
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quinolone antibiotics
Thu D. M.
Pham
a,
Zyta M.
Ziora
 b and
Mark A. T.
Blaskovich
*b
b and
Mark A. T.
Blaskovich
*b
aSchool of Chemistry & Molecular Biosciences, The University of Queensland, Brisbane, QLD 4072, Australia
bInstitute for Molecular Bioscience, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: m.blaskovich@uq.edu.au
First published on 28th June 2019
Abstract
The quinolone antibiotics arose in the early 1960s, with the first examples possessing a narrow-spectrum of activity with unfavorable pharmacokinetic properties. Over time, the development of new quinolone antibiotics has led to improved analogues with an expanded spectrum and high efficacy. Nowadays, quinolones are widely used for treating a variety of infections. Quinolones are broad-spectrum antibiotics that are active against both Gram-positive and Gram-negative bacteria, including mycobacteria, and anaerobes. They exert their actions by inhibiting bacterial nucleic acid synthesis through disrupting the enzymes topoisomerase IV and DNA gyrase, and by causing breakage of bacterial chromosomes. However, bacteria have acquired resistance to quinolones, similar to other antibacterial agents, due to the overuse of these drugs. Mechanisms contributing to quinolone resistance are mediated by chromosomal mutations and/or plasmid gene uptake that alter the topoisomerase targets, modify the quinolone, and/or reduce drug accumulation by either decreased uptake or increased efflux. This review discusses the development of this class of antibiotics in terms of potency, pharmacokinetics and toxicity, along with the resistance mechanisms which reduce the quinolones' activity against pathogens. Potential strategies for future generations of quinolone antibiotics with enhanced activity against resistant strains are suggested.
1. Introduction
The quinolones are a family of antibiotics containing a bicyclic core structure related to the compound 4-quinolone (Fig. 1).1 Since their discovery in the early 1960s, they have gained increasing importance as key therapies to treat both community-acquired and severe hospital-acquired infections.2 The first quinolone antibiotic is generally considered to be nalidixic acid, which was reported in 1962 as part of a series of 1-alkyl-1,8-naphthyridines prepared at the Sterling-Winthrop Research Institute.3 However, a 2015 perspective that examined the origins of quinolone antibiotics in greater detail points out that the author of the 1962 publication (George Lesher) described the isolation of-chloro-1-ethyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid in the late 1950s as a by-product of chloroquine synthesis, with modest antibacterial activity leading to further work on analogues, including nalidixic acid.1 Around the same time, Imperial Chemical Industries (ICI) published patent applications with antibacterial quinolones, including a 6-fluoroquinolone.1 Nalidixic acid is a narrow-spectrum agent against enteric bacteria used for treating uncomplicated urinary tract infections (UTIs).4 During the 1970s–1980s, the coverage of the quinolone class was expanded significantly by the breakthrough development of fluoroquinolones, which show a much broader spectrum of activity and improved pharmacokinetics compared to the first-generation quinolone.5 Those fluoroquinolones, such as ciprofloxacin and ofloxacin, are active against both Gram-negative and Gram-positive pathogens; importantly, they are also active against the causative agent of tuberculosis, Mycobacterium tuberculosis. Quinolones have been favoured as antibiotics for more than five decades because of their high potency, broad spectrum of activity, favorable bioavailability, convenient formulations, and high serum concentrations, as well as a comparatively low incidence of side effects.6 Quinolones are widely prescribed for several different types of human infections,7 with side effects including gastrointestinal reactions, CNS reactions, genotoxicity, phototoxicity, and some minor adverse effects.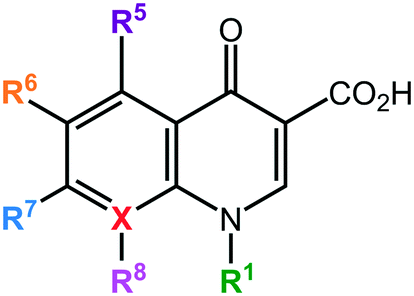 | ||
| Fig. 1 Core structure of quinolone antibiotics. There are 6 important positions for modifications to improve the activity of the drug: R1, R5, R6, R7, R8, and X. X = C defines quinolones, X = N defines naphthyridones. | ||
The quinolone class of antibiotics inhibits the DNA synthesis of bacteria by disrupting the bacterial topoisomerase type II; inhibiting the catalytic activity of DNA gyrase and topoisomerase IV.8 These two enzymes are critical bacterial enzymes that regulate the chromosomal supercoiling required for DNA synthesis.9 Over time, quinolone resistance has become a serious problem among many emerging resistant pathogens.10 The mutations generated by the bacteria against quinolones are generally located on the target enzyme binding sites in DNA gyrase and topoisomerase IV.11 In addition, resistance to this class of antibiotics can be obtained by acquisition of a resistant plasmid from other sources in the environment through horizontal transfer, leading to the rapid spread of resistance.12
This review discusses the current knowledge of the development process of quinolones on how structural modifications in the evolving generations have mediated improvements in terms of potency, pharmacokinetics, and toxicity. It also summarizes the relevant knowledge of mode of actions and resistance. Lastly, the review examines future strategies to improve the activity of this class and overcome the resistance.
2. Development of the quinolones
The prototypical quinolone, nalidixic acid (technically a naphthyridone), was discovered in the 1960s as a by-product during the synthesis of anti-malarial quinine compounds.3 It was soon found to act by inhibiting the activity of bacterial topoisomerase type II enzymes, inhibiting the bacterial replication.13 In 1967, nalidixic acid was approved for clinical treatment for urinary tract infections (UTIs) caused by Gram-negative bacteria.4 However, its use was limited because of the narrow spectrum of activity, low serum concentrations achieved, high inhibitory concentration required, and several adverse effects.4 It was not until the 1980s that improved analogues were made, when the need for new treatments of diarrhea and UTIs caused by resistant Shigella and Escherichia coli led the attention of researchers to improve the activity and optimize the toxicity of the quinolones.Many researchers have studied the structure–activity relationships of quinolone antibiotics. Fig. 1 presents the core structure of the basic quinolones with two major groups developed from it: quinolones and naphthyridones, which can be identified by the ‘X’ position. A carbon atom at the X position defines the quinolones, while a nitrogen atom at the X position defines the naphthyridones.14 Based on their spectrum of activity, quinolones are classified into four generations.15 The development of quinolones from generation to generation to obtain broader spectrum activity has proceeded by addition of different substituents into different position on the pharmacophore. Table 1 presents a summary of the quinolone development process.
| Generation | Name | Structure | Antimicrobial spectrum | Modifications | Comment |
|---|---|---|---|---|---|
| 1 | Nalidixic acid |

|
Gram-negative organisms (except Pseudomonas species) | N at X8 position = naphthyridone | First molecule to be discovered in quinolone class |
| 2a | Enoxacin |
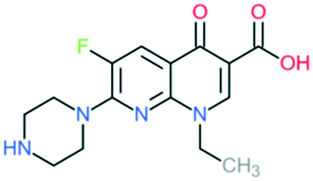
|
All Gram-negative pathogens and some atypical pathogens (including Mycoplasma pneumoniae and Chlamydia pneumoniae) | Addition of (1) piperazine to C7 position, and (2) –F to C6 position | (1) Improves activity against Gram-negative organisms (inhibits the efflux mechanism) |
| Norfloxacin |
|
Addition of (1) piperazine to C7 position (quinolone), and (2) –F to C6 position | (1) Improves bioavailability, side effects | ||
| Improves activity against Gram-negative organisms (inhibits the efflux mechanism) | |||||
| Ciprofloxacin |
|
Addition of (1) piperazine to C7 position, (2) –F to C6 position, and (3) cyclopropyl at the N1 position | (1) Improves anti-Gram-negative activity | ||
| (2) Increases potency | |||||
| 2b | Ofloxacin (L-isomer = levofloxacin) |
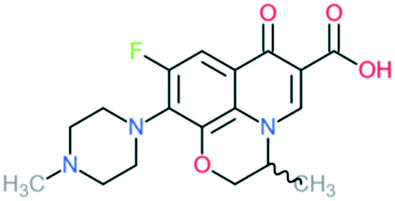
|
All Gram-negative pathogens and some Gram-positive bacteria (including Staphylococcus aureus, except Streptococcus pneumoniae) and some atypical organisms | Addition of (1) methylated piperazine to C7 position and (2) –OCH2 at C8 position | (1) Increases anti-Gram-positive activity |
| (2) Increases anti-Gram-positive activity, tissue penetration, half-life | |||||
| (3) L-Isomer is 4-fold more active | |||||
| Lomefloxacin |
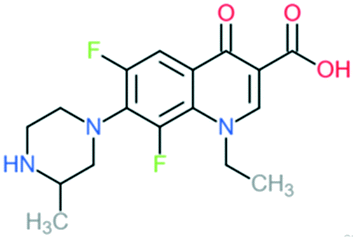
|
Addition of (1) methylated piperazine to C7 position and (2) –F at C8 position | (1) Increases anti-Gram-positive activity | ||
| (2) Increases anti-Gram-positive activity, tissue penetration, half-life | |||||
| 3 | Sparfloxacin |
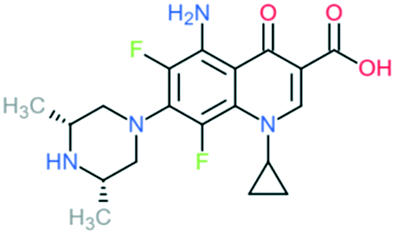
|
Retains the activity of second-generation drugs and possesses expanded Gram-positive coverage (penicillin-sensitive and penicillin-resistant S. pneumoniae) and improved activity against atypical pathogens | Addition of (1) dimethylated piperazine to C7 position, (2) –F at C6 and C8 positions, (3) –NH2 at C5 position, and (4) cyclopropyl ring at N1 position | (1) Increases anti-Gram-positive activity |
| (2) Increases anti-Gram-positive activity, tissue penetration, half-life | |||||
| (3) Improves activity against Gram-positive pathogens | |||||
| (4) Improves potency of the drug | |||||
| Grepafloxacin |
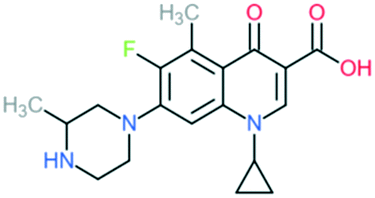
|
Addition of (1) methylated piperazine to C7 position, (2) –CH3 at C5 position, and (3) cyclopropyl ring at N1 position | (1) Improves anti-Gram-positive activity | ||
| (2) Improves anti-Gram-positive activity compared to ciprofloxacin | |||||
| (3) Improves potency of the drug | |||||
| Clinafloxacin |
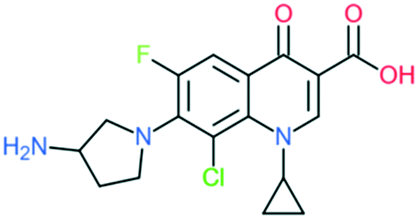
|
Addition of (1) 3-aminopyrrolidin-1-yl group to C7 position, (2) –Cl at C8 position, and (3) cyclopropyl ring at N1 position | (1) Improves anti-Gram-positive activity and overcomes physical disadvantages | ||
| (2) Improve anti-Gram-positive activity, tissue penetration, half-life | |||||
| (3) Improves potency of the drug | |||||
| Gatifloxacin |
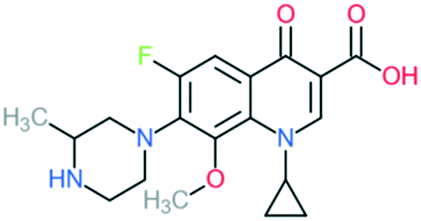
|
Addition of (1) methylated piperazine group to C7 position, (2) methoxy group at C8 position, and (3) cyclopropyl ring at N1 position | (1) Improves anti-Gram-positive activity | ||
| (2) Improves anti-Gram-positive activity, tissue penetration, half-life | |||||
| (3) Improves potency of the drug | |||||
| 4 | Moxifloxacin |
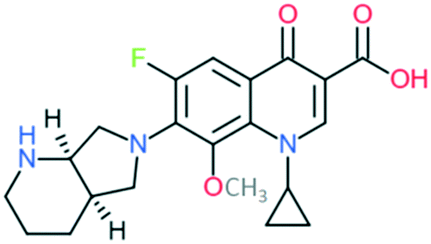
|
Covers all the activities of third generation drugs and extra anaerobic activity | Addition of (1) azabicyclo group to C7 position, (2) –OCH3 at C8 position, and (3) cyclopropyl ring at N1 position | (1) Improves anti-Gram-positive activity but may result in low water solubility and oral bioavailability |
| (2) Improves anti-Gram-positive activity, tissue penetration, half-life | |||||
| (3) Improves potency of the drug | |||||
| Gemifloxacin |

|
Addition of (1) 3-methoximine-4-aminomethyl-pyrrolidin-1-yl group to C7 position and (2) cyclopropyl ring at N1 position | (1) Improves anti-Gram-positive activity and overcomes the physical disadvantages compared with pyrrolidine group alone | ||
| (2) Improves potency of the drug | |||||
| Trovafloxacin |
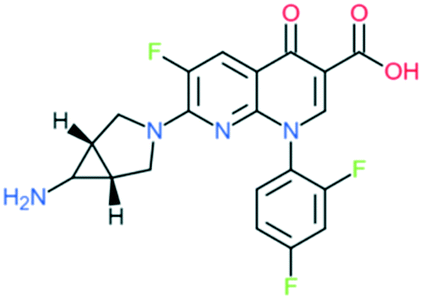
|
Addition of (1) amine-substituted bicyclic pyrrolidin-1-yl group to C7 position and (2) 2,4-difluorophenyl group at N1 position | (1) Improve anti-Gram-positive activity | ||
| (2) Improves potency and activity against anaerobes | |||||
| Garenoxacin |

|
Addition of (1) azabicyclo group to C7 position, (2) cyclopropyl group at N1, and (3) difluoromethyl ether group at C8 position | (1) Significantly improves anti-Gram-positive activity (lipophilicity and half-lives) | ||
| (2) Improves potency of the drug | |||||
| (3) Improves anti-Gram-positive activity |
2.1. Development in activity
The first-generation quinolone activity was limited to only Gram-negative organisms, excluding Pseudomonas species.16 Shortly after the clinical introduction of nalidixic acid, it was found to cause rapid resistance development in a number of organisms, reducing its effectiveness17 and leading to investigations to discover analogues with improved properties.The first second-generation quinolone, flumequine, exemplified the discovery that a key modification, adding a fluorine (F) atom at the R6 position, could significantly improve the spectrum of activity.18 This change dramatically increased the quinolone activity, since almost all quinolone antibiotics have been designated as fluoroquinolones, with the exception of the most recent compounds from the fourth generation. Other fluoroquinolones from the second generation include enoxacin, norfloxacin, and ciprofloxacin, which were able to inhibit all Gram-negative organisms, including Pseudomonas species.19 In addition to the fluoro substituent, these drugs were further modified by addition of a piperazine ring to the R7 position and addition of a cyclopropyl group to the R1 position. The R7 piperazine ring improved the Gram-negative potency,20 while the cyclopropyl group was found to improve the overall activity of the compounds.21 This combination made ciprofloxacin the most active compound among the early compounds of the second generation and made it the first choice used against Pseudomonas aeruginosa today.22 Subsequent development of the second generation produced analogues with activity against some Gram-positive bacteria, including Staphylococcus aureus but not Streptococcus pneumoniae, and some atypical organisms (Mycoplasma pneumoniae and Chlamydia pneumoniae).23 The presence of an alkylated piperazine group at the R7 position, as in ofloxacin, marked the first modifications that help inhibit Gram-positive organisms.24 The addition of an –OCH3 substituent to the R8 position of the latter group also helped to improve Gram-positive activity.25 Of all compounds in this latter group (2b), ofloxacin is considered as the most powerful as it combines all the new substituents and it is now still being used for clinical treatment. Ofloxacin is a chiral molecule and its L-isomer is the only active compound, which is known as levofloxacin. It was proposed to have 4-fold higher activity compared with ofloxacin and is also more active than ciprofloxacin in treating some strains.26,27
With the synthesis of fleroxacin, the quinolones entered their third generation. The improvements of this generation included addition of alkylated piperazine and pyrrolidinyl groups to the R7 position, and –NH2, –OH, and –CH3 groups to the R5 position to the pharmacophore. The cyclopropyl group at the R1 position and the –OCH3 group at position R8 were kept unchanged from the second generation. The third generation also added new substituents, such as a chloro group (Cl) at the R8 position; this was verified to improve the anti-Gram-positive activity of the drug.25 Among all modifications at this position, 8-methoxyquinolone was shown to surpass other compounds in activity and spectrum. The improvement is best exemplified by comparing grepafloxacin and gatifloxacin; the MIC90 of gatifloxacin (8-MeO) improved significantly compared with that of grepafloxacin (8-H) (Table 2). These modifications expanded the Gram-positive activity of the third generation, including penicillin-sensitive and penicillin-resistant S. pneumoniae, while the activity against atypical bacteria was also increased. While a piperazine group in the second generation improved the Gram-negative activity, the alkylated form of this group added to the Gram-positive activity of the fluoroquinolone compounds. A pyrrolidinyl group in this position showed the same improvement as the alkylated piperazine group.28 Manipulation of the group at the R5 position was shown to increase the activity against Gram-positive organisms.26,29 The antibacterial potency improvement mediated by substitution at this position was found to increase in the order –CH3, –OH, –NH2, respectively.30 All the modifications (positions R8, R5, and R7) presented in this third generation were designed to improve the activity against Gram-positive bacteria. Among these modifications, manipulation at the R7 has generally been the most effective. It can be observed by comparing the MIC90 of these compounds. Clinafloxacin is described to possess the most potential among these third-generation drugs, with a methylated pyrrolidinyl group at R7 and chlorine at C8. The MIC90 of clinafloxacin is the lowest in this group (Table 2). There are similarities between the structures of ciprofloxacin and sparfloxacin, but addition of –NH2 at R5 and alkylation of the piperazine group make the potency of sparfloxacin better than that of ciprofloxacin (Table 2). It is similar in the case of grepafloxacin, with the –CH3 substituted.
| MIC90 (mg L−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gram-negative pathogens | Gram-positive pathogens | |||||||||
| E. coli | P. aeruginosa | Klebsiella spp. | B. fragilis | Haemophilus influenzae | S. aureus | S. pneumoniae | Group A Streptococci | Enterococcus spp. | Clostridium perfringens | |
| Nalidixic acid | 8 | >64 | 16 | >64 | 2 | >64 | >64 | >64 | >64 | >64 |
| Enoxacin | 0.25 | >64 | 2 | >64 | 0.12 | 2 | 64 | >64 | 8 | >64 |
| Norfloxacin | 0.12 | 2 | 0.5 | >64 | 0.06 | 1 | 16 | 4 | 4 | ND |
| Ciprofloxacin | 0.03 | 1 | 0.25 | 16 | 0.03 | 1 | 2 | 1 | 4 | 0.5 |
| Ofloxacin | 0.12 | 4 | 0.5 | 16 | 0.03 | 0.5 | 2 | 2 | 2 | 1 |
| Lomefloxacin | 0.06 | 2 | 0.25 | ND | 0.06 | 2 | 4 | 4 | 4 | ND |
| Sparfloxacin | 0.06 | 4 | 0.5 | 4 | 0.03 | 0.12 | 0.5 | 1 | 2 | 0.25 |
| Grepafloxacin | 0.06 | 8 | 0.12 | 8 | 0.01 | 0.12 | 0.25 | 1 | 4 | 1 |
| Clinafloxacin | 0.01 | 0.5 | 0.03 | 0.25 | 0.01 | 0.06 | 0.12 | 0.06 | 0.25 | 0.12 |
| Gatifloxacin | 0.06 | 4 | 0.25 | 1 | 0.03 | 0.25 | 0.25 | 0.25 | 1 | 0.5 |
| Moxifloxacin | 0.06 | 8 | 0.12 | 1 | 0.06 | 0.06 | 0.12 | 0.25 | 2 | 0.25 |
| Gemifloxacin | 0.03 | 4 | 0.25 | ND | 0.06 | 0.06 | 0.03 | 0.06 | 4 | ND |
| Trovafloxacin | 0.06 | 1 | 0.25 | 0.25 | 0.01 | 0.03 | 0.12 | 0.25 | 1 | 0.25 |
| Garenoxacin | 0.06 | 16 | 0.5 | 1 | 0.03 | 0.03 | 0.12 | 0.25 | 0.5 | 0.25 |
The spectrum of activity of fourth-generation compounds covers all the criteria of the third generation with the addition of activity against anaerobic organisms.23 The presence of nitrogen (N) at the R8 position is responsible for the improved activity against anaerobes,31 while a 2,4-difluorophenyl group at the N position improves the overall potency of the drug.24 This modification can be seen from the structures of moxifloxacin, gemifloxacin, and trovafloxacin (Table 1). Other modifications are addition of an azabicyclic group and a bulky side chain on the pyrrolidine group at the R7 position and addition of a difluoromethyl ether group at the R8 position, which all improve the Gram-positive activity.21 The azabicyclic group at the R7 position produced the highest potency against the Gram-positive bacteria, as demonstrated by comparing the potency and structure between moxifloxacin and gatifloxacin. These two compounds have an otherwise similar structure, differing only at the R7 position. The azabicyclic group in moxifloxacin substantially improves Gram-positive potency compared with gatifloxacin (Table 2).
As discussed above, the development of the structure–activity relationship of quinolones through successive generations can be summarized in Fig. 2.
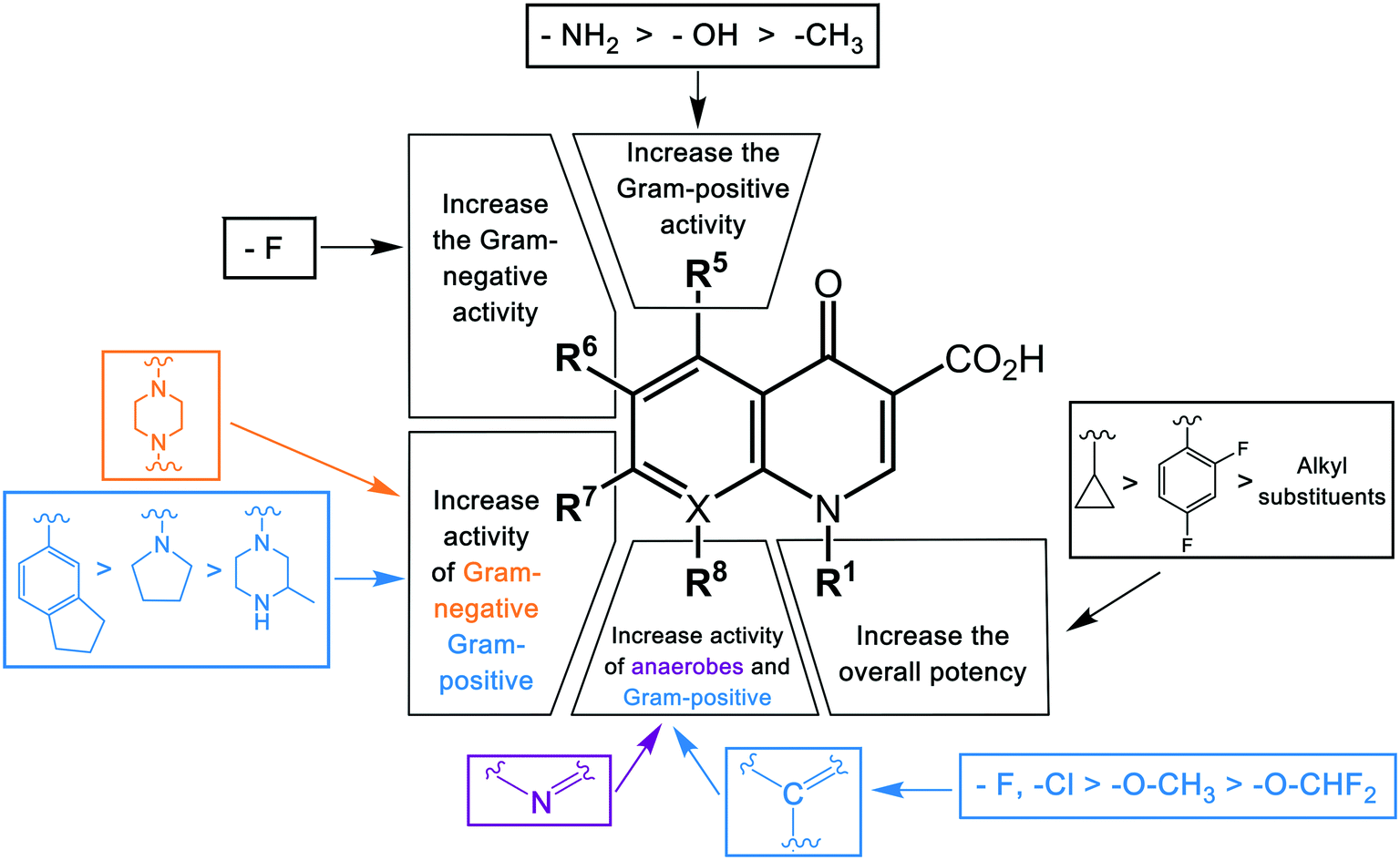 | ||
| Fig. 2 The structure–activity relationships (SAR) of quinolones. The antibacterial activity of quinolones is improved by modifications of different substituents in different positions. The color of the groups in the bracket correlates with the type of activities. | ||
2.2. Development in pharmacokinetics (PK) and pharmacodynamics (PD)
The development of quinolones in terms of pharmacokinetics and pharmacodynamics relates to improvements in metabolism, elimination, and transportation, leading to improved antibiotic dosing strategies to enhance the efficacy and prevention of resistant mutations. Use of the very first quinolone agent, nalidixic acid, was limited because it had low serum levels; therefore, it was used as a urinary agent only.50 The modifications in the structure of later generations of quinolones led to improved oral absorption as well as larger area under the curve (AUC) and/or maximum serum concentrations (Cmax) compared to nalidixic acid.5 Those modifications also produced longer elimination half-lives, which permitted once-daily dosing for some agents of the second generation and all agents of later generations (Table 3). Since most of the earlier quinolones had low serum levels and moderate potency, they required frequent doses, with the once-daily dosing of latter agents resulting not only from better exposure but also from their significantly enhanced potency. They also had better tissue penetration.5 There is no trend in the extent of protein binding related to the structural modifications. This parameter varies between agents, with some <30% (norfloxacin, lomefloxacin, and gatifloxacin) and others >80% (nalidixic acid, trovafloxacin, and garenoxacin). Over time, changes in the metabolism of quinolones were observed; although earlier quinolones were primarily eliminated by metabolism and renal clearance, later quinolones were modified to become non-renal clearance agents (sparfloxacin, moxifloxacin, gemifloxacin, trovafloxacin, and garenoxacin) (Table 3).| Quinolone | Dose (g) (frequency per day) | C max (mg L−1) | AUC (mg h L−1) | Half-life (h) | Protein binding (%) | Elimination route |
|---|---|---|---|---|---|---|
| Nalidixic acid | 1 (×4) | Variable | Variable | 1.5 | 90 | Renal |
| Enoxacin | 0.6 (×1) | 3.7 | 29 | 2 | 60 | Renal |
| Norfloxacin | 0.4 (×2) | 1.5 | 10 | 3 | 15 | Renal hepatic |
| Ciprofloxacin | 0.75 (×2) | 3.5 | 30 | 4 | 40 | Renal and enteral |
| Ofloxacin | 0.4 (×2) | 4.8 | 64 | 6 | 40 | Renal |
| Lomefloxacin | 0.4 (×1) | 2.8 | 26 | 8 | 10 | Renal |
| Sparfloxacin | 0.4 (×1) | 1.0 | 20 | 18 | 40 | Renal |
| Grepafloxacin | 0.4 (×1) | 1.4 | 14 | 14 | 50 | Hepatic |
| Clinafloxacin | 0.2 (×2) | 1.6 | 18 | 6 | 40 | Renal |
| Gatifloxacin | 0.4 (×1) | 3.8 | 33 | 12 | 20 | Renal |
| Moxifloxacin | 0.4 (×1) | 3.1 | 30 | 13 | 50 | Hepatic |
| Gemifloxacin | 0.32 (×1) | 1.0 | 9 | 7 | 60 | Renal and other |
| Trovafloxacin | 0.3 (×1) | 2.5 | 40 | 12 | 85 | Hepatic |
| Garenoxacin | 0.4 (×1) | 5.8 | 59 | 15 | 87 | Renal and other |
The quinolones show concentration-dependent killing (CDK) with persistent post-antibiotic effect (PAE),53 and the therapeutic outcomes of this group are based on either the AUC/MIC ratio or the Cmax/MIC ratio. Clearly, a high AUC or Cmax value combined with low MIC is ideal for increasing the ratio and thereby improving the efficacy. For decades, it was debated as to which ratio best indicated the microbiological and clinical outcomes of quinolones.30 It was not until the alarming rise in resistance to ciprofloxacin when treating infections with common low-dose regimens54–56 that large-scale clinical studies were conducted to define the PD parameters for predicting efficacy. According to several studies, the second-generation quinolones did not obtain a high Cmax/MIC ratio,28 with the AUC/MIC ratio more accurately reflecting their efficacy. It was shown that an AUC/MIC ratio of >125 indicated the best therapeutic outcomes, and any agents with a Cmax/MIC ratio lower than 4 indicated sub-optimal outcomes.57,58 However, it is still uncertain what the minimum acceptable AUC/MIC ratio is. Some researchers have proposed that an AUC/MIC ratio of 25 is appropriate for use in mild infections and immunocompetent patients, while a value of ≥100 is needed for serious infections and immunocompromised patients.59
While the AUC/MIC ratio is used to determine the microbiological outcome of quinolone treatment, the Cmax/MIC ratio has been determined to be a factor for preventing the emergence of resistance to quinolones.30 A higher Cmax is preferable for lower resistance occurrence. Many in vitro studies showed that a low AUC/MIC ratio will increase the selection of resistant mutants, even if this ratio is clinically effective for the infections.60–62 Combined with the Cmax/MIC ratio, a “mutant prevention concentration” (MPC) was developed for prevention of resistance. It is the concentration necessary to prevent the growth of the least susceptible, single-step mutants, with 1010 bacteria incubated in the presence of different increasing concentrations of the antibiotics. The MPC is the concentration in which there is no observation of growth of that bacteria.63 This MPC is used to prevent resistance during therapy, suggesting a minimum serum concentration to be achieved. This target was used during the development of the third generation of quinolones (gatifloxacin, gemifloxacin, moxifloxacin); they exert lower MPC values than the earlier quinolones when used against Streptococcus pneumoniae.64,65 Accordingly, the MPC of ciprofloxacin for Pseudomonas aeruginosa is lower than that of levofloxacin.66
Key structural modifications for improving the pharmacokinetics of quinolones are presented at the R5, R6, R7, and R8 positions (Fig. 3), which result in longer elimination half-life, better tissue penetration, increased volume distribution, and better bioavailability. The addition of an amino group at R5 increased the quinolones' lipophilicity,67 which can be seen from the structure of sparfloxacin. The fluorine substituent at position R6 proved to facilitate penetration into the bacterial cell68 and also improve the volume of distribution of the drug. This improvement was observed during the development of the second-generation of quinolones and was retained until the latest agent of the fourth generation, garenoxacin. The addition of substituents at the R7 position mediated the improvement of the half-life and bacterial tissue penetration.5 The azabicyclic group and piperazine group at R7 extended the agents' half-life to >10 h by increasing the lipophilicity.69,70 Another substituent at this position is the pyrrolidine rings; while this modification is critical for enhancing the potency of quinolones; it was reported to be associated with unfavorable water solubility and oral bioavailability.71 To overcome these physical properties, subsequent generations of quinolones introduced a methyl group into the rings, which can be seen from the examples of gemifloxacin and trovafloxacin.71 Furthermore, the alkylation of the rings at the R7 position increased the elimination half-life and bioavailability of the agents. The addition of a methyl group to the piperazine rings significantly increased the elimination half-life of ofloxacin, lomefloxacin, sparfloxacin, grepafloxacin, and gatifloxacin compared to enofloxacin, norfloxacin, and ciprofloxacin, which have only the piperazine group in the structure (Table 3). Alkylation at the R8 position was shown to increase the elimination half-life and also increase the tissue penetration of the agents.72 Moxifloxacin and gatifloxacin are examples of these modifications. The latest key modification is a methoxy group at this position, which lowered the development of resistance to quinolones.
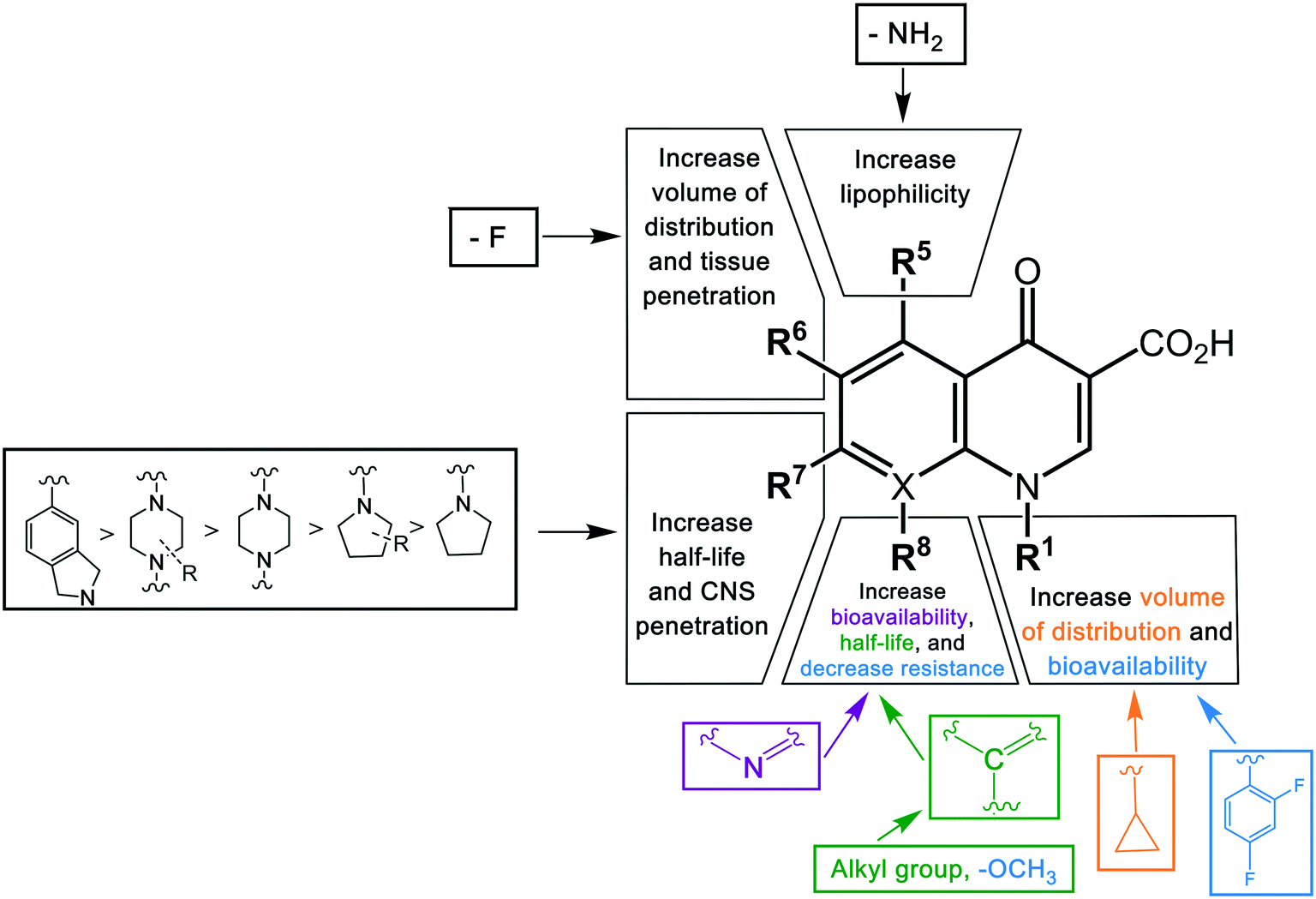 | ||
| Fig. 3 The structure–pharmacokinetic relationship of quinolones. The pharmacokinetics of quinolones is improved by modifications of different substituents in different positions. The color of the groups correlates with the color of a pharmacokinetic property. | ||
2.3. Development in toxicological data
The most common adverse effects of the quinolones are gastrointestinal effects and arthralgia (or joint pain), which are associated with the structural feature of the quinolone pharmacophore.30 Due to concerns of these primary adverse effects, this class is limited for prescription to pediatric patients.73 In addition to these class-related toxicological disadvantages, earlier quinolones were limited in their clinical use due to several unwanted adverse effects, with some mild but frequent, and others rare but severe. Those disadvantages were reported to be dependent on the substituents in different positions on the pharmacophore and specific to particular agents (Table 4). QTc prolongation was reported to occur in patients using sparfloxacin and grepafloxacin.5 QTc prolongation can lead to cardiac arrhythmias.74 Phototoxicity was observed when using clinafloxacin and sparfloxacin.75 Tendon rupture, nerve damage, and fluoroquinolone-associated disability syndrome has been reported for most fluoroquinolones when they are used for a long-term period, and these side effects are proposed to be potentially permanent.76 Other effects include haematological toxicity with temafloxacin,77 hepatitis with trovafloxacin,78 and hypoglycaemia effects with clinafloxacin and gatifloxacin.79–81 Immunological side effects were seen in a number of agents, as were central nervous system (CNS) effects and genotoxicity (Table 4). The genotoxicity of quinolones is only seen in some fluoroquinolones when exposed to ultraviolet (UV) light, such as lomefloxacin, ciprofloxacin, and moxifloxacin. They were reported to be toxic and mutagenic following the reaction with human topoisomerase IIα in the presence of UV radiation.156 Some of these effects have led to quinolones being withdrawn from the market. Over time, the toxicity of quinolones has been reduced by structural modifications, and the latest agent (garenoxacin) has proved to have little adverse toxicological data.5 The safety profile of quinolones is being updated constantly, since some life-threatening adverse effects, such as aortic rupture and dissection caused by exposure to fluoroquinolones, have recently attracted additional warnings by the FDA in 2018. It is advised that fluoroquinolones are not to be used for patients with an aortic aneurysm, or the elderly, and only as a last-line defense.82| Side effect | Agent | Frequency |
|---|---|---|
| Gastrointestinal effects | Sparfloxacin, grepafloxacin | >10% |
| Others | 2–8% | |
| Arthralgia effects | Sprafloxacin, levofloxacin, grepafloxacin ≫ others | 0.5–2% |
| CNS effects | Trovafloxacin | 2–11% dizziness |
| Levofloxacin | 0.026% confusion, alteration in mentation and effect | |
| Phototoxicity | Clinafloxacin, sparfloxacin | >10% |
| Others | <2.5% | |
| Genotoxicity | Lomefloxacin, moxifloxacin, ciprofloxacin | |
| QTc prolongation | Grepafloxacin, sparfloxacin | 2.9% |
| Haematological effect | Temafloxacin | 0.02% thrombocytopenia, haemolysis, and renal failure |
| Hepatic eosinophilia effect | Trovafloxacin | 0.006% |
| Grepafloxacin | 12–16% transaminase elevation | |
| Others | <3% | |
| Pulmonary interstitial eosinophilia | Gemifloxacin | |
| Immunological side effect | Gemifloxacin | |
| Hypoglycaemia | Clinafloxacin, gatifloxacin | |
| CYP 450 inhibition | Enoxacin, clinafloxacin > ciprofloxacin > lomefloxacin, ofloxacin > levofloxacin, sparfloxacin, gatifloxacin, moxifloxacin |
The substituent at the R1 position was shown to be related to the inhibition of cytochrome P450, with cyclopropyl and the alkyl groups at this position affected more than when substituted with a 2,4-difluorophenyl group. Other modifications leading to cytochrome P450 interactions were the replacement of the carbon atom with nitrogen at the X position, and the addition of a bulky side chain into the X8 of quinolones. Genotoxicity was shown to occur in agents with –NH2 and –CH3 substituents at the R5 position, fluorine (F) at the R6 and R8 positions, and chlorine (Cl) at the R8 position.5 Another specific structural change associated with the genotoxicity was modifications of the group at position 7, with a decrease in severe effects by pyrrolidinyl, piperazine, and alkyl groups, respectively.30
Phototoxicity is an adverse effect caused by the accumulation of susceptible drugs in the skin where they can be activated by exposure to sunlight, causing damage to the skin.90 This was observed in agents with an –NH2 group at the R5 position and fluorine (F) or chlorine (Cl) at the R8 position.30 Quinolones possessing this adverse effect include lomefloxacin, sparfloxacin, and clinafloxacin. Central nervous system (CNS) reactions including dizziness, insomnia, and headache have been induced by some quinolones.91 This adverse effect has been shown to be associated with the inhibition of GABA receptors, a major inhibitory neurotransmitter, and was observed in agents with additional groups at position R7.92 In contrast to the genotoxicity effect in this position, the degree of CNS effect increased in the reverse order, with alkyl > piperazine > pyrrolidinyl group. This highlights the difficulties in optimizing substituents against multiple parameters, with favorable changes in some properties balanced by increased detrimental outcomes in other properties. A summary of the structure–toxicity relationship of quinolones is shown in Fig. 4.
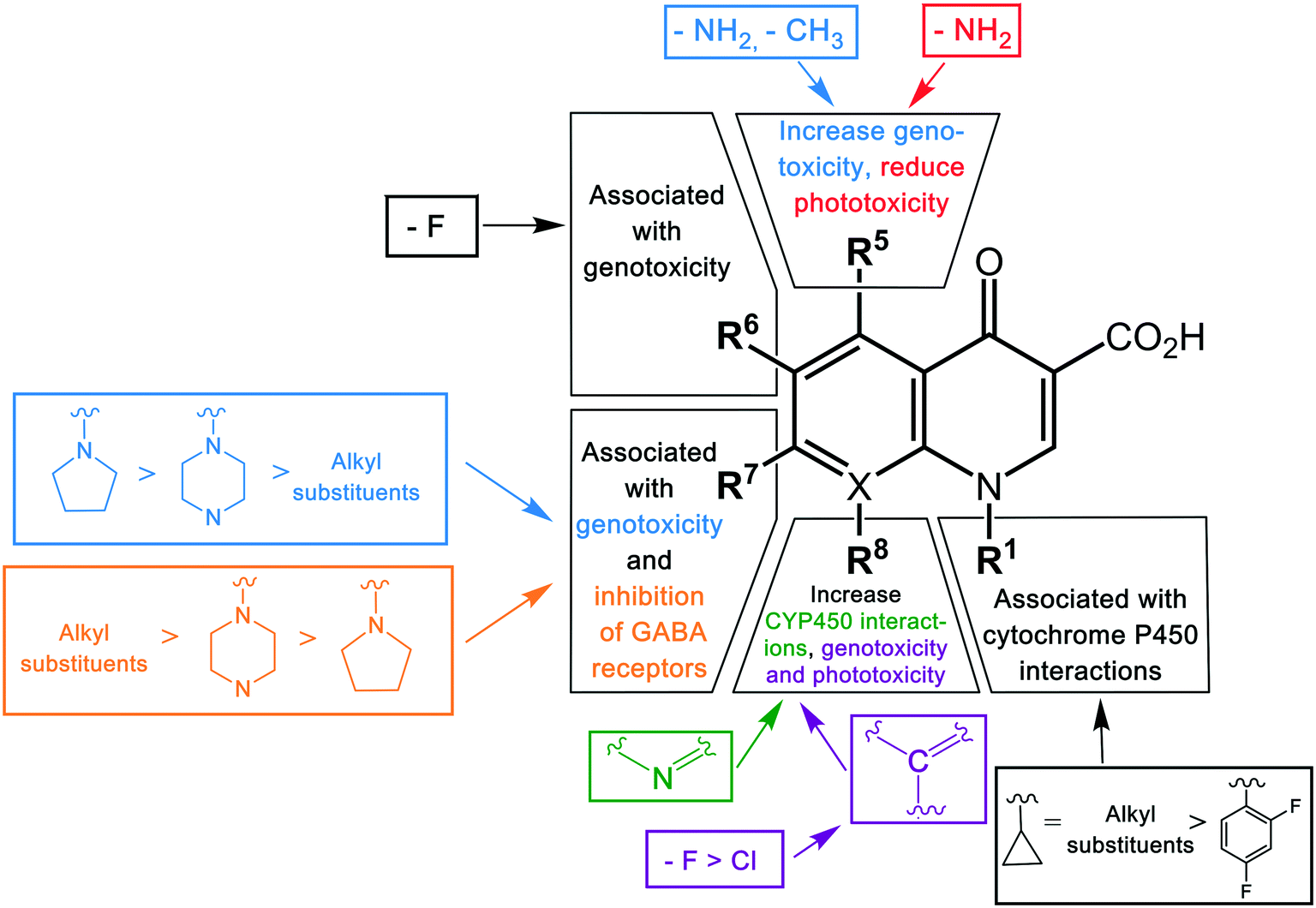 | ||
| Fig. 4 The structure–toxicity relationship of quinolones. The toxicity of quinolones is altered by modifications of different substituents in different positions. The color of the groups in the bracket correlates with the type of toxicity. | ||
2.4. Conclusion
Studies of structure–activity, structure–pharmacokinetics, and structure–toxicity relations of quinolones have enabled a better understanding of different modifications to the core structure to offer the best manipulation for combining clinical efficacy, reduced toxicity, and safety. The best substituents in each position include cyclopropyl at position R1, fluorine at position R6, a pyrrolidine, piperazine, or azabicyclic group at position R7, and a methoxy group at position R8. Due to the genotoxicity of the fluoroquinolone class found to be associated with the fluorine at position R7, studies have been focused on the development of the fluoroquinolones. Optimizing the activity gained from other substituents (R5, R1) to offset the loss in activity by removing the fluorine has led to garenoxacin, the lead example of the fourth generation of quinolones.2.5. Clinical indications and trend of quinolone use
Quinolones are antibiotics with broad-spectrum activity and a variety of clinical indications. However, the increasing rates of resistance are leading to a re-adjustment of strategies and usage for this antibiotic class,160 particularly to reduce the risk for selection of resistance. In recent years, fluoroquinolones have been the primary agents for treating urinary tract infections (UTIs) and infections of the digestive tract and respiratory system.161 Excessive prescriptions of quinolones have led to the rapid development of quinolone resistance, leading to a loss in effectiveness of this class. Australia restricted the use of quinolones in humans through a national pharmaceutical subsidy scheme and did not allow the use of quinolones in food-producing animals, leading to low rates of resistance compared to other countries.158 Globally, high levels of resistance to E. coli, the predominant cause of UTIs, has led to the replacement of quinolones by third-generation cephalosporin for this indication.93 Fluoroquinolones are still the mainstay for treatment of typhoid caused by Salmonella owing to resistance to previous first-line agents, such as ampicillin, chloramphenicol and trimethoprim/sulfamethoxazole. However, resistance to quinolones in Salmonella is increasingly being reported in the Americas, South Asia, Southeast Asia and especially sub-Saharan Africa, and Salmonella is now ranked as a high-priority pathogen for the research and development of new antibiotics.94,95 For respiratory tract infections, especially community-acquired pneumonia, quinolones are not recommended for first-line treatment but are reserved for serious cases. To reduce the development of fluoroquinolone resistance it is recommended to limit the use of this class in the treatment of patients with less severe infections, patients with prolonged hospitalized stay and patients with chronic, recurrent disease. Unfortunately, quinolones are also widely used in veterinary and husbandry practice which can lead to serious resistance through a variety of routes.96 High rates of resistance have been seen in food-borne pathogens such as Campylobacter, E. coli, and Salmonella from isolates from the United States (19%) and Spain (>80%).97 As mentioned earlier, resistance of these strains is much lower in Australia, where this class is not approved for animal use. This supports the argument that quinolone usage in animals increases selection of resistance.97Clinical use of the fluoroquinolones is also restricted by the known and suspected toxicities of this class in specific populations, such as pregnant and breast-feeding women and pediatric and elderly patients. Although it is a powerful antibiotic for treating children's infections such as diarrhoea or Gram-negative meningitis, the toxicity (arthralgia) of this class combined with the issues of increasing resistance have reduced its use for this group.73 The recently reported potential for aortic rupture and dissection side effects also raises safety issues for the use of quinolones in elderly patients, where they may lead to serious bleeding or even death.76,82 In addition, there are possible teratogenic and mutagenic effects, so prescription of fluoroquinolones for pregnant and breast-feeding women is limited.98
Despite these increasing concerns of quinolone resistance and toxicity, the use of this class of antibiotics still remains high as it is effective for serious infections.99 The development of novel quinolone or quinolone-like agents with improved properties is still desirable, particularly if these new agents are only used to treat appropriate types of patients and used in an educated fashion.
3. Mode of action
Quinolones kill bacteria by interfering with DNA synthesis and inhibiting their replication pathway.100 During DNA synthesis, double-stranded DNA needs to unwind into two single-stranded structures to be used as the template, allowing the transcription complexes to proceed and complementary base pairing to occur.101 This unwinding process is done by the bacterial topoisomerase II type enzymes, DNA gyrase and DNA topoisomerase IV.101 Quinolones exert their action by inhibiting these enzymes, thereby stopping the synthesis process.3.1. Quinolone target: DNA gyrase and topoisomerase IV
The two enzymes responsible for DNA synthesis in bacteria are DNA gyrase and topoisomerase IV.101 These enzymes take part in controlling the amount of DNA under- and over-winding, and remove the topological stress of the bacterial chromosome.102 Under normal conditions, DNA is highly condensed, so prior to replication it must be unwound to separate the two strands and provide the template for transcription. During the unwinding, as the replication forks move forward, super positive helical twists in the DNA are created in front of them.101 For the replication to proceed, the DNA topoisomerase type II enzyme removes this helical twist by cutting the DNA backbone at the double strand 4 bp apart to generate a 5′-overhang, which helps in the process of synthesizing and separating the daughter chromosomes.102Although the two topoisomerase type II enzymes were reported to have similar functions and structure, there are some different physiological functions between them (Table 5). DNA gyrase uses energy in the hydrolysis of ATP to introduce negative supercoils into the DNA, resulting in the condensation of the chromosomes.103 In the absence of ATP, it causes relaxation of the DNA, thereby relieving the topological stress accumulated ahead of replication forks mediating the replication process.104 In contrast, topoisomerase IV is unable to introduce the negative supercoils. It can only relax the positive supercoils by binding to the crossovers between two interlinked daughter cells and removing knots.104 As a result, its primary function is associated in decatenating daughter chromosomes for the separation of two daughter cells.
| Topoisomerase IV | DNA gyrase |
|---|---|
| Main target in most Gram-positive bacteria | Main target in most Gram-negative bacteria |
| Decatenates DNA for separation into daughter cells during DNA replication | Removes positive super helical twists in the DNA ahead of replication |
| Can act as topoisomerase IV in organisms that lack Top IV (such as M. tuberculosis, T. pallidum, H. pylori) |
Genetic studies on E. coli strains found that the primary target of quinolone is gyrase, and the second targeted enzyme is topoisomerase IV.105 This is consistent with other findings that show a higher amount of gyrase-DNA cleavage complexes when incubating E. coli with different quinolones.106 On the other hand, a study by Pan and his colleague in 1996 showed that in the case of Streptococcus pneumoniae, the primary target of ciprofloxacin was topoisomerase.107 From this analysis, it was proposed that topoisomerase IV is the primary target for quinolones in Gram-positive strains, and DNA gyrase is the primary target for Gram-negative bacteria. However, several researchers have shown that this is untrue in many cases, with examples of DNA gyrase as the primary target in some Gram-positive bacteria (e.g. gyrase is the primary target for Staphylococcus aureus in treatments with norfloxacin), and conversely that topoisomerase IV is also a primary target in some Gram-negative bacteria. Moreover, subsequent studies showed that different quinolones have a different primary target for a particular strain.109–111 Therefore, investigations should be conducted on a species-by-species and drug-by-drug basis for detailed evaluation.
DNA gyrase and topoisomerase IV are A2B2 heterotetramer enzymes including two pairs of identical GyrA/GyrB and ParC/ParE in Gram-negative or GlrA/GlrB in Gram-positive species.112 GyrA and ParC or GlrA contain an active site tyrosine residue, which is involved in the breakage/reunion of the DNA. GyrB and ParE or GlrB contain the ATPase domain and the TOPRIM domain, which are involved in the energy transduction for DNA cleavage and ligation.113 The differences in the physiological functions between DNA gyrase and topoisomerase IV are due to the difference in the C-terminal region of these enzymes.113 The C-termini of GyrA and ParC (GrlA) associating with the topological recognition are not well conserved. The addition of a CTD region in the A subunits allows DNA gyrase to generate supercoils in DNA, which cannot be modulated by topoisomerase IV114 (Fig. 5).
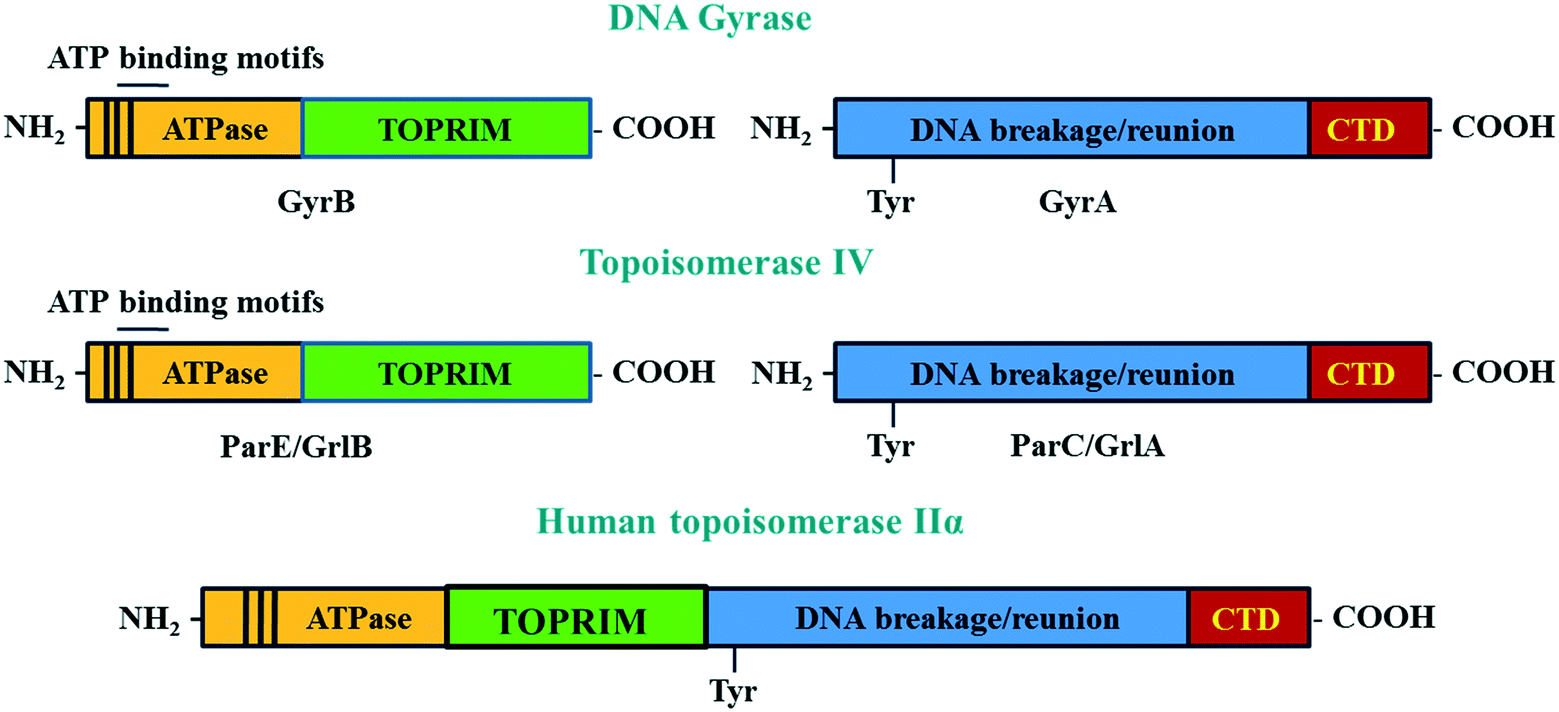 | ||
| Fig. 5 The structure of DNA gyrase and topoisomerase IV and human topoisomerase IIα. The GyrB and its equivalent domain on topoisomerase IV (ParE/GrlB) are responsible for hydrolyzing ATP during the cleavage/ligation process. The GyrA contains the tyrosine active site, which takes part in the breakage/reunion of the chromosomes. The CTD region, which is only observed in the GyrA but not in ParC/GrlA, is involved in topology recognition. Unlike two distinct domains seen in bacterial enzymes, the two subunits A and B of human topoisomerase IIα are fused together to form the homodimer enzyme. | ||
Although there is similarity in the sequence of human topoisomerase type II, IIα and IIβ with that of bacterial enzymes, quinolones have been shown to not affect the action of human enzymes. This is because the A and B subunits of human enzymes have fused during evolution, and so function as homodimers.115 This structure is different from the heterotetramers of bacterial enzymes. The differences in structure of bacterial and human topoisomerase II are shown in Fig. 5.
3.2. Quinolone action
During replication, gyrase and topoisomerase IV generate double-stranded breaks in the DNA to relax the super positive twists.100 This complex includes binding of the enzymes to the DNA, and is called the DNA cleavage complex. Quinolones bind to the enzyme–DNA complexes, rather than the target enzymes alone, thereby inhibiting the replication process and leading to cell death of the bacteria. There are two basic actions reported for the quinolones leading to cell death, via the DNA inhibition and/or via activation of the bacterial DNA stress response.116Quinolones bind to the DNA cleavage complex at the cleavage-ligation active site in a non-covalent manner. Two molecules of quinolones are required for this binding. The formation of the drug–DNA cleavage complex at both cleaved scissile bonds leads to the accumulation of DNA replication machinery at the replication forks.117 Due to their intercalation, quinolones strengthen the stable state of the cleavage complex by acting as a physical block to the ligation, resulting in bacteriostasis with low concentrations of quinolone and bactericidal activity with lethal concentrations.118 Moreover, when the DNA tracking systems collide with these drug–DNA cleavage complexes, permanent chromosome breaks are generated, triggering the DNA stress response.157 This activates RecA protein and promotes self-cleavage of the LexA repressor, thus de-repressing the expression of SOS response genes.119 Therefore, preventing the induction of SOS response leads to an inability to repair DNA breakage. The increase in the DNA breakages combines with the disabled SOS system to augment the bactericidal potency of quinolones (Fig. 6).119 Some studies have reported that reducing the number of targeted enzymes reduces the activity of quinolones. Moreover, the results from a study on the contribution of the reactive oxygen species to the quinolone-mediated bactericidal action showed that death was mainly a protein synthesis-dependent mechanism.120 The bactericidal activity of quinolones is potentially due to both the inhibition of DNA synthesis and the subsequent bacterial response through stress-induced protein expression. However, while the primary target of quinolone antibiotics is clear, the underlying molecular mechanism of activity leading to death is still unclear, as with many classes of antibiotics.
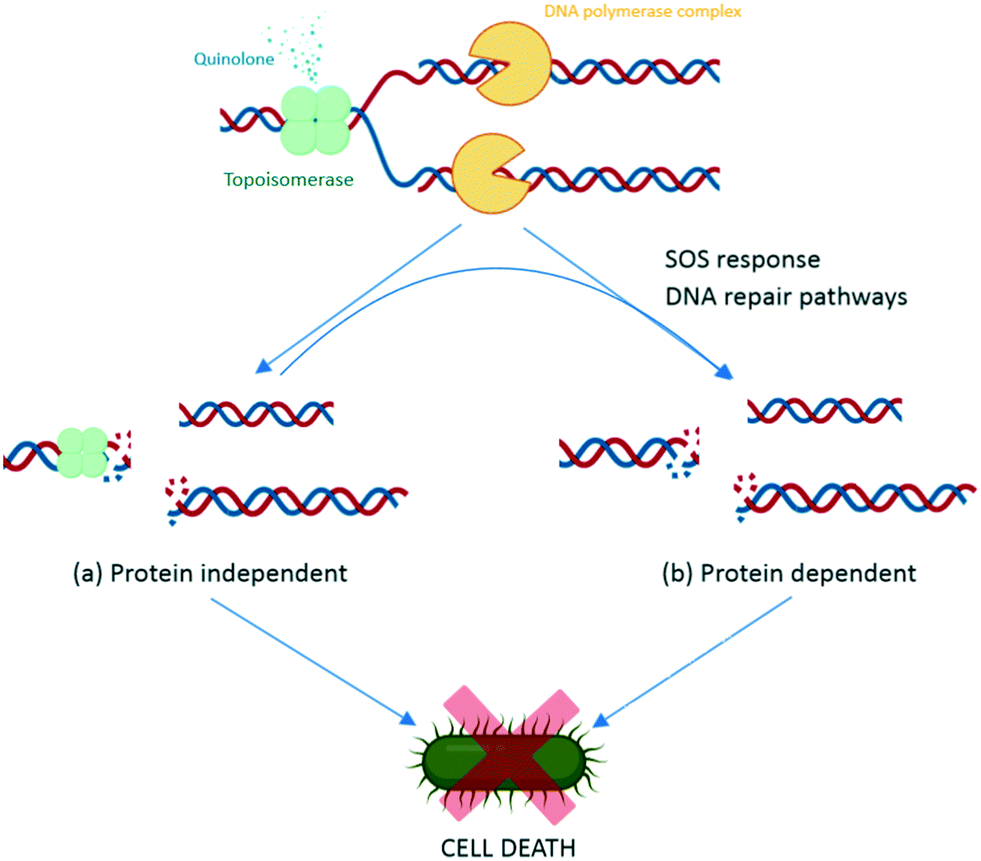 | ||
| Fig. 6 Intracellular action of quinolones. Quinolones bind to the DNA–enzyme cleavage complex at the cleavage-ligation active site. This binding creates a steady-state concentration of cleavage complexes and disrupts the replication process, which causes collision of the stabilized cleavage complexes with the DNA replication systems (replication fork, transcription complexes, and tracking systems) leading to chromosomal breaks (a). In response to this damage, SOS response and other DNA repair pathways are activated, resulting in subsequent action of the SOS system, such as extended cell filaments by expression of LexA repressor and programmed cell death by activation of toxin–antitoxin modules (b). | ||
Another action of quinolones is inhibiting the activity of the bacterial topoisomerase II. Following inhibition of the ligation of the enzymes, they also disrupt the catalytic functions of the enzymes. Therefore, this adds to the overall toxicity of the quinolones by acting as a catalytic inhibitor.
3.3. Enzyme–quinolone interactions
Recent studies including structural and functional analysis studies have shown that the binding of quinolones to bacterial topoisomerase type II enzymes is via a water–metal ion bridge.121 This interaction is mediated by a noncatalytic Mg2+ ion coordinated with four water molecules, forming a bridge for hydrogen bonds between the bound quinolone and the active site serine and acidic residues. The interaction sites on the quinolones are located on the R3/R4 keto acid of the drug pharmacophore (Fig. 7), supporting the tolerance for structural development with substituents at positions R1, R7, and R8 that lie on the opposite side of the molecule from this position. In contrast, alterations at R5, R6, and R2 are likely to disrupt this interaction due to their proximity.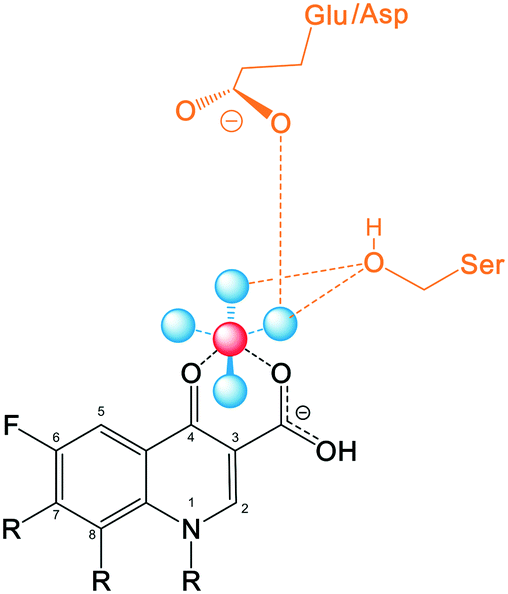 | ||
| Fig. 7 A simplified diagram of the water–metal ion bridge between a fluoroquinolone and topoisomerase IV – DNA cleavage complex. Fluoroquinolone (black) binds via a non-catalytic Mg2+ ion (red) through four water molecules (blue) that fill out the coordination sphere of the Mg2+ ion, interacting with the side chains of the serine and acidic residues (yellow). | ||
This structure also explains why the quinolones do not interfere with the human topoisomerase type II enzymes, mediating the safety of this class of drug, as the human enzyme does not have the serine and acidic residues which are essential for anchoring the water–metal ion bridge.122
4. Mode of resistance
The emergence of resistance to the quinolones is becoming a critical issue that is limiting the use of this class of antibiotics. Mechanisms of resistance are classified into two different categories, based on mutations in the bacteria genes (mutation in genes encoding the DNA gyrase and topoisomerase IV targets, or other genomic alterations that affect antibiotic accumulation) versus acquisition of resistance genes from other sources (plasmid acquisition from the environment or other resistant strains, with multiple pathways of resistance).4.1. Mutation
| Species | gyrA | gyrB | parC | parE |
|---|---|---|---|---|
| E. coli | Tyr50Phe | Asp426Asn | Ala56Thr | Leu416Phe |
| Ala51Val | Lys447Glu | Ser57Thr | Ile444Phe | |
| Ala67Ser | Ser429Asn | Asp69Glu | Leu445His/Ile | |
| Gly87Cys | Gly78Asp | Ser458Ala/Pro/Thr/Trp | ||
| Ser80Arg/Ile | Ser80Arg/Ile | Glu460Asp/Lys | ||
| Gly81Asp/Cys | Ser83Leu | Ile464Phe | ||
| Asp82Gly | Glu84Ala/Gly/Lys/Val | Ile529Leu | ||
| Ser83Ala/Ile/Leu/Trp/Tyr/Val | Cys107Trp | |||
| Ala84Pro/Val | ||||
| Asp87Asn/Glu/Gly/His/Tyr/Val | Ala108Thr/Val | |||
| Gln106Arg/His | ||||
| Ala119Glu | ||||
| Ala196Glu | ||||
| Arg237His | ||||
| Salmonella spp. | Ala67Pro | Tyr420Cys | Glu21Lys | Glu453Gly |
| Asp72Gly | Gly434Leu | Thr57Ser | Ser458Pro | |
| Val73Ile | Gly435Ala/Glu/Val | Thr66Ile | Glu459Thr | |
| Gly81Asp/Gly | Arg437Leu | Gly72Cys | His461Tyr | |
| Ser83Ala/Leu/Phe/Thr/Tyr | Gly447Cys | Gly78Asp | Gly468Cys | |
| Asp87Asn/Gly/Lys/Tyr | Ser464Phe/Tyr | Ser80Arg/Ile | Ser493Phe | |
| Leu98Val | Glu466Asp | Glu84Gly/Lys | Ala498Thr | |
| Ala119Glu/Ser/Val | Ala468Glu | Phe115Ser | Arg507Ile | |
| Ala131Gly | Leu470Met | Ala141Ser | Val512Gly | |
| Glu133Gly | Lys514Asn | |||
| Glu139Ala | ||||
| Proteus mirabilis | Ser83Arg/Ile | Ser464Phe/Tyr | Gly78Asp | — |
| Glu87Lys | Glu466/Asp | Ser80Arg/Ile | ||
| Addition of lysine between K455 and A456 | ||||
| Capnocytophaga spp. | Gly80Asn/Asp | — | Met55Ile | Gly377Asp |
| Asp81Gly | Glu101Gln | Lys410Gln | ||
| Ser82Phe/Tyr | Ile502Xaa | |||
| Thr82Ile | Thr503Xaa | |||
| Asp86Tyr | Phe504Xle | |||
| Phe508Xaa | ||||
| Phe509Xaa | ||||
| GLu511Xaa | ||||
| Glu515Asp | ||||
| Clostridium perfringens | Gly81Cys | Ala431Ser | Asp11Tyr | V637 |
| Asp82Asn | 172V | Val22Phe | Glu486Lys | |
| Ser83Leu | Asp88Tyr | |||
| Asp87Tyr | Ser89Ile | |||
| Ala119Glu | Asp93Tyr | |||
| Ala131Ser | ||||
| Val196Phe | ||||
| Asp502Tyr | ||||
| S. aureus | Ser84Ala/Leu | Val28Ala | Ile45Met | Gly78Ser |
| Ser85Pro | Ile56Ser | Ser80Phe/Tyr | Gly107Ser | |
| Glu88Lys/Gly | Gln66Lys | Glu84Lys | Arg136Gly | |
| Val248Glu | GLy85Ser | Pro144Ser | Asn139Ser | |
| Gly255Arg | Asp89Gly | Ile233Val | Ser230Gly | |
| Ala457Thr | Ile102Ser/Thr | Ser267GLy | Val327Ile | |
| Asp483Glu | Ser128Leu | Arg372His | HLu422Asp | |
| Asp495Asn | Arg144Ser/Ile | Arg400Cys | His478Tyr | |
| Glu594Gly | Thr173Ala | Glu404Gly | Gly530Asp | |
| Val598Ile | Glu317Asp | Tyr410Phe | Glu596Asp | |
| Ser668Ala | Asp437Asn | Phe521Tyr | Val609Leu | |
| Val712Ile | Arg458Gln | Phe594Tyr | ||
| Thr818Val | Gly491Asp | Asp641Asn | ||
| Arg837His | Glu568Lys | Lys650Arg | ||
| Asp856Glu | Val656Ile | |||
| Asn860Thr | Ala688Val | |||
| Glu886Asp | Met694Val | |||
| S. pneumoniae | Ala17Thr | Val432Asp | Ser52Gly | Asp435Asn |
| Gly54Val | Asp435Asn/Glu/Ile | Gly77Glu | Pro454Ser | |
| Val71Ile | Glu474Lys | Asp78Asn | Ile460Val | |
| Asp80Ala | Ser79Phe/Tyr | Glu474Lys | ||
| Ser81Phe/Tyr | Asp83Ala/Asn/Gly/Tyr | |||
| Ser83Phe/Tyr | Asn91Asp | |||
| Glu85Gly/Lys | Gly128Asp | |||
| Glu87Lys/Gln | Gly135Asp | |||
| Trp93Ser | Lys137Asn/Asp | |||
| Ala142Ser |
The degree of resistance caused by mutation of a single amino acid in the enzymes varies among bacterial species and quinolones. Different bacterial strains have different primary targets for quinolones, thereby having different relative sensitivities to a given quinolone. Resistant clinical isolates indicate that a single target-site gene mutation on either of the two enzymes results in an 8–16-fold increase in resistance.126 A single target-site mutation in both DNA gyrase and topoisomerase IV results in increasing the levels of resistance. Sequential mutations in both target enzymes in clinical isolates increases the resistance up to 100-fold.128 Some bacteria, such as Mycobacterium tuberculosis, Treponema pallidum, and Helicobacter pylori, have only the DNA gyrase enzyme.
Many quinolone-resistant strains do not have mutations in the enzymatic target QRDR139 and were less susceptible to unrelated compounds, such as cyclohexane, salicylate, and tetracycline, proving that resistance is associated with broad-spectrum efflux activity.140 The multiple antibiotic resistance (mar) gene is known to cause tolerance to a variety of compounds.141 Mutation of this gene leads to both over-expression of the acrAB efflux pump and reduced expression of OmpF (outer membrane protein F) porin. MarA, a positive regulator of acrAB transcription, can be induced either by mutation of the mppA gene or by exposure to salicylate and tetracycline. Thereby, salicylate and tetracycline may stimulate quinolone resistance. Moreover, MarA prevents translation of OmpF and activates the expression of OmpX, which is a porin expression down-regulator, thus reducing the expression of a variety of porins, such as OmpC, OmpD, OmpD, OmpF, LamB, and Tsx.142,143
Another gene that contributes to the resistance against quinolones and other antibacterial agents is the nfxB gene, which confers alterations in expression of functional OmpF at the cell surface OmpF, thereby decreasing quinolone entry.144 In addition, modifications of OmpA, a β-barrel protein associated with the integrity of the cell envelope or acting as a porin, depending on the species, may lead to reduced quinolone susceptibility,4 as can changes in SoxRS regulons resulting from bacterial adaption to superoxide stress.140
Quinolone resistance associated with efflux pumps include modification of the major facilitator superfamily (MFS) of Gram-positive bacteria or the resistance–nodulation–division (RND) family, multiple antibiotic and toxin extrusion (MATE), and ATP-binding cassette (ABC) of Gram-negative bacteria.142 Mutations of efflux systems can alter their specificity for quinolones or cause upregulation.
4.2. Acquisition of resistance plasmids
In addition to resistance caused by mutations in the bacterial genome, quinolone resistance also occurs via plasmid-mediated mechanisms. Plasmids carrying the quinolone resistance genes can cause serious clinical issues, with 10–250-fold decreases in susceptibility.145 The transmission of these resistance plasmids is through horizontal transfer from bacteria to bacteria as well as vertical transfer from generation to generation. Three reported gene families are involved in this plasmid-mediated quinolone resistance (Table 7). They reduce the bacterial susceptibility to quinolones and mediate the selection of mutants promoting treatment failure.| qnr gene | |
|---|---|
| qnrS | DNA mimics |
| qnrB | Decreases binding of enzymes to DNA → lowering the enzyme targets on the chromosome |
| qnrC | |
| qnrD | Binds to the enzymes and inhibits the quinolones from entering the cleavage complexes |
| qnrVC | |
| aac(6′)-Ib-cr include 2 mutations | |
| Trp102Arg | Variant of aminoglycoside acetyltransferase |
| Asp179Tyr | Acetylates the unsubstituted N of the C7 piperazine ring → decreases drug activity |
| Plasmid-mediated quinolone efflux pumps | |
| OqxAB | Increases efflux pump activity |
| QepA | Decreases susceptibility to hydrophilic quinolones |
The first plasmid-encoded protein is Qnr, a pentapeptide repeat family protein.146 These proteins are folded into a right-hand quadrilateral β-helix shape and dimerize to format rod-like structure with a size, shape, and electrostatic surface mimicking that of β-form DNA.147 More than 100 variants have been discovered in clinical isolates, which are classified into 6 subfamilies (qnrA, qnrB, qnrC, qnrD, qnrS, and qnrVC).146 The qnr gene has been reported to originate from the chromosomes of many aquatic bacteria; with qnrA originally from Shewanella algae, qnrB from Citrobacter spp., qnrC, qnrS, and qnrVC from Vibrio spp., and qnrD and qnrE from Enterobacter spp.148 These Qnr proteins compete with DNA binding to the enzymes, thereby inhibiting the quinolone from entering the cleavage complexes and reducing the number of double-stranded breaks on the chromosomes, resulting in reduced quinolone toxicity to the chromosomes.
The second plasmid-encoded protein involved in quinolone resistance is AAC(6′)-Ib-cr, a derivative of aminoglycoside acetyltransferase that has Trp102Arg and Asp179Tyr mutations.149 These two unique mutations distinguish this variant enzyme from other ACC(6′)-Ib enzymes, and leads to specific targeting of quinolones with an amine on the piperazinyl ring skeleton, such as ciprofloxacin, norfloxacin, and enoxacin. The enzyme acetylated the unsubstituted nitrogen of the R7 piperazine ring, thereby decreasing the quinolone activity. Both mutations are necessary for this specific enzyme action, with the Trp102Arg mutation positioning the Asp179Tyr tyrosine aromatic ring for optimal interaction with the quinolone, anchoring it in place.149
The third family associated with quinolone resistance is the plasmid-mediated quinolone efflux pumps including OqxAB and QepA. OqxAB is a multidrug-resistant efflux pump encoded by conjugative plasmid pOLA52 found in E. coli strains isolated from swine manure. It was recently detected in human clinical isolates of E. coli and K. pneumoniae. Bacteria with this oqxAB gene were 8- to 16-fold less susceptible to nalidixic acid and ciprofloxacin, respectively. This efflux pump not only mediates low-level quinolone resistance but also helps bacteria to survive under low concentration of quinolones, thus facilitating the subsequent development of higher level resistance.149 Another novel plasmid-mediated quinolone efflux pump is QepA, which is encoded by pHPA plasmid found in clinical isolates of E. coli from Japan. It is an efflux pump of the major facilitator family that decreases bacterial susceptibility to hydrophilic quinolones.11 These multidrug-resistant efflux pump encoded genes do not directly cause high levels of resistance to quinolones, but can facilitate the development of mutations to topoisomerase enzymes by allowing the bacteria to adapt to low concentrations in quinolones inside the bacteria.
5. Future development of quinolone antibiotics
A number of other quinolones have recently been approved or are under advanced clinical development (Table 8).150–152 The C7 substituents of classical quinolones lack a strong basic group, so the quinolones have weak acidity. Basicity can enhance their activity in acidic environments, including phagolysosomes, inflammatory cells related to infection sites such as skin, soft tissue, vagina, and urinary tract. Studies by Kocsis and his colleagues have reviewed four potential substituents at the R7 position that could increase the activity of the compounds in an acidic environment including 3-hydroxy-1-azetidinyl (delafloxacin, FDA approved in June 2017), (3E)-3-(2-amino-1-fluoroethylidene)piperidinyl (avarofloxacin, ceased development after phase 3), pyrrolo-oxazinyl (finafloxacin, FDA approved 2014), and (8E)-8-methoxyimino-2,6-diazaspiro[3.4]octan (zabofloxacin, phase 3)153 (Fig. 8).| Compound | Development phase | Developer | Chemical structure |
|---|---|---|---|
| Delafloxacin | Approved | Melinta (via Wakunaga Pharmaceutical and Rib-X Pharmaceutical) |
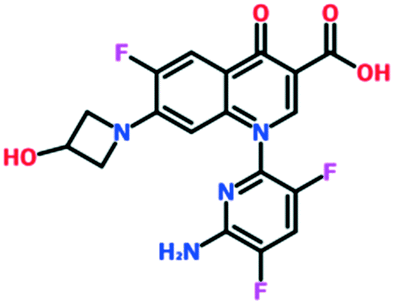
|
| Finafloxacin | Approved for otic suspension | MerLion Pharmaceuticals Pte Ltd. |
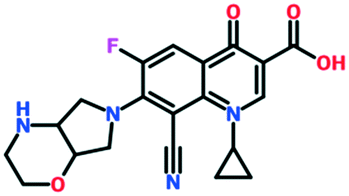
|
| Ozenoxacin | Approved in Japan (2015), topical cream approved in USA/Canada for impetigo | Maruho Co (via Toyama Chemical) in Japan, Ferrer Internacional in Europe, Medimetriks in USA |
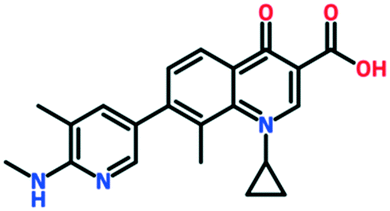
|
| Avarofloxacin | Completed phase 3, development halted | Furiex Pharmaceuticals |

|
| Cadazolid | Completed phase 3, development halted 2018 | Johnson & Johnson (via Actelion) |
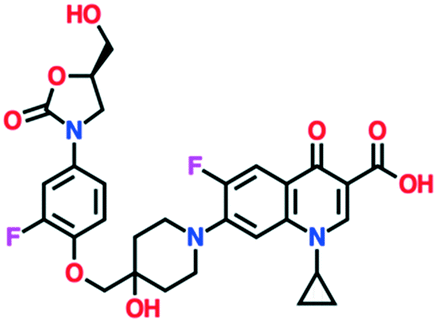
|
| Zabofloxacin | Phase 3 | Dong Wha Pharmaceuticals/Pacific Beach BioSciences |
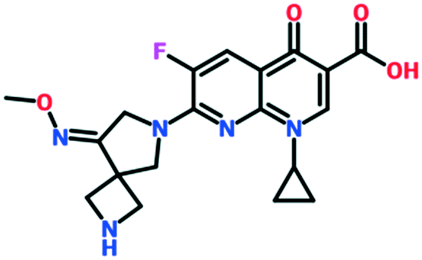
|
| Lascufloxacin | Phase 3 completed | Kyorin Pharmaceutical Co. Ltd. |
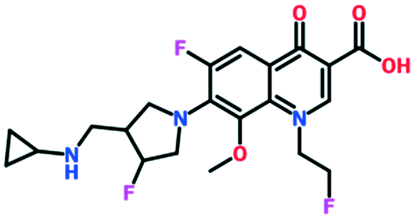
|
| Nemonoxacin | Phase 2 (marketed in Russia, Taiwan, China, as Taigexyn) | TaiGen Biotechnology Co. Ltd. |
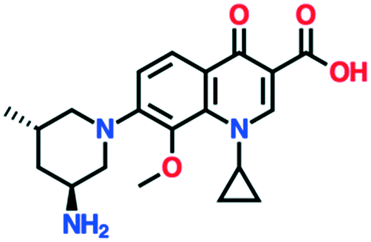
|
| OPS-2071 | Phase 2 | Otsuka Pharmaceutical Co. Ltd. | Unknown |
| Levonadifloxacin (WCK 771) + alalevonadifloxacin (WCK 23491, oral prodrug) | Phase 2 | Wockhardt Ltd. |
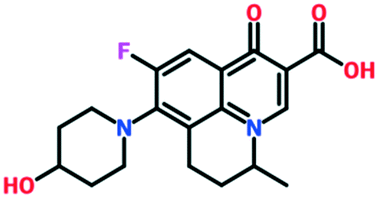
|
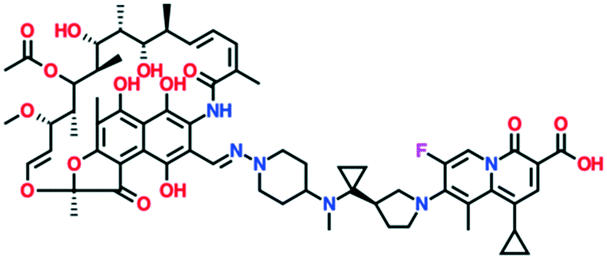
| TNP-2092 (a rifamycin–quinolizinone hybrid) | Phase 1 | TenNor Therapeutics |
|
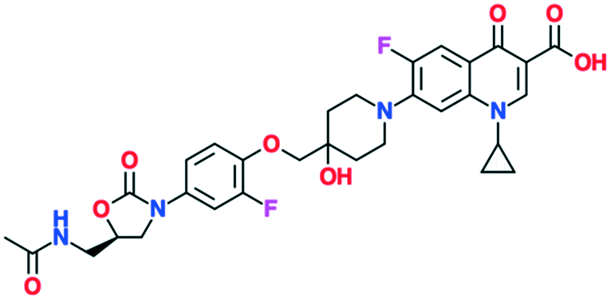
| MCB3837 (oxazolidinone–quinolone hybrid) | Phase 1 | Deinove SA (formerly Morphochem AG) |
|
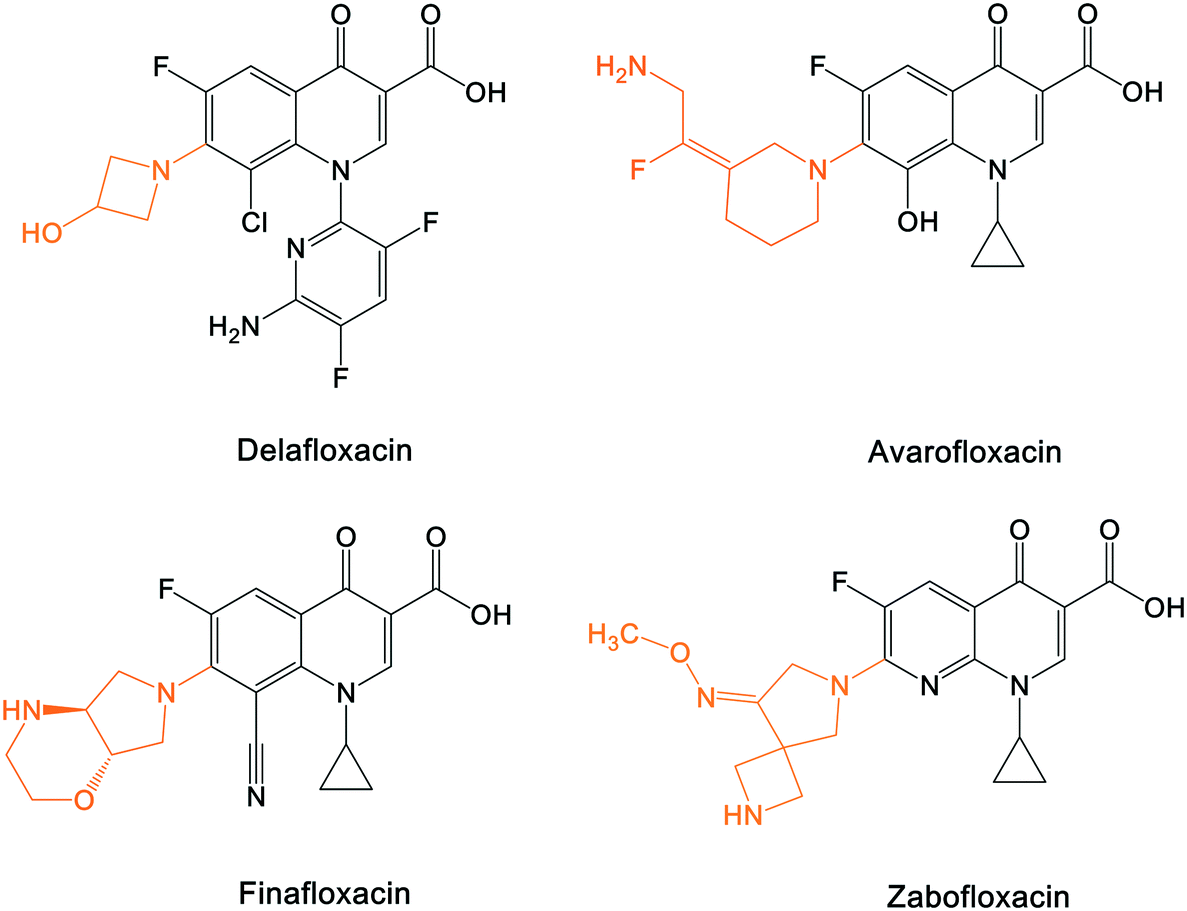 | ||
| Fig. 8 The structure of novel quinolones and their C7 substituents. The varied C7 substituents are presented in orange. | ||
These novel quinolones have been found to result in improved potency and safety profile and decreased toxicity compared with classical quinolones.
A number of other quinolones are in various stages of development (Table 8).150–152 Several of these are hybrid antibiotics, in which the quinolone scaffold is attached to either rifamycin (TNP-2092) or an oxazolidinone (MCB3837) to increase the spectrum of activity. A related fluoroquinolone–oxazolidinone hybrid, cadazolid, was developed by Actelion and progressed through phase III trials for Clostridium difficile infections, but after Actelion was acquired by Johnson & Johnson in June 2017 further development was discontinued in April 2018 after meeting the endpoint in only one of two trials.
As shown in the description of quinolone resistance, the main resistance mutations are located on the DNA gyrase and topoisomerase IV enzymes, which disrupt the quinolones from the cleavage complexes. Therefore, several researchers have focused on developing novel agents that could overcome this resistance. While many attempts have been made, the most recent studies have attempted to modify the structure to find a new binding site on the enzymes, distinct from the water–metal ion bridge. Based on this approach, quinazolinediones have been proposed as a new class of antimicrobial compound. They possess a similar structure to quinolones but do not contain the keto acid moiety that associates to the water–metal ion bridge interactions154 (Fig. 7). The keto acid is replaced by an R2 carbonyl that can bind to the enzyme's conserved arginine residue by a hydrogen bond (Fig. 9).108 This binding seems to overcome the resistance generated against quinolones; but the hydrogen bond interaction was shown to be weaker than the metal-ion interaction of quinolones. However, QnrA has been reported to reduce susceptibility to quinazoline-2,4-diones.159 Other studies on quinazolinediones have demonstrated that some agents in the quinazolinedione class with a 3′-(aminomethyl)pyrrolidinyl as R7 substituent produce stronger binding to the bacterial topoisomerase type II enzymes.155 However, this substituent also targets the human type IIα enzymes, leading to toxicity and therefore cannot be used for human therapy. Future studies should invest in identifying novel C7 substituents for quinazolinediones that are selective for bacterial over human enzymes. It is possible that the types of basic C7 substituents found in the new fluoroquinolones recently approved or under development could be applied to the quinazolinedione scaffold (Fig. 9).
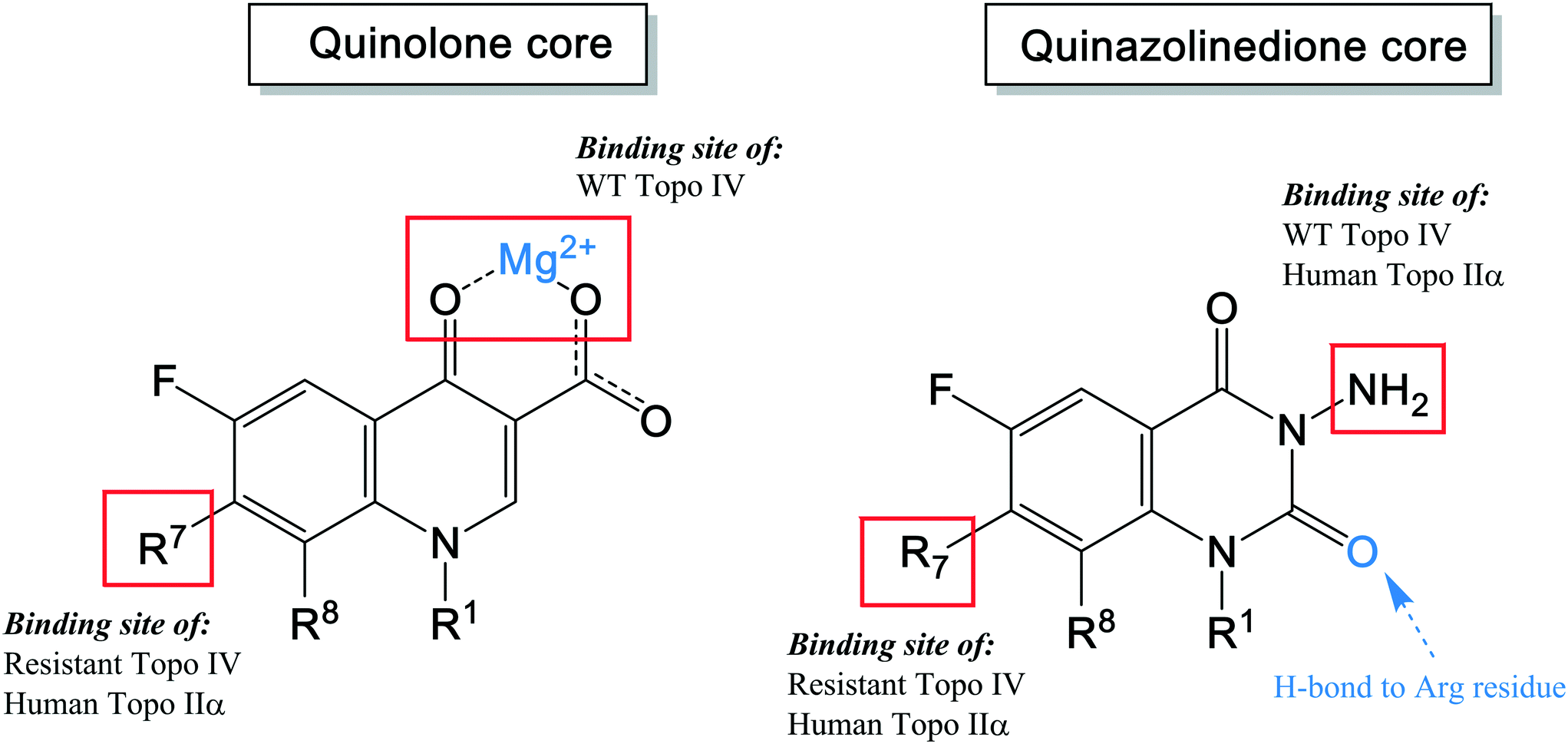 | ||
| Fig. 9 The pharmacophore structure of quinolones and quinazolinediones showing different potential binding sites. | ||
6. Conclusions
Quinolones are a class of synthetic bactericidal antibiotics with broad-spectrum activity, which can inhibit both Gram-negative and Gram-positive bacteria, including anaerobes. They exert their activity by binding to the bacterial topoisomerase type II enzymes, interfering with the DNA synthesis pathway. Binding to the cleavage complex occurs via a water–metal ion bridge, which links the keto carbonyl group of quinolone indirectly to the serine and acidic residue of the enzymes mediated by a Mg2+ ion.As with other antibiotics, this class is faced with a rapid increase in global levels of resistance, either through self-generating mutations or via plasmid-mediated acquisition. The main genomic mutations occur by altering the enzyme target active site serine residue, which accounts for more than 90% of the mutant pool. Quinazolinediones, a related structural class of antibiotics, do not rely on this critical interaction for their binding to the same enzyme, and so have potential for further development if potency can be improved and human enzyme interactions reduced.
The quinolones can be modified at the R1, R6, R7, and R8 (and, less commonly, R5) positions to optimize activity, pharmacokinetics, and toxicity. The best substituents at each position are a cyclopropyl group at R1, a fluorine at R6, an azabicyclic group at R7, and a methoxy group at R8. Although the fluorine at position R6 significantly improves quinolone activity, current research is focused on its removal because it is related to genotoxicity. The reduction in potency on its removal can be compensated for by using alternative substituents at R5 (amine, –NH2) and R1 (cyclopropyl or 2,4-difluorophenyl), which can give the same potency as the fluorine at R6 but with reduced toxicity.
It seems clear that improvements in activity and anti-resistant properties are still possible, and new generations of quinolones can still contribute to the effective treatment of bacterial infections, as reflected by the number of new analogues in the clinical pipeline.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
MATB is supported in part by Wellcome Trust Strategic Grant WT1104797/Z/14/Z and NHMRC Development grant APP1113719.References
- G. S. Bisacchi, J. Med. Chem., 2015, 58, 4874–4882 CrossRef CAS
.
- H. Liu and S. G. Mulholland, Am. J. Med., 2005, 118, 14–20 CrossRef PubMed
- G. Y. Lesher, E. J. Froelich, M. D. Gruett, J. H. Bailey and R. P. Brundage, J. Med. Pharm. Chem., 1962, 5, 1063–1065 CrossRef CAS
- A. M. Emmerson, J. Antimicrob. Chemother., 2003, 51, 13–20 CrossRef CAS
- M. D. Adjei, J. Deck, T. M. Heinze, J. P. Freeman, A. J. Williams and J. B. Sutherland, J. Ind. Microbiol. Biotechnol., 2006, 34, 219–224 CrossRef
- M. I. Andersson, J. Antimicrob. Chemother., 2003, 51, 1–11 CrossRef CAS
- L. A. Mitscher, Chem. Rev., 2005, 105, 559–592 CrossRef CAS
- V. Anderson and N. Osheroff, Curr. Pharm. Des., 2001, 7, 337–353 CrossRef CAS
- C. D. Lima and A. Mondragón, Structure, 1994, 2, 559–560 CrossRef CAS
- D. Hooper, Emerging Infect. Dis., 2001, 7, 337–341 CrossRef CAS
- K. J. Aldred, S. A. Mcpherson, C. L. Turnbough, R. J. Kerns and N. Osheroff, Nucleic Acids Res., 2013, 41, 4628–4639 CrossRef CAS
- D. C. Hooper and G. A. Jacoby, Ann. N. Y. Acad. Sci., 2015, 1354, 12–31 CrossRef CAS
- L. J. V. Piddock, M. Johnson, V. Ricci and S. L. Hill, Antimicrob. Agents Chemother., 1998, 42, 2956–2960 CrossRef CAS
- G. S. Tillotson, J. Med. Microbiol., 1996, 44, 320–324 CrossRef CAS PubMed
- K. Naber and D. Adam, Int. J. Antimicrob. Agents, 1998, 10, 255–257 CrossRef CAS
- J. M. Blondeau, Surv. Ophthalmol., 2004, 49(Suppl 2), S73–78 CrossRef
- M. Gellert, K. Mizuuchi, M. H. Odea, T. Itoh and J.-I. Tomizawa, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4772–4776 CrossRef CAS
- P. Pharma, A. Jain and S. Jain, Acta Pol. Pharm., 2009, 66, 587–604 Search PubMed
-
G. L. Patrick, in An introduction to Medicinal Chemistry, Oxford University Press, Oxford, United Kingdom, 2003, pp. 379–435 Search PubMed
- D. L. V. Caekenberghe and S. R. Pattyn, Antimicrob. Agents Chemother., 1984, 25, 518–521 CrossRef
- L. R. Peterson, Clin. Infect. Dis., 2001, 33(Suppl 3), S180–186 CrossRef CAS PubMed
- A. A. E. Solh and A. Alhajhusain, J. Antimicrob. Chemother., 2009, 64, 229–238 CrossRef
- K. Naber and D. Adam, Int. J. Antimicrob. Agents, 1998, 10, 255–257 CrossRef CAS
- J. M. Domagala, J. Antimicrob. Chemother., 1994, 33, 685–706 CrossRef CAS
- B. Ledoussal, J. K. Almstead and C. P. Flaim, presented in part at the Program and abstracts of the 39th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, 1999 Search PubMed.
- A. P. Macgowan, J. Antimicrob. Chemother., 1999, 43, 345–349 CrossRef CAS
- K. P. Fu, S. C. Lafredo, B. Foleno, D. M. Isaacson, J. F. Barrett, A. J. Tobia and M. E. Rosenthale, Antimicrob. Agents Chemother., 1992, 36, 860–866 CrossRef CAS PubMed
- A. Bryskier, Clin. Infect. Dis., 1999, 29, 632–658 CrossRef CAS
- T. Yoshida, Y. Yamamoto, H. Orita, M. Kakiuchi, Y. Takahashi, M. Itakura, N. Kado, K. Mitani, S. Yasuda, H. Kato and Y. Itoh, Chem. Pharm. Bull., 1996, 44, 1074–1085 CrossRef CAS PubMed
- F. V. Bambeke, J.-M. Michot, J. V. Eldere and P. Tulkens, Clin. Microbiol. Infect., 2005, 11, 256–280 CrossRef PubMed
- G. S. Tillotson, J. Med. Microbiol., 1996, 44, 320–324 CrossRef CAS PubMed
- D. A. Leigh, S. Tait and B. Walsh, J. Antimicrob. Chemother., 1991, 27, 589–598 CrossRef CAS
- M. N. Lowe and H. M. Lamb, Drugs, 2000, 59, 1137–1147 CrossRef CAS PubMed
- A. Dalhoff, Expert Opin. Invest. Drugs, 1999, 8, 123–137 CrossRef CAS
- D. S. Reeves, M. J. Bywater and H. A. Holt, J. Antimicrob. Chemother., 1984, 14, 7–17 CrossRef
- S. W. B. Newsom, J. Antimicrob. Chemother., 1984, 13, 25–31 CrossRef CAS
- R. Grüneberg, D. Felmingham, M. Ohare, M. J. Robbins, K. Perry, R. A. Wall and G. L. Ridgway, J. Antimicrob. Chemother., 1988, 22, 9–19 CrossRef
- A. King and I. Phillips, J. Antimicrob. Chemother., 1986, 18, 1–20 CrossRef CAS
- B. Wiedemann and P. Heisig, J. Antimicrob. Chemother., 1997, 40, 19–25 CrossRef CAS
- B. A. Cunha, S. M. Qadri, Y. Ueno, E. A. Walters and P. Domenico, J. Antimicrob. Chemother., 1997, 39, 29–34 CrossRef CAS PubMed
- R. Wise, J. P. Ashby and J. M. Andrews, Antimicrob. Agents Chemother., 1988, 32, 1251–1256 CrossRef CAS PubMed
- A. P. Macgowan, Expert Opin. Invest. Drugs, 1999, 8, 181–199 CrossRef CAS
- C. M. Perry, J. A. B. Balfour and H. M. Lamb, Drugs, 1999, 58, 683–696 CrossRef CAS
- X. Zhao, J.-Y. Wang, C. Xu, Y. Dong, J. Zhou, J. Domagala and K. Drlica, Antimicrob. Agents Chemother., 1998, 42, 956–958 CrossRef CAS
- V. Cecchetti, A. Fravolini, M. C. Lorenzini, O. Tabarrini, P. Terni and T. Xin, J. Med. Chem., 1996, 39, 436–445 CrossRef CAS
- A. Berrington, Int. J. Med. Microbiol., 2001, 291, 237–239 CrossRef CAS PubMed
- R. B. Muijsers and B. Jarvis, Drugs, 2002, 62, 967–973 CrossRef CAS
- L. C. Parish, H. B. Routh, B. Miskin, J. Fidelholtz, P. Werschler and A. Heyd, Int. J. Clin. Pract., 2000, 54, 497–503 CAS
- J. M. Entenza, Y. A. Que, J. Vouillamoz, M. P. Glauser and P. Moreillon, Antimicrob. Agents Chemother., 2001, 45, 3076–3083 CrossRef CAS
-
M. Farrington, Antibacterial drugs, in Clinical Pharmacology, ed. N. P. Bennett, J. M. Brown and P. Sharma, Churchill linvingstone, USA, 2012, pp. 173–190 Search PubMed
- R. Wolf, R. Eberl, A. Dunky, N. Mertz, T. Chang, J. R. Goulet and J. Latts, J. Antimicrob. Chemother., 1984, 14, 63–69 CrossRef
- H. Stass, J. Antimicrob. Chemother., 1999, 43, 83–90 CrossRef CAS
-
D. Hooper, Quinolones, in Principles and Practice of Infectious Diseases, ed. G. L. Mandell, R. Douglas and J. E. Bennett, Churchill Linvingstone, USA, 2000, pp. 404–423 Search PubMed
- C. A. Peloquin, Arch. Intern. Med., 1989, 149, 2269–2273 CrossRef CAS
- L. Lee, A. M. Padula and R. C. Kimbrough, N. Engl. J. Med., 1991, 325, 520–521 CrossRef
- D. E. Nix, M. F. Sands and C. A. Peloquin, Am. J. Med., 1987, 82, 352–356 CAS
- A. Forrest, D. E. Nix, C. H. Ballow, T. F. Goss, M. C. Birmingham and J. J. Schentag, Antimicrob. Agents Chemother., 1993, 37, 1073–1081 CrossRef CAS
- S. L. Preston, G. L. Drusano, A. L. Berman, C. L. Fowler, A. T. Chow, B. Dornseif, V. Reichl, J. Natarajan and M. Corrado, JAMA, 1998, 279, 125 CrossRef CAS PubMed
- M. Jacobs, Clin. Microbiol. Infect., 2001, 7, 589–596 CrossRef CAS
- J. K. Thomas, A. Forrest, S. M. Bhavnani, J. M. Hyatt, A. Cheng, C. H. Ballow and J. J. Schentag, Antimicrob. Agents Chemother., 1998, 42, 521–527 CrossRef CAS
- A. A. Firsov, J. Antimicrob. Chemother., 2002, 50, 533–539 CrossRef CAS PubMed
- T. Gumbo, A. Louie, M. R. Deziel, L. M. Parsons, M. Salfinger and G. L. Drusano, J. Infect. Dis., 2004, 190, 1642–1651 CrossRef CAS
- X. Zhao and K. Drlica, J. Infect. Dis., 2002, 185, 561–565 CrossRef
- G. P. Allen, G. W. Kaatz and M. J. Rybak, Antimicrob. Agents Chemother., 2003, 47, 2606–2614 CrossRef CAS
- H. J. Smith, M. Walters, T. Hisanaga, G. G. Zhanel and D. J. Hoban, Antimicrob. Agents Chemother., 2004, 48, 3954–3958 CrossRef CAS
- D. Croisier, M. Etienne, E. Bergoin, P.-E. Charles, C. Lequeu, L. Piroth, H. Portier and P. Chavanet, Antimicrob. Agents Chemother., 2004, 48, 1699–1707 CrossRef CAS
- J. M. Domagala, J. Antimicrob. Chemother., 1994, 33, 685–706 CrossRef CAS
- T. D. Gootz and K. E. Brighty, Med. Res. Rev., 1996, 16, 433–486 CrossRef CAS
- P. Ball, Int. J. Clin. Pract., 2000, 54, 329–332 CAS
- K. E. Brighty and T. D. Gootz, J. Antimicrob. Chemother., 1997, 39, 1–14 CrossRef CAS
- M. N. Lowe and H. M. Lamb, Drugs, 2000, 59, 1137–1147 CrossRef CAS
- Y. Dong, C. Xu, X. Zhao, J. Domagala and K. Drlica, Antimicrob. Agents Chemother., 1998, 42, 2978–2984 CrossRef CAS
- K. Patel and J. L. Goldman, J. Clin. Pharmacol., 2016, 56, 1060–1075 CrossRef CAS PubMed
- E. Cornett, M. B. Novitch, A. D. Kaye, C. A. Pann, H. S. Bangalore, G. Allred, M. Bral, P. K. Jhita and A. M. Kaye, Postgrad. Med., 2017, 129, 715–724 CrossRef
- T. D. Gootz and K. E. Brighty, Med. Res. Rev., 1996, 16, 433–486 CrossRef CAS
- J. Marchant, Nature, 2018, 555, 431–433 CrossRef CAS
- M. D. Blum, D. J. Graham and C. A. Mccloskey, Clin. Infect. Dis., 1994, 18, 946–950 CrossRef CAS
- H. J. Chen, K. J. Bloch and J. A. Maclean, N. Engl. J. Med., 2000, 342, 359–360 CrossRef CAS
- E. Rubinstein, Chemotherapy, 2001, 47, 3–8 CrossRef CAS PubMed
- R. Stahlmann, Toxicol. Lett., 2002, 127, 269–277 CrossRef CAS
- C. W. Stratton, Clin. Drug Invest., 1998, 15, 19–34 CrossRef CAS
- S. Singh and A. Nautiyal, Am. J. Med., 2017, 130, 1449–1457.e1449 CrossRef CAS
- S. Childs, Infectious in Urology, 2000, 13, 3–10 Search PubMed
- P. Ball, L. Mandell, Y. Niki and G. Tillotson, Drug Saf., 1999, 21, 407–421 CrossRef CAS
- Factive, Gemifloxacin, 2002.
- R. Stahlmann, Toxicol. Lett., 2002, 127, 269–277 CrossRef CAS
- H. Lode and E. Rubinstein, mBio, 2003, 407–419 Search PubMed
- J. Bertino and D. Fish, Clin. Ther., 2000, 22, 798–817 CrossRef CAS
- D. J. Menzies, P. A. Dorsainvil, B. A. Cunha and D. H. Johnson, Am. J. Med., 2002, 113, 232–234 CrossRef
- K. Shimoda, Toxicol. Lett., 1998, 95, 19 CrossRef
- W. Christ, J. Antimicrob. Chemother., 1990, 26, 219–225 CrossRef CAS
- J. Kawakami, K. Yamamoto, A. Asanuma, K. Yanagisawa, Y. Sawada and T. Iga, Toxicol. Appl. Pharmacol., 1997, 145, 246–254 CrossRef CAS
- M. R. Bidell, M. Palchak, J. Mohr and T. P. Lodise, Antimicrob. Agents Chemother., 2016, 60, 3170–3173 CrossRef CAS
- W. L. Cuypers, J. Jacobs, V. Wong, E. J. Klemm, S. Deborggraeve and S. Van Puyvelde, Microb. Genomics, 2018, 4, e000195 Search PubMed
- N. Schellack, E. Bronkhorst, C. Maluleka, L. Hunt, P. Srinivas, W. Grootboom, D. Goff, P. Naicker, T. Modau and O. Babarinde, S. Afr. J. Infect. Dis., 2017, 33, 54–56 Search PubMed
- H. H. Liu, Curr. Ther. Res., 2004, 65, 225–238 CrossRef CAS
- P. Collignon, M. J. Iovine, A. Gupta, R. V. Tauxe and F. J. Angulo, Emerging Infect. Dis., 2005, 11, 1789–1792 CrossRef
- E. Yefet, R. Salim, B. Chazan, H. Akel, S. Romano and Z. Nachum, Obstet. Gynecol. Surv., 2014, 69, 681–694 CrossRef
- Z. S. Almalki, X. Yue, Y. Xia, P. R. Wigle and J. J. Guo, Pharmacoecon. Open, 2016, 1, 123–131 CrossRef
- D. C. Hooper, Drugs, 1995, 49, 10–15 CrossRef CAS
- F. Jacob, S. Brenner and F. Cuzin, Cold Spring Harbor Symp. Quant. Biol., 1963, 28, 329–348 CrossRef CAS
- V. Anderson and N. Osheroff, Curr. Pharm. Des., 2001, 7, 337–353 CrossRef CAS
- C. Levine, H. Hiasa and K. J. Marians, Biochim. Biophys. Acta, Gene Struct. Expression, 1998, 1400, 29–43 CrossRef CAS
- P. M. Hawkey, J. Antimicrob. Chemother., 2003, 51, 29–35 CrossRef CAS
- A. B. Khodursky, E. L. Zechiedrich and N. R. Cozzarelli, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 11801–11805 CrossRef CAS PubMed
- S. Aedo and Y.-C. Tse-Dinh, Antimicrob. Agents Chemother., 2012, 56, 5458–5464 CrossRef CAS
- X. S. Pan, J. Ambler, S. Mehtar and L. M. Fisher, Antimicrob. Agents Chemother., 1996, 40, 2321–2326 CrossRef CAS
- B. Fournier, X. Zhao, T. Lu, K. Drlica and D. C. Hooper, Antimicrob. Agents Chemother., 2000, 44, 2160–2165 CrossRef CAS
- X. S. Pan and L. M. Fisher, Antimicrob. Agents Chemother., 1997, 41, 471–474 CrossRef CAS
- X. S. Pan and L. M. Fisher, Antimicrob. Agents Chemother., 1998, 42, 2810–2816 CrossRef CAS
- J. J. Champoux, Annu. Rev. Biochem., 2001, 70, 369–413 CrossRef CAS
- E. M. Tretter and J. M. Berger, J. Biol. Chem., 2012, 287, 18636–18644 CrossRef CAS
- E. M. Tretter and J. M. Berger, J. Biol. Chem., 2012, 287, 18645–18654 CrossRef CAS
- P. Forterre and D. Gadelle, Nucleic Acids Res., 2009, 37, 679–692 CrossRef CAS
- K. J. Aldred, R. J. Kerns and N. Osheroff, Biochemistry, 2014, 53, 1565–1574 CrossRef CAS
- L. S. Redgrave, S. B. Sutton, M. A. Webber and L. J. Piddock, Trends Microbiol., 2014, 22, 438–445 CrossRef CAS
- D. C. Hooper, Drugs, 1999, 58, 6–10 CrossRef CAS
- T. T. Qin, H. Q. Kang, P. Ma, P. P. Li and L. Y. Huang, Ann. Transl. Med., 2015, 3, 358 Search PubMed
- X. Wang, X. Zhao, M. Malik and K. Drlica, J. Antimicrob. Chemother., 2010, 65, 520–524 CrossRef CAS
- C. Sissi, E. Perdonà, E. Domenici, A. Feriani, A. J. Howells, A. Maxwell and M. Palumbo, J. Mol. Biol., 2001, 311, 195–203 CrossRef CAS
- K. J. Aldred, H. A. Schwanz, G. Li, S. A. Mcpherson, C. L. Turnbough, R. J. Kerns and N. Osheroff, ACS Chem. Biol., 2013, 8, 2660–2668 CrossRef CAS
- K. Drlica, H. Hiasa, R. Kerns, M. Malik, A. Mustaev and X. Zhao, Curr. Top. Med. Chem., 2009, 9, 981–998 CrossRef CAS
- K. L. Hopkins, R. H. Davies and E. J. Threlfall, Int. J. Antimicrob. Agents, 2005, 25, 358–373 CrossRef CAS
- L. B. Price, A. Vogler, T. Pearson, J. D. Busch, J. M. Schupp and P. Keim, Antimicrob. Agents Chemother., 2003, 47, 2362–2365 CrossRef CAS
- S. K. Morgan-Linnell, L. B. Boyd, D. Steffen and L. Zechiedrich, Antimicrob. Agents Chemother., 2008, 53, 235–241 CrossRef PubMed
- L. S. Redgrave, S. B. Sutton, M. A. Webber and L. J. Piddock, Trends Microbiol., 2014, 22, 438–445 CrossRef CAS
- K. Hiramatsu, M. Igarashi, Y. Morimoto, T. Baba, M. Umekita and Y. Akamatsu, Int. J. Antimicrob. Agents, 2012, 39, 478–485 CrossRef CAS
- D. C. Hooper and G. A. Jacoby, Cold Spring Harbor Perspect. Med., 2016, 6(9), a025320 CrossRef
- Y. T. Wang, M. F. Lee and C. F. Peng, Biomarkers Genomic Med., 2014, 6, 79–83 CrossRef
- C. M. Sanfilippo, C. K. Hesje, W. Haas and T. W. Morris, Chemotherapy, 2011, 57, 363–371 CrossRef CAS
- M. Fujimoto-Nakamura, H. Ito, Y. Oyamada, T. Nishino and J.-I. Yamagishi, Antimicrob. Agents Chemother., 2005, 49, 3810–3815 CrossRef CAS
- K. Hirose, A. Hashimoto, K. Tamura, Y. Kawamura, T. Ezaki, H. Sagara and H. Watanabe, Antimicrob. Agents Chemother., 2002, 46, 3249–3252 CrossRef CAS
- E. Ehrmann, A. Jolivet-Gougeon, M. Bonnaure-Mallet and T. Fosse, J. Antimicrob. Chemother., 2017, 72, 2208–2212 CrossRef CAS
- F. Rafii, M. Park and J. S. Novak, Antimicrob. Agents Chemother., 2005, 49, 488–492 CrossRef CAS
- L. M. Weigel, G. J. Anderson and F. C. Tenover, Antimicrob. Agents Chemother., 2002, 46, 2582–2587 CrossRef CAS
- H. Ito, H. Yoshida, M. Bogaki-Shonai, T. Niga, H. Hattori and S. Nakamura, Antimicrob. Agents Chemother., 1994, 38, 2014–2023 CrossRef CAS
- N. Oizumi, S. Kawabata, M. Hirao, K. Watanabe, S. Okuno, T. Fujiwara and M. Kikuchi, J. Infect. Chemother., 2001, 7, 191–194 CrossRef CAS
- X. S. Pan and L. M. Fisher, Antimicrob. Agents Chemother., 1998, 42, 2810–2816 CrossRef CAS
- E. Cambau, S. Matrat, X. S. Pan, R. R. D. Bettoni, C. Corbel, A. Aubry, C. Lascols, J. Y. Driot and F. M. Fisher, J. Antimicrob. Chemother., 2009, 63, 443–450 CrossRef CAS
- L. Mcmurry, FEMS Microbiol. Lett., 1998, 166, 305–309 CrossRef CAS
- J. D. Goldman, D. G. White and S. B. Levy, Antimicrob. Agents Chemother., 1996, 40, 1266–1269 CrossRef CAS
- S. Correia, P. Poeta, M. Hébraud, J. L. Capelo and G. Igrejas, J. Med. Microbiol., 2017, 66, 551–559 CrossRef CAS
- A. Dalhoff, Interdiscip. Perspect. Infect. Dis., 2012, 2012, 1–37 CrossRef
- D. C. Hooper, J. S. Wolfson, K. S. Souza, C. Tung, G. L. Mchugh and M. N. Swartz, Antimicrob. Agents Chemother., 1986, 29, 639–644 CrossRef CAS
- J. Strahilevitz, G. A. Jacoby, D. C. Hooper and A. Robicsek, Clin. Microbiol. Rev., 2009, 22, 664–689 CrossRef CAS
- J. H. Tran and G. A. Jacoby, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5638–5642 CrossRef CAS
- M. W. Vetting, S. S. Hegde, M. Wang, G. A. Jacoby, D. C. Hooper and J. S. Blanchard, J. Biol. Chem., 2011, 286, 25265–25273 CrossRef CAS
- M. B. Sánchez, A. Hernández, J. M. Rodríguez-Martínez, L. Martínez-Martínez and J. L. Martínez, BMC Microbiol., 2008, 8, 148 CrossRef
- M. W. Vetting, C. H. Park, S. S. Hegde, G. A. Jacoby, D. C. Hooper and J. S. Blanchard, Biochemistry, 2008, 47, 9825–9835 CrossRef CAS
-
World Health Organization, 2017, http://www.who.int/medicines/areas/rational_use/antibacterial_agents_clinical_development/en/ Search PubMed
-
The Pew Charitable Trust, 2014, https://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development Search PubMed
- M. S. Butler, M. A. Blaskovich and M. A. Cooper, J. Antibiot., 2016, 70, 3–24 CrossRef
- B. Kocsis, J. Domokos and D. Szabo, Ann. Clin. Microbiol. Antimicrob., 2016, 15, 34 CrossRef
- N. German, M. Malik, J. D. Rosen, K. Drlica and R. J. Kerns, Antimicrob. Agents Chemother., 2008, 52, 3915–3921 CrossRef CAS
- A. Mustaev, M. Malik, X. Zhao, N. Kurepina, G. Luan, L. M. Oppengard, H. Hiasa, K. R. Marks, R. J. Kerns, J. M. Berger and K. Drlika, J. Biol. Chem., 2014, 113, 529–539 Search PubMed
- C. E. Perrone, Toxicol. Sci., 2002, 69, 16–22 CrossRef CAS
- K. Drlica, M. Malik, R. J. Kerns and X. Zhao, Antimicrob. Agents Chemother., 2008, 52, 385–392 CrossRef CAS
- A. C. Cheng, J. Turnidge, P. Collignon, D. Looke, M. Barton and T. Gottlieb, Emerging Infect. Dis., 2012, 18, 1453–1460 CrossRef CAS
- G. A. Jacoby, M. A. Corcoran and D. C. Hooper, Antimicrob. Agents Chemother., 2015, 59, 6689–6695 CrossRef CAS
- A. Dalhoff, Interdiscip. Perspect. Infect. Dis., 2012, 2012, 1–37 CrossRef
-
A. S. Wagman and M. P. Wentland, Comprehensive Medicinal Chemistry II, 2007, vol. 7, pp. 567–596 Search PubMed
| This journal is © The Royal Society of Chemistry 2019 |