
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of N-acyl amide natural products using a versatile adenylating biocatalyst†
Piera M.
Marchetti‡
 ,
Shona M.
Richardson‡
,
Noor M.
Kariem
and
Dominic J.
Campopiano
*
,
Shona M.
Richardson‡
,
Noor M.
Kariem
and
Dominic J.
Campopiano
*
EastCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: Dominic.Campopiano@ed.ac.uk
First published on 31st May 2019
Abstract
Natural products are secondary metabolites produced by many different organisms such as bacteria, fungi and plants. These biologically active molecules have been widely exploited for clinical application. Here we investigate TamA, a key enzyme from the biosynthetic pathway of tambjamine YP1, an acylated bipyrrole that is produced by the marine microorganism Pseudoalteromonas tunicata. TamA is a didomain enzyme composed of a catalytic adenylation (ANL) and an acyl carrier protein (ACP) domain that together control the fatty acid chain length of the YP1. Here we show that the TamA ANL domain alone can be used to generate a range of acyl adenylates that can be captured by a number of amines thus leading to the production of a series of fatty N-acyl amides. We exploit this biocatalytic promiscuity to produce the recently discovered class of N-acyl histidine amide natural products from Legionella pneumophila.
Introduction
Many plants, bacteria and fungi, as well as other organisms, produce small molecule secondary metabolites with potent biological activity. The 2015 Nobel Prize in Physiology or Medicine was awarded for natural product (NP) research, recognising their impact in human health.1 These important NPs (avermectin and artemisinin) have been successful in the treatment of diseases including river blindness and malaria. NPs, derivatives or mimics constituted 60% of newly approved small molecule drugs in the period between 1981 and 2014.2–4 With the advent of the genomics era it is predicted that even more NPs from plants and microbes will be found. However, many of these NPs will require derivatisation to improve activity, stability or uptake.5The large family of pyrrole NPs have gained recent interest due to their incorporation in many different biologically active molecules. For example the structure, function and biosynthesis of the prodiginine family of alkaloids have attracted great attention due to their potential uses in medical chemistry.6 They can contain two, three or four pyrrole rings and one interesting subset are the bipyrrole members of the tambjamine class. They are thought to be involved in chemical defense mechanisms but since they also display killing activity, they have been explored for their potential therapeutic value.7 Tambjamine YP1 was extracted from the anti-biofouling marine bacterium Pseudoalteromonas tunicata and was found to have antimicrobial, antimalarial and cytotoxic activity.7–14 Its biosynthetic pathway is encoded in an operon (the tam cluster) consisting of 19 genes, 11 of which have proposed biosynthetic functions involved in the formation of a key 4-methoxy-2,2′-bipyrrole carbaldehyde (MBC) intermediate and attachment of a fatty amine tail, (Fig. 1).14,15 Included in this pathway is the gene encoding the predicted AMP-ligating enzyme TamA, which consists of both a catalytic adenylating (ANL) N-terminal domain and a C-terminal acyl carrier protein (ACP).
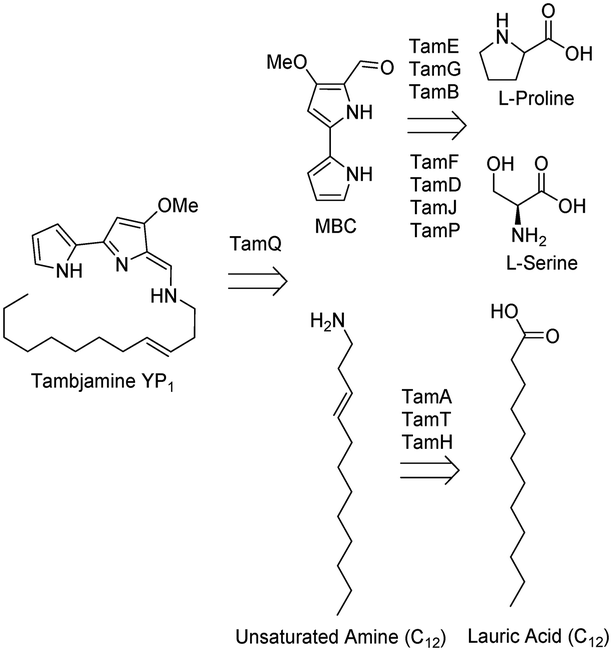 | ||
| Fig. 1 The tambjamine YP1 retrobiosynthetic pathway, showing the two convergent routes (MBC and C12 amine) involved in biosynthesis and ligation. Highlighted are the 11 enzymes encoded in the tam cluster that catalyse reactions in the pathway. | ||
Adenylating enzymes use ATP as a mechanism to activate the fatty acid in two independent half reactions (Fig. 2). The first half reaction is attack of the deprotonated carboxylic acid on the α-phosphate of ATP creating an acyl-adenylate intermediate which is driven by liberation of the PPi product. The incoming nucleophile can subsequently attack the tightly bound acyl-adenylate intermediate, releasing adenosine monophosphate (AMP), which is the driving force for the second half reaction. This approach is often used by nature to combine carboxylic acids with a range of nucleophiles to produce esters, amides, and thioesters.16,17 The adenylating superfamily contains a series of amide bond synthetases (ABS) which catalyse the formation of various amide-containing NPs.18 Included in this family are the firefly luciferases involved in bioluminescence19 and the acyl-CoA synthetases20 which channel carboxylic acids into their respective metabolic pathway. Also, the recently discovered McbA, from Marinactinospora thermotolerans, has been shown to catalyse amide bond formation on a broad range of β-carboline substrates which can be exploited for the sustainable synthesis of amides by the pharmaceutical industry.21,22 These enzymes illustrate the vast potential of the adenylate-forming enzymes for biocatalysis. Recently we showed that TamA plays a key role in tambjamine YP1 biosynthesis; the ANL domain selects and activates the fatty acid substrate and delivers it to its ACP domain as a thioester. This ACP-bound product is the substrate for downstream processing to the corresponding amines (Fig. 1). We also discovered that as well as activating the C12 lauric acid found in YP1, TamA could also use C6–C14 substrates suggesting that it displays some substrate flexibility.23
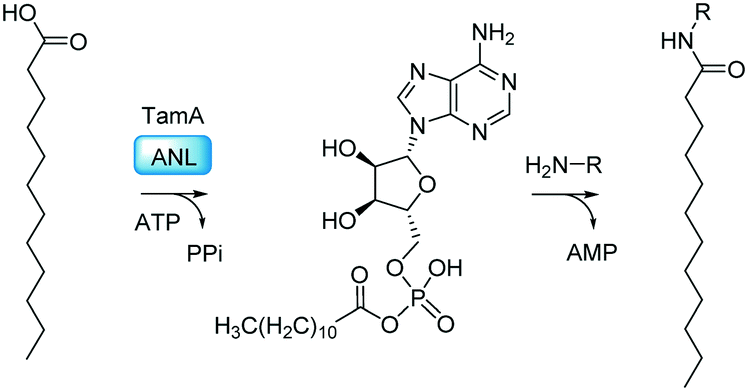 | ||
| Fig. 2 The TamA ANL domain catalyses the ATP-dependent formation of the C12-adenylate intermediate. This reacts with an amine (H2N-R) to generate the N-acyl amide. | ||
An interesting family of recently discovered biocatalysts are the carboxylic acid reductases (CARs) which catalyse the conversion of carboxylic acids to aldehydes. A number of different CAR isoforms have been characterised but they are all composed of three domains; ANL, carrier protein (CP) and NADPH-dependent reductase (R), which select, activate and reduce a broad range of different acids.24–27 Four members of the CAR family have recently been investigated by Flitsch and coworkers to produce a number of primary, secondary and tertiary amides.28 Since ANL domain enzymes are gaining attention we sought to explore the synthetic capability of TamA. We found that the TamA ANL domain alone could be used to couple various fatty acids with several amino acids to generate the corresponding N-acyl amides. We then applied this to the production of the recently discovered class of N-acyl amide NPs from Legionella pneumophila. The versatility of the TamA ANL domain suggests it is a useful addition to the biocatalytic toolbox.
Results and discussion
Since adenylating enzymes activate carboxylic acids for nucleophilic attack, once the TamA ANL domain generated the key acyl-adenylate intermediate from the fatty acid substrate and ATP, we began by exploring whether this could be captured with a range of nucleophilic amines. We wished to use only the catalytic TamA ANL domain so this was prepared by deleting the C-terminal ACP domain and a short segment thought to be the inter-domain linker sequence (Fig. S1†). In the absence of an X-ray crystal structure of TamA, construction of an active TamA ANL domain was guided by sequence alignment with the fatty acid AMP ligase (FAAL) from Legionella pneumophila and a homology model which predicts that the domain spans residues (1-571) in the N-terminus (Fig. S2 and S3†).29 This also suggests the residues that are most likely involved in the acyl chain length specificity (I195, F199 and F266). We prepared four constructs of different lengths with N-terminal His-tags and based on solubility, the shortest version (TamA ANL576) was isolated in good yield and used in all subsequent biocatalysis experiments (Fig. S4†). Gel filtration chromatography (GFC) and LC ESI-MS analysis of the TamA ANL domain confirmed the protein was monomeric with a deconvoluted mass of 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 653 Da ± 1 Da consistent with the predicted mass of 64651 Da (Fig. S5†). We also isolated full length TamA according to Marchetti et al. for comparative analysis (Fig. S6†).23
653 Da ± 1 Da consistent with the predicted mass of 64651 Da (Fig. S5†). We also isolated full length TamA according to Marchetti et al. for comparative analysis (Fig. S6†).23
With the TamA ANL domain in hand we began by exploring primary amide formation with a range of long chain fatty acids (C2–C16) and ammonia, following established reaction conditions.23,28 The amide products were detected by LC ESI-MS with an increase in retention time corresponding to an increasing chain length (Fig. 3 and S7†). Under these conditions TamA ANL generates the amide products from the corresponding C9–C14 acids which is similar to the chain length specificity that we observed for the full length TamA didomain.23
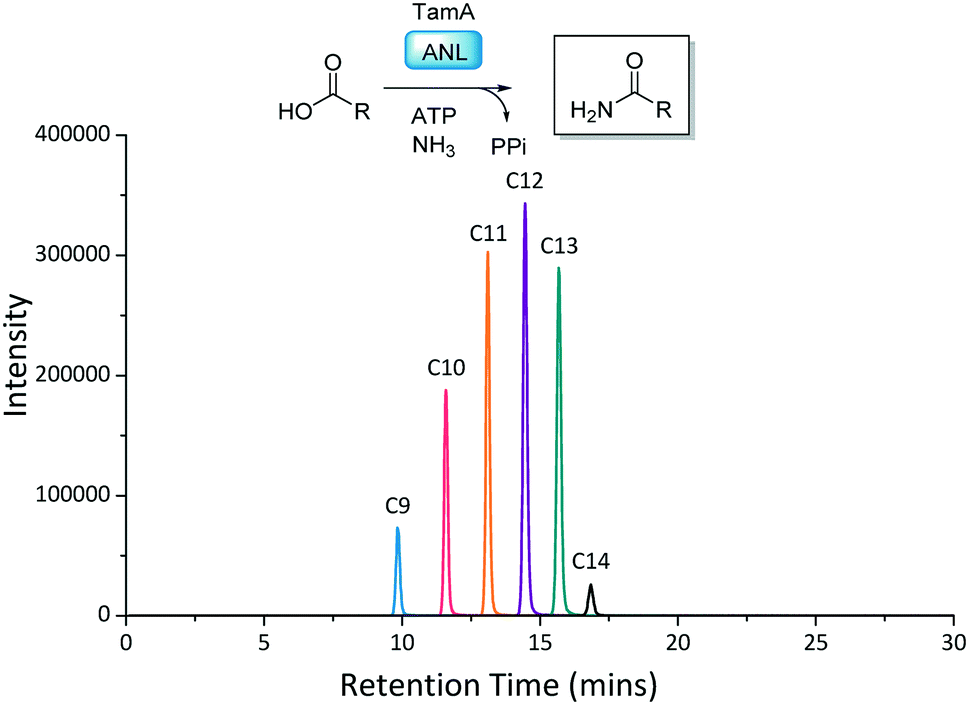 | ||
| Fig. 3 Reaction scheme and LC ESI-MS extracted ion chromatograms (EICs) for each of the amide products from the ANL catalysed reactions of (C2–C16) fatty acids with ATP and ammonia. | ||
We then selected the C12 acid as a substrate to investigate the amine scope of the TamA ANL-catalysed reaction. We picked enthanolamine and benzylamine, as well as amino acids to test whether the enzyme could be used to generate the corresponding N-acyl amino acid secondary amides. We focussed on L- and D-histidine for three reasons; firstly, to prepare the N-acyl histidine targets which have recently been discovered in L. pneumophila, a human pathogen.30 These metabolites are upregulated during the L. pneumophila infection cycle and could regulate human signalling pathways, analogously to other identified acyl amides. Secondly, the C12 N-acyl histidine has also been investigated for its surfactant properties.31 Therefore these molecules are important for studying human-pathogen interactions as well as having biotechnology applications. Since they are not commercially available, a biocatalytic synthesis of these NPs would be of interest. Finally, we also used both enantiomers of histidine to explore the enantioselectivity and mechanism of the acyl adenylate intermediate amine capture reaction. The TamA ANL was incubated with the C12 acid, ATP and amine and LC ESI-MS analysis of the products revealed that each amide target was present (Fig. 4a–h and S8†). It is important to note that the L- and D-histidine amides were produced in almost equal amounts (Fig. 4g and h).
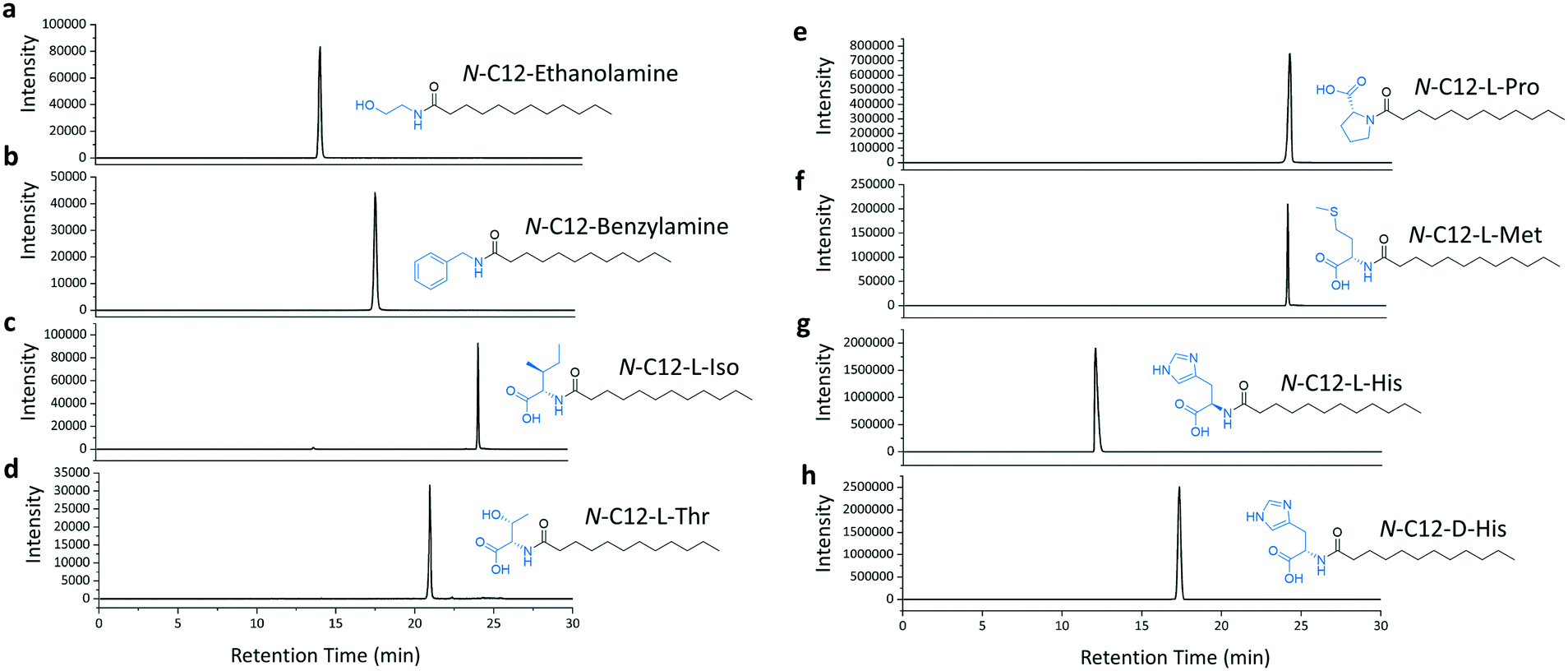 | ||
| Fig. 4 Reaction scheme and LC ESI-MS extracted ion chromatograms (EICs) for each of the amide products from the ANL catalysed reactions of C12 fatty acid with ATP and a series of amines (a and b) and amino acids (c–h). | ||
This lack of enantioselectivity strongly suggests that the second step amide formation is not enzyme catalysed (Fig. 2). The mechanism of adenylate capture was recently explored by Flitsch and colleagues by using altered forms of the three-domain CAR enzymes. The CAR CP domain was inactivated by mutation of a key Ser689 residue that is post-translationally modified to the 4′phosphopantetheine (4′-PP) form. Similarly, a truncated CAR with a deleted R domain was also prepared. The resulting CARs lacking either a functional CP or R domain were still active towards amide synthesis.28 The authors used this as evidence to support the hypothesis that amide bond formation resulted via amine capture of the adenylate intermediate – either still bound to the enzyme or released into solution.
The N-C12-L-histidine derivative can be chemically synthesised using the coupling of the acid chloride with L-histidine31 or from a His-loaded Wang resin (for the C6, C8 and C10 L-His versions).30 We used the acid chloride method and the NMR and MS analysis of our isolated product is consistent with published data, although we noted technical issues with solvent solubility (Fig. S9†).31 We also probed the synthetic utility of the TamA ANL biocatalyst by carrying out a time-dependent study of the formation of the N-C12-L-histidine target. After 10 minutes the product was observed LC ESI-MS and by 2 hours the reaction was complete (no further change was observed between 2 and 24 hours) (Fig. 5).
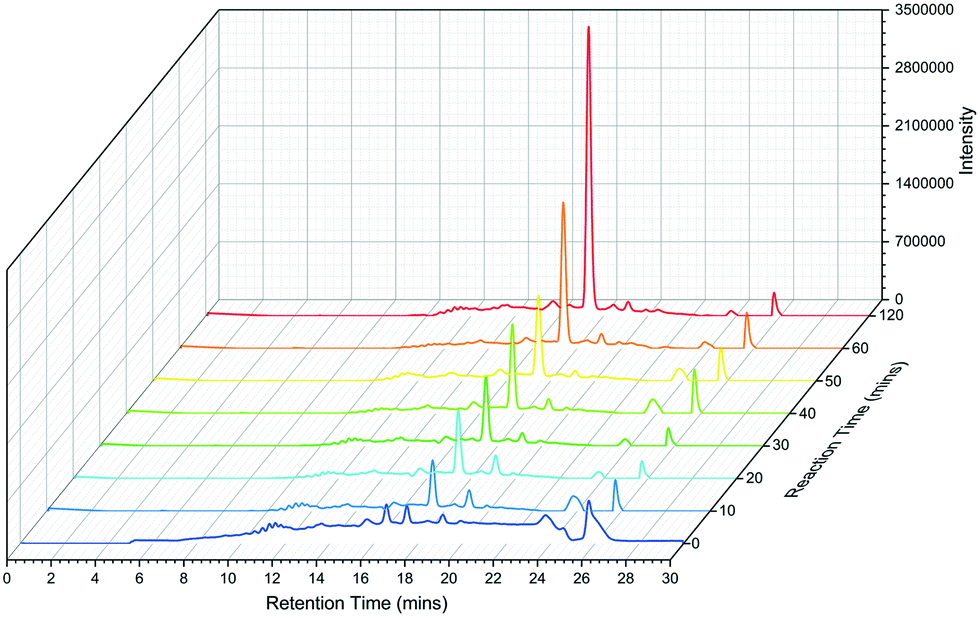 | ||
| Fig. 5 LC ESI-MS extracted ion chromatograms (EICs) of TamA ANL catalysed N-dodecanoyl-L-histidine formation over a 2 hour period. | ||
Product conversion was determined using HPLC analysis and a calibration curve was formed using varying concentrations of the chemically synthesised N-C12-L-histidine (Fig. S10 and S11†). The reaction was then stopped at varying points and was analysed by HPLC. This analysis showed a maximum product conversion of 12.7% observed after 24 hours, although this time-dependent analysis revealed the reaction was complete after 2 hours. This modest biocatalytic conversion from the C12 acid compares with the 5% yield we achieved with the chemical route in our hands using the C12 acid-chloride and L-histidine (see ESI† methods). A larger scale biotransformation was achieved by scaling up the TamA ANL reaction (100 ml buffer + 4% DMSO) and careful addition of the enzyme (1 mg at a time) over 5 hours. A crude yield of 29 mg (86%) of the N-C12-L-histidine product was achieved (Fig. S12†) but product insolubility prevented HPLC purification. The reaction with full length TamA was also analysed for comparison and a similar conversion of 12.5% was observed. However the instability of the full length enzyme caused problems with the enzyme precipitating after a short period of time. In contrast the truncated TamA ANL remains soluble throughout the reaction, therefore deleting the ACP domain offers increased stability of the biocatalyst.
We then combined the acyl-chain promiscuity of the TamA ANL domain with acyl-adenylate amino capture to generate the C6, C8 and C10 derivatives of the N-acyl histidines (Fig. 6 and S13†). The LC ESI-MS data clearly shows the production of these N-acyl histidines. However, the peak intensity decreases with decreasing chain length suggesting that even though TamA ANL does accept a range of chain lengths, it still exhibits a preference for the natural substrate lauric acid.
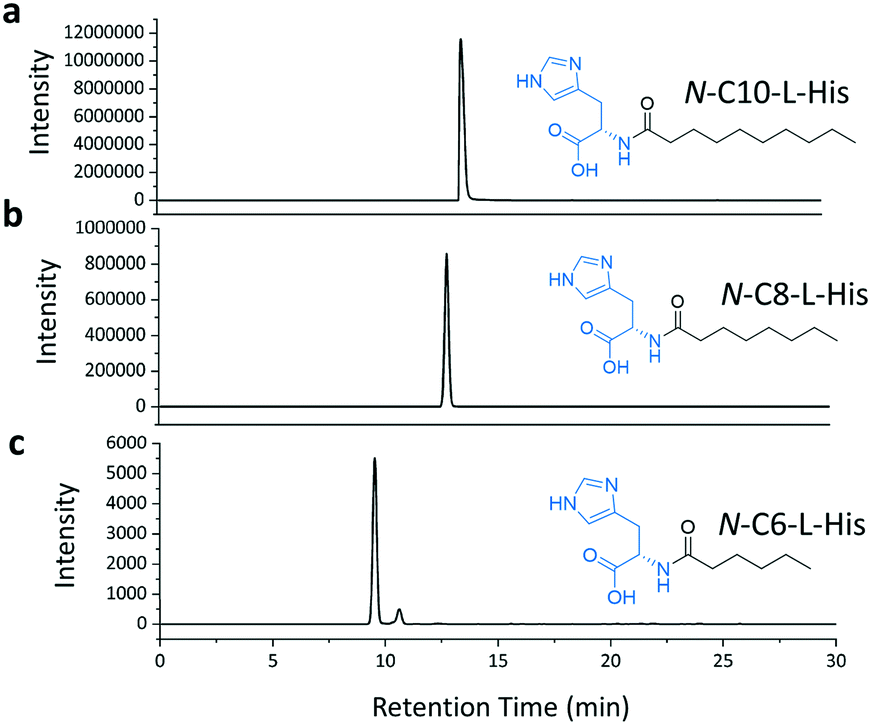 | ||
| Fig. 6 LC ESI-MS extracted ion chromatograms (EICs) for each of the amide products from the ANL catalysed reactions of C10 (a), C8 (b) and C6 (c) fatty acids with ATP and L-histidine. | ||
Conclusion
The ANL superfamily of enzymes play an important role in NP biosynthesis by catalysing the activation of a range of fatty acids. Here we have shown that the TamA ANL domain is a useful ABS biocatalyst for the production of a range of fatty acyl amides including the L. pneumophila N-acyl histidines. The use of both L- and D-amino acid substrates has revealed the uncatalysed nature of the capture of the acyl-adenylate intermediate by amine nucleophiles. Further work to identify the residues involved in the TamA ANL substrate specificity should allow targeted engineering of the enzyme to broaden its synthetic utility.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PMM was supported by a BBSRC Doctoral Training Grant “EastBio” (BB/J01446X/1). SMR was funded the Derek Stewart Charitable Trust and the University of Edinburgh. MS data were acquired on an instrument funded by the Engineering and Physical Sciences Research Council (EPSRC, EP/K039717/1). Also, special thanks to Catherine Connolly who helped with the HPLC analysis.References
- B. Shen, Cell, 2015, 163, 1297–1300 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- K. C. Morrison and P. J. Hergenrother, Nat. Prod. Rep., 2014, 31, 6–14 RSC.
- D. X. Hu, D. M. Withall, G. L. Challis and R. J. Thomson, Chem. Rev., 2016, 116, 7818–7853 CrossRef CAS PubMed.
- D. M. Pinkerton, M. G. Banwell, M. J. Garson, N. Kumar, M. O. de Moraes, B. C. Cavalcanti, F. W. A. Barros and C. Pessoa, Chem. Biodiversity, 2010, 7, 1311–1324 CrossRef CAS PubMed.
- B. Carte and D. J. Faulkner, J. Org. Chem., 1983, 48, 2314–2318 CrossRef CAS.
- M. S. Melvin, D. C. Ferguson, N. Lindquist and R. A. Manderville, J. Org. Chem., 1999, 64, 6861–6869 CrossRef CAS PubMed.
- M. S. Melvin, K. E. Wooton, C. C. Rich, G. R. Saluta, G. L. Kucera, N. Lindquist and R. A. Manderville, J. Inorg. Biochem., 2001, 87, 129–135 CrossRef CAS PubMed.
- B. C. Cavalcanti, H. V. N. Junior, M. H. R. Seleghim, R. G. S. Berlinck, G. M. A. Cunha, M. O. Moraes and C. Pessoa, Chem.-Biol. Interact., 2008, 174, 155–162 CrossRef CAS PubMed.
- L. N. Aldrich, S. L. Stoops, B. C. Crews, L. J. Marnett and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2010, 20, 5207–5211 CrossRef CAS PubMed.
- M. Kotev, P. Manuel-Manresa, E. Hernando, V. Soto-Cerrato, M. Orozco, R. Quesada, R. Perez-Tomas and V. Guallar, J. Chem. Inf. Model., 2017, 57, 2089–2098 CrossRef CAS PubMed.
- C. Holmstrom, S. James, B. A. Neilan, D. C. White and S. Kjelleberg, Int. J. Syst. Bacteriol., 1998, 48, 1205–1212 CrossRef CAS PubMed.
- C. Burke, T. Thomas, S. Egan and S. Kjelleberg, Environ. Microbiol., 2007, 9, 814–818 CrossRef CAS PubMed.
- S. Schmelz and J. H. Naismith, Curr. Opin. Struct. Biol., 2009, 19, 666–671 CrossRef CAS PubMed.
- A. M. Gulick, ACS Chem. Biol., 2009, 4, 811–827 CrossRef CAS PubMed.
- J. Pitzer and K. Steiner, J. Biotechnol., 2016, 235, 32–46 CrossRef CAS PubMed.
- E. Conti, N. P. Franks and P. Brick, Structure, 1996, 4, 287–298 CrossRef CAS PubMed.
- J. M. Ellis, J. L. Frahm, L. O. Li and R. A. Coleman, Curr. Opin. Lipidol., 2010, 21, 212–217 CrossRef CAS PubMed.
- C. Ji, Q. Chen, Q. Li, H. Huang, Y. Song, J. Ma and J. Ju, Tetrahedron Lett., 2014, 55, 4901–4904 CrossRef CAS.
- M. Petchey, A. Cuetos, B. Rowlinson, S. Dannevald, A. Frese, P. W. Sutton, S. Lovelock, R. C. Lloyd, I. J. S. Fairlamb and G. Grogan, Angew. Chem., Int. Ed., 2018, 57, 11584–11588 CrossRef CAS PubMed.
- P. M. Marchetti, V. Kelly, J. P. Simpson, M. Ward and D. J. Campopiano, Org. Biomol. Chem., 2018, 16, 2735–2740 RSC.
- M. Winkler, Curr. Opin. Chem. Biol., 2018, 43, 23–29 CrossRef CAS PubMed.
- W. Finnigan, A. Thomas, H. Cromar, B. Gough, R. Snajdrova, J. P. Adams, J. A. Littlechild and N. J. Harmer, ChemCatChem, 2017, 9, 1005–1017 CrossRef CAS PubMed.
- D. Gahloth, M. S. Dunstan, D. Quaglia, E. Klumbys, M. P. Lockhart-Cairns, A. M. Hill, S. R. Derrington, N. S. Scrutton, N. J. Turner and D. Leys, Nat. Chem. Biol., 2017, 13, 975–981 CrossRef CAS PubMed.
- G. Qu, J. Guo, D. Yang and Z. Sun, Green Chem., 2018, 20, 777–792 RSC.
- A. J. L. Wood, N. J. Weise, J. D. Frampton, M. S. Dunstan, M. A. Hollas, S. R. Derrington, R. C. Lloyd, D. Quaglia, F. Parmeggiani, D. Leys, N. J. Turner and S. L. Flitsch, Angew. Chem., Int. Ed., 2017, 56, 14498–14501 CrossRef CAS PubMed.
- Z. Zhang, R. Zhou, J. M. Sauder, P. J. Tonge, S. K. Burley and S. Swaminathan, J. Mol. Biol., 2011, 406, 313–324 CrossRef CAS PubMed.
- T. Torring, S. R. Shames, W. Cho, C. R. Roy and J. M. Crawford, ChemBioChem, 2017, 18, 638–646 CrossRef CAS PubMed.
- S. Lei, J. Zhang, J. R. Wang and J. B. Huang, Langmuir, 2010, 26, 4288–4295 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section with enzyme isolation, biocatalysis methods, HPLC, MS and NMR analysis. See DOI: 10.1039/c9md00063a |
| ‡ These authors contributed equally to the paper. |
| This journal is © The Royal Society of Chemistry 2019 |