
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFgaPT2, a biocatalytic tool for alkyl-diversification of indole natural products†
Chandrasekhar
Bandari‡
,
Erin M.
Scull‡
,
Tejaswi
Bavineni
,
Susan L.
Nimmo
,
Eric D.
Gardner
,
Ryan C.
Bensen
,
Anthony W.
Burgett
and
Shanteri
Singh
 *
*
Department of Chemistry and Biochemistry, University of Oklahoma, Stephenson Life Sciences Research Center, 101 Stephenson Parkway, Norman, Oklahoma 73019, USA. E-mail: shanteri.singh@ou.edu
First published on 21st June 2019
Abstract
Aromatic prenyltransferases from natural product biosynthetic pathways display relaxed specificity for their aromatic substrates. While a growing body of evidence suggests aromatic prenyltransferases to be more tolerant towards their alkyl-donor substrates, most studies aimed at probing their donor-substrate specificity are limited to only a small set of alkyl pyrophosphate donors, restricting their broader utility as biocatalysts for synthetic applications. Here, we assess the donor substrate specificity of an L-tryptophan C4-prenyltransferase, also known as C4-dimethylallyltryptophan synthase, FgaPT2 from Aspergillus fumigatus, using an array of 34 synthetic unnatural alkyl-pyrophosphate analogues, and demonstrate FgaPT2 can catalyze the transfer of 25 of the 34 non-native alkyl groups from their corresponding synthetic alkyl-pyrophosphate analogues at N1, C3, C4 and C5 position of tryptophan in a normal and reverse manner. The kinetic studies and regio-chemical analysis of the alkyl-L-tryptophan products suggest that the alkyl-donor transfer by FgaPT2 is a function of the stability of the carbocation and the steric factors in the active site of the enzyme. Further, to demonstrate the biocatalytic utility of FgaPT2, this study also highlights the FgaPT2-catalyzed synthesis of a small set of alkyl-diversified indolocarbazole analogues. These results reveal FgaPT2 to be more tolerant to diverse non-native alkyl-donor substrates beyond their known acceptor substrate promiscuity and set the stage for its development as a novel biocatalytic tool for the differential alkylation of natural products for drug discovery and other synthetic applications.
Introduction
Prenylation is one of the important late-stage modifications found in nature,1–5 which can enhance the lipophilicity of compounds and alter the binding specificity of the biological targets.6,7 Aromatic prenyltransferases are a subset of prenyltransferases (PTs) involved in the biosynthesis of natural products (NPs), which catalyze the transfer of a C5, C10, or C15 isoprenoid chain from the corresponding prenyl pyrophosphate donors to an electron-rich aromatic acceptor such as an indole or phenolic molecule. Prenylated NPs display a diverse spectrum of biological activities, including anti-inflammatory,8,9 antibacterial,10–12 anticancer,13,14 and antiviral15,16 (Fig. 1). Due to the high structural complexity and chemical reactivity of many NPs, purely synthetic methods for analogous alkylation reactions are unavailable due to poor regio- and chemo-specificity. Therefore, aromatic PTs show great promise as useful biocatalysts for the production and diversification of bioactive NPs due to their broad substrate scope and remarkable regiospecificity.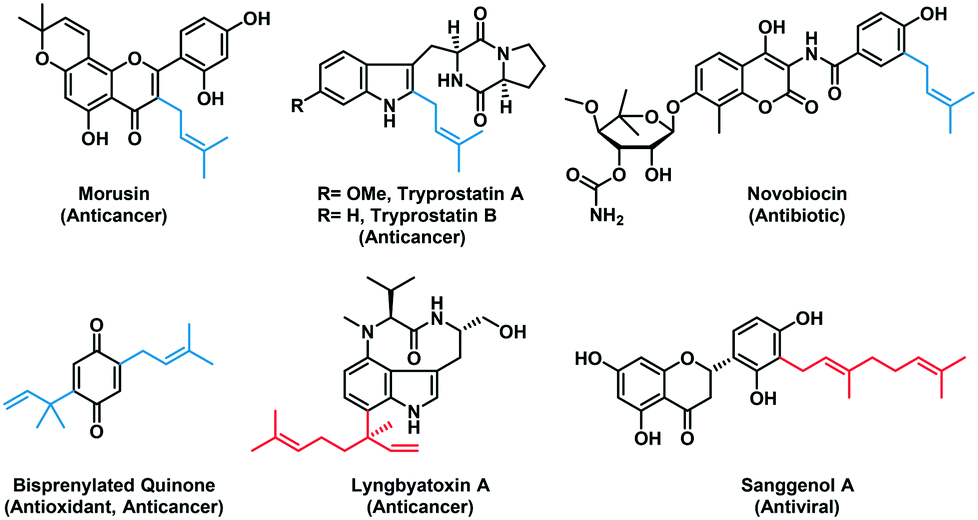 | ||
| Fig. 1 Representative biologically active prenylated natural products in which the prenyl moieties are derived from DMAPP (blue) or GPP (red) through PT-catalyzed prenylation reaction. | ||
Dimethylallyltryptophan synthases (DMATSs) are a subclass of aromatic PTs, responsible for the regioselective prenylation of tryptophan moieties using dimethylallyl pyrophosphate (DMAPP) as the prenyl donor. DMATS are known to have relaxed acceptor substrate specificities2,17 that use DMAPP as the preferred alkyl donor. DMATS capable of selectively alkylating all seven positions of the indole ring have been biochemically characterized.2,17–19 Further, prenylation can take place in a normal or reverse fashion, referring to C1′ or C3′ attachment of DMAPP, respectively.17,19 Currently, crystal structures of several aromatic PTs are available in the Protein Data Bank (PDB), which have enabled a better understanding of substrate recognition and catalytic mechanisms. All these structures have revealed aromatic PTs to have a common structural motif known as the “ABBA PT” fold, made up of a 10-stranded antiparallel β-barrel surrounded by α-helices. This β-barrel contains a hydrophobic reaction chamber capable of binding both donor and acceptor substrates to carry out the prenyl transfer reaction.20–29
FgaPT2 from Aspergillus fumigatus is a C4-DMATS that catalyzes the normal prenylation of L-tryptophan (L-Trp) using DMAPP. This prenylation is the first committed step in the biosynthetic pathway of the ergot alkaloid, fumigaclavine C (Fig. 2A).30 The reaction proceeds through the generation of an allylic carbocation via pyrophosphate dissociation, followed by electrophilic aromatic substitution at C4 of L-Trp to generate an arenium intermediate.31 The subsequent deprotonation at C4 restores the aromaticity, generating 4-dimethylallyltryptophan (4-DMAT).31,32 The crystal structure of FgaPT2 in complex with L-Trp and the unreactive substrate analogue, dimethylallyl S-thiolopyrophosphate (DMASPP), shows the Michaelis complex, where the plane of the dimethylallyl group is stacked against the plane of the indole ring, with C4 of L-Trp at a distance of 3.8 Å from C1′ of DMASPP, and C3 of L-Trp at a distance of 3.5 Å from C3′ of DMASPP.20 This structure also revealed that Lys174 is positioned properly to function as a base in the final re-aromatization step.20 Subsequent mutational studies of FgaPT2 revealed that the K174A mutant produced a C3 reverse prenylated product.33 This data, along with the high electron density at C3, support the possibility of initial reverse prenylation at C3, followed by a Cope-rearrangement to produce the final C4 prenylated product of L-Trp.33 Later reports using C4 substituted L-Trp substrates that resulted in five different regio-isomers of dimethylallyl L-Trp (N1, C3, C5, C6, and C7) strengthened the argument of direct prenylation at C4 of L-Trp by FgaPT2.34
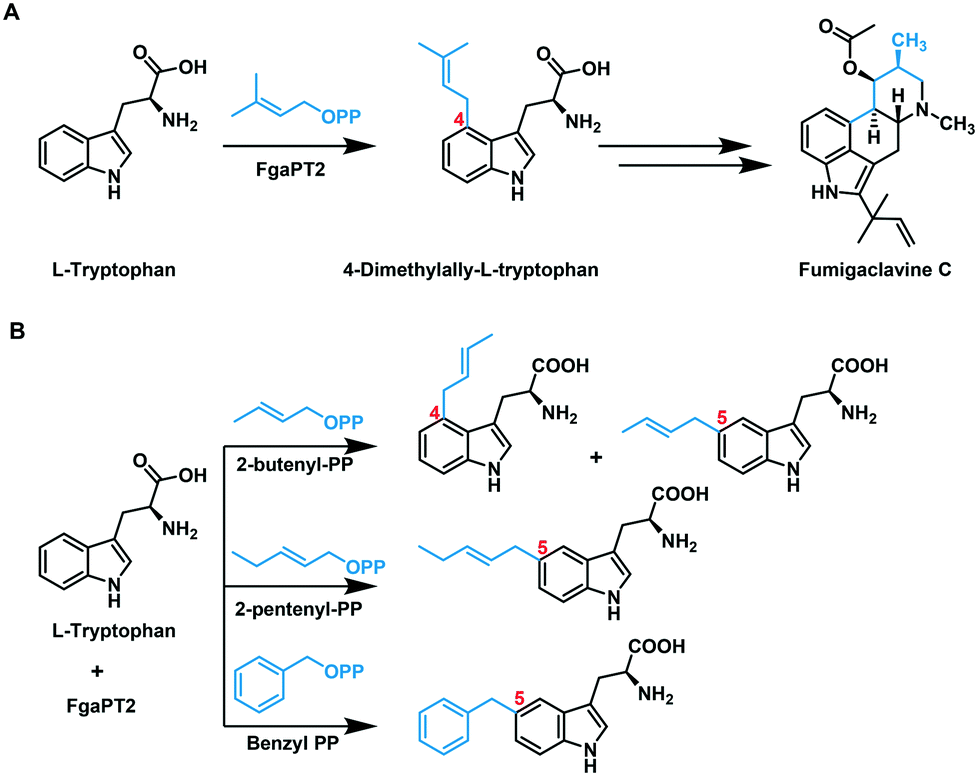 | ||
| Fig. 2 (A) Reaction catalyzed by FgaPT2 in the biosynthetic pathway of fumigaclavine C.30 (B) Previously known FgaPT2 catalyzed alkylation reaction using unnatural alkyl-pyrophosphate donors.41,42 The regio-chemistry of the alkyl group transfer on L-tryptophan has been labelled in red. | ||
Several studies have revealed that FgaPT2 has a broad and diverse acceptor substrate scope, including substituted tryptophan, L-Trp containing cyclic dipeptides, isomers of L-tyrosine, indolocarbazoles, and phenolic molecules.35–40 A few alternative alkyl donors have also been reported with FgaPT2 including the C10 geranyl pyrophosphate (GPP), 2-butenyl pyrophosphate, 2-pentenyl pyrophosphate, and benzyl pyrophosphate.41,42 These studies revealed that GPP was poorly accepted by FgaPT2,30 while 2-butenyl pyrophosphate produced a mixture of C4 and C5 alkylated products, and the bulkier 2-pentenyl and benzyl pyrophosphates produced C5 alkylated tryptophan products (Fig. 2B).41,42 Our recent study of the tyrosine-O-prenyltransferase, SirD, demonstrated its ability to accept 16 of the 21 diverse unnatural allylic donors (1–21 in Fig. 3A).43 This prompted our investigation of the alkyl-donor substrate scope of FgaPT2 to assess its potential use as a biocatalytic tool for differential alkylation of indole-containing NPs.
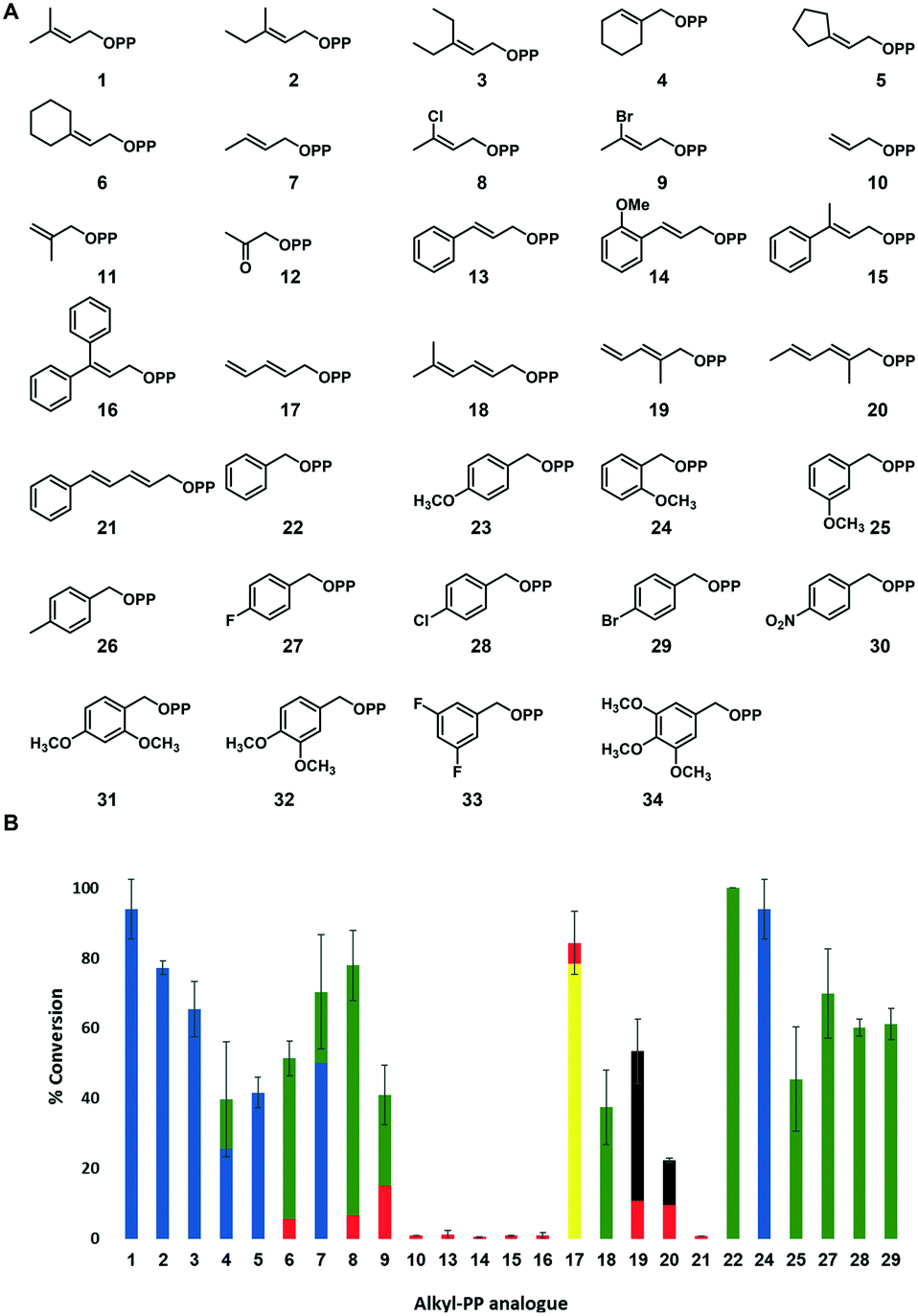 | ||
| Fig. 3 (A) Library of synthetic alkyl-PP analogues assessed in FgaPT2-catalyzed reactions with L-Trp. (B) The percent conversion of alkyl-PP analogues to the corresponding alkyl-L-Trp catalyzed by FgaPT2 based upon RP-HPLC. The standard deviation for the total product conversion was obtained from two experimental replicates (see ESI† Table S7 for the exact values used to generate this figure). Reactions were performed with 1.2 mM alkyl-PP analogue, 1 mM L-Trp, 5 μM FgaPT2, 25 mM Tris, 5 mM MgCl2, and 50 mM KCl at pH 7.5 for 16 h at 35 °C. No product formation was observed in the absence of FgaPT2 or the alkyl-PP analogue. The blue, green, yellow, and black colors in the bar diagram denote C4, C5, C3, and N1 alkyl-L-Trp regio-isomers as confirmed by NMR, whereas the red color represents the alkyl-L-Trp isomer with an undetermined regio-chemistry. | ||
Herein, we report the alkyl donor substrate scope of FgaPT2 using our library of 34 synthetic allylic and benzylic alkyl donors with varying carbon chain lengths and substituents (Fig. 3A). Our systematic evaluation of alkyl pyrophosphate (alkyl-PP) donors revealed FgaPT2 can utilize 25 out of the 34 diverse alkyl-PP analogues tested. The subsequent structural characterization of alkyl-diversified L-Trp analogues revealed that FgaPT2 predominantly catalyzes the normal alkylation of C4 and C5, and less frequently, the normal or reverse prenylation of C3 and N1 of L-Trp. Furthermore, we were able to demonstrate the potential utility of FgaPT2 as a chemoenzymatic late stage diversification tool by producing a library of alkylated indolocarbazole analogues. These analogues were assayed for cytotoxic activity against the human colon cancer cell line HCT-116.
Results and discussion
Synthesis of alkyl-PP analogues
In the present study, 34 unnatural alkyl-PP analogues (1–34 in Fig. 3A) were evaluated for their potential as alkyl donors for the FgaPT2 catalyzed alkylation with L-Trp. These alkyl-PP analogues include 21 previously reported allylic analogues (1–21),43 a benzyl analogue, 22, and 12 new benzyl-substituted alkyl-PP analogues (23–34). The syntheses of all analogues were carried out using previously reported methods.44–46 All synthetic alkyl-PP analogues were purified and confirmed by high-resolution electrospray ionization (ESI) mass spectroscopy (HRMS) (see ESI† Table S2) and 1H and 31P NMR spectroscopy. Of the 34 alkyl-PP analogues, the natural alkyl donor, dimethylallyl (1), 2-butenyl (7) and benzyl (22) were previously reported to be substrates for FgaPT2 catalyzed reaction with L-Trp (Fig. 2B)41,42 and were included here for comparison purposes. Several factors were considered while designing these alkyl-PP analogues (Fig. 3A) including the effect of the carbon chain length (10 < 7 < 1 < 2 < 3 < 6 < 13) to explore the steric factors in the active site and the electronic nature of the substituents (1, versus8 and 9) to understand their potential utility in transferring diverse functional groups. We also included conjugated diene analogues (17–21) to grasp the contribution of carbocation resonance stabilization, in addition to introducing methyl groups at different positions (7, 1, 2, 10, 11, 18–20) to explore the methyl group effect. The benzylic pyrophosphate analogues (22–34) were tested to explore the inductive and resonance contribution of substituents. The length of the carbon chain in the linear direction of the designed alkyl-PP analogues was kept <10, to be consistent with the ability of FgaPT2 catalyzed reaction, which barely accepted a C10 GPP as a substrate.30In vitro activity assays with alkyl-PP analogues
In order to assess the ability of FgaPT2 to accept diverse unnatural alkyl-PP analogues, the recombinant FgaPT2 was overproduced in Escherichia coli BL21(DE3) cells transformed with the codon-optimized synthetic FgaPT2 gene in a pET28a vector. The resulting FgaPT2 with N-terminal His6-fusion protein was purified to homogeneity via Ni-NTA affinity chromatography as described previously,28 yielding ∼40 mg L−1 pure N-terminal His6-fusion protein. Standard uniform assay conditions (1.2 mM alkyl-PP analogue, 1 mM L-Trp, 5 μM FgaPT2, 25 mM Tris, 5 mM MgCl2, 50 mM KCl, pH 7.5, 16 h at 35 °C) were adopted to facilitate the initial assessment of the donor substrate specificity of FgaPT2 with synthetic alkyl-PP analogues. Negative controls were included to ensure that the product formation was enzymatic. Production of alkyl-L-Trp derivatives was determined by the difference in the retention time of L-Trp and alkyl-L-Trp products using a reverse phase high-pressure liquid chromatography (RP-HPLC) endpoint assay with subsequent confirmation by HRMS coupled with liquid chromatography (LC) analysis for all positive reactions (see ESI† Table S3) to afford complete analysis of the desired product.The initial assessment of FgaPT2 catalyzed alkylation of L-Trp with 34 synthetic alkyl-PP analogues based upon RP-HPLC of analytical scale reaction is illustrated in Fig. 3B, wherein 25 of the 34 synthetic analogues (Fig. 3A) in FgaPT2 catalyzed reaction produced the corresponding alkyl-L-Trp. All products were subsequently confirmed to be mono-alkylated products from HRMS coupled with liquid chromatography (LC) analysis (see ESI† Table S3). Of the 25 positive reactions, 12 led to appreciable (>50%) conversion, an additional seven led to moderate (20–50%) conversion, while six offered detectable product formation (<5%) under the conditions described (Fig. 3B, see ESI,† Fig. S2). Analysis of HPLC and HRMS results suggested more than one product for eight (Fig. 3B, 4, 6–9, 17, 19, 20) of the 19 analytical scale reactions with product yield >20% (Fig. 3B, 1–9, 17–20, 22, 24, 25, 27–29). Notably, the 3-carbon allyl pyrophosphate 10 was accepted as a substrate although with a very low conversion rate of 1%, resulting in the formation of allyl-L-Trp. However, 11 with a methyl group substituted at the β-carbon of allyl pyrophosphate was not accepted by FgaPT2. No product formation was observed for 12 that has a carbonyl group at β-carbon. The bulky cinnamyl analogue (13) and its derivatives (14–16, and 21) produced only a trace amount of the alkylated product with a low yield (<2%). In addition, while o-methoxy (24), m-methoxy (25) and p-halogen (27–29) substituted benzyl analogues were accepted by FgaPT2, introduction of bulky groups at the para-position, including p-methoxy (23), p-methyl (26), p-nitro (30) and di- and tri-substituted benzyl analogues (31–34), did not serve as substrates for FgaPT2 catalyzed reaction with L-Trp due to the potential steric hindrance in the active site of the enzyme. These results suggest the FgaPT2 catalyzed reaction with L-Trp is sterically limited by bulky substitution on the alkyl-donor.
Kinetic studies and structural analysis
Michaelis–Menten kinetic parameters for all 19 alkyl-PP analogues with product conversion >20% (Table 1) and for L-Trp with representative alkyl-PP analogues (see ESI† Table S1) were studied. The kinetic parameters, rates (kcat) and Michaelis constants (KM) were determined from a nonlinear regression fit of initial velocities versus the concentration of the alkyl-PP analogue by GraphPad Prism 5.04. Interestingly, the KM value of L-Trp for FgaPT2 with various alkyl-PP donors did not change drastically with the nature of the alkyl donors and remained within the range of 0.20 ± 0.08 mM (see ESI† Table S1). Consistent with our previous observations with tyrosine-O-prenyltransferase SirD,43 the alkyl-PP analogues that contained groups to enhance the stability of the carbocation via increased conjugation and resonance stabilization correlated with the increased turnover of the product. For example, the kcat values of the reaction correlated with conjugation by a methyl group in the order 1 > 2 > 3 > 7 (Table 1). In addition, the KM values increased with the increased length of the carbon chain in the order 1 < 2 ≅ 3 ≅ 5 < 6 and 17 < 18 ≅ 19 ≅ 20. Removing one methyl group of 1 decreased the efficiency (kcat/KM) of 7 88-fold compared to 1. In addition, changing one or both methyl groups of 1 to an ethyl group (2 and 3) lowered the enzyme efficiency 17–20 times compared to 1. Similarly, the kcat values of the reaction correlated with resonance stabilization in the order 17 > 7; 22 > 4; 22 > 27 > 28 > 29. Furthermore, a 9-fold higher kcat value of 22 compared to 4 suggests that the resonance stabilization of the carbocation by the benzylic group contributes to the rate of the reaction. However, the kcat values of the cyclohexyl (4) and benzyl analogues (22) were nearly 310- and 36-fold lower than the kcat value of 1. When an electron withdrawing group such as chloro (8) or bromo (9) replaced one of the methyl groups of 1, the efficiency of the enzyme was lowered nearly 150–250-fold compared to the natural alkyl donor, 1, which explains the importance of the methyl group in stabilizing the carbocation. Surprisingly, the dienyl analogues (17–20) displayed relatively higher rates with just ∼2-fold lower kcat values compared to the natural alkyl donor, 1, implying that the extended resonance stabilization of the carbocation favors the FgaPT2 catalyzed reaction. However, the KM values of the dienyl analogues were 5–10-fold higher than the KM value of the natural alkyl donor, 1 and the trend of KM values correlated with the increasing length of the carbon chain (C5–C7), in the order 17 < 19 < 20.| Alkyl-PP analogue | K cat (min−1) | K M (mM) | k cat/KM (mM−1 min−1) | Relative catalytic efficiencya |
|---|---|---|---|---|
| a Relative catalytic efficiency is defined as the percent kcat/KM when compared to 1. | ||||
| 1 | 50 ± 1 | 0.14 ± 0.01 | 349 ± 28 | 100 |
| 2 | 9.9 ± 0.4 | 0.48 ± 0.06 | 21 ± 3 | 5.88 |
| 3 | 6.5 ± 0.1 | 0.37 ± 0.02 | 17 ± 1 | 5.04 |
| 4 | 0.160 ± 0.006 | 0.16 ± 0.02 | 1.0 ± 0.1 | 0.28 |
| 5 | 5.04 ± 0.18 | 0.31 ± 0.03 | 16 ± 2 | 4.48 |
| 6 | 3.2 ± 0.4 | 1.4 ± 0.2 | 2.2 ± 0.4 | 0.64 |
| 7 | 0.57 ± 0.02 | 0.14 ± 0.01 | 3.9 ± 0.4 | 1.14 |
| 8 | 1.3 ± 0.1 | 0.53 ± 0.08 | 2.5 ± 0.4 | 0.70 |
| 9 | 0.47 ± 0.01 | 0.35 ± 0.03 | 1.3 ± 0.1 | 0.38 |
| 17 | 26 ± 1 | 0.66 ± 0.06 | 39 ± 4 | 10.92 |
| 18 | 28 ± 1 | 1.1 ± 0.1 | 26 ± 3 | 7.14 |
| 19 | 20.2 ± 0.9 | 1.3 ± 0.1 | 16 ± 2 | 4.34 |
| 20 | 16.9 ± 2 | 1.39 ± 0.2 | 12 ± 2 | 3.39 |
| 22 | 1.36 ± 0.02 | 0.22 ± 0.01 | 6.2 ± 0.3 | 1.74 |
| 24 | 1.93 ± 0.05 | 0.57 ± 0.04 | 3.4 ± 0.2 | 0.95 |
| 25 | 0.170 ± 0.005 | 0.73 ± 0.06 | 0.23 ± 0.02 | 0.064 |
| 27 | 0.276 ± 0.004 | 0.4 ± 0.02 | 0.69 ± 0.04 | 0.19 |
| 28 | 0.196 ± 0.004 | 0.43 ± 0.03 | 0.46 ± 0.03 | 0.13 |
| 29 | 0.134 ± 0.003 | 0.32 ± 0.03 | 0.42 ± 0.04 | 0.12 |
In order to understand the structural basis for the alkyl group specificity, we analyzed the alkyl-binding site within the crystal structure of FgaPT2 (PDB: 3I4X),20 where the pyrophosphate moiety of DMASPP is anchored in the active site by hydrogen bonding interaction between the oxygen atom of phosphate groups and the amine side chains of arginine and lysine residues (Arg100, Arg257, Arg404, Lys187 and Lys259) (Fig. 4). The alkyl group is encaged within the hydrophobic cavity formed by the side chains of Tyr189, Tyr191, Leu245, Tyr261, Met328, Tyr345, Tyr398, and Tyr413 residues (Fig. 4). The carbocation formed during the reaction is stabilized via π-interactions with 2 aromatic rings: substrate tryptophan on one side and Tyr345 on the other side.20 A closer analysis of the active site revealed that the the methyl groups of DMASPP are within 3.6 Å and 4.4 Å from the side chain of Met328 and Tyr398, respectively (Fig. 4). Therefore, considering the C–C bond length is about ∼1.5 Å, any additional increase in size of the alkyl group beyond the length of DMAPP will have potential steric clash with Met328 and/or Tyr398. This explains the higher KM values seen in the case of alkyl-PP analogues with a longer carbon chain, especially the dienyl analogues (17, 18, 19, and 20) which have a good catalytic ability and increased KM values. Similarly, the C2 and C3 of DMASPP are in the range of 3.7–4.5 Å from the side chain of Tyr345 and Tyr261 (Fig. 4). Therefore, the C2-methyl substitution in 19 and 20 will have additional steric constraints from the side chains of Tyr345 and Tyr261 and potentially explains the lowered catalytic efficiency.
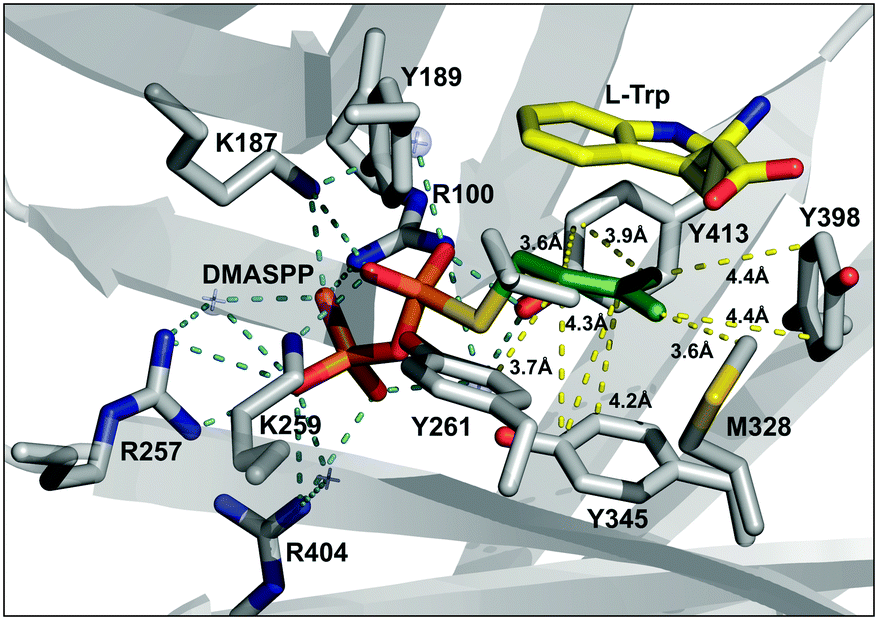 | ||
| Fig. 4 The active site of the FgaPT2 structure (PDB: 3I4X)20 displaying distances between DMASPP and the active site residues, where pyrophosphate interactions and alkyl group distances within the active site are colored cyan and yellow, respectively. | ||
Among the benzylic analogues, the mono-substituted analogues (24, 25, 27, 28, and 29) displayed similar KM values, which were 2–3-fold higher than the KM value of the benzyl analogue, 22. The 10-fold higher kcat value of o-methoxy (24) compared to m-methoxy (25) suggests that benzylic cation stabilization by an o-methoxy group increases the efficiency of the FgaPT2 catalyzed alkylation reaction. Among the para-substituted halogens, the kcat values followed the order benzyl (22) > p-fluorobenzyl (27) > p-chloro (28) ≅ p-bromo (29), suggesting a combination of steric factors and possible stabilization of the carbocation of the 27 carbocation via back donation of the non-bonded electron pairs of the fluorine atom to the 27 carbocation through effective 2p–2p orbital interaction. The observed kcat values of para-substituted halogens were ∼5–10-fold lower than the kcat value of 22, which is consistent with the inductive effect of the halogens. Surprisingly, while the para-substituted halogens were accepted by FgaPT2, p-methoxy and p-methyl were not substrates for FgaPT2 catalyzed reaction even with their ability to contribute to the stabilization of the benzylic cation, likely due to the steric factors in the active site of the enzyme. These results emphasize the fact that the FgaPT2 catalyzed Friedel–Craft alkylation reaction proceeds via a stabilized carbocation,41 and the steric factor and the orientation of the alkyl group in the active site have a major role in the ability of FgaPT2 to utilize alkyl donors.
Despite the fact that the present analysis suggests the steric factor to be a major contributing aspect to the low or no turn-over of alkyl donors containing long carbon chains or bulky aromatic groups, a more definite answer requires in-depth mutational studies or 3D structural studies of FgaPT2 in the presence of diverse unnatural analogues. Remarkably, all amino acids involved in the shielding of the dimethylallyl carbocation are conserved among the DMATS enzymes. Therefore, the conclusions for the alkyl-PP substrate specificity drawn from the present study of FgaPT2 are likely to be valid for other DMATS orthologs as well. Overall, while the active site of wild-type FgaPT2 is optimized for the efficient transfer of the dimethylallyl group, future structure-based engineering is expected to produce enzyme variants with enhanced efficiency and altered substrate scope.
Regio-chemistry of the alkyl-tryptophan products
To assess the regio-specificity of FgaPT2 catalyzed alkyl transfer, the 19 analytical scale reactions with an alkyl-tryptophan yield >20% (reaction involving 1–9, 17–20, 22, 24, 25, 27–29, Fig. 3B) were scaled up. Subsequently, all alkyl-L-Trp analogues (35–55 in Fig. 5) were purified via RP-HPLC, and 1H and 2D NMR experiments were carried out in deuterated DMSO or methanol. While eight of the 19 alkyl-transfer reactions involving alkyl donors 4, 6–9, 17, 19 and 20 yielded two or more mono-alkylated products based on RP-HPLC and HRMS analysis, regio-specific alkyl-group attachment of minor products of reactions involving 6, 8, 9, 17, 19, and 20 could not be attained due to insufficient intensity of NMR signals. The regiospecificity of 21 alkyl-tryptophan analogues was determined using 1H and 2D NMR analysis. The detailed NMR analysis of 21 alkyl-tryptophan analogues revealed that FgaPT2 is able to catalyze alkyl transfer at four different positions (N1, C3, C4, and C5) of tryptophan using unnatural alkyl donors, resulting in normal alkylation at C4 and C5 and normal or reverseC5′ alkylation at C3 and N1 of tryptophan (Fig. 5). Among the 21 alkyl-tryptophan analogues characterized by NMR (see ESI† Table S5), the alkyl group of seven were at C4 (derived from 1–5, 7, and 24), 11 at C5 (derived from 4, 6–9, 18, 22, 25, 27–29), one at C3 (derived from 17) forming hexahydro-pyrroloindoline product, and two were at the N1 (derived from 19 and 20) position of tryptophan, respectively (see ESI† Fig. S1 and S4).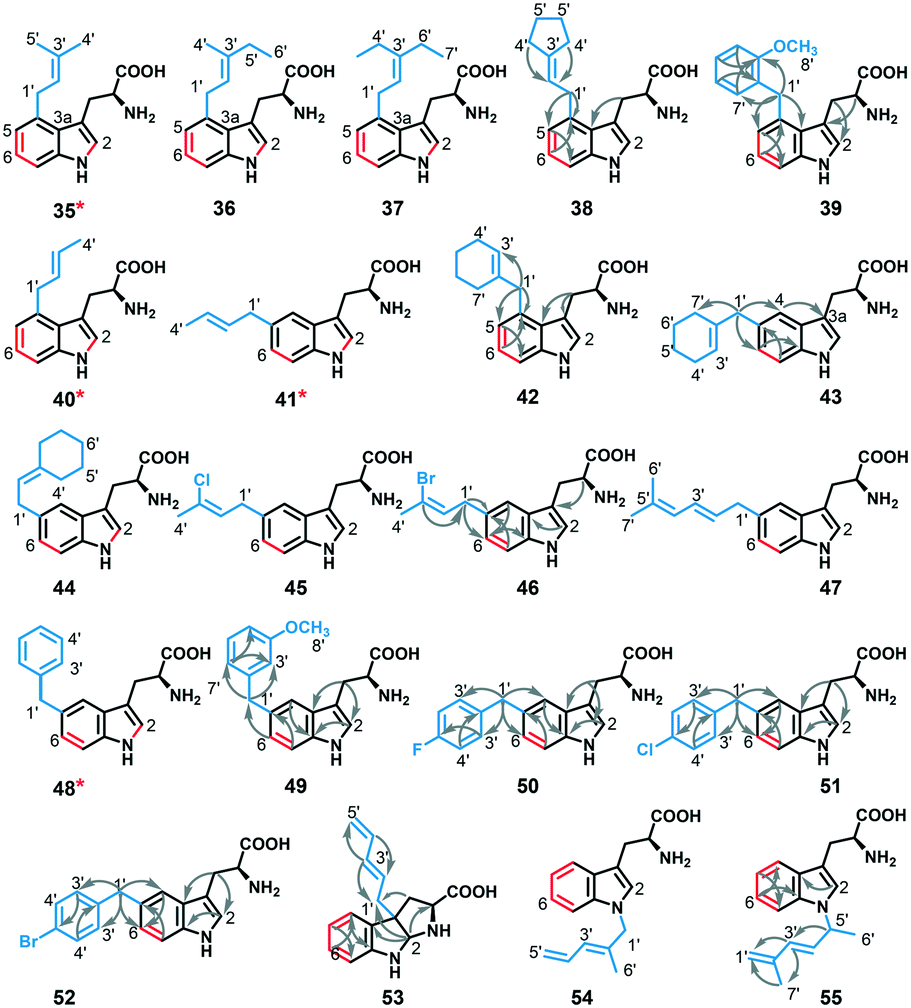 | ||
| Fig. 5 NMR characterized structures of isolated alkyl-tryptophan products obtained from FgaPT2 reaction with L-Trp and alkyl-PP analogues. The red colored bonds represent the COSY correlation between the protons across the bond and arrows represent HMBC correlations of representative compounds, respectively (see the ESI† for NMR Table S5). * represents known compounds. | ||
Intriguingly, apart from the strict C4 alkylation of tryptophan by FgaPT2 using the natural alkyl donor 1, the unnatural alkyl donors, 2, 3 and 5, which contained an ethyl group (2 and 3) in place of one or both methyl group/s of 1, or the similarly sized conformationally restricted cyclopentyl analogue (5) selectively added the alkyl group at the C4 position of tryptophan. Alkyl-PP analogues (4 and 7) with a smaller alkyl chain in the linear direction compared to 1 resulted in a mixture of C4 and C5 regio-isomers, and alkyl-PP analogues with a methyl group of 1 replaced with a halogen (8 and 9) or a larger alkyl chain (6 and 18) predominantly produced the C5 alkylated product. While most of the unnatural benzylic analogues (22, 25, 27, 28, and 29) produced selectively C5 alkylation of tryptophan, the o-methoxy benzyl analogue (24) produced a single C4-alkylated tryptophan. Careful analysis of the C4 alkylating analogues (1, 2, 3, 5, and 24) suggests that C4 substitution is preferred when the carbon chain length is less than 6 while C3′ is also disubstituted to facilitate stacking interaction with the indole ring. Therefore, the active site steric factors in combination with improper stacking interaction of the alkyl group with the indole ring conceivably displaces the carbocation resulting in direct addition of the alkyl group to C5 of tryptophan, as seen for 11 of the C5-alkyl-Trp products. The addition of alkyl groups at the C5 position of tryptophan by most of the alkyl-PP analogues is consistent with the orientation of DMASPP with respect to the indole ring in the active site of the FgaPT2 crystal structure,20 where C5 of tryptophan is just 3.5 Å from the C1′ of the alkyl group compared to a distance of 3.8 Å between C4 of tryptophan and the C1′ of the alkyl group (Fig. 6).
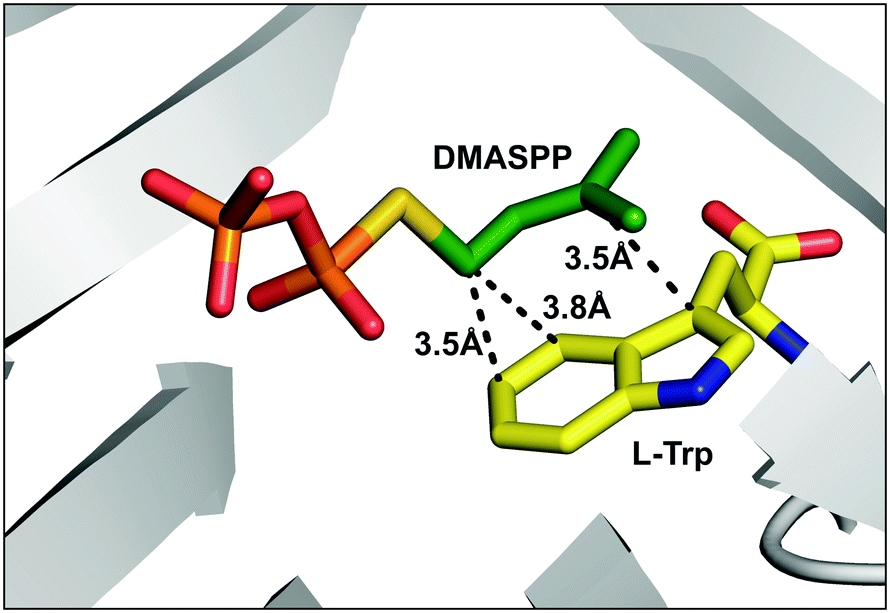 | ||
| Fig. 6 The active site of the FgaPT2 structure (PDB: 3I4X)20 in complex with L-Trp and DMASPP displaying distances between C1′ of DMASPP and C4 and C5 of L-Trp, and C3′ of DMASPP and C3 of L-Trp. | ||
Among the dienyl analogues, except for 18, which alkylated the C5 position due to possible steric hindrance in the active site, the rest of the dienyl analogues (17, 19, and 20) produced C3, N1-normal, and N1-reverseC5′ alkylation. While in all three cases alkylation can be explained as direct addition of the alkyl group to the respective position (via carbocations A- or C-form for 17 (Fig. 7A), A-form for 19 (Fig. 7B) and C-form for 20 (Fig. 7C), respectively), an alternative explanation is that these products could have arisen from the attachment of C3′ of the alkyl group (B-form in Fig. 7) at C3 of tryptophan followed by a rearrangement. Importantly, due to the conjugated double bonds in analogues 17, 19 and 20, there are three possible resonance structures for the carbocation (Fig. 7, A-, B- and C-form), with the divinyl B-form being the most stabilized resonance form. As seen from the Michaelis complex within the crystal structure of FgaPT2, the C3 of tryptophan is just 3.5 Å from C3′ of DMASPP (Fig. 6), and thus, it is likely that the stable ‘B-form’ adds at the reactive C3 position of tryptophan due to the carbocation stability and its proximity to C3 of tryptophan. Subsequently, the C3 reverse alkylated iminium intermediate undergoes a [1,3] alkyl shift in the presence of 17 to yield a normal or reverseC5′ C3 alkylation product of 17 (Fig. 7A), both of which are structurally indistinguishable. Similarly, the C3 reverse alkylated iminium intermediate undergoes an aza-Cope rearrangement in the presence of 19 and 20 to yield an N1 normal and N1 reverseC5′ alkylation product with 19 and 20, respectively (Fig. 7B and C). Especially in the case of 19 and 20, the direct addition to N1 of the indole ring (using A-form for 19 and C-form for 20) seems unlikely as the indole nitrogen is very poorly nucleophilic when compared to the C3 position. Thus, these product studies involving C4, C5, C3, and N1 alkyl-tryptophan analogues suggest that the the regioselectivity of tryptophan alkylation by FgaPT2 depends primarily on the nature of the alkyl donor. Engineering studies of FgaPT2 along with structural characterization using a combination of acceptor and non-native donor analogues are underway to understand the molecular description responsible for FgaPT2 regioselectivity.
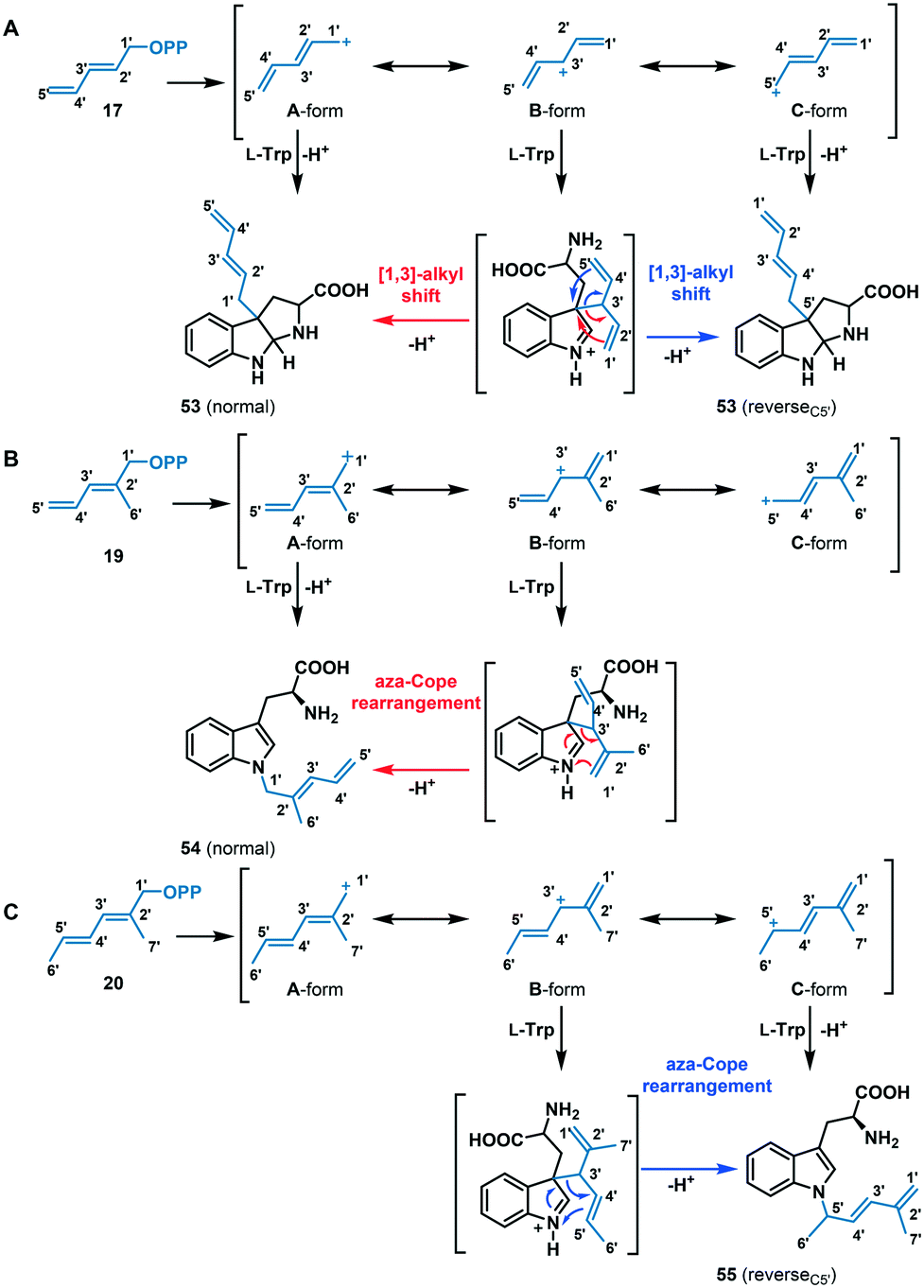 | ||
| Fig. 7 Proposed mechanism for FgaPT2 catalyzed alkyl-transfer reaction of tryptophan with (A) 17, (B) 19 and (C) 20. | ||
Alkyl-diversification of indolocarbazole, 7-hydroxy-K252c (56)
To subsequently assess the practical utility of FgaPT2 for alkyl-diversification of NPs, indolocarbazole 56 was selected as the representative candidate, which was previously demonstrated to be taken by FgaPT2 with the native alkyl-donor, 1.38 The synthesis of 56 was carried out using previously reported methods.47 Standard assay conditions (1.2 mM alkyl-PP analogue, 1 mM 7-hydroxy-K252c, 5 μM FgaPT2, 25 mM Tris, 5 mM MgCl2, 50 mM KCl, pH 7.5, 16 h at 35 °C) were adopted to facilitate the assessment of the donor substrate specificity of FgaPT2 using synthetic alkyl-PP analogues (Fig. 3A) with 56. From this pilot study, six of the 34 synthetic alkyl-PP analogues led to the production of the corresponding alkylated indolocarbazoles in moderate yields (≥15%, Fig. 8 and ESI† Fig. S3). The product yield decreased with the size of the alkyl chain, in the order 1 > 2 > 3 and 5 > 6, respectively. Surprisingly, the dienyl analogues with comparable carbon chain lengths as alkenyl analogues did not serve as substrates for FgaPT2 catalyzed reaction with 56. Subsequently, all six analytical scale reactions were scaled-up and purified via RP-HPLC for the investigation of regio-chemical alkyl substitution on 56 (Fig. 8, ESI† Tables S4 and S6). The HRMS analysis of FgaPT2 catalyzed reaction with 56 using 1, 2, 3, 4, 5 or 6 as alkyl donors provided the mass corresponding to the mono-alkylated product of 56 (see ESI† Table S4). Based on the detailed analysis of 1H, COSY, 1H-13C HSQC, and 1H-13C HMBC spectra, all six compounds confirmed the C3-normal alkylation of 56 (see ESI† Table S6). Thus, FgaPT2 catalyzes regio-specific C3-alkylation of 56 in the presence of natural and unnatural alkyl-donors. The C3 position of 56 structurally corresponds to the C5 position of tryptophan. These results, along with the C5-alkylation of L-Trp using most of the unnatural alkyl-PP donors, imply that the preferred position for FgaPT2 catalyzed alkyl transfer is the C5 position of the indole ring.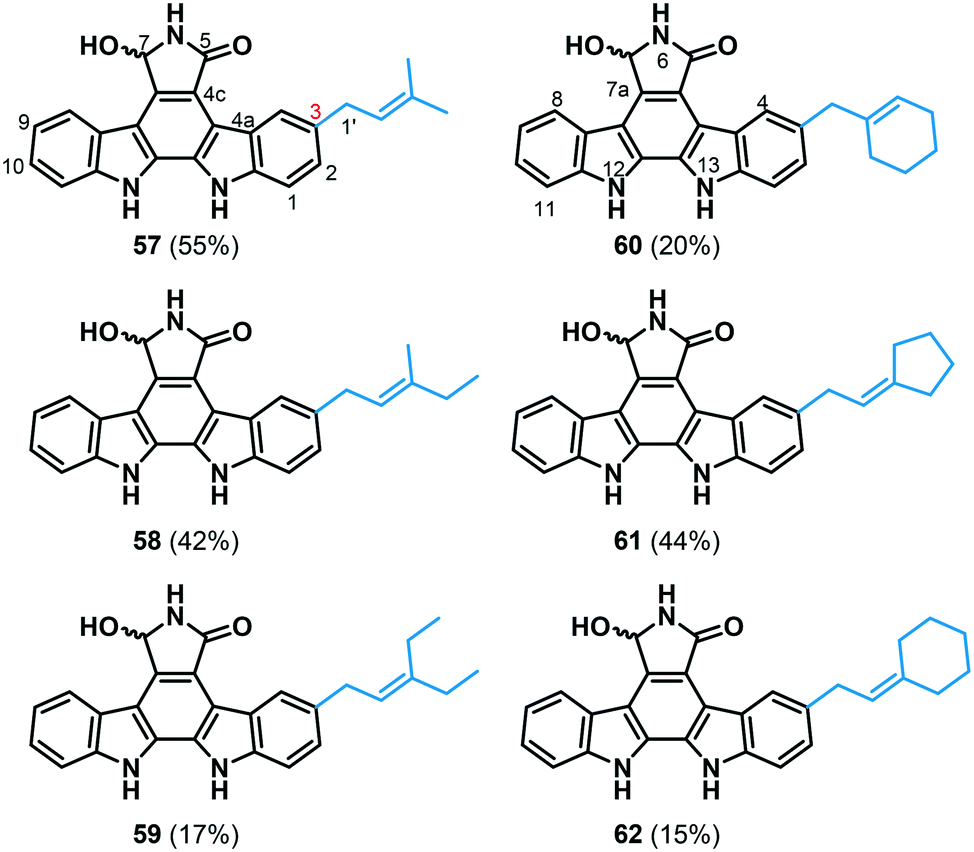 | ||
| Fig. 8 FgaPT2-catalyzed alkyl-diversification of 7-hydroxy-indolocarbazole (56). NMR characterized structures of pure isolated alkyl-7-hydroxy-indolocarbazole products (57–62) of the enzymatic reaction of 56 with alkyl-PP analogues. The percent conversion in the standard analytical scale reaction for each compound is enclosed in parentheses (see the ESI† for NMR, Table S6). | ||
Finally, the cytotoxic activity of compounds 56–62 was studied in human colon carcinoma (HCT-116) cells. While the C3-alkyl substituted analogues of 56 displayed 3–6-fold diminished cytotoxicity in HCT116 (Table 2), generation of these analogues demonstrated the biocatalytic utility of FgaPT2 in differential alkylation of indole-containing natural products. Notably, this is the first report on DMATS-catalyzed differential alkylation of a natural product, and it clearly sets the stage for the utility of FgaPT2 for differential alkylation of other natural products.
| Compound number | 56 | 57 | 58 | 59 | 60 | 61 | 62 |
|---|---|---|---|---|---|---|---|
| IC50 (μM) | 11 ± 1 | 62 ± 12 | 38 ± 1 | 35 ± 1 | 61 ± 23 | 58 ± 6 | 31 ± 1 |
Conclusions
In conclusion, this work has revealed that FgaPT2 is a highly permissive enzyme and accepts diverse allylic and benzylic unnatural alkyl-donor substrates. While FgaPT2 can transfer alkyl groups at C4, C5, N1 and C3 positions of L-Trp, the C5 position is the preferred alkylation position for FgaPT2 with unnatural alkyl donors. Further, our studies involving the synthesis of differently alkylated indolocarbazole highlight the utility of FgaPT2 for broader applications such as alkyl-diversification of indole-containing natural products for drug discovery and other synthetic applications. Future enzyme mutational studies along with substrate engineering is expected to drive the development of more efficient and selective FgaPT2-based biocatalysts.Experimental
General materials
Unless otherwise stated, all chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA), Acros (New Jersey, USA), Alfa-Aesar (Ward Hill, MA, USA) or TCI (Portland, OR, USA) and were of reagent grade or better. The PD-10 column and Ni-NTA superflow columns were purchased from GE Healthcare (Piscataway, NJ). The HCT-116 cell line was purchased from ATTC (ATCC® CCL-247™).General methods
All reactions were conducted in oven- or flame-dried glassware under a nitrogen atmosphere with anhydrous solvents, unless otherwise noted. Reactions were monitored by thin-layer chromatography (TLC) (EMD Millipore Corp, Billerica, MA, USA) and visualization was accomplished with UV light (254 nm) followed by staining with phosphomolybdic acid solution or anisaldehyde solution and heating. Flash column chromatography was performed using ACS grade solvents and silica gel (SiliCycle Inc, P60, particle size 40–63 μm). HPLC was conducted using an Agilent 1220 system equipped with a DAD detector.General procedure of synthesis of alkyl-PP analogues
The procedures for the synthesis of alkyl-PP analogues 1–4, 6–8, and 10–21 and phosphorylation using bis(triethylammonium) phosphate (TEAP) and tris(tetrabutylammonium) pyrophosphate (TBAPP) have been reported previously.432-Cyclopentylideneethyl pyrophosphate (5)
The title product was obtained as an ivory solid (33%) from 2-cyclopentylideneethan-1-ol48 using TEAP. TLC (iPrOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NH4OH:H2O 7:2:1 v/v): Rf = 0.35; 1H NMR (400 MHz, deuterium oxide) δ 5.56 (tt, J = 7.3, 2.4 Hz, 1H), 4.45 (t, J = 7.0 Hz, 2H), 2.40–2.20 (m, 4H), 1.65 (dq, J = 20.8, 6.7 Hz, 4H). 31P NMR (162 MHz, deuterium oxide) δ −7.73 (d, J = 22.0 Hz), −10.51 (d, J = 21.9 Hz). HR-ESI-MS (m/z): [M − H]− calcd for C7H13O7P2−, 271.0142; observed, 271.0148.
:NH4OH:H2O 7:2:1 v/v): Rf = 0.35; 1H NMR (400 MHz, deuterium oxide) δ 5.56 (tt, J = 7.3, 2.4 Hz, 1H), 4.45 (t, J = 7.0 Hz, 2H), 2.40–2.20 (m, 4H), 1.65 (dq, J = 20.8, 6.7 Hz, 4H). 31P NMR (162 MHz, deuterium oxide) δ −7.73 (d, J = 22.0 Hz), −10.51 (d, J = 21.9 Hz). HR-ESI-MS (m/z): [M − H]− calcd for C7H13O7P2−, 271.0142; observed, 271.0148.
(Z)-3-Bromobut-2-en-1-yl pyrophosphate (9)
The title product was obtained as a white solid (74%) from crude (Z)-3-bromo-1-chlorobut-2-ene (prepared from (Z)-3-bromo-but-2-en-1-ol49 using the procedure described for (Z)-3-chloro-1-chlorobut-2-ene50) using TBAPP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.45; 1H NMR (400 MHz, deuterium oxide) δ 5.99 (td, J = 6.2, 1.4 Hz, 1H), 4.53–4.40 (m, 2H), 2.29 (q, J = 1.3 Hz, 3H). 31P NMR (162 MHz, deuterium oxide) δ −7.28 (d, J = 21.9 Hz), −10.61 (d, J = 22.0 Hz). HR-ESI-MS (m/z): [M − H]− calcd for C4H8BrO7P2−, 308.8934, observed, 308.8932.
Benzyl pyrophosphate (22)
22 was obtained as a white solid (78%) from benzylbromide using TBAPP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.4; 1H NMR (400 MHz, deuterium oxide) δ 7.42–7.36 (m, 2H), 7.35–7.23 (m, 3H), 4.88 (d, J = 6.6 Hz, 2H); 31P NMR (162 MHz, deuterium oxide) δ −8.10 (d, J = 21.5 Hz), −10.80 (d, J = 21.5 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C7H9O7P2−, 266.9824; observed, 266.9828.
p-Methoxybenzyl pyrophosphate (23)
23 was obtained as a white solid (82%) from p-methoxybenzyl bromide using TBAPP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.39; 1H NMR (400 MHz, deuterium oxide) δ 7.31 (d, J = 8.5 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 4.78 (d, J = 6.3 Hz, 2H), 3.70 (s, 3H); 31P NMR (162 MHz, deuterium oxide) δ −8.54 (d, J = 21.1 Hz), −10.89 (d, J = 21.1 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C8H11O8P2−, 296.9929; observed, 296.9939.
o-Methoxybenzyl pyrophosphate (24)
24 was obtained as a white solid (32%) from o-methoxybenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.39; 1H NMR (400 MHz, deuterium oxide) δ 7.32 (dd, J = 7.5, 1.7 Hz, 1H), 7.21 (td, J = 7.9, 1.7 Hz, 1H), 6.95–6.74 (m, 2H), 4.83 (d, J = 6.8 Hz, 2H), 3.69 (s, 3H); 31P NMR (162 MHz, deuterium oxide) δ −8.45 (d, J = 21.4 Hz), −10.76 (d, J = 21.2 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C8H11O8P2−, 296.9929; observed, 296.9927.
m-Methoxybenzyl pyrophosphate (25)
25 was obtained as a white solid (30%) from m-methoxybenzyl using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.39; 1H NMR (400 MHz, deuterium oxide) δ 7.25 (t, J = 8.0 Hz, 1H), 6.99 (d, J = 2.6 Hz, 2H), 6.84 (dd, J = 8.2, 2.5 Hz, 1H), 4.85 (d, J = 6.6 Hz, 2H); 31P NMR (162 MHz, deuterium oxide) δ −7.37 (d, J = 22.5 Hz), −10.75 (d, J = 21.7 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C8H11O8P2−, 296.9929; observed, 296.9926.
p-Methylbenzyl pyrophosphate (26)
26 was obtained as a white solid (32%) from p-methylbenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.39; 1H NMR (400 MHz, deuterium oxide) δ 7.22 (d, J = 7.9 Hz, 2H), 7.10 (d, J = 7.8 Hz, 2H), 4.77 (d, J = 6.3 Hz, 2H), 2.16 (s, 3H); 31P NMR (162 MHz, deuterium oxide) δ −6.95 (d, J = 22.6 Hz), −10.81 (d, J = 21.8 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C8H11O7P2−, 280.9980; observed, 280.9978.
p-Fluorobenzyl pyrophosphate (27)
27 was obtained as a white solid (29%) from p-fluorobenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.4; 1H NMR (400 MHz, deuterium oxide) δ 7.37 (dd, J = 8.2, 5.7 Hz, 2H), 7.02 (t, J = 8.7 Hz, 2H), 4.85 (dd, J = 17.1, 6.8 Hz, 2H); 31P NMR (162 MHz, deuterium oxide) δ −7.64 (d, J = 22.3 Hz), −10.85 (d, J = 21.6 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C7H8FO7P2−, 284.9729; observed, 284.9728.
p-Chlorobenzyl pyrophosphate (28)
28 was obtained as a white solid (35%) from p-chlorobenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.41; 1H NMR (400 MHz, deuterium oxide) δ 7.40–7.06 (m, 4H), 4.79 (d, J = 6.8 Hz, 2H); 31P NMR (162 MHz, deuterium oxide) δ −7.77 (d, J = 21.7 Hz), −10.90 (d, J = 21.9 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C7H8ClO7P2−, 300.9434; observed, 300.9428.
p-Bromobenzyl pyrophosphate (29)
29 was obtained as a white solid (72%) from p-bromobenzyl bromide using TBAPP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.41; 1H NMR (400 MHz, deuterium oxide) δ 7.40 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H), 4.77 (d, J = 6.9 Hz, 2H); 31P NMR (162 MHz, deuterium oxide) δ −10.38 (d, J = 21.4 Hz), −11.25 (d, J = 20.7 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C7H8BrO7P2−, 344.8929; observed, 344.8927.
p-Nitrobenzyl pyrophosphate (30)
30 was obtained as a brown solid (28%) from p-nitrobenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.37; 1H NMR (400 MHz, deuterium oxide) δ 8.05 (d, J = 8.5 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 4.91 (d, J = 7.4 Hz, 2H); 31P NMR (162 MHz, deuterium oxide) δ −7.29 (d, J = 21.9 Hz), −10.78 (d, J = 21.7 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C7H8NO9P2−, 311.9674; observed, 311.9677.
2,4-Dimethoxybenzyl pyrophosphate (31)
31 was obtained as a white solid (30%) from 2,4-dimethoxybenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.35; 1H NMR (400 MHz, deuterium oxide) δ 7.28 (d, J = 9.0 Hz, 1H), 6.50 (dd, J = 4.5, 2.1 Hz, 2H), 4.79 (d, J = 6.3 Hz, 2H), 3.71 (s, 3H), 3.70 (s, 3H); 31P NMR (162 MHz, deuterium oxide) δ −6.58 (d, J = 21.9 Hz), −10.58 (d, J = 21.7 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C9H13O9P2−, 327.0035; observed, 327.0048.
3,4-Dimethoxybenzyl pyrophosphate (32)
32 was obtained as a white solid (35%) from 3,4-dimethoxybenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.35; 1H NMR (400 MHz, deuterium oxide) δ 7.06 (d, J = 1.8 Hz, 1H), 6.98–6.87 (m, 2H), 4.79 (d, J = 6.2 Hz, 2H), 3.74 (s, 3H), 3.72 (s, 3H); 31P NMR (162 MHz, deuterium oxide) δ −7.20 (d, J = 21.7 Hz), −10.83 (d, J = 21.8 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C9H13O9P2−, 327.0035; observed, 327.0027.
3,5-Difluorobenzyl pyrophosphate (33)
33 was obtained as a white solid (28%) from 3,5-difluorobenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.37; 1H NMR (400 MHz, deuterium oxide) δ 6.94 (d, J = 7.5 Hz, 2H), 6.75 (t, J = 9.8 Hz, 1H), 4.84 (d, J = 7.3 Hz, 2H). 31P NMR (162 MHz, deuterium oxide) δ −6.99 (d, J = 21.9 Hz), −10.82 (dd, J = 18.2, 10.6 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C7H7F2O7P2−, 302.9635; observed, 302.9629.
3,4,5-Trimethoxybenzyl pyrophosphate (34)
34 was obtained as a white solid (31%) from 3,4,5-trimethoxybenzyl alcohol using TEAP. TLC (iPrOH:NH4OH:H2O 7:2:1 v/v): Rf = 0.35; 1H NMR (400 MHz, deuterium oxide) δ 6.76 (d, J = 2.0 Hz, 2H), 4.80 (dd, J = 6.4, 1.7 Hz, 2H), 3.75 (s, 6H), 3.64 (d, J = 1.9 Hz, 3H). 31P NMR (162 MHz, deuterium oxide) δ −7.38 (d, J = 21.6 Hz), −10.88 (d, J = 21.9 Hz); HR-ESI-MS (m/z): [M − H]− calcd for C10H15O10P2−, 357.0140; observed, 357.0135.
Determination of structures
High-resolution mass spectrometry (HRMS) and liquid chromatography mass spectrometry (LCMS) were conducted on an Agilent 6545-QTOF W/1290 HPLC mass spectrometer at the University of Oklahoma Department of Chemistry and Biochemistry. NMR spectra were obtained on Varian VNMRS 400 or 500 MHz instruments at the NMR facility of the Department of Chemistry and Biochemistry of the University of Oklahoma using 99.9% DMSO-d6 with 0.05% v/v TMS or 99.9% D2O or 99.8% MeOH-d4 and 99.9% acetone-d6 (Cambridge Isotope Laboratories, MA, USA). 1H, 13C, and 31P chemical shifts were referenced to internal solvent resonances. Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), m (multiplet), and br (broad). Chemical shifts are reported in parts per million (ppm) and coupling constants J are given in Hz. All NMR spectra were recorded at ambient temperature and processed using MestReNova software.In vitro FgaPT2 assay
The recombinant FgaPT2 was overproduced in Escherichia coli BL21(DE3) cells transformed with the codon-optimized synthetic FgaPT2 gene in a pET28a vector. The resulting FgaPT2 with N-terminal His6-fusion protein was purified to homogeneity via Ni-NTA affinity chromatography as described previously.28In vitro FgaPT2 reactions were conducted in a volume of 20 μl with 1.2 mM alkyl-PP analogue (1–34), 1 mM L-Trp or 7-hydroxy-K252c and 5 μM purified FgaPT2 in 25 mM Tris buffer pH 7.5, 5 mM MgCl2, and 50 mM KCl, incubated at 35 °C for 16 h. Reactions were quenched by adding an equal volume of methanol followed by centrifugation (10000 × g for 15 min) to remove the precipitated protein, and product formation for each reaction was subsequently analyzed by RP-HPLC using method A or method C (Fig. S2 and S3†). Negative controls included reaction in the absence of an enzyme or alkyl-PP analogue. For each reaction, the percent yield was based upon the integration of species at 254 nm and calculated by dividing the integrated area of the product by the sum of the integrated areas of the product and/or and the remaining substrate. All putative products were subsequently confirmed by HRMS in positive (+) and/or negative (−) mode (see general methods) (Table S3†). Kinetic parameters were assessed using 1.2 mM L-Trp and variable alkyl-PP analogues (0.1–6 mM). Assays were conducted in triplicate and all rates were confirmed to be linear. The kinetic curves were fit to Michaelis–Menton kinetics using Prism 5.04 (GraphPad Software, Inc. La Jolla, CA 92037 USA). The reactions with >20% product yield were performed in 2–5 mL volume under standard conditions, products were purified by semi-prep HPLC using method B or method D and the putative new products were confirmed by NMR (Tables S5 and S6†).
HPLC method A
To monitor FgaPT2 reactions of L-tryptophan, analytical reverse-phase (RP) HPLC was conducted employing a Gemini-NX C-18 (5 μm, 4.6 mm × 250 mm) column (Phenomenex, Torrance, California, USA) [gradient of 10% B to 25% B over 10 min, 25% B to 100% B over 15 min, 100% B for 3 min, 100% B to 10% B over 0.1 min, 10% B for 7 min (A = ddH2O with 0.1% TFA; B = acetonitrile); flow rate = 1 mL min−1; A254]. The reaction was monitored by means of the retention time difference between the starting material and product.HPLC method B
Semi-preparative RP HPLC was conducted using a Gemini-NX C-18 (5 μm, 10 mm × 250 mm) column (Phenomenex, Torrance, California, USA) to purify the L-Trp analogues [gradient of 10% B to 25% B over 10 min, 25% B to 100% B over 15 min, 100% B for 3 min, 100% B to 10% B over 0.1 min, 10% B for 7 min (A = ddH2O with 0.1% TFA; B = acetonitrile); flow rate = 2 mL min−1; A254].HPLC method C
To monitor FgaPT2 reactions of 7-hydroxy-K252c, analytical reverse-phase (RP) HPLC was conducted employing a Gemini-NX C-18 (5 μm, 4.6 mm × 250 mm) column (Phenomenex, Torrance, California, USA) [gradient of 20% B to 50% B over 6 min, 50% B to 100% B over 16 min, 100% B for 2 min, 100% B to 10% B over 0.1 min, 10% B for 6 min (A = ddH2O with 0.1% TFA; B = acetonitrile); flow rate = 1 mL min−1; A254]. The reaction was monitored by means of the retention time difference between the starting material and product.HPLC method D
Semi-preparative RP HPLC was conducted using a Gemini-NX C-18 (5 μm, 10 mm × 250 mm) column (Phenomenex, Torrance, California, USA) to purify the 7-hydroxy-K252c analogues [gradient of 20% B to 60% B over 10 min, 60% B to 100% B over 10 min, 100% B over 3 min, 100% B to 20% B over 0.1 min, 20% B for 7 min (A = ddH2O with 0.1% TFA; B = acetonitrile); flow rate = 2 mL min−1; A254].The biological activity of alkyl-7-hydroxy-K252c analogues
HCT-116 cells were seeded out in 96-well plates containing 75 μl of McCoy's media at a cell number of 2.0e3 per well. Following a 24 h incubation, 25 μl of medium and 20 μl of CellTiter-Blue (Promega) were added per well to one 96-well plate and incubated at 37 °C for 90 min. Fluorescence was detected (544 nm excitation; 590 nm emission) on a plate reader (GloMax Explorer, Promega) to establish the baseline viability at the time of treating. Compounds were serially diluted in the medium at 4X concentration and delivered to the cells in a volume of 25 μl. After a 48 h incubation, CellTiter-Blue was added for 90 min, and fluorescence was measured as previously described. Growth relative to untreated cells was calculated and expressed as IC50 values on GraphPad Prism software utilizing the four-parameter-dose–response curve. Experiments were run in duplicate (n = 2) utilizing Taxol as an internal control.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by University of Oklahoma start-up funding and by the National Institutes of Health grant AI141950. We gratefully acknowledge the University of Oklahoma, Department of Chemistry and Biochemistry NMR and Mass Spectrometry facility for analytical support. Thanks to J. S. Thorson for graciously providing the FgaPT2 expression strain.Notes and references
- P. H. Wang, A. N. Khusnutdinova, F. Luo, J. Xiao, K. Nemr, R. Flick, G. Brown, R. Mahadevan, E. A. Edwards and A. F. Yakunin, Cell Chem. Biol., 2018, 25, 560–570e566 CrossRef CAS PubMed.
- J. Winkelblech, A. Fan and S. M. Li, Appl. Microbiol. Biotechnol., 2015, 99, 7379–7397 CrossRef CAS PubMed.
- S.-M. Li, Nat. Prod. Rep., 2010, 27, 57–78 RSC.
- O. Saleh, Y. Haagen, K. Seeger and L. Heide, Phytochemistry, 2009, 70, 1728–1738 CrossRef CAS PubMed.
- M. Tello, T. Kuzuyama, L. Heide, J. P. Noel and S. B. Richard, Cell. Mol. Life Sci., 2008, 65, 1459–1463 CrossRef CAS PubMed.
- M. Wang and P. J. Casey, Nat. Rev. Mol. Cell Biol., 2016, 17, 110–122 CrossRef CAS PubMed.
- B. Botta, A. Vitali, P. Menendez, D. Misiti and G. Delle Monache, Curr. Med. Chem., 2005, 12, 717–739 CrossRef PubMed.
- V. Brezani, K. Smejkal, J. Hosek and V. Tomasova, Curr. Med. Chem., 2018, 25, 1094–1159 CrossRef CAS PubMed.
- S. N. Sunassee and M. T. Davies-Coleman, Nat. Prod. Rep., 2012, 29, 513–535 RSC.
- M. Steffensky, A. Mühlenweg, Z.-X. Wang, S.-M. Li and L. Heide, Antimicrob. Agents Chemother., 2000, 44, 1214–1222 CrossRef CAS PubMed.
- N. Tanaka, Y. Yano, Y. Tatano and Y. Kashiwada, Org. Lett., 2016, 18, 5360–5363 CrossRef CAS PubMed.
- X. Chen, E. Mukwaya, M. S. Wong and Y. Zhang, Pharm. Biol., 2014, 52, 655–660 CrossRef CAS PubMed.
- A. Wolf-Rainer, Curr. Med. Chem., 2018, 25, 123–140 Search PubMed.
- T. Usui, M. Kondoh, C. B. Cui, T. Mayumi and H. Osada, Biochem. J., 1998, 333(Pt 3), 543–548 CrossRef CAS PubMed.
- U. Grienke, M. Richter, E. Walther, A. Hoffmann, J. Kirchmair, V. Makarov, S. Nietzsche, M. Schmidtke and J. M. Rollinger, Sci. Rep., 2016, 6, 27156 CrossRef CAS PubMed.
- C. Sanna, M. Scognamiglio, A. Fiorentino, A. Corona, V. Graziani, A. Caredda, P. Cortis, M. Montisci, E. R. Ceresola, F. Canducci, F. Poli, E. Tramontano and F. Esposito, PLoS One, 2018, 13, e0195168 CrossRef PubMed.
- A. Fan, J. Winkelblech and S. M. Li, Appl. Microbiol. Biotechnol., 2015, 99, 7399–7415 CrossRef CAS PubMed.
- M. E. Tanner, Nat. Prod. Rep., 2015, 32, 88–101 RSC.
- C. T. Walsh, ACS Chem. Biol., 2014, 9, 2718–2728 CrossRef CAS PubMed.
- U. Metzger, C. Schall, G. Zocher, I. Unsold, E. Stec, S. M. Li, L. Heide and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309–14314 CrossRef CAS PubMed.
- M. Jost, G. Zocher, S. Tarcz, M. Matuschek, X. Xie, S. M. Li and T. Stehle, J. Am. Chem. Soc., 2010, 132, 17849–17858 CrossRef CAS PubMed.
- J. Wang, C. C. Chen, Y. Yang, W. Liu, T. P. Ko, N. Shang, X. Hu, Y. Xie, J. W. Huang, Y. Zhang and R. T. Guo, Biochem. Biophys. Res. Commun., 2018, 495, 1782–1788 CrossRef CAS PubMed.
- C. P. Wong, T. Awakawa, Y. Nakashima, T. Mori, Q. Zhu, X. Liu and I. Abe, Angew. Chem., Int. Ed., 2018, 57, 560–563 CrossRef CAS.
- T. Awakawa, T. Mori, Y. Nakashima, R. Zhai, C. P. Wong, M. L. Hillwig, X. Liu and I. Abe, Angew. Chem., Int. Ed., 2018, 57, 6810–6813 CrossRef CAS PubMed.
- X. Liu, M. L. Hillwig, L. M. Koharudin and A. M. Gronenborn, Chem. Commun., 2016, 52, 1737–1740 RSC.
- J. M. Schuller, G. Zocher, M. Liebhold, X. Xie, M. Stahl, S. M. Li and T. Stehle, J. Mol. Biol., 2012, 422, 87–99 CrossRef CAS PubMed.
- X. Yu, G. Zocher, X. Xie, M. Liebhold, S. Schutz, T. Stehle and S. M. Li, Chem. Biol., 2013, 20, 1492–1501 CrossRef CAS PubMed.
- S. I. Elshahawi, H. Cao, K. A. Shaaban, L. V. Ponomareva, T. Subramanian, M. L. Farman, H. P. Spielmann, G. N. Phillips, Jr., J. S. Thorson and S. Singh, Nat. Chem. Biol., 2017, 13, 366–368 CrossRef CAS PubMed.
- T. Mori, L. Zhang, T. Awakawa, S. Hoshino, M. Okada, H. Morita and I. Abe, Nat. Commun., 2016, 7, 10849 CrossRef CAS PubMed.
- I. A. Unsold and S. M. Li, Microbiology, 2005, 151, 1499–1505 CrossRef PubMed.
- J. C. Gebler, A. B. Woodside and C. D. Poulter, J. Am. Chem. Soc., 1992, 114, 7354–7360 CrossRef CAS.
- L. Y. Luk and M. E. Tanner, J. Am. Chem. Soc., 2009, 131, 13932–13933 CrossRef CAS PubMed.
- L. Y. Luk, Q. Qian and M. E. Tanner, J. Am. Chem. Soc., 2011, 133, 12342–12345 CrossRef CAS PubMed.
- J. D. Rudolf, H. Wang and C. D. Poulter, J. Am. Chem. Soc., 2013, 135, 1895–1902 CrossRef CAS PubMed.
- N. Steffan, I. A. Unsold and S. M. Li, ChemBioChem, 2007, 8, 1298–1307 CrossRef CAS PubMed.
- N. Steffan and S. M. Li, Arch. Microbiol., 2009, 191, 461–466 CrossRef CAS PubMed.
- X. Yu, X. Xie and S. M. Li, Appl. Microbiol. Biotechnol., 2011, 92, 737–748 CrossRef CAS PubMed.
- X. Yu, A. Yang, W. Lin and S.-M. Li, Tetrahedron Lett., 2012, 53, 6861–6864 CrossRef CAS.
- A. Fan, X. Xie and S. M. Li, Org. Biomol. Chem., 2015, 13, 7551–7557 RSC.
- P. Mai, G. Zocher, L. Ludwig, T. Stehle and S.-M. Li, Adv. Synth. Catal., 2016, 358, 1639–1653 CrossRef CAS.
- M. Liebhold, X. Xie and S. M. Li, Org. Lett., 2012, 14, 4882–4885 CrossRef CAS.
- M. Liebhold and S. M. Li, Org. Lett., 2013, 15, 5834–5837 CrossRef CAS.
- C. Bandari, E. M. Scull, J. M. Masterson, R. H. Q. Tran, S. B. Foster, K. M. Nicholas and S. Singh, ChemBioChem, 2017, 18, 2323–2327 CrossRef CAS PubMed.
- G. Popják, J. L. Rabinowitz and J. M. Baron, Biochem. J., 1969, 113, 861–868 CrossRef PubMed.
- R. K. Keller and R. Thompson, J. Chromatogr. A, 1993, 645, 161–167 CrossRef CAS.
- V. J. Davisson, A. B. Woodside, T. R. Neal, K. E. Stremler, M. Muehlbacher and C. D. Poulter, J. Org. Chem., 1986, 51, 4768–4779 CrossRef CAS.
- M. M. Faul, L. L. Winneroski and C. A. Krumrich, J. Org. Chem., 1998, 63, 6053–6058 CrossRef CAS.
- P. Szcześniak, M. Pieczykolan and S. Stecko, J. Org. Chem., 2016, 81, 1057–1074 CrossRef PubMed.
- R. J. Armstrong, C. García-Ruiz, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 786–790 CrossRef CAS PubMed.
- N. A. Heaps and C. D. Poulter, J. Org. Chem., 2011, 76, 1838–1843 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: These include HPLC chromatograms, HRMS, NMR table, and NMR spectra for all new compounds. See DOI: 10.1039/c9md00177h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |