the tetralin and benzocycloheptane groups, as measured by luciferase assay, was confirmed by determining viral RNA and NS5A protein levels. As the compounds showed higher activity against HCV genotype 4, Huh7.5-4a replicon cells were used for validating activity. Specifically, cells were treated with serial dilutions of compounds 17a or 18, or the solvent DMSO (control) and viral RNA was quantified by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) whereas NS5A was evaluated by indirect immunofluorescence. We observed that compounds 17a and 18 caused a reduction in HCV RNA replication (Fig. 5A), with EC50s similar to those calculated on the basis of virus-derived luciferase activity (Table 1). Consistent results were obtained by indirect immunofluorescence analysis of HCV NS5A in Huh7.5-4a replicon cells treated with compound 18, which were best visible at high compound concentration (Fig. 5B). Nuclei were stained with propidium iodide (PI) as a cell viability control.
 | ||
| Fig. 5 Inhibitory effect of compounds on HCV RNA levels and protein expression in a subgenomic HCV Con1 replicon assay. A) Quantification of (+) strand HCV RNA by RT-qPCR in Huh7.5-4a replicon cells treated with serial dilutions of compounds 17a, 18 or the solvent DMSO. Values from compound-treated cells are expressed as percentage of those obtained with cells that received the solvent (control). mRNA levels of the housekeeping gene YWHAZ were used for normalization. B) Indirect immunofluorescence for NS5A (left panels) in Huh7.5-4a replicon cells, treated with serial dilutions of compound 18 or the solvent DMSO. Nuclei were stained with propidium iodide (PI; middle panels). Merged images are shown at the right. Bar, 50 μm. | ||
Activity of the compounds against DENV. Taking into account the broad activity of the compounds against different HCV genotypes, they were also tested against the closely related DENV. Both HCV and DENV belong to the same family (Flaviviridae) and express metalloenzymes, which are potential targets of our compounds. To examine the anti-DENV activity of the non-substituted analogues (16, 17 and 18), we determined viral replication-derived luciferase activity in Huh7-D2 stable cell line containing the subgenomic replicon of DENV serotype 2 (strain 16681). Interestingly, the order of activity obtained with 16, 17 and 18 was the same when they were tested against DENV. Compound 16 exhibited the lowest inhibitory activity (EC50 = 96.34 μM), which was progressively increased along with the bulkiness of the spiro-substituent, resulting in an EC50 value of 16.29 μM for analogue 18.
Activity of the compounds against T. brucei. Acetohydroxamic acid derivatives exhibited considerable trypanocidal activity against T. brucei, with EC50s ranging from 0.013 to 1.91 μM. Compounds 18b and 17b were the most potent against African trypanosomes with low nanomolar EC50 values (13 nM and 50 nM respectively, Table 2), while hydroxamates 16b, 17b and 18b were also significantly active against T. cruzi epimastigotes, with EC50 values in the low micromolar range.
Changing the indane ring in structure 16 with the tetralin ring decreased the activity, with the EC50 value of 17 being 1.5-fold higher (EC50 = 0.71 μM). Somewhat surprisingly, the replacement of indane by benzocycloheptane led to compound 18, which was almost equipotent to lead compound 16 (EC5016 = 0.44 μM; EC5018 = 0.40 μM). These molecules appeared to confer a beneficial effect on the trypanocidal potency, with respect to the structurally related cycloheptane spiro-substituted 2,6-DKP analogue II (EC50 = 1.87 μM),57 indicating that the fused bicyclic systems that were used as substituents at position 4 of the hydantoin core were a favorable structural modification.
Methyl substitution on the amide nitrogen atom of the respective 2,4-diketoimidazolidine residue of the parent compound 18 led to a 2-fold increase in potency for the benzocycloheptane substituted analogue 18a. On the other hand, incorporation of a methyl substituent in 16 and 17, lowered the activity observed in the N-methylated analogues 16a and 17a, with EC50 values 1.22 μM and 1.91 μM respectively. They were almost 3 times less potent than the corresponding NH-analogues (Table 2).
Addition of the bulky hydrophobic benzyl substituent to the same nitrogen atom of the hydantoin scaffold yielded analogues that were extremely potent against T. brucei, as exemplified by compounds 16b, 17b and 18b (Table 2). Their activities were respectively 4.5, 14 and 31 times higher than the parent compounds 16, 17 and 18. All the N-benzylated analogues retained high potency, with EC50 values ranging between 13 and 101 nM (Table 2). It is noteworthy that the incorporation of a benzyl group in the corresponding non-substituted precursors 16, 17 and 18 progressively increased activity against T. brucei in vitro. This enhanced potency of analogues 16b, 17b and 18b reflects the strongly favorable stereoelectronic and hydrophobic effects exerted by the benzyl substituent in the binding site.
The cytotoxicity of all target compounds against mammalian cells was determined using the rat skeletal myoblast L6 cell line. Notably, the non-substituted analogues 16, 17 and 18 displayed very low cytotoxicity against mammalian cells, having CC50s in the 125–419 μM range, thus resulting in remarkable selectivity indices, which varied from 313 (18) to 609 (16). In general, as in the case of HCV, it was observed that the incorporation of an alkyl substituent onto the amide nitrogen atom progressively increased the cytotoxicity of the compounds, with the exception of compound 16a (CC50 = 550 μM, SI 451). Finally, the acetohydroxamic acid derivative 18b, which had the highest activity against T. brucei (EC50 = 0.013 μM) displayed a very good selectivity (SI 604), despite the benzyl substitution.
Activity of the compounds against T. cruzi. Compounds were also tested against cultured T. cruzi epimastigotes, with EC50 values at submicromolar to micromolar levels (0.49–27.6 μM) (Table 2). Acetohydroxamic acid derivatives with a substituent on the amide nitrogen atom were able to inhibit parasite growth, an effect that was not observed with the non-substituted derivatives. More specifically, compounds 16, 17 and 18 exhibited no activity at the highest concentration tested (30 μM), whereas the N-methylated analogues showed medium inhibitory activity (EC5017a = 27.6 μM; EC5018a = 10.6 μM), with the only exception being analogue 16a. It is of great importance that the incorporation of a benzyl group resulted in unexpectedly potent analogues. The EC50s obtained ranged from 0.49 μM for the bulkier analogue 18b to 1.70 μM for the indane analogue 16b, indicating the positive effect of increasing the bulkiness and lipophilicity of the compounds, a pattern also observed in T. brucei sp. Interestingly, activity of the N-benzylated analogue 18b was 21.5-fold higher than the N-methylated counterpart 18a. For the respective tetralin analogue 17b, this increase in potency was 29 times higher. Finally, the absence of the acetohydroxamic acid moiety was detrimental for the activity against bloodstream form T. brucei and T. cruzi epimastigotes (Table 2), suggesting that the hydroxamic acid unit is essential also for the trypanocidal activity.
Concerning the physicochemical parameters of the prepared analogues, enhanced lipophilicity quantified by calculated logD values seems to be important to their bioactivity, and in most cases beneficial in terms of their capacity as drug leads. Conversely, pKa values of the various analogues do not demonstrate significant difference and thus they do not appear to contribute to the observed structure–activity relationships.
Biological evaluation indicates that metalloproteins may constitute the targets of the acetohydroxamic acid-based analogues described herein. However, binding and pulldown assays will be helpful to fully rationalize the observed bioactivity and determine the exact molecular targets of the novel analogues, thus facilitating structure-based design of metalloenzyme inhibitors.
Conclusions
In summary, despite the small overlapping degree between the starting frameworks I and II, we have successfully designed and synthesized compounds with dual activity, by merging two distinct scaffolds into a single chemical entity. The framework combination strategy involved the integration of structural elements from indole–flutimide analogues, exhibiting antiviral potency, with spiro carbocyclic 2,6-DKP acetohydroxamic acid-based trypanocidal agents, in order to achieve both activities into a single lead molecule. The novel bicyclic substituted hydantoin analogues not only exhibited individually improved antiviral and antiparasitic activity compared to analogues I and II, but also demonstrated exceptional inhibitory activity against different HCV genotypes (1b, 3a, 4a) and DENV, as well the two Trypanosoma species (T. brucei, T. cruzi). Structure–activity relationships deduced from the biological results suggest that the increased size of the spiro-substituent generally favors inhibitory activity towards the targets of interest. Moreover, activity is enhanced upon increasing the lipophilicity of the compounds, particularly via introduction of a methyl- or benzyl-substituent at the amide nitrogen atom of the hydantoin ring. In fact, the results suggest that increasing the bulkiness of the substituent improves rather than retards activity. Along with the lipophilicity, the acetohydroxamic acid moiety, capable of chelating active site metal ion(s), is an indispensable component of this class of inhibitors, since removal of this functional group from the imidic nitrogen atom caused a dramatic loss of activity. Finally, the remarkable selectivity indices observed for most of the tested analogues indicate that the novel scaffold of an appropriately substituted hydantoin ring and a metal binding moiety may be the key to unlock multiple pathogen inhibition, as a successful strategy for producing multifunctional drugs.Experimental
Chemistry
General experimental procedures
:1 (40 mL) was hydrogenated for 3 h at 40–45 °C and 50–55 psi pressure, in the presence of Pd/C (10 wt%) as a catalyst. The catalyst was filtered off, washed with portions of hot MeOH (3 × 15 mL) and the combined filtrates were evaporated under reduced pressure to yield the corresponding carboxylic acids 10–12, 10a–12a and 10b–12b.
Method A. To a solution of the appropriate carboxylic acid (10, 12, 12b) (2.0 mmol, 1.0 eq.) in 20 mL dry CH2Cl2/dry DMF (4
:1), EDCI·HCl (2.4 mmol, 1.2 eq.), HOBt (2.4 mmol, 1.2 eq., 80% monohydrate), O-benzylhydroxylamine hydrochloride (2.4 mmol, 1.2 eq.) and TEA (11.6 mmol, 5.8 eq.) were added sequentially. The reaction mixture was stirred for 40 h, at 30–35 °C under argon. After removal of CH2Cl2 under reduced pressure, ice-water was added (40 mL) and the mixture was extracted with AcOEt (4 × 40 mL). The combined organic layers were washed with H2O (3 × 70 mL) and brine (2 × 70 mL), dried over anh. Na2SO4 and evaporated in vacuo. The crude residue was purified by column chromatography on silica gel using a mixture of CH2Cl2/AcOEt and AcOEt as eluents to afford the desired amides (13, 15, 15b respectively).
Method B. The appropriate carboxylic acid (11, 10a–12a, 10b, 11b) (2.0 mmol, 1.0 eq.) was treated with CDI (2.4 mmol, 1.2 eq.) in dry THF (26 mL) and the mixture was stirred for 1 h, at 28–30 °C under argon. Then, O-benzylhydroxylamine hydrochloride (2.4 mmol, 1.2 eq.) and TEA (3.64 mmol, 1.82 eq.) were added successively, and the stirring was continued for 24 h, at 28–30 °C under Ar. After removal of the solvent in vacuo, the reaction mixture was quenched with ice-water (40 mL) and extracted with AcOEt (4 × 40 mL). The combined organic phases were washed with H2O (3 × 70 mL) and brine (2 × 70 mL), dried with anh. Na2SO4 and evaporated under reduced pressure. The crude residue was purified by column chromatography on silica gel using a mixture of CH2Cl2/AcOEt and AcOEt as eluents to afford the desired amides (14, 13a–15a, 13b, 14b respectively).
:1 (40 mL). After 3 h of shaking under an atmosphere of 50–55 psi hydrogen, at 40–45 °C, the catalyst was removed by filtration and washed with hot MeOH (3 × 15 mL). The combined filtrates were concentrated to dryness under reduced pressure to afford the title acetohydroxamic acid analogues (16–18, 16a–18a and 16b–18b respectively).
:1 (24 mL), following the previously described general procedure for the preparation of carboxylic acids. Evaporation of the solvents under reduced pressure afforded the title compound 10 as a white crystalline solid (310 mg, almost quantitative yield); mp 237–239 °C (from AcOEt/n-pentane), Rf = 0.06 (AcOEt). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.22 (dt, 1H, J1 = 13.3 Hz, J2 = 7.8 Hz, H2′), 2.55 (dt, 1H, J1 = 13.2 Hz, J2 = 6.5 Hz, H2′), 3.05 (t, 2H, J = 7.2 Hz, H3′), 4.07–4.15 (q, AB, 2H, JAB = 17.5 Hz, NCH2COOH), 7.19 (d, 1H, J = 7.5 Hz, H7′), 7.26 (tdd, 1H, J1 = 7.0 Hz, J2 = 1.9 Hz, J3 = 1.0 Hz, H6′), 7.33 (td, 1H, J1 = 7.5 Hz, J2 = 1.2 Hz, H5′), 7.34–7.36 (m, 1H, H4′), 8.84 (s, 1H, H3), 13.12 (br s, 1H, NCH2COOH); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 29.7 (C3′), 36.0 (C2′), 39.1 (NCH2COOH), 70.8 (C1′/C4), 123.1 (C7′), 125.0 (C4′), 127.0 (C6′), 129.0 (C5′), 141.0 (C7a′), 143.9 (C3a′), 155.2 (C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 168.7 (NCH2COOH), 175.2 (C5O). Found: C, 60.08; H, 4.70; N, 10.79. Calc. for C13H12N2O4: C, 60.00; H, 4.65; N, 10.76%.
:1) following the general procedure for the preparation of O-benzyl hydroxamates (Method A). After removal of CH2Cl2 under reduced pressure, the reaction mixture was poured into 20 mL ice-water and extracted with AcOEt (4 × 20 mL). The combined organic layers were washed with H2O (3 × 35 mL) and brine (2 × 35 mL), dried over Na2SO4 and concentrated to dryness in vacuo. The oily pale-yellow residue was purified by column chromatography on silica gel eluting first with CH2Cl2/AcOEt 8:1, 5:1 and then AcOEt to give the corresponding O-benzyl hydroxamate 13 as a white foamy solid, which strongly binds the eluting solvents. Removal of the entrapped solvents upon drying under high vacuum and treatment with n-pentane at 0 °C gave 13 as a white crystalline solid (260 mg, 71%); mp 154–156 °C (AcOEt/n-pentane), Rf = 0.06 (from CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.22 (dt, 1H, J1 = 13.5 Hz, J2 = 7.7 Hz, H2′), 2.57 (dt, 1H, J1 = 13.1 Hz, J2 = 6.6 Hz, H2′), 3.05 (t, 2H, J = 7.1 Hz, H3′), 3.92–4.01 (q, AB, 1.55H, JAB = 16.3 Hz, NCH2CO), 4.25 (br s, 0.3H, NCH2CO), 4.81, 4.86 (s + s, 2H, OCH2Ph), 7.25–7.30 (m, 2H, H6′, H7′), 7.31–7.35 (m, 2H, H4′, H5′), 7.35–7.47 (m, 5H, H2′′, H3′′, H4′′, H5′′, H6′′), 8.81 (s, 1H, H3), 11.02 (s, 0.1H, CONHOCH2Ph), 11.39 (s, 0.7H, CONHOCH2Ph); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 29.7 (C3′), 36.0 (C2′), 38.0 (NCH2CO), 70.8 (C1′/C4), 77.0 (OCH2Ph), 123.3 (C7′), 124.9 (C4′), 127.0 (C6′), 128.3 (C3′′, C4′′, C5′′), 128.8 (C2′′, C6′′), 129.0 (C5′), 135.8 (C1′′), 141.1 (C7a′), 143.9 (C3a′), 155.3 (C2O), 163.6 (CONHOCH2Ph), 175.4 (C5O). Found: C, 65.69; H, 5.30; N, 11.53. Calc. for C20H19N3O4: C, 65.74; H, 5.24; N, 11.50%.
:1, 22 mL) was hydrogenated following the general procedure described above for the preparation of acetohydroxamic acids. Evaporation of the solvents in vacuo yielded the title compound 16 as a white foamy solid, which strongly binds the aforementioned solvents. Removal of the entrapped solvents upon drying under high vacuum gave 16 as a glass solid, which was further crystallized upon treatment with n-pentane-dry Et2O 5:1 under ice-cooling (150 mg, almost quantitative yield); mp 179–182 °C (from AcOEt/n-pentane-dry Et2O), Rf = 0.29 (AcOEt). This compound appeared in the 1H and 13C NMR spectra as a mixture of E/Z conformers. 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.22 (dt, 1H, J1 = 13.4 Hz, J2 = 7.5 Hz, H2′), 2.56 (ddd, 1H, J1 = 13.4 Hz, J2 = 7.5 Hz, J3 = 6.0 Hz, H2′), 3.04 (ddd, 2H, J1 = 8.2 Hz, J2 = 6.5 Hz, J3 = 2.4 Hz, H3′), 3.91–4.00 (q, AB, 1.5H, JAB = 16.5 Hz, NCH2CO, E-isomer), 4.20–4.29 (q, AB, 0.45H, JAB = 17.5 Hz, NCH2CO, Z-isomer), 7.23–7.35 (complex m, 4H, H4′, H5′, H6′, H7′), 8.73 (s, 0.1H, H3), 8.76, 8.77 (s + s, 0.9H, H3), 8.95 (s, 0.6H, CH2CONHOH, E-isomer), 9.33 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.29 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.72 (s, 0.6H, CH2CONHOH, E-isomer); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 29.8 (C3′), 36.0, 36.1 (C2′), 38.0 (NCH2CO, E-isomer), 38.5 (NCH2CO, Z-isomer), 70.8 (C1′/C4), 123.4 (C7′), 124.9 (C4′), 127.0 (C6′), 129.0 (C5′), 141.3 (C7a′), 143.9 (C3a′), 155.4 (C2O, E-isomer), 155.6 (C2O, Z-isomer), 163.3 (NCH2CO, E-isomer), 168.7 (NCH2CO, Z-isomer), 175.5 (C5O, E-isomer), 175.7 (C5O, Z-isomer). Found: C, 56.78; H, 4.81; N, 15.30. Calc. for C13H13N3O4: C, 56.72; H, 4.75; N, 15.27%.
:1), following the general procedure for the preparation of carboxylic acids. After removal of the solvent under reduced pressure, the title compound 10a was obtained as a white crystalline solid (240 mg, almost quantitative yield); mp 203–205 °C (melted gradually from 198 °C) (AcOEt/n-pentane), Rf = 0.24 (AcOEt). 1H NMR (400.13 MHz, MeOD-d4) δ (ppm) 2.40 (ddd, 1H, J1 = 13.9 Hz, J2 = 8.4 Hz, J3 = 6.8 Hz, H2′), 2.56 (ddd, 1H, J1 = 14.0 Hz, J2 = 8.3 Hz, J3 = 5.7 Hz, H2′), 2.69 (s, 3H, NCH3), 3.07–3.22 (m, 2H, H3′), 4.21–4.34 (q, AB, 2H, JAB = 17.6 Hz, NCH2COOH), 5.13 (br s, 1H, NCH2COOH), 7.20 (d, 1H, J = 7.3 Hz, H7′), 7.22–7.29 (m, 1H, H6′), 7.31–7.38 (m, 2H, H4′, H5′); 13C NMR (50.32 MHz, MeOD-d4) δ (ppm) 25.4 (NCH3), 31.4 (C3′), 33.5 (C2′), 40.4 (NCH2COOH), 76.5 (C1′/C4), 124.5 (C7′), 126.4 (C4′), 128.4 (C6′), 130.9 (C5′), 139.0 (C7a′), 146.4 (C3a′), 156.6 (C2O), 170.3 (NCH2COOH), 176.6 (C5O). Found: C, 61.29; H, 5.21; N, 10.28. Calc. for C14H14N2O4: C, 61.31; H, 5.15; N, 10.21%.
:1, 10:1 and AcOEt as eluents to afford the corresponding O-benzyl hydroxamate 13a as a colorless oil, which was further crystallized to a white crystalline solid by treatment with n-pentane under ice-cooling (210 mg, 76%); mp 108–110 °C (from AcOEt/n-pentane-dry Et2O), Rf = 0.21 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.41 (ddd, 1H, J1 = 14.4 Hz, J2 = 8.5 Hz, J3 = 6.7 Hz, H2′), 2.48 (ddd, 1H, J1 = 14.3 Hz, J2 = 8.3 Hz, J3 = 6.0 Hz, H2′), 2.62 (s, 3H, NCH3), 3.05–3.16 (m, 2H, H3′), 3.96–4.06 (q, AB, 1.6H, JAB = 16.2 Hz, NCH2CO), 4.27 (br s, 0.3H, NCH2CO), 4.81, 4.86 (s + s, 2H, OCH2Ph), 7.22 (d, 1H, J = 7.6 Hz, H7′), 7.29 (t, 1H, J = 7.2 Hz, H6′), 7.33–7.47 (m, 7H, H4′, H5′, H2′′, H3′′, H4′′, H5′′, H6′′), 11.06 (s, 0.1H, CONHOCH2Ph), 11.40 (s, 0.7H, CONHOCH2Ph); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 24.7 (NCH3), 30.0 (C3′), 32.1 (C2′), 38.4 (NCH2CO), 74.3 (C1′/C4), 77.0 (OCH2Ph), 123.4 (C7′), 125.2 (C4′), 127.2 (C6′), 128.3 (C3′′, C4′′, C5′′), 128.8 (C2′′, C6′′), 129.5 (C5′), 135.7 (C1′′), 138.0 (C7a′), 144.7 (C3a′), 155.3 (C2O), 163.6 (CONHOCH2Ph), 175.4 (C5O). Found: C, 66.54; H, 5.61; N, 11.00. Calc. for C21H21N3O4: C, 66.48; H, 5.58; N, 11.08%.
:1 (17 mL) was subjected to catalytic hydrogenolysis in exactly the same procedure described above for the preparation of the final compounds. Removal of the solvents under reduced pressure provided the acetohydroxamic acid 16a as a white foamy solid. Crystallization of this product upon treatment with n-pentane-dry Et2O under ice-cooling gave a white crystalline solid (120 mg, almost quantitative yield); mp melted gradually from 158 °C to 162 °C (AcOEt/n-pentane-dry Et2O), Rf = 0.22 (from AcOEt). This compound exhibited distinct peaks attributed to each of the two E/Z conformers in the 1H and 13C NMR spectra. 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.40 (ddd, 1H, J1 = 14.1 Hz, J2 = 8.5 Hz, J3 = 4.2 Hz, H2′), 2.47 (ddd, 1H, J1 = 14.1 Hz, J2 = 8.2 Hz, J3 = 4.0 Hz, H2′), 2.61 (s, 3H, NCH3), 3.04–3.16 (m, 2H, H3′), 3.95–4.04 (q, AB, 1.5H, JAB = 16.1 Hz, NCH2CO, E-isomer), 4.24–4.33 (q, AB, 0.5H, JAB = 17.3 Hz, NCH2CO, Z-isomer), 7.23 (d, 1H, J = 7.8 Hz, H7′), 7.27 (td, 1H, J1 = 7.3 Hz, J2 = 2.0 Hz, H6′), 7.34–7.40 (m, 2H, H4′, H5′), 8.98 (s, 0.7H, CH2CONHOH, E-isomer), 9.36 (s, 0.23H, NCH2CONHOH, Z-isomer), 10.34 (s, 0.23H, NCH2CONHOH, Z-isomer), 10.75 (s, 0.7H, CH2CONHOH, E-isomer); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 24.8 (NCH3), 30.1 (C3′), 32.1 (C2′), 38.5 (NCH2CO, E-isomer), 39.0 (NCH2CO, Z-isomer), 74.3 (C1′/C4), 123.5, 123.6 (C7′), 125.2 (C4′), 127.2 (C6′), 129.5 (C5′), 138.2 (C7a′), 144.7 (C3a′), 154.6 (C2O, E-isomer), 154.7 (C2O, Z-isomer), 163.2 (NCH2CO, E-isomer), 168.6 (NCH2CO, Z-isomer), 174.6 (C5O, E-isomer), 174.8 (C5O, Z-isomer). Found: C, 58.23; H, 5.26; N, 14.50. Calc. for C14H15N3O4: C, 58.13; H, 5.23; N, 14.53%.
:1). 1H NMR (600.11 MHz, CDCl3) δ (ppm) 2.05 (ddd, 1H, J1 = 14.2 Hz, J2 = 8.9 Hz, J3 = 5.5 Hz, H2′), 2.52 (ddd, 1H, J1 = 13.8 Hz, J2 = 9.1 Hz, J3 = 5.6 Hz, H2′), 2.84 (ddd, 1H, J1 = 16.0 Hz, J2 = 8.9 Hz, J3 = 5.6 Hz, H3′), 3.07 (ddd, 1H, J1 = 15.9 Hz, J2 = 9.1 Hz, J3 = 5.5 Hz, H3′), 3.81 (d, 1H, J = 15.9 Hz, NCHHPh), 4.41–4.49 (q, AB, 2H, JAB = 17.3 Hz, NCH2COO), 4.82 (d, 1H, J = 15.9 Hz, NCHHPh), 5.20–5.26 (q, AB, 2H, JAB = 12.1 Hz, OCH2Ph), 7.06 (d, 1H, J = 7.7 Hz, H7′), 7.10–7.13 (m, 2H, H2Bz, H6Bz), 7.14 (td, 1H, J1 = 7.4 Hz, J2 = 1.3 Hz, H6′), 7.21–7.27 (complex m, 3H, H3Bz, H4Bz, H5Bz), 7.28 (dt, 1H, J1 = 7.5 Hz, J2 = 1.1 Hz, H4′), 7.31 (td, 1H, J1 = 7.4 Hz, J2 = 1.1 Hz, H5′), 7.34–7.41 (m, 5H, H2′′, H3′′, H4′′, H5′′, H6′′); 13C NMR (150.9 MHz, CDCl3) δ (ppm) 30.7 (C3′), 33.9 (C2′), 40.1 (NCH2COO), 44.1 (NCH2Ph), 67.9 (COOCH2Ph), 76.0 (C1′/C4), 124.0 (C7′), 125.4 (C4′), 127.7 (C2Bz, C6Bz), 127.8 (C6′), 128.6 (C3Bz, C5Bz), 128.7 (C2′′, C6′′, C4Bz), 128.76 (C4′′), 128.84 (C3′′, C5′′), 130.0 (C5′), 135.0 (C1′′), 137.5 (C1Bz), 137.9 (C7a′), 145.1 (C3a′), 155.7 (C2O), 167.3 (COOCH2Ph), 175.0 (C5O). Found: C, 73.66; H, 5.55; N, 6.30. Calc. for C27H24N2O4: C, 73.62; H, 5.49; N, 6.36%.
:1) following the procedure described for the preparation of the above-mentioned carboxylic acids. The desired compound 10b was afforded as a glass solid, which was further crystallized upon treatment with n-pentane-dry Et2O (5:1) under ice-cooling to give a white crystalline solid (316 mg, almost quantitative yield); mp 144–147 °C (from AcOEt/n-pentane), Rf = 0.22 (AcOEt). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.14 (ddd, 1H, J1 = 14.1 Hz, J2 = 8.7 Hz, J3 = 5.6 Hz, H2′), 2.44 (ddd, 1H, J1 = 14.5 Hz, J2 = 8.9 Hz, J3 = 5.9 Hz, H2′), 2.93 (ddd, 1H, J1 = 15.8 Hz, J2 = 8.7 Hz, J3 = 5.9 Hz, H3′), 3.00 (ddd, 1H, J1 = 15.0 Hz, J2 = 8.9 Hz, J3 = 5.6 Hz, H3′), 3.96 (d, 1H, J = 16.3 Hz, NCHHPh), 4.19–4.27 (q, AB, 2H, JAB = 17.4 Hz, NCH2COOH), 4.58 (d, 1H, J = 16.3 Hz, NCHHPh), 7.07–7.14 (m, 3H, H7′, H2Bz, H6Bz), 7.15–7.26 (complex m, 4H, H6′, H3Bz, H4Bz, H5Bz), 7.30–7.35 (m, 2H, H4′, H5′), 13.33 (br s, 1H, NCH2COOH); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 30.0 (C3′), 33.0 (C2′), 40.1 (NCH2COOH), 43.2 (NCH2Ph), 74.9 (C1′/C4), 123.6 (C7′), 125.2 (C4′), 127.1 (C6′, C2Bz, C4Bz, C6Bz), 128.2 (C3Bz, C5Bz), 129.5 (C5′), 137.6 (C1Bz), 138.3 (C7a′), 144.7 (C3a′), 155.1 (C2O), 168.7 (NCH2COOH), 174.3 (C5O). Found: C, 68.60; H, 5.14; N, 8.13. Calc. for C20H18N2O4: C, 68.56; H, 5.18; N, 8.00%.
:1, 10:1 and AcOEt as eluents to give the corresponding O-benzyl hydroxamate 13b as a glass solid. Crystallization of this material upon treatment with a n-pentane-dry Et2O mixture (5:1) gave a white crystalline solid (310 mg, 82%); mp melted gradually from 60 °C to 78 °C (from AcOEt/n-pentane-dry Et2O), Rf = 0.41 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.13 (ddd, 1H, J1 = 14.0 Hz, J2 = 8.6 Hz, J3 = 5.3 Hz, H2′), 2.45 (ddd, 1H, J1 = 14.5 Hz, J2 = 8.9 Hz, J3 = 6.0 Hz, H2′), 2.93 (ddd, 1H, J1 = 15.8 Hz, J2 = 8.6 Hz, J3 = 6.0 Hz, H3′), 2.99 (ddd, 1H, J1 = 14.9 Hz, J2 = 8.8 Hz, J3 = 5.4 Hz, H3′), 3.94 (d, 1H, J = 16.3 Hz, NCHHPh), 4.04–4.13 (q, AB, 1.65H, JAB = 16.2 Hz, NCH2CO), 4.35 (br s, 0.35H, NCH2CO), 4.58 (d, 1H, J = 16.2 Hz, NCHHPh), 4.82, 4.89 (s + s, 2H, OCH2Ph), 7.13 (d, 1H, J = 7.2 Hz, H2Bz, H6Bz), 7.16–7.26 (m, 5H, H6′, H7′, H3Bz, H4Bz, H5Bz), 7.30–7.35 (m, 2H, H4′, H5′), 7.35–7.49 (m, 5H, H2′′, H3′′, H4′′, H5′′, H6′′), 11.09 (s, 0.15H, CONHOCH2Ph), 11.46 (s, 0.75H, CONHOCH2Ph); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 30.0 (C3′), 33.0 (C2′), 38.6 (NCH2CO), 43.1 (NCH2Ph), 74.9 (C1′/C4), 77.0 (OCH2Ph), 123.8 (C7′), 125.1 (C4′), 127.1 (C6′, C2Bz, C4Bz, C6Bz), 128.2 (C3Bz, C5Bz), 128.3 (C2′′, C4′′, C6′′), 128.8 (C3′′, C5′′), 129.5 (C5′), 135.7 (C1′′), 137.6 (C1Bz), 138.4 (C7a′), 144.7 (C3a′), 155.2 (C2O), 163.5 (CONHOCH2Ph), 174.5 (C5O). Found: C, 71.25; H, 5.60; N, 9.32. Calc. for C27H25N3O4: C, 71.19; H, 5.53; N, 9.22%.
:1 (22 mL) according to the previously described procedure for the synthesis of hydroxamate analogues. Concentration to dryness of the solvents under reduced pressure yielded the title compound 16b as a white crystalline solid (200 mg, almost quantitative yield); mp melted gradually from 174 °C to 181 °C (from AcOEt/n-pentane-dry Et2O), Rf = 0.49 (AcOEt). In the 1H and 13C NMR spectra of this compound, double set of characteristic peaks are distinguished for each of the two E/Z conformers. 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.13 (ddd, 1H, J1 = 13.9 Hz, J2 = 8.6 Hz, J3 = 5.4 Hz, H2′), 2.44 (ddd, 1H, J1 = 13.9 Hz, J2 = 8.9 Hz, J3 = 6.1 Hz, H2′), 2.92 (ddd, 1H, J1 = 16.0 Hz, J2 = 8.5 Hz, J3 = 6.3 Hz, H3′), 2.98 (ddd, 1H, J1 = 14.4 Hz, J2 = 8.9 Hz, J3 = 5.4 Hz, H3′), 3.93 (d, 1H, J = 16.3 Hz, NCHHPh), 4.04–4.12 (q, AB, 1.5H, JAB = 16.0 Hz, NCH2CO, E-isomer), 4.32–4.40 (q, AB, 0.5H, JAB = 17.4 Hz, NCH2CO, Z-isomer), 4.57 (dd, 1H, J1 = 16.3 Hz, J2 = 8.8 Hz, NCHHPh), 7.12 (d, 1H, J = 6.7 Hz, H2Bz, H6Bz), 7.15–7.25 (complex m, 5H, H6′, H7′, H3Bz, H4Bz, H5Bz), 7.29–7.34 (m, 2H, H4′, H5′), 9.03 (s, 0.68H, CH2CONHOH, E-isomer), 9.41 (s, 0.22H, NCH2CONHOH, Z-isomer), 10.37 (s, 0.22H, NCH2CONHOH, Z-isomer), 10.80 (s, 0.68H, CH2CONHOH, E-isomer); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 30.0 (C3′), 32.9, 33.0 (C2′), 38.6 (NCH2CO, E-isomer), 39.1 (NCH2CO, Z-isomer), 43.1 (NCH2Ph), 74.9 (C1′/C4), 123.9 (C7′), 125.0 (C4′), 127.1 (C6′, C2Bz, C4Bz, C6Bz), 128.2 (C3Bz, C5Bz), 129.5 (C5′), 137.6 (C1Bz), 138.5 (C7a′), 144.7 (C3a′), 155.3 (C2O, E-isomer), 155.5 (C2O, Z-isomer), 163.2 (NCH2CO, E-isomer), 168.6 (NCH2CO, Z-isomer), 174.6 (C5O, E-isomer), 174.8 (C5O, Z-isomer). Found: C, 65.79; H, 5.18; N, 11.55. Calc. for C20H19N3O4: C, 65.74; H, 5.24; N, 11.50%.
O), 168.7 (NCH2COOH), 175.2 (C5O). Found: C, 60.08; H, 4.70; N, 10.79. Calc. for C13H12N2O4: C, 60.00; H, 4.65; N, 10.76%.
:1) following the general procedure for the preparation of O-benzyl hydroxamates (Method A). After removal of CH2Cl2 under reduced pressure, the reaction mixture was poured into 20 mL ice-water and extracted with AcOEt (4 × 20 mL). The combined organic layers were washed with H2O (3 × 35 mL) and brine (2 × 35 mL), dried over Na2SO4 and concentrated to dryness in vacuo. The oily pale-yellow residue was purified by column chromatography on silica gel eluting first with CH2Cl2/AcOEt 8:1, 5:1 and then AcOEt to give the corresponding O-benzyl hydroxamate 13 as a white foamy solid, which strongly binds the eluting solvents. Removal of the entrapped solvents upon drying under high vacuum and treatment with n-pentane at 0 °C gave 13 as a white crystalline solid (260 mg, 71%); mp 154–156 °C (AcOEt/n-pentane), Rf = 0.06 (from CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.22 (dt, 1H, J1 = 13.5 Hz, J2 = 7.7 Hz, H2′), 2.57 (dt, 1H, J1 = 13.1 Hz, J2 = 6.6 Hz, H2′), 3.05 (t, 2H, J = 7.1 Hz, H3′), 3.92–4.01 (q, AB, 1.55H, JAB = 16.3 Hz, NCH2CO), 4.25 (br s, 0.3H, NCH2CO), 4.81, 4.86 (s + s, 2H, OCH2Ph), 7.25–7.30 (m, 2H, H6′, H7′), 7.31–7.35 (m, 2H, H4′, H5′), 7.35–7.47 (m, 5H, H2′′, H3′′, H4′′, H5′′, H6′′), 8.81 (s, 1H, H3), 11.02 (s, 0.1H, CONHOCH2Ph), 11.39 (s, 0.7H, CONHOCH2Ph); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 29.7 (C3′), 36.0 (C2′), 38.0 (NCH2CO), 70.8 (C1′/C4), 77.0 (OCH2Ph), 123.3 (C7′), 124.9 (C4′), 127.0 (C6′), 128.3 (C3′′, C4′′, C5′′), 128.8 (C2′′, C6′′), 129.0 (C5′), 135.8 (C1′′), 141.1 (C7a′), 143.9 (C3a′), 155.3 (C2O), 163.6 (CONHOCH2Ph), 175.4 (C5O). Found: C, 65.69; H, 5.30; N, 11.53. Calc. for C20H19N3O4: C, 65.74; H, 5.24; N, 11.50%.
:1, 22 mL) was hydrogenated following the general procedure described above for the preparation of acetohydroxamic acids. Evaporation of the solvents in vacuo yielded the title compound 16 as a white foamy solid, which strongly binds the aforementioned solvents. Removal of the entrapped solvents upon drying under high vacuum gave 16 as a glass solid, which was further crystallized upon treatment with n-pentane-dry Et2O 5:1 under ice-cooling (150 mg, almost quantitative yield); mp 179–182 °C (from AcOEt/n-pentane-dry Et2O), Rf = 0.29 (AcOEt). This compound appeared in the 1H and 13C NMR spectra as a mixture of E/Z conformers. 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.22 (dt, 1H, J1 = 13.4 Hz, J2 = 7.5 Hz, H2′), 2.56 (ddd, 1H, J1 = 13.4 Hz, J2 = 7.5 Hz, J3 = 6.0 Hz, H2′), 3.04 (ddd, 2H, J1 = 8.2 Hz, J2 = 6.5 Hz, J3 = 2.4 Hz, H3′), 3.91–4.00 (q, AB, 1.5H, JAB = 16.5 Hz, NCH2CO, E-isomer), 4.20–4.29 (q, AB, 0.45H, JAB = 17.5 Hz, NCH2CO, Z-isomer), 7.23–7.35 (complex m, 4H, H4′, H5′, H6′, H7′), 8.73 (s, 0.1H, H3), 8.76, 8.77 (s + s, 0.9H, H3), 8.95 (s, 0.6H, CH2CONHOH, E-isomer), 9.33 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.29 (s, 0.2H, NCH2CONHOH, Z-isomer), 10.72 (s, 0.6H, CH2CONHOH, E-isomer); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 29.8 (C3′), 36.0, 36.1 (C2′), 38.0 (NCH2CO, E-isomer), 38.5 (NCH2CO, Z-isomer), 70.8 (C1′/C4), 123.4 (C7′), 124.9 (C4′), 127.0 (C6′), 129.0 (C5′), 141.3 (C7a′), 143.9 (C3a′), 155.4 (C2O, E-isomer), 155.6 (C2O, Z-isomer), 163.3 (NCH2CO, E-isomer), 168.7 (NCH2CO, Z-isomer), 175.5 (C5O, E-isomer), 175.7 (C5O, Z-isomer). Found: C, 56.78; H, 4.81; N, 15.30. Calc. for C13H13N3O4: C, 56.72; H, 4.75; N, 15.27%.
:1), following the general procedure for the preparation of carboxylic acids. After removal of the solvent under reduced pressure, the title compound 10a was obtained as a white crystalline solid (240 mg, almost quantitative yield); mp 203–205 °C (melted gradually from 198 °C) (AcOEt/n-pentane), Rf = 0.24 (AcOEt). 1H NMR (400.13 MHz, MeOD-d4) δ (ppm) 2.40 (ddd, 1H, J1 = 13.9 Hz, J2 = 8.4 Hz, J3 = 6.8 Hz, H2′), 2.56 (ddd, 1H, J1 = 14.0 Hz, J2 = 8.3 Hz, J3 = 5.7 Hz, H2′), 2.69 (s, 3H, NCH3), 3.07–3.22 (m, 2H, H3′), 4.21–4.34 (q, AB, 2H, JAB = 17.6 Hz, NCH2COOH), 5.13 (br s, 1H, NCH2COOH), 7.20 (d, 1H, J = 7.3 Hz, H7′), 7.22–7.29 (m, 1H, H6′), 7.31–7.38 (m, 2H, H4′, H5′); 13C NMR (50.32 MHz, MeOD-d4) δ (ppm) 25.4 (NCH3), 31.4 (C3′), 33.5 (C2′), 40.4 (NCH2COOH), 76.5 (C1′/C4), 124.5 (C7′), 126.4 (C4′), 128.4 (C6′), 130.9 (C5′), 139.0 (C7a′), 146.4 (C3a′), 156.6 (C2O), 170.3 (NCH2COOH), 176.6 (C5O). Found: C, 61.29; H, 5.21; N, 10.28. Calc. for C14H14N2O4: C, 61.31; H, 5.15; N, 10.21%.
:1, 10:1 and AcOEt as eluents to afford the corresponding O-benzyl hydroxamate 13a as a colorless oil, which was further crystallized to a white crystalline solid by treatment with n-pentane under ice-cooling (210 mg, 76%); mp 108–110 °C (from AcOEt/n-pentane-dry Et2O), Rf = 0.21 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.41 (ddd, 1H, J1 = 14.4 Hz, J2 = 8.5 Hz, J3 = 6.7 Hz, H2′), 2.48 (ddd, 1H, J1 = 14.3 Hz, J2 = 8.3 Hz, J3 = 6.0 Hz, H2′), 2.62 (s, 3H, NCH3), 3.05–3.16 (m, 2H, H3′), 3.96–4.06 (q, AB, 1.6H, JAB = 16.2 Hz, NCH2CO), 4.27 (br s, 0.3H, NCH2CO), 4.81, 4.86 (s + s, 2H, OCH2Ph), 7.22 (d, 1H, J = 7.6 Hz, H7′), 7.29 (t, 1H, J = 7.2 Hz, H6′), 7.33–7.47 (m, 7H, H4′, H5′, H2′′, H3′′, H4′′, H5′′, H6′′), 11.06 (s, 0.1H, CONHOCH2Ph), 11.40 (s, 0.7H, CONHOCH2Ph); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 24.7 (NCH3), 30.0 (C3′), 32.1 (C2′), 38.4 (NCH2CO), 74.3 (C1′/C4), 77.0 (OCH2Ph), 123.4 (C7′), 125.2 (C4′), 127.2 (C6′), 128.3 (C3′′, C4′′, C5′′), 128.8 (C2′′, C6′′), 129.5 (C5′), 135.7 (C1′′), 138.0 (C7a′), 144.7 (C3a′), 155.3 (C2O), 163.6 (CONHOCH2Ph), 175.4 (C5O). Found: C, 66.54; H, 5.61; N, 11.00. Calc. for C21H21N3O4: C, 66.48; H, 5.58; N, 11.08%.
:1 (17 mL) was subjected to catalytic hydrogenolysis in exactly the same procedure described above for the preparation of the final compounds. Removal of the solvents under reduced pressure provided the acetohydroxamic acid 16a as a white foamy solid. Crystallization of this product upon treatment with n-pentane-dry Et2O under ice-cooling gave a white crystalline solid (120 mg, almost quantitative yield); mp melted gradually from 158 °C to 162 °C (AcOEt/n-pentane-dry Et2O), Rf = 0.22 (from AcOEt). This compound exhibited distinct peaks attributed to each of the two E/Z conformers in the 1H and 13C NMR spectra. 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.40 (ddd, 1H, J1 = 14.1 Hz, J2 = 8.5 Hz, J3 = 4.2 Hz, H2′), 2.47 (ddd, 1H, J1 = 14.1 Hz, J2 = 8.2 Hz, J3 = 4.0 Hz, H2′), 2.61 (s, 3H, NCH3), 3.04–3.16 (m, 2H, H3′), 3.95–4.04 (q, AB, 1.5H, JAB = 16.1 Hz, NCH2CO, E-isomer), 4.24–4.33 (q, AB, 0.5H, JAB = 17.3 Hz, NCH2CO, Z-isomer), 7.23 (d, 1H, J = 7.8 Hz, H7′), 7.27 (td, 1H, J1 = 7.3 Hz, J2 = 2.0 Hz, H6′), 7.34–7.40 (m, 2H, H4′, H5′), 8.98 (s, 0.7H, CH2CONHOH, E-isomer), 9.36 (s, 0.23H, NCH2CONHOH, Z-isomer), 10.34 (s, 0.23H, NCH2CONHOH, Z-isomer), 10.75 (s, 0.7H, CH2CONHOH, E-isomer); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 24.8 (NCH3), 30.1 (C3′), 32.1 (C2′), 38.5 (NCH2CO, E-isomer), 39.0 (NCH2CO, Z-isomer), 74.3 (C1′/C4), 123.5, 123.6 (C7′), 125.2 (C4′), 127.2 (C6′), 129.5 (C5′), 138.2 (C7a′), 144.7 (C3a′), 154.6 (C2O, E-isomer), 154.7 (C2O, Z-isomer), 163.2 (NCH2CO, E-isomer), 168.6 (NCH2CO, Z-isomer), 174.6 (C5O, E-isomer), 174.8 (C5O, Z-isomer). Found: C, 58.23; H, 5.26; N, 14.50. Calc. for C14H15N3O4: C, 58.13; H, 5.23; N, 14.53%.
:1). 1H NMR (600.11 MHz, CDCl3) δ (ppm) 2.05 (ddd, 1H, J1 = 14.2 Hz, J2 = 8.9 Hz, J3 = 5.5 Hz, H2′), 2.52 (ddd, 1H, J1 = 13.8 Hz, J2 = 9.1 Hz, J3 = 5.6 Hz, H2′), 2.84 (ddd, 1H, J1 = 16.0 Hz, J2 = 8.9 Hz, J3 = 5.6 Hz, H3′), 3.07 (ddd, 1H, J1 = 15.9 Hz, J2 = 9.1 Hz, J3 = 5.5 Hz, H3′), 3.81 (d, 1H, J = 15.9 Hz, NCHHPh), 4.41–4.49 (q, AB, 2H, JAB = 17.3 Hz, NCH2COO), 4.82 (d, 1H, J = 15.9 Hz, NCHHPh), 5.20–5.26 (q, AB, 2H, JAB = 12.1 Hz, OCH2Ph), 7.06 (d, 1H, J = 7.7 Hz, H7′), 7.10–7.13 (m, 2H, H2Bz, H6Bz), 7.14 (td, 1H, J1 = 7.4 Hz, J2 = 1.3 Hz, H6′), 7.21–7.27 (complex m, 3H, H3Bz, H4Bz, H5Bz), 7.28 (dt, 1H, J1 = 7.5 Hz, J2 = 1.1 Hz, H4′), 7.31 (td, 1H, J1 = 7.4 Hz, J2 = 1.1 Hz, H5′), 7.34–7.41 (m, 5H, H2′′, H3′′, H4′′, H5′′, H6′′); 13C NMR (150.9 MHz, CDCl3) δ (ppm) 30.7 (C3′), 33.9 (C2′), 40.1 (NCH2COO), 44.1 (NCH2Ph), 67.9 (COOCH2Ph), 76.0 (C1′/C4), 124.0 (C7′), 125.4 (C4′), 127.7 (C2Bz, C6Bz), 127.8 (C6′), 128.6 (C3Bz, C5Bz), 128.7 (C2′′, C6′′, C4Bz), 128.76 (C4′′), 128.84 (C3′′, C5′′), 130.0 (C5′), 135.0 (C1′′), 137.5 (C1Bz), 137.9 (C7a′), 145.1 (C3a′), 155.7 (C2O), 167.3 (COOCH2Ph), 175.0 (C5O). Found: C, 73.66; H, 5.55; N, 6.30. Calc. for C27H24N2O4: C, 73.62; H, 5.49; N, 6.36%.
:1) following the procedure described for the preparation of the above-mentioned carboxylic acids. The desired compound 10b was afforded as a glass solid, which was further crystallized upon treatment with n-pentane-dry Et2O (5:1) under ice-cooling to give a white crystalline solid (316 mg, almost quantitative yield); mp 144–147 °C (from AcOEt/n-pentane), Rf = 0.22 (AcOEt). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.14 (ddd, 1H, J1 = 14.1 Hz, J2 = 8.7 Hz, J3 = 5.6 Hz, H2′), 2.44 (ddd, 1H, J1 = 14.5 Hz, J2 = 8.9 Hz, J3 = 5.9 Hz, H2′), 2.93 (ddd, 1H, J1 = 15.8 Hz, J2 = 8.7 Hz, J3 = 5.9 Hz, H3′), 3.00 (ddd, 1H, J1 = 15.0 Hz, J2 = 8.9 Hz, J3 = 5.6 Hz, H3′), 3.96 (d, 1H, J = 16.3 Hz, NCHHPh), 4.19–4.27 (q, AB, 2H, JAB = 17.4 Hz, NCH2COOH), 4.58 (d, 1H, J = 16.3 Hz, NCHHPh), 7.07–7.14 (m, 3H, H7′, H2Bz, H6Bz), 7.15–7.26 (complex m, 4H, H6′, H3Bz, H4Bz, H5Bz), 7.30–7.35 (m, 2H, H4′, H5′), 13.33 (br s, 1H, NCH2COOH); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 30.0 (C3′), 33.0 (C2′), 40.1 (NCH2COOH), 43.2 (NCH2Ph), 74.9 (C1′/C4), 123.6 (C7′), 125.2 (C4′), 127.1 (C6′, C2Bz, C4Bz, C6Bz), 128.2 (C3Bz, C5Bz), 129.5 (C5′), 137.6 (C1Bz), 138.3 (C7a′), 144.7 (C3a′), 155.1 (C2O), 168.7 (NCH2COOH), 174.3 (C5O). Found: C, 68.60; H, 5.14; N, 8.13. Calc. for C20H18N2O4: C, 68.56; H, 5.18; N, 8.00%.
:1, 10:1 and AcOEt as eluents to give the corresponding O-benzyl hydroxamate 13b as a glass solid. Crystallization of this material upon treatment with a n-pentane-dry Et2O mixture (5:1) gave a white crystalline solid (310 mg, 82%); mp melted gradually from 60 °C to 78 °C (from AcOEt/n-pentane-dry Et2O), Rf = 0.41 (CH2Cl2/AcOEt 8:1). 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.13 (ddd, 1H, J1 = 14.0 Hz, J2 = 8.6 Hz, J3 = 5.3 Hz, H2′), 2.45 (ddd, 1H, J1 = 14.5 Hz, J2 = 8.9 Hz, J3 = 6.0 Hz, H2′), 2.93 (ddd, 1H, J1 = 15.8 Hz, J2 = 8.6 Hz, J3 = 6.0 Hz, H3′), 2.99 (ddd, 1H, J1 = 14.9 Hz, J2 = 8.8 Hz, J3 = 5.4 Hz, H3′), 3.94 (d, 1H, J = 16.3 Hz, NCHHPh), 4.04–4.13 (q, AB, 1.65H, JAB = 16.2 Hz, NCH2CO), 4.35 (br s, 0.35H, NCH2CO), 4.58 (d, 1H, J = 16.2 Hz, NCHHPh), 4.82, 4.89 (s + s, 2H, OCH2Ph), 7.13 (d, 1H, J = 7.2 Hz, H2Bz, H6Bz), 7.16–7.26 (m, 5H, H6′, H7′, H3Bz, H4Bz, H5Bz), 7.30–7.35 (m, 2H, H4′, H5′), 7.35–7.49 (m, 5H, H2′′, H3′′, H4′′, H5′′, H6′′), 11.09 (s, 0.15H, CONHOCH2Ph), 11.46 (s, 0.75H, CONHOCH2Ph); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 30.0 (C3′), 33.0 (C2′), 38.6 (NCH2CO), 43.1 (NCH2Ph), 74.9 (C1′/C4), 77.0 (OCH2Ph), 123.8 (C7′), 125.1 (C4′), 127.1 (C6′, C2Bz, C4Bz, C6Bz), 128.2 (C3Bz, C5Bz), 128.3 (C2′′, C4′′, C6′′), 128.8 (C3′′, C5′′), 129.5 (C5′), 135.7 (C1′′), 137.6 (C1Bz), 138.4 (C7a′), 144.7 (C3a′), 155.2 (C2O), 163.5 (CONHOCH2Ph), 174.5 (C5O). Found: C, 71.25; H, 5.60; N, 9.32. Calc. for C27H25N3O4: C, 71.19; H, 5.53; N, 9.22%.
:1 (22 mL) according to the previously described procedure for the synthesis of hydroxamate analogues. Concentration to dryness of the solvents under reduced pressure yielded the title compound 16b as a white crystalline solid (200 mg, almost quantitative yield); mp melted gradually from 174 °C to 181 °C (from AcOEt/n-pentane-dry Et2O), Rf = 0.49 (AcOEt). In the 1H and 13C NMR spectra of this compound, double set of characteristic peaks are distinguished for each of the two E/Z conformers. 1H NMR (600.11 MHz, DMSO-d6) δ (ppm) 2.13 (ddd, 1H, J1 = 13.9 Hz, J2 = 8.6 Hz, J3 = 5.4 Hz, H2′), 2.44 (ddd, 1H, J1 = 13.9 Hz, J2 = 8.9 Hz, J3 = 6.1 Hz, H2′), 2.92 (ddd, 1H, J1 = 16.0 Hz, J2 = 8.5 Hz, J3 = 6.3 Hz, H3′), 2.98 (ddd, 1H, J1 = 14.4 Hz, J2 = 8.9 Hz, J3 = 5.4 Hz, H3′), 3.93 (d, 1H, J = 16.3 Hz, NCHHPh), 4.04–4.12 (q, AB, 1.5H, JAB = 16.0 Hz, NCH2CO, E-isomer), 4.32–4.40 (q, AB, 0.5H, JAB = 17.4 Hz, NCH2CO, Z-isomer), 4.57 (dd, 1H, J1 = 16.3 Hz, J2 = 8.8 Hz, NCHHPh), 7.12 (d, 1H, J = 6.7 Hz, H2Bz, H6Bz), 7.15–7.25 (complex m, 5H, H6′, H7′, H3Bz, H4Bz, H5Bz), 7.29–7.34 (m, 2H, H4′, H5′), 9.03 (s, 0.68H, CH2CONHOH, E-isomer), 9.41 (s, 0.22H, NCH2CONHOH, Z-isomer), 10.37 (s, 0.22H, NCH2CONHOH, Z-isomer), 10.80 (s, 0.68H, CH2CONHOH, E-isomer); 13C NMR (150.9 MHz, DMSO-d6) δ (ppm) 30.0 (C3′), 32.9, 33.0 (C2′), 38.6 (NCH2CO, E-isomer), 39.1 (NCH2CO, Z-isomer), 43.1 (NCH2Ph), 74.9 (C1′/C4), 123.9 (C7′), 125.0 (C4′), 127.1 (C6′, C2Bz, C4Bz, C6Bz), 128.2 (C3Bz, C5Bz), 129.5 (C5′), 137.6 (C1Bz), 138.5 (C7a′), 144.7 (C3a′), 155.3 (C2O, E-isomer), 155.5 (C2O, Z-isomer), 163.2 (NCH2CO, E-isomer), 168.6 (NCH2CO, Z-isomer), 174.6 (C5O, E-isomer), 174.8 (C5O, Z-isomer). Found: C, 65.79; H, 5.18; N, 11.55. Calc. for C20H19N3O4: C, 65.74; H, 5.24; N, 11.50%.
Biology
X using the Prism 5.0 software (GraphPad Software Inc.).
X using the Prism 5.0 software (GraphPad Software Inc.).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. G. would like to thank the European Federation for Medicinal Chemistry (EFMC) and the editors of the journal for their kind invitation. This work was partially supported by the General Secretariat for Research and Technology (grant KRIPIS II-MIS5002486), by the International Pasteur Network grant ACIP 18-2015 and by the Empirikion Foundation. We would like to thank the State Scholarships Foundation for providing a Ph.D fellowship to E. F. (MIS-5003404) and V. P. (MIS-5000432). We are grateful to Prof. Ralf Bartenschlager (Heidelberg University, Germany) for kindly providing the Huh5-2 cell line and Prof. Charles M. Rice for kindly providing Huh7.5 cells, replicon plasmids S52-SG (Feo) (AII), ED43-SG (Feo) (VYG) and pD2-hRUPac, as well as the 9E10 HCV NS5A-specific antibody.Notes and references
- World Health Organization, Hepatitis C, http://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed March 2019) Search PubMed.
- P. Simmonds, P. Becher, J. Bukh, E. A. Gould, G. Meyers, T. Monath, S. Muerhoff, A. Pletnev, R. Rico-Hesse, D. B. Smith and J. T. Stapleton, J. Gen. Virol., 2017, 98, 2–3 CrossRef CAS PubMed.
- R. Bartenschlager, V. Lohmann and F. Penin, Nat. Rev. Microbiol., 2013, 11, 482–496 CrossRef CAS PubMed.
- D. Moradpour, F. Penin and C. M. Rice, Nat. Rev. Microbiol., 2007, 5, 453–463 CrossRef CAS PubMed.
- S. Ramirez and J. Bukh, Antiviral Res., 2018, 158, 264–287 CrossRef CAS PubMed.
- J. P. Messina, I. Humphreys, A. Flaxman, A. Brown, G. S. Cooke, O. G. Pybus and E. Barnes, Hepatology, 2015, 61, 77–87 CrossRef PubMed.
- V. Soriano, M. G. Peters and S. Zeuzem, Clin. Infect. Dis., 2009, 48, 313–320 CrossRef PubMed.
- VOSEVI (sofosbuvir, velpatasvir, and voxilaprevir) tablets: FDA Highlights of Prescibing Information, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209195s000lbl.pdf, (accessed 7 May 2019) Search PubMed.
- DAKLINZA (daclatasvir) tablets: FDA Highlights of Prescribing Information, https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/206843s006lbl.pdf, (accessed 7 May 2019) Search PubMed.
- SOVALDI (sofosbuvir) tablets: FDA Highlights of Prescribing Information, https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/204671s002lbl.pdf, (accessed 7 May 2019) Search PubMed.
- VIEKIRA PAK (ombitasvir, paritaprevir and ritonavir tablets; dasabuvir tablets): FDA Highlights of Prescibing Information, https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206619lbl.pdf, (accessed 7 May 2019) Search PubMed.
- The American Association for the Study of Liver Disease, http://www.hcvguidelines.org (accessed March 2019) Search PubMed.
- J. M. Pawlotsky, F. Negro, A. Aghemo, M. Berenguer, O. Dalgard, G. Dusheiko, F. Marra, M. Puoti and H. Wedemeyer, J. Hepatol., 2018, 69, 461–511 CrossRef PubMed.
- T. Asselah, P. Marcellin and R. F. Schinazi, Liver Int., 2018, 38(Suppl 1), 7–13 CrossRef PubMed.
- V. Gimeno-Ballester, M. Buti, R. San Miguel, M. Riveiro and R. Esteban, J. Viral Hepatitis, 2017, 24, 904–916 CrossRef CAS PubMed.
- C. Hézode, P. Lebray, V. De Ledinghen, F. Zoulim, V. Di Martino, N. Boyer, D. Larrey, D. Botta-Fridlund, C. Silvain, H. Fontaine, L. D'Alteroche, V. Leroy, M. Bourliere, I. Hubert-Fouchard, D. Guyader, I. Rosa, E. Nguyen-Khac, L. Fedchuk, R. Akremi, Y. Bennai, A. Filipovics, Y. Zhao and J. P. Bronowicki, Liver Int., 2017, 37, 1314–1324 CrossRef PubMed.
- L. Cuypers, F. Ceccherini-Silberstein, K. Van Laethem, G. Li, A. M. Vandamme and J. K. Rockstroh, Rev. Med. Virol., 2016, 26, 408–434 CrossRef CAS PubMed.
- H. Barth, World J. Hepatol., 2015, 7, 725–737 CrossRef PubMed.
- J. D. Stanaway, D. S. Shepard, E. A. Undurraga, Y. A. Halasa, L. E. Coffeng, O. J. Brady, S. I. Hay, N. Bedi, I. M. Bensenor, C. A. Castañeda-Orjuela, T. W. Chuang, K. B. Gibney, Z. A. Memish, A. Rafay, K. N. Ukwaja, N. Yonemoto and C. J. L. Murray, Lancet Infect. Dis., 2016, 16, 712–723 CrossRef PubMed.
- A. S. Leong, K. T. Wong, T. Y. Leong, P. H. Tan and P. Wannakrairot, Semin. Diagn. Pathol., 2007, 24, 227–236 CrossRef PubMed.
- S. R. Hadinegoro, J. L. Arredondo-Garcia, M. R. Capeding, C. Deseda, T. Chotpitayasunondh, R. Dietze, H. I. Muhammad Ismail, H. Reynales, K. Limkittikul, D. M. Rivera-Medina, H. N. Tran, A. Bouckenooghe, D. Chansinghakul, M. Cortes, K. Fanouillere, R. Forrat, C. Frago, S. Gailhardou, N. Jackson, F. Noriega, E. Plennevaux, T. A. Wartel, B. Zambrano, M. Saville and CYD-TDV Dengue Vaccine Working Group, N. Engl. J. Med., 2015, 373, 1195–1206 CrossRef CAS PubMed.
- J. G. Low, E. E. Ooi and S. G. Vasudevan, J. Infect. Dis., 2017, 215, S96–S102 CrossRef CAS PubMed.
- R. Bartenschlager and S. Miller, Future Microbiol., 2008, 3, 155–165 CrossRef CAS PubMed.
- M. S. Diamond and T. C. Pierson, Cell, 2015, 162, 488–492 CrossRef CAS PubMed.
- M. Stempniak, Z. Hostomska, B. R. Nodes and Z. Hostomsky, J. Virol., 1997, 71, 2881–2886 CAS.
- D. N. Frick, S. Banik and R. S. Rypma, J. Mol. Biol., 2007, 365, 1017–1032 CrossRef CAS PubMed.
- P. J. Lim, U. Chatterji, D. Cordek, S. D. Sharma, J. A. Garcia-Rivera, C. E. Cameron, K. Lin, P. Targett-Adams and P. A. Gallay, J. Biol. Chem., 2012, 287, 30861–30873 CrossRef CAS PubMed.
- S. Bressanelli, L. Tomei, F. A. Rey and R. De Francesco, J. Virol., 2002, 76, 3482–3492 CrossRef CAS PubMed.
- R. B. Davidson, J. Hendrix, B. J. Geiss and M. McCullagh, PLoS Comput. Biol., 2018, 14, e1006103 CrossRef PubMed.
- T. L. Yap, T. Xu, Y. L. Chen, H. Malet, M. P. Egloff, B. Canard, S. G. Vasudevan and J. Lescar, J. Virol., 2007, 81, 4753–4765 CrossRef CAS PubMed.
- World Health Organization, Trypanosomiasis, human African (sleeping sickness), https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed March 2019) Search PubMed.
- E. Bottieau and J. Clerinx, Infect. Dis. Clin. North Am., 2019, 33, 61–77 CrossRef PubMed.
- S. Varikuti, B. K. Jha, G. Volpedo, N. M. Ryan, G. Halsey, O. M. Hamza, B. S. McGwire and A. R. Satoskar, Front. Microbiol., 2018, 9, 2655 CrossRef PubMed.
- P. G. E. Kennedy and J. Rodgers, Front. Immunol., 2019, 10, 39 CrossRef PubMed.
- C. H. Baker and S. C. Welburn, Trends Parasitol., 2018, 34, 818–827 CrossRef PubMed.
- R. J. Wall, E. Rico, I. Lukac, F. Zuccotto, S. Elg, I. H. Gilbert, Y. Freund, M. R. K. Alley, M. C. Field, S. Wyllie and D. Horn, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9616–9621 CrossRef CAS PubMed.
- F. Chappuis, Lancet, 2018, 391, 100–102 CrossRef.
- World Health Organization, Chagas disease (American trypanosomiasis)https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis (accessed March 2019) Search PubMed.
- A. Rassi, J. M. de Rezende, A. O. Luquetti and A. Rassi Jr, in American Trypanosomiasis Chagas Disease: One Hundred Years of Research, ed. J. Telleria and M. Tibayrenc, Elsevier, Amsterdam (Netherlands), 2nd edn, 2017, vol. 28, pp. 653–686 Search PubMed.
- J. Bermudez, C. Davies, A. Simonazzi, J. P. Real and S. Palma, Acta Trop., 2016, 156, 1–16 CrossRef CAS PubMed.
- Q. P. Wang, T. Kawahara and D. Horn, Mol. Microbiol., 2010, 77, 1237–1245 CrossRef CAS PubMed.
- M. D. Urbaniak, A. Crossman, T. Chang, T. K. Smith, D. M. F. van Aalten and M. A. J. Ferguson, J. Biol. Chem., 2005, 280, 22831–22838 CrossRef CAS PubMed.
- G. F. Mercaldi, H. M. Pereira, A. T. Cordeiro, P. A. M. Michels and O. H. Thiemann, FEBS J., 2012, 279, 2012–2021 CrossRef CAS PubMed.
- C. Gong, A. Martins and S. Shuman, J. Biol. Chem., 2003, 278, 50843–50852 CrossRef CAS PubMed.
- S. R. Wilkinson, S. R. Prathalingam, M. C. Taylor, A. Ahmed, D. Horn and J. M. Kelly, Free Radical Biol. Med., 2006, 40, 198–209 CrossRef CAS PubMed.
- S. K. Menzies, L. B. Tulloch, G. J. Florence and T. K. Smith, Parasitology, 2016, 145, 175–183 CrossRef PubMed.
- S. Wyllie and A. H. Fairlamb, Semin. Cell Dev. Biol., 2011, 22, 271–277 CrossRef CAS PubMed.
- D. R. de Menezes, C. M. Calvet, G. C. Rodrigues, M. C. de Souza Pereira, I. R. Almeida, A. P. de Aguiar, C. T. Supuran and A. B. Vermelho, J. Enzyme Inhib. Med. Chem., 2016, 31, 964–973 CrossRef PubMed.
- A. A. Zuma and W. D. Souza, Future Sci. OA, 2018, 4, FSO325 CrossRef PubMed.
- M. D. Urbaniak, A. S. Capes, A. Crossman, S. O'Neill, S. Thompson, I. H. Gilbert and M. A. J. Ferguson, Carbohydr. Res., 2014, 387, 54–58 CrossRef CAS PubMed.
- G. U. Ebiloma, E. O. Balogun, E. J. Cueto-Díaz, H. P. de Koning and C. Dardonville, Med. Res. Rev., 2019 DOI:10.1002/med.21560.
- P. Smith, C. K. Ho, Y. Takagi, H. Djaballah and S. Shuman, mBio, 2016, 7, e00058 CAS.
- E. Giannakopoulou, V. Pardali and G. Zoidis, Future Med. Chem., 2018, 10, 1283–1285 CrossRef CAS PubMed.
- A. Y. Chen, R. N. Adamek, B. L. Dick, C. V. Credille, C. N. Morrison and S. M. Cohen, Chem. Rev., 2018, 119, 1323–1455 CrossRef PubMed.
- L. Riccardi, V. Genna and M. De Vivo, Nat. Rev. Chem., 2018, 2, 100–112 CrossRef CAS.
- G. Zoidis, E. Giannakopoulou, A. Stevaert, E. Frakolaki, V. Myrianthopoulos, G. Fytas, P. Mavromara, E. Mikros, R. Bartenschlager, N. Vassilaki and L. Naesens, Med. Chem. Commun., 2016, 7, 447–456 RSC.
- C. Fytas, G. Zoidis, N. Tzoutzas, M. C. Taylor, G. Fytas and J. M. Kelly, J. Med. Chem., 2011, 54, 5250–5254 CrossRef CAS PubMed.
- G. Zoidis, A. Tsotinis, A. Tsatsaroni, M. C. Taylor, J. M. Kelly, A. Efstathiou, D. Smirlis and G. Fytas, Chem. Biol. Drug Des., 2018, 91, 408–421 CrossRef CAS PubMed.
- E. Giannakopoulou, V. Pardali, I. Skrettas and G. Zoidis, ChemistrySelect, 2019, 4, 3195–3198 CrossRef CAS.
- C. Fytas, G. Zoidis and G. Fytas, Tetrahedron, 2008, 64, 6749–6754 CrossRef CAS.
- J. M. Vrolijk, A. Kaul, B. E. Hansen, V. Lohmann, B. L. Haagmans, S. W. Schalm and R. Bartenschlager, J. Virol. Methods, 2003, 110, 201–209 CrossRef CAS PubMed.
- Marvin was used for determining logD, Marvin 5.12.2, 2013, ChemAxon (http://www.chemaxon.com) Search PubMed.
- Schrödinger Release 2019–1: Jaguar pKa, Schrödinger, LLC, New York, NY, 2019 Search PubMed.
- A. D. Bochevarov, M. A. Watson, J. R. Greenwood and D. M. Philipp, J. Chem. Theory Comput., 2016, 12, 6001–6019 CrossRef CAS PubMed.
- H. S. Yu, M. A. Watson and A. D. Bochevarov, J. Chem. Inf. Model., 2018, 58, 271–286 CrossRef CAS PubMed.
- J. J. Klicić, R. A. Friesner, S. Y. Liu and W. C. Guida, J. Phys. Chem. A, 2002, 106, 1327–1335 CrossRef.
- M. Saeed, T. K. Scheel, J. M. Gottwein, S. Marukian, L. B. Dustin, J. Bukh and C. M. Rice, Antimicrob. Agents Chemother., 2012, 56, 5365–5373 CrossRef CAS PubMed.
- N. Lougiakis, E. Frakolaki, P. Karmou, N. Pouli, P. Marakos, V. Madan, R. Bartenschlager and N. Vassilaki, Chem. Biol. Drug Des., 2017, 90, 352–367 CrossRef CAS PubMed.
- J. Dufner-Beattie, A. O'Guin, S. O'Guin, A. Briley, B. Wang, J. Balsarotti, R. Roth, G. Starkey, U. Slomczynska, A. Noueiry, P. D. Olivo and C. M. Rice, Antimicrob. Agents Chemother., 2014, 58, 3399–3410 CrossRef PubMed.
- A. Kaul, I. Woerz, P. Meuleman, G. Leroux-Roels and R. Bartenschlager, J. Virol., 2007, 81, 13168–13179 CrossRef CAS PubMed.
- N. Vassilaki, P. Friebe, P. Meuleman, S. Kallis, A. Kaul, G. Paranhos-Baccalà, G. Leroux-Roels, P. Mavromara and R. Bartenschlager, J. Virol., 2008, 82, 11503–11515 CrossRef CAS PubMed.
- M. C. Taylor, A. P. McLatchie and J. M. Kelly, Mol. Microbiol., 2013, 89, 420–432 CrossRef CAS PubMed.
- G. Kendall, A. F. Wilderspin, F. Ashall, M. A. Miles and J. M. Kelly, EMBO J., 1990, 9, 2751–2758 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9md00200f |
| ‡ Principal investigator of the study on Trypanosoma species. |
| § Principal investigator of the study on Flaviviridae viruses. |
| ¶ Principal investigator of the design and synthesis of the compounds. |
| This journal is © The Royal Society of Chemistry 2019 |