
Pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines to develop functionalized ligands to target adenosine receptors: fluorescent ligands as an example†
Stephanie
Federico
 *a,
Enrico
Margiotta
b,
Silvia
Paoletta‡
b,
Sonja
Kachler
c,
Karl-Norbert
Klotz
c,
Kenneth A.
Jacobson
d,
Giorgia
Pastorin
e,
Stefano
Moro
b and
Giampiero
Spalluto
a
*a,
Enrico
Margiotta
b,
Silvia
Paoletta‡
b,
Sonja
Kachler
c,
Karl-Norbert
Klotz
c,
Kenneth A.
Jacobson
d,
Giorgia
Pastorin
e,
Stefano
Moro
b and
Giampiero
Spalluto
a
aDepartment of Chemical and Pharmaceutical Sciences, University of Trieste, Via Licio Giorgeri 1, 34127 Trieste, Italy. E-mail: sfederico@units.it
bMolecular Modeling Section (MMS), Dipartimento di Scienze del Farmaco, Università degli Studi di Padova, Via F. Marzolo 5, 35131 Padova, Italy
cInstitut für Pharmakologie und Toxicologie, Universität Würzburg, Versbacher Straße 9, 97078 Würzburg, Germany
dLaboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland 20892-0810, USA
eDepartment of Pharmacy, National University of Singapore, 3 Science Drive 3, 117543 Singapore
First published on 18th February 2019
Abstract
A series of adenosine receptor antagonists bearing a reactive linker was developed. Functionalization of these derivatives is useful to easily obtain multi-target ligands, receptor probes, drug delivery systems, and diagnostic or theranostic systems. The pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine scaffold was chosen as a pharmacophore for the adenosine receptors. It was substituted at the 5 position with reactive linkers of different lengths. Then, these compounds were used to synthesise probes for the adenosine receptors by functionalization with a fluorescent moiety. Both series of compounds were evaluated for their binding at the four adenosine receptor subtypes. Different affinity and selectivity profiles were observed towards hA1, hA2A and hA3 adenosine receptors. In particular, fluorescent compounds behave as dual hA2A/hA3 ligands. Computational studies suggested different binding modes for developed compounds at the three receptors. Both molecular docking and supervised molecular dynamics (SuMD) simulations confirmed that the preferred binding mode at the single receptor was driven by the substitution present at the 5 position. Obtained results rationalized the compounds' binding profile at the adenosine receptors and pave the way for the development of more potent conjugable and conjugated ligands targeting these membrane receptors.
Introduction
Functionalization of a ligand is a strategy useful in the design of receptor probes,1,2 drug delivery systems3–5 and multitarget ligands.6 In particular, a pharmacophore is conjugated to a function bearing moiety by means of a linker, which is incorporated at an insensitive site on the pharmacophore structure.7 For example, receptor probes could be obtained by introducing a fluorophore,8 or a lipid could be used to gain specific characteristics useful in delivering the active molecule at a target tissue.9Adenosine receptors (ARs) are family A G protein-coupled receptors (GPCRs) involved in a number of physiological and pathological conditions that make them promising therapeutic targets for several diseases. In particular, four distinct AR subtypes have been identified so far: A1, A2A, A2B and A3 ARs.10 Each subtype has a specific distribution in the human body, which confers to their ligands particular potential therapeutic applications, such as in: pain conditions (A1 and A3 agonists),11 myocardial perfusion imaging (A2A agonists),12 cardiac and renal conditions (A1 antagonists),13 neurodegenerative diseases (A2A antagonists),14 cancer immunotherapy (A2A antagonists),14 inflammatory bowel disease (A2B antagonists)15 and glaucoma (A3 antagonists).16 It is clear that a pharmacophore, with tunable affinity towards AR subtypes might be useful in order to easily obtain theranostic agents, drug delivery systems or multivalent ligands for these receptors. The pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine (PTP) is a well-known scaffold (Fig. 1) from which many potent AR antagonists have been derived. Among these, preladenant (1) is an A2AAR antagonist under clinical development to treat neurodegeneration and other pathological conditions.17 As depicted in Fig. 1, the PTP scaffold could be simultaneously substituted at different positions, which determines a desired specific affinity and selectivity profiles for the targets. In addition, we have extensive experience on this scaffold.18–21 For these reasons, in this work we have developed conjugable AR antagonists bearing a PTP scaffold (2–19). In order to understand the structural requirements crucial to retain the affinity and selectivity towards ARs, we have investigated these antagonists both from a pharmacological and computational perspective. In a previous study, these compounds were functionalized with a fluorophore leading to fluorescent receptor probes (20–25), which we have reported here, with their full characterization, as an example of application of our strategy.22,23
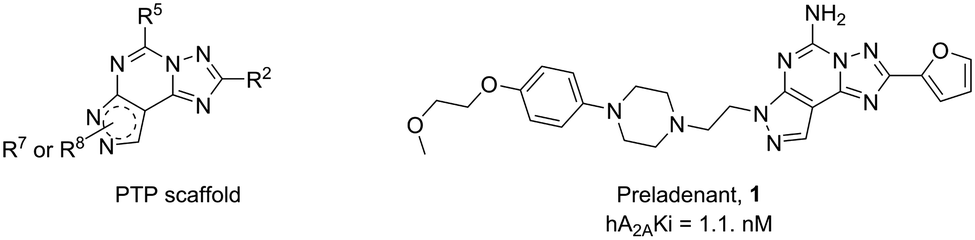 | ||
| Fig. 1 General structure of the pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidine (PTP) nucleus and structure of the hA2AAR antagonist preladenant (1). | ||
Results and discussion
Development of conjugable adenosine receptor ligands
In order to obtain a molecule that is easily functionalized with the desired function-bearing moiety, we decided to use the PTP scaffold and to introduce the linker at the 5 position. In fact, in our previous AR docking studies, we obtained data supporting that alkylamino substituents at the 5 position of PTP point away from the binding pocket of the ARs, thus becoming a suitable molecular site for modification.20 The rationale is that the PTP scaffold lies inside the binding cleft of the receptor to provide high affinity and selectivity towards the target, while the linker could bridge between the pharmacophore and a function-bearing moiety in the extracellular side of the membrane.In order to investigate the effect on the AR affinity, we decided to design nine homologous derivatives bearing diamino linkers of different lengths, from a hydrazino linker to longer diaminopolyglycolic chains. These chains are versatile because one amino group could be reacted with the AR ligand and the other with the detection moiety. In fact, there are several groups that could react with amines, such as carboxylic acids or their activated derivatives, isocyanates and isothiocyanates.
 | ||
| Scheme 1 Synthesis of diamino-linked PTP 2–19. Reagents and conditions: a) appropriated mono-Boc-protected-diamino linker, TEA, ethanol, 110 °C, sealed tube, 2 h; b) tert-butyl piperazine-1-carboxylate, TEA, ethanol, 110 °C, sealed tube, 2 h; c) ethyl acetate-HCl, rt, 1 h. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Compd | X | R | hA1a (Ki nM) | hA2Ab (Ki nM) | hA2Bc (Ki nM) | hA3d (Ki nM) |
| a Displacement of specific [3H]-CCPA binding at hA1ARs expressed in CHO cells (n = 3–6). b Displacement of specific [3H]-NECA binding at hA2AARs expressed in CHO cells (n = 3–6). c Inhibition of the NECA-stimulated adenylyl cyclase in CHO cells (n = 3–6) expressing hA2BARs. d Displacement of specific [3H]-HEMADO binding at hA3ARs expressed in CHO cells (n = 3–6). Data are expressed as geometric means with 95% confidence limits. | ||||||
| 2, WS86 | — | Boc | 7820 (6510–9380) | 3900 (3630–4200) | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
173 (124–241) |
| 3 | –CH2CH2– | Boc | 2550 (1740–3760) | 624 (467–834) | >10000 |
553 (470–651) |
| 4, WS88 | –CH2CH2CH2– | Boc | 1770 (1430–2200) | 195 (129–295) | >3000 | 32.1 (21–49.1) |
| 5 | –CH2CH2CH2CH2– | Boc | 1750 (1310–2340) | 313 (256–383) | 11500 (8590–15300) |
84.4 (79.5–89.7) |
| 6 | –CH2CH2CH2CH2CH2– | Boc | 2580 (1690–3940) | 346 (299–401) | 3000 | 129 (98.1–171) |
| 7, WS91 | –CH2CH2OCH2CH2OCH2CH2– | Boc | 12200 (10700–13800) |
1260 (995–1590) | >10000 |
585 (438–781) |
| 8 | –CH2CH2CH2O(CH2CH2O)2CH2CH2CH2– | Boc | 8480 (5960–12060) |
674 (568–799) | >10000 |
264 (136–515) |
| 9 | — | Boc | 4070 (3470–4770) | 2170 (1980–2370) | ∼10000 |
856 (696–1053) |
| 10 | — | Boc | >100000 |
21400 (18900–24300) |
>10000 |
737 (505–1077) |
| 11 | — | H × HCl | 7100 (4870–10300) |
233 (128–422) | >10000 |
174 (126–241) |
| 12 | –CH2CH2– | H × HCl | 2500 (1480–4230) | 756 (454–1260) | >10000 |
2400 (1938–2971) |
| 13 | –CH2CH2CH2– | H × HCl | 1440 (1290–1610) | 253 (186–344) | >10000 |
3784 (3031–4723) |
| 14 | –CH2CH2CH2CH2– | H × HCl | 1220 (1080–1370) | 169 (108–263) | >3000 | 4205 (3715–4760) |
| 15, WS98 | –CH2CH2CH2CH2CH2– | H × HCl | 778 (608–994) | 52.6 (45.1–61.4) | >3000 | 2246 (2139–2359) |
| 16 | –CH2CH2OCH2CH2OCH2CH2– | H × HCl | 5110 (4830–5400) | 314 (256–387) | >10000 |
2662 (2461–2880) |
| 17 | –CH2CH2CH2O(CH2CH2O)2CH2CH2CH2– | H × HCl | 1660 (1350–2050) | 348 (269–450) | >10000 |
312 (264–368) |
| 18 | — | H × HCl | 5520 (3530–8620) | 682 (610–762) | >10000 |
2914 (1714–4955) |
| 19 | — | H × HCl | >100000 |
12900 (10400–15900) |
>10000 |
8646 (7293–10250) |
In particular, in the Boc-protected series (2–10) we observed low affinity towards hA1AR: long or bulky linkers (7–10) were not tolerated at this receptor, while simple alkyl chains lead to Ki values in the range of 1.7–2.6 μM (i.e.7, hA1AR Ki = 12200 nM vs.4, hA1AR Ki = 1770 nM). Better results were obtained at the hA2AAR, where the affinity follows the same trend observed at the hA1AR. Nevertheless, most of the compounds displayed affinities in the submicromolar range, with a propyl linker providing the best Ki value (4, hA2AAR Ki = 195 nM). On the other hand, Boc-protected derivatives were quite good A3AR antagonists (2–10, hA3AR Ki = 32.1–856 nM). In particular, a clear hA3AR affinity enhancement was observed comparing derivatives 3 and 4 (WS88), which bear an ethyl and a propyl chain, respectively (3, hA3AR Ki = 553 nM vs.4, hA3AR Ki = 32.1 nM). Elongation of the linker caused a progressive decrease of hA3AR affinity (i.e.5, hA3AR Ki = 84.4 nM, 7, hA3AR Ki = 585 nM) as also observed when constrained in a piperazine ring (10, hA3AR Ki = 737 nM). With a Ki value of 32.1 nM, compound 4 was the most potent derivative towards the hA3AR, and it showed 55- and 6-fold selectivity against hA1 and hA2A ARs, respectively. Thus, it was the analogue that displayed the best balance between affinity and selectivity. The most selective compound for the hA3AR was the piperazine derivative 10, which displayed a >136- and 29-fold higher affinity value at the hA3AR, with respect to hA1 and hA2A ARs, respectively, but it showed an affinity in the high nanomolar range towards hA3AR (Ki = 737 nM). The other compounds of this subseries displayed a common selectivity profile at the ARs that was very low against hA2AAR (hA2A/hA3 = 1.1–3.7) and higher towards hA1AR (hA1/hA3 = 20–55). A different behaviour was observed when the shorter hydrazine linker was present at the 5 position of PTP. In fact, first of all, with compound 2 (WS86) we gained hA3AR affinity with respect to the ethyl compound 3 (3, hA3AR Ki = 553 nM vs.2, hA3AR Ki = 173 nM). Moreover, this compound exhibited one of the best selectivity profiles against both hA2AAR (22.5-fold) and hA1AR (45-fold).
However, when the Boc group was removed (leading to the free amino series 11–19) affinity at the hA1AR was not particularly affected (e.g.9, hA1AR Ki = 4070 nM vs.18, hA1AR Ki = 5520 nM). Conversely, at the hA3AR we observed a pronounced detrimental effect (e.g.9, hA3AR Ki = 856 nM vs.18, hA3AR Ki = 2914 nM) and a simultaneous increase of hA2AAR affinity (e.g.9, hA2AAR Ki = 2170 nM vs.18, hA2AAR Ki = 682 nM). As a consequence, in general, free amino compounds could be considered hA2AAR antagonists. Among these, an exception was seen with compounds 11 and 17, which showed similar Ki values towards hA2A and hA3 ARs (11, hA2AAR Ki = 233 nM, hA3AR Ki = 174 nM; 17, hA2AAR Ki = 348 nM, hA3AR Ki = 312 nM). Thus, they could be considered as dual hA2A–hA3 AR antagonists. Interestingly, it seems that for hA3AR, with the hydrazine linker compounds (2, 11) the presence of the Boc group did not influence the affinity at this receptor (2, hA3AR Ki = 173 nM vs.11, hA3AR Ki = 174 nM), while it had a strong effect on the hA2AAR (2, hA2AAR Ki = 3900 nM vs.11, hA2AAR Ki = 233 nM). Notably, observing data for compounds bearing oxygen-containing chains (16, 17), the longer {[(propoxy)-ethoxy]-ethoxy}-propyl linker conferred higher hA3AR affinity than the diethoxyethyl linker (17, hA3AR Ki = 312 nM vs.16, hA3AR Ki = 2662 nM). A similar behaviour was observed, although to a less extent, in Boc-protected compounds (8, hA3AR Ki = 264 nM vs.7, hA3AR Ki = 585 nM). Concerning the structure–activity relationship (SAR) at the hA2AAR, an affinity enhancement was observed upon chain elongation from 2 to 5 methylene units that decreased when longer or bulkier linkers were present at the 5 position. Compound 15 displayed the best hA2AAR affinity of the series, with a Ki value of 52.6 nM and a 15- and 43-fold selectivity against hA1 and hA3 ARs, respectively.
Longer linkers, which should extend into the extracellular part of the receptor, gave especially low affinity compounds and variable selectivity profiles. This observation suggested that the linker had a very crucial role in the recognition process. In order to better understand this behaviour, we performed computational studies on the ligand–receptor recognition process.
Conjugation with a fluorescent moiety
In order to prove the effectiveness of our strategy, we have reported here an example of application where amino-linked compounds 11, 13–17 were conjugated with fluorescein isothiocyanate to afford fluorescent conjugates 20–25.22 Fluorescent ligands could be used to probe the ARs serving as pharmacological tools. As an alternative to radioligands, such fluorescent probes could be useful both in the drug discovery process and in physiological or pathophysiological studies at cellular and tissue levels.24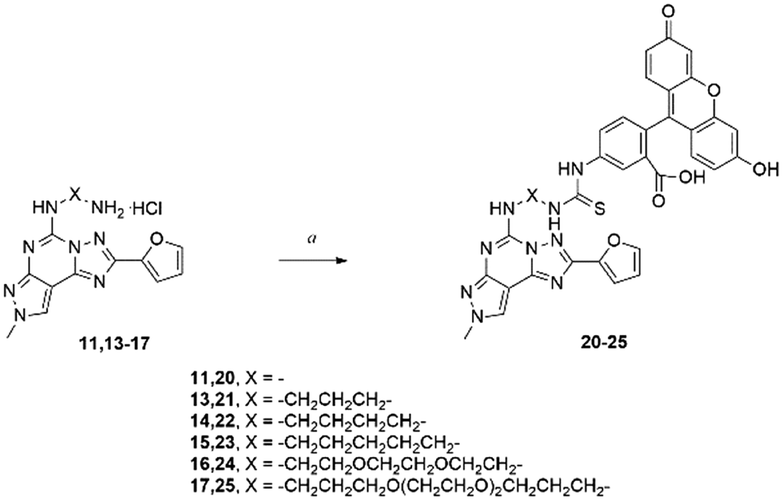 | ||
| Scheme 2 Synthesis of fluorescent probes 20–25.22 Reagents and conditions: a) FITC, TEA, DMF, rt, 48 h. | ||
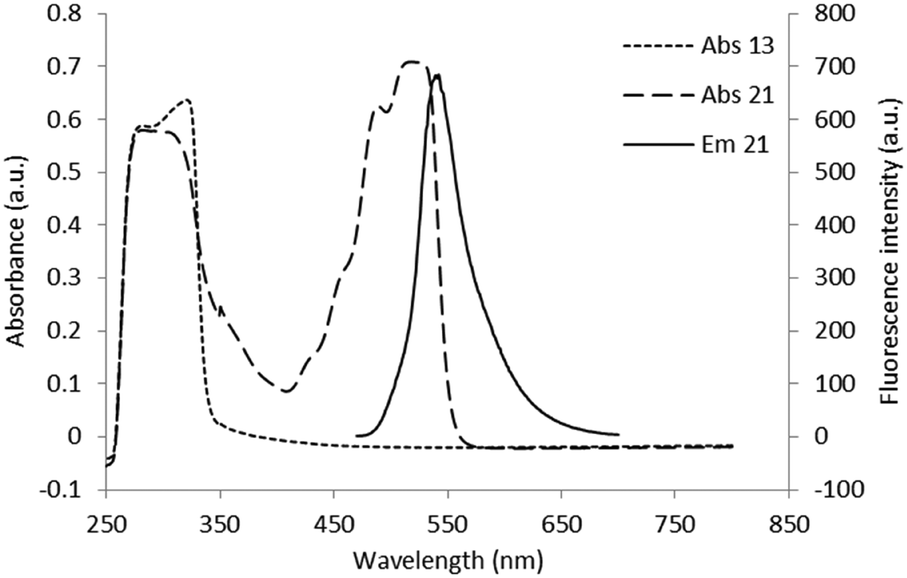 | ||
| Fig. 2 Superimposition of absorbance spectra of compound 13 (10−3 M) and of its fluorescent derivative 21 (10−4 M) and emission spectra of compound 21 (10−5 M). | ||
| Compd | X | hA1b (Ki nM) | hA2Ac (Ki nM) | hA3d (Ki nM) |
|---|---|---|---|---|
| a All data are from ref. 22. b Displacement of specific [3H]-RPIA binding at hA1ARs expressed in CHO cells (n = 3–5). c Displacement of specific [3H]-CGS21680 binding at hA2AARs expressed in HEK293 cells (n = 3–5). d Displacement of specific [125I]I-AB-MECA binding at hA3AR expressed in CHO cells (n = 3–5). Duplicate determination. Data are expressed as geometric means with 95% confidence limits. | ||||
| 20 | — | >10000 |
2480 (1640–3320) | 2390 (1070–3710) |
| 21 | –CH2CH2CH2– | >10000 |
460 (340–580) | 720 (580–860) |
| 22 | –CH2CH2CH2CH2– | >10000 |
1750 (810–2690) | 5660 (4440–6880) |
| 23 | –CH2CH2CH2CH2CH25– | 9500d | 920 (700–1140) | 780d |
| 24 | –CH2CH2OCH2CH2OCH2CH2– | >10000 |
>10000 |
9390 (5930–12850) |
| 25 | –CH2CH2CH2O(CH2CH2O)2CH2CH2CH2– | >10000 |
6010 (5170–6850) | 7090 (6350–7830) |
The low affinity of these compounds highlights again the crucial role of the linker in the recognition process, suggesting that the PTP core alone is not sufficient to provide the desired affinity and selectivity at the hARs. The introduction between the PTP core and the linker of a substituent able to achieve a specific affinity for one of the receptors could enhance the affinity and selectivity of the conjugated compounds.
Computational studies
Molecular docking simulations were performed with the aim to rationalize the affinity data and SAR, as described in detail in the Experimental section. According to our recently published work,25 all molecular docking studies have been carried out with a sodium ion and its first hydration shell in all AR structures.As previously reported, also in this case, regardless of the nature of the receptor subtype considered, docking simulations suggested, for all ligands belonging to the PTP class, three different and alternative binding modes (BM) in their orthosteric binding sites.20 With a specific reference to the position assumed by the furyl moiety, the binding modes differ for the orientation of the ring along the main axis of the binding cavity (see details in Fig. 3): in particular, BM1 and BM3 orient the furyl ring down compared to the extracellular environment (for BM1, alternative up orientation is also retrieved), while BM2 orients it towards the TM2. Interestingly, the three binding modes guarantee the stabilizing interaction with Asn (6.55) and with other hydrophobic residues, such as Phe (EL2). The relative BM sampling frequency strongly depends critically on the nature of the substituent at the 5-position of the PTP scaffold, suggesting that the specific substituents there can modulate the receptor potency and/or receptor subtype selectivity. Considering this specific PTP series, docking studies suggested that BM2 is the most frequently sampled for any receptor, even if BM3 is its most relevant alternative on the hA3AR. In Fig. 4–6 are shown the interaction energy fingerprint (IEF) maps of both electrostatic and hydrophobic contributions, calculated for each sampled pose on all three receptor subtypes.
 | ||
| Fig. 3 The binding modes observed for the PTP scaffold during molecular docking simulations. | ||
 | ||
| Fig. 4 A) IE map of the electrostatic interaction between each compound (y-axis) and each residue (x-axis) of hA1AR. The strength of the interaction is represented by a colorimetric scale going from blue to red for negative to positive values. B) IE map representing the hydrophobic interaction between each compound (y-axis) and each residue (x-axis) of hA1AR. The strength of the interaction is represented by a colorimetric scale going from white to dark green for low to high values. | ||
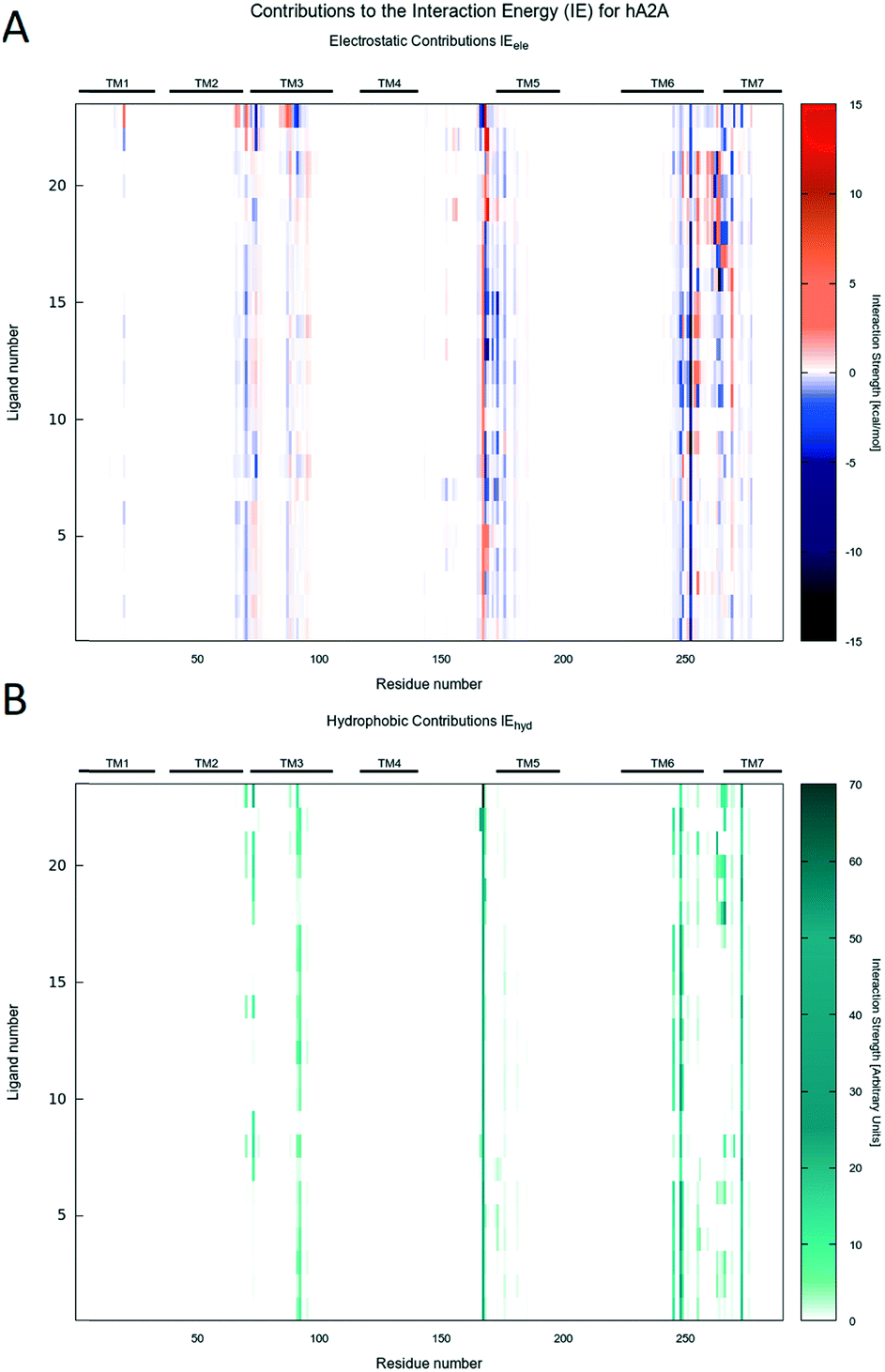 | ||
| Fig. 5 A) IE map of the electrostatic interaction between each compound (y-axis) and each residue (x-axis) of hA2AAR. The strength of the interaction is represented by a colorimetric scale going from blue to red for negative to positive values. B) IE map representing the hydrophobic interaction between each compound (y-axis) and each residue (x-axis) of hA2AAR. The strength of the interaction is represented by a colorimetric scale going from white to dark green for low to high values. | ||
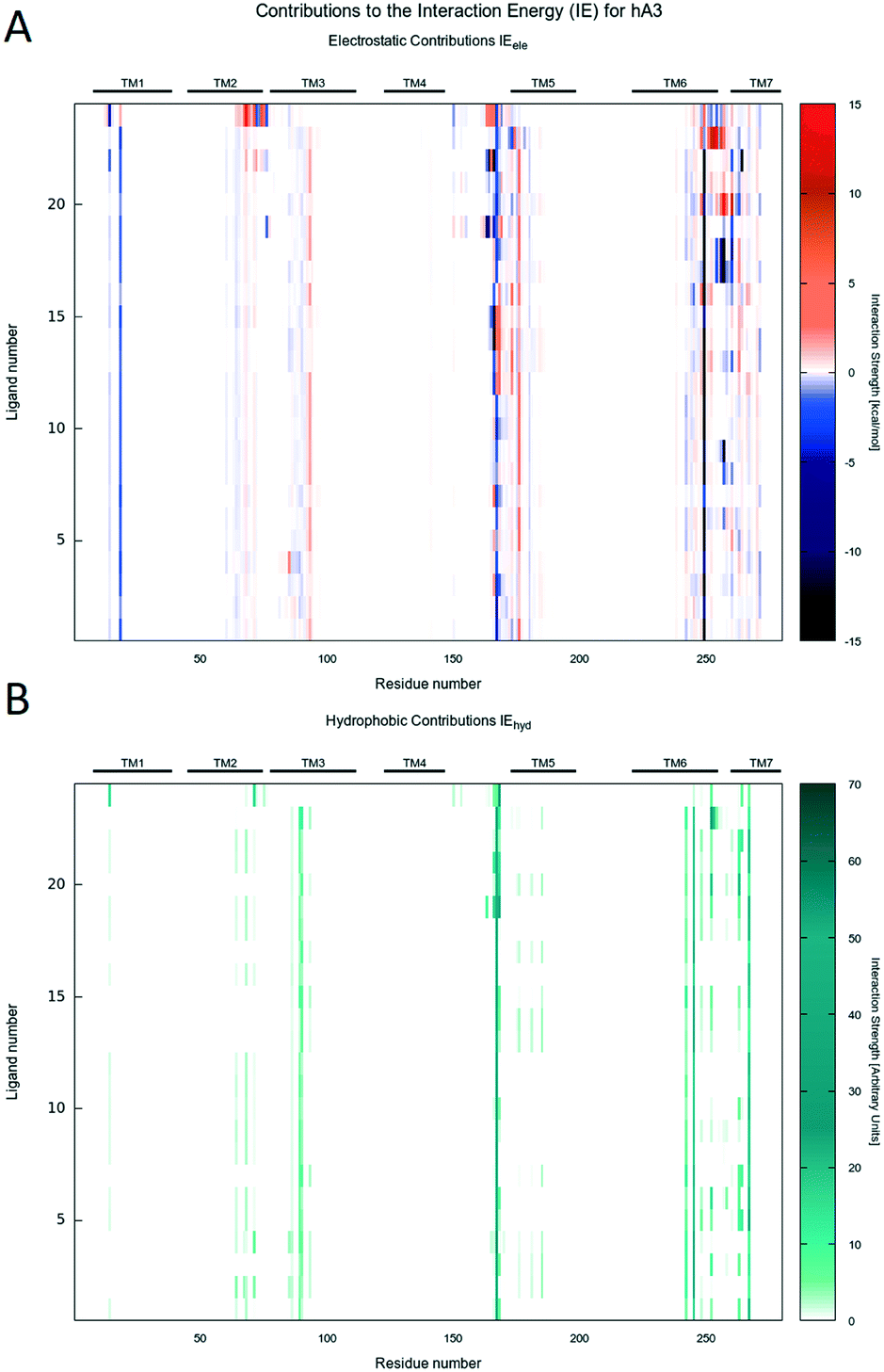 | ||
| Fig. 6 A) IE map of the electrostatic interaction between each compound (y-axis) and each residue (x-axis) of hA3AR. The strength of the interaction is represented by a colorimetric scale going from blue to red for negative to positive values. B) IE map representing the hydrophobic interaction between each compound (y-axis) and each residue (x-axis) of hA3AR. The strength of the interaction is represented by a colorimetric scale going from white to dark green for low to high values. | ||
Interestingly, regardless of the nature of the receptor subtype considered, almost any analyzed PTP analogue can be accommodated in the orthosteric site by the canonical binding pattern, already annotated for other AR antagonists: these include the hydrogen bonds and the aromatic hydrophobic π–π interactions with Asn (6.55) and Phe (EL2), respectively. Results can be related to the structural and sequence diversities between receptors. Indeed, favorable electrostatics are detected along the EL1 and TM2 for ligands docked to the hA2AAR, while, for the hA1AR, some repulsion is achieved at the TM3, i.e. non-conserved residues play a major role.
To analyze in more detail the interactive scheme of some representative ligands belonging to this class of antagonists, per-residue interaction energy profiles (electrostatic, van der Waals and hydrophobic contributions) were calculated for compounds 4 (WS88), 7 (WS91), 15 (WS98) and 23, and shown in Fig. 7–10. Conserved and non-conserved residues were selected for each receptor subtype to better appreciate their contributions in the stabilizing ligand–receptor binding state. Moreover, docking poses are also collected in Videos S1–S3 (ESI†), together with electrostatic and hydrophobic contributions of peculiar residues.
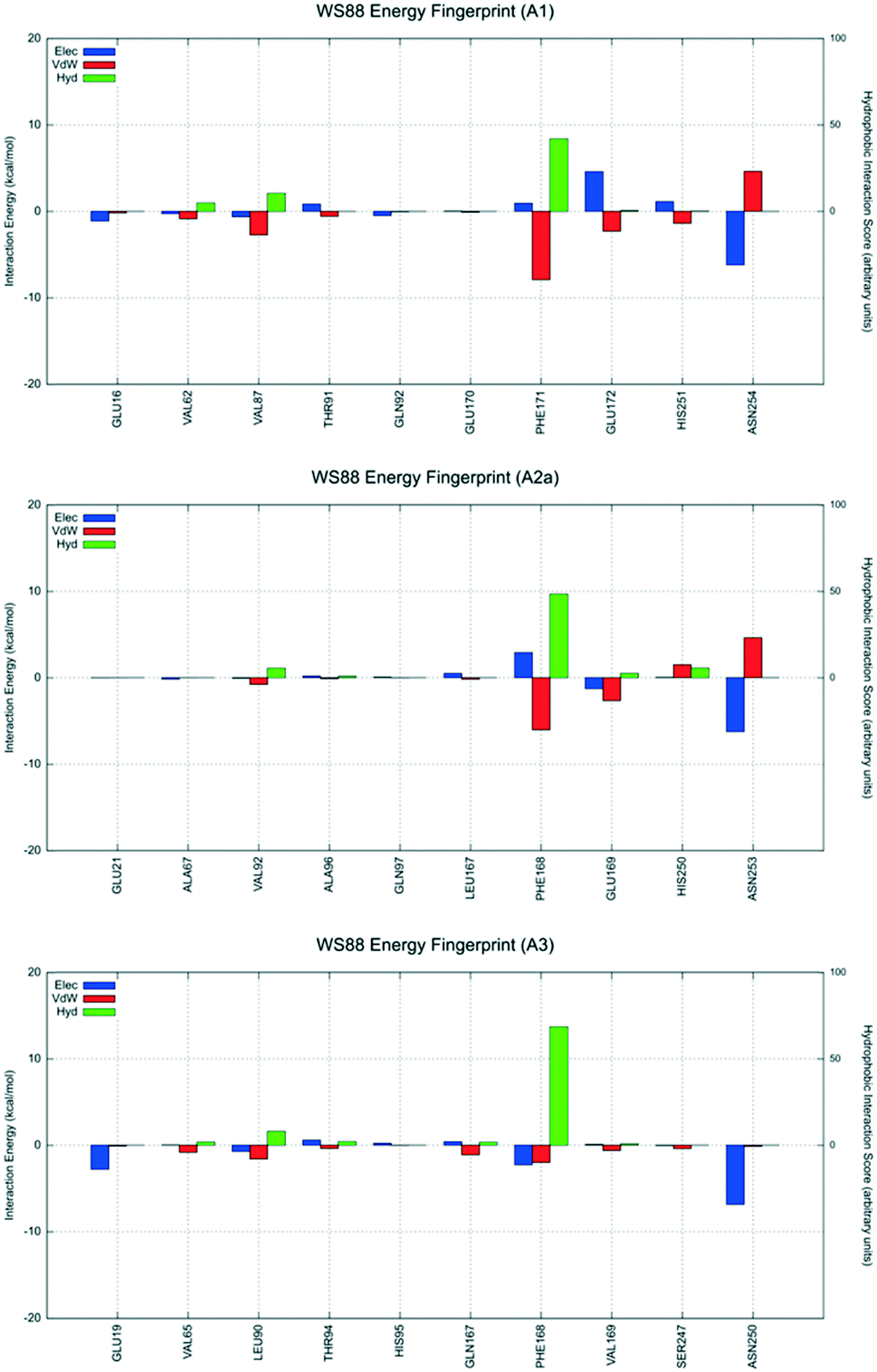 | ||
| Fig. 7 Interaction energy histograms of compound 4 (WS88) for conserved and non-conserved residues of any receptor subtype. | ||
 | ||
| Fig. 8 Interaction energy histograms of compound 7 (WS91) for conserved and non-conserved residues of any receptor subtype. | ||
 | ||
| Fig. 9 Interaction energy histograms of compound 15 (WS98) for conserved and non-conserved residues of any receptor subtype. | ||
 | ||
| Fig. 10 Interaction energy histograms of compound 23 for conserved and non-conserved residues of any receptor subtype. | ||
The first analyzed antagonist was derivative 4, the most potent compound of the series towards the hA3AR (Ki = 32.1 nM), despite its modest selectivity (hA1/hA3 = 55.3 and hA2A/hA3 = 6.1). Interestingly, derivative 4 preferably recognizes the orthosteric binding site of the hA1AR using the interactive scheme BM1up, the hA2A subtype using the interactive scheme BM3, and the hA3 using the interactive scheme BM1down. As shown in Fig. 7, these three different binding modes are characterized by three different interaction energy profiles and the analysis shows that the most favorable interactive scheme is for subtype hA3 engaging strong hydrophobic contacts with the conserved Phe168 (EL2), with the non-conserved Leu90 (3.32), and an electrostatically favorable interaction with Glu19 (1.39), without showing relevant repulsive van der Waals contributions. Conversely, an unfavorable van der Waals term is detected when derivative 4 is docked to both hA1 and hA2A ARs, mediated by Asn254 and Asn253, respectively (6.55).
The second analyzed antagonist is derivative 7, a moderately selective compound for the hA3AR, especially when compared to the hA1AR (hA1/hA3 = 135.7 and hA2A/hA3 = 2.1). This antagonist preferably recognizes the orthosteric binding site of the hA1AR using the interactive scheme BM2, and the hA2A and hA3 using the interactive scheme BM3. Interestingly, unless for Glu172 (EL2) and Val87 (3.32), the analysis of the interaction energy profiles indicates a very modest stabilizing interaction network compared to the hA3AR. At the hA2AAR, the ligand interacts unfavorably with His250 (6.52). The most favorable profile is observed at the hA3AR.
Derivative 15 is the highest affinity compound of the series at the hA2AAR (Ki = 52.6 nM). Nevertheless, it shows affinity constants in high nanomolar (Ki = 778 nM) and micromolar ranges (Ki = 2246 nM), for the hA1 and hA3 ARs, respectively. Derivative 15 preferably recognizes the orthosteric binding site of the hA1 and hA2AARs using the interactive scheme BM2, but interactive scheme BM3 is used at the hA3AR. For this ligand, at the hA1AR, several stabilizing interactions were detectable, in particular with Phe171 (EL2), Glu172 (EL2) and Asn254 (6.55), as shown in Fig. 9. This stabilizing network of interactions is also present at the hA2A AR, even if the intensity of each interaction energy term is lower than that measured at the hA1AR. To appropriately interpret the lower affinity determined experimentally against the hA3 subtype, it is possible to invoke the crucial repulsive interactions detected for Val169 (EL2) and Phe168 (EL2), together with highly unfavorable van der Waals contributions by Asn250 (6.55). Involvement of these residues is likely to be critical for affinity.
The final analyzed antagonist is derivative 23, the most representative compound among the fluorescent ligands of the series, and the most selective towards hA3AR. As expected, BM2 is the only interactive scheme sampled for each receptor subtypes, allowing a better comparison (Fig. 10). As already described, this specific binding mode allows us to orient the substituent that carries the fluorescent probe towards the extracellular environment. Unfortunately, in this case, considering the relevant volumetric properties of these ligands and the fact that their receptor recognition involves not only the orthosteric binding sites but also relevant portions of the extracellular loops (in particular, the EL2), the analysis of the interaction energy profiles is not very helpful to interpret the experimental selectivity profile. However, it is emphasized that all the most crucial interactions with the conserved Phe (EL2) and Asn (6.55) are preserved. Moreover, the amino acid sequences and the architectures of EL2 play an important role in controlling both the selectivity profiles and ligand affinities of the fluorescent analogues against all receptor subtypes.
Finally, to support or negate the hypothesis suggested by docking studies concerning the presence of different and alternative ligand binding modes (BM) inside the receptor orthosteric binding site, we decided to carry out a supervised molecular dynamics (SuMD) study with the aim to explore the possible ligand–receptor recognition pathways (from an unbound state to the orthosteric bound state) and to verify if the final states reached by supervised trajectories converge to the most populated poses obtained by molecular docking. For this specific scope, we decided to use the smaller PTP analogue of this series, derivative 2 (WS86), which showed a moderate affinity against the hA3AR (Ki = 173 nM) and a reasonable selective profile (hA1/hA3 = 45.2 and hA2A/hA3 = 22.5). Docking simulations suggested that derivative 2 preferably recognizes the orthosteric binding site of the hA1AR using the interactive scheme BM1up; the hA2AAR used the interactive scheme BM3, whereas the hA3 used two distinct interactive schemes BM1down and BM2. As already reported for all previously analyzed antagonists, these four different binding modes are characterized by four different interaction energy profiles and the analysis shows that the most favorable interactive scheme for subtype hA3AR is the one in which the ligand preferably adopts the interactive scheme BM1down, establishing the already described stabilizing interaction with the conserved Phe168 (EL2) and Asn250 (6.55) (Fig. 11).
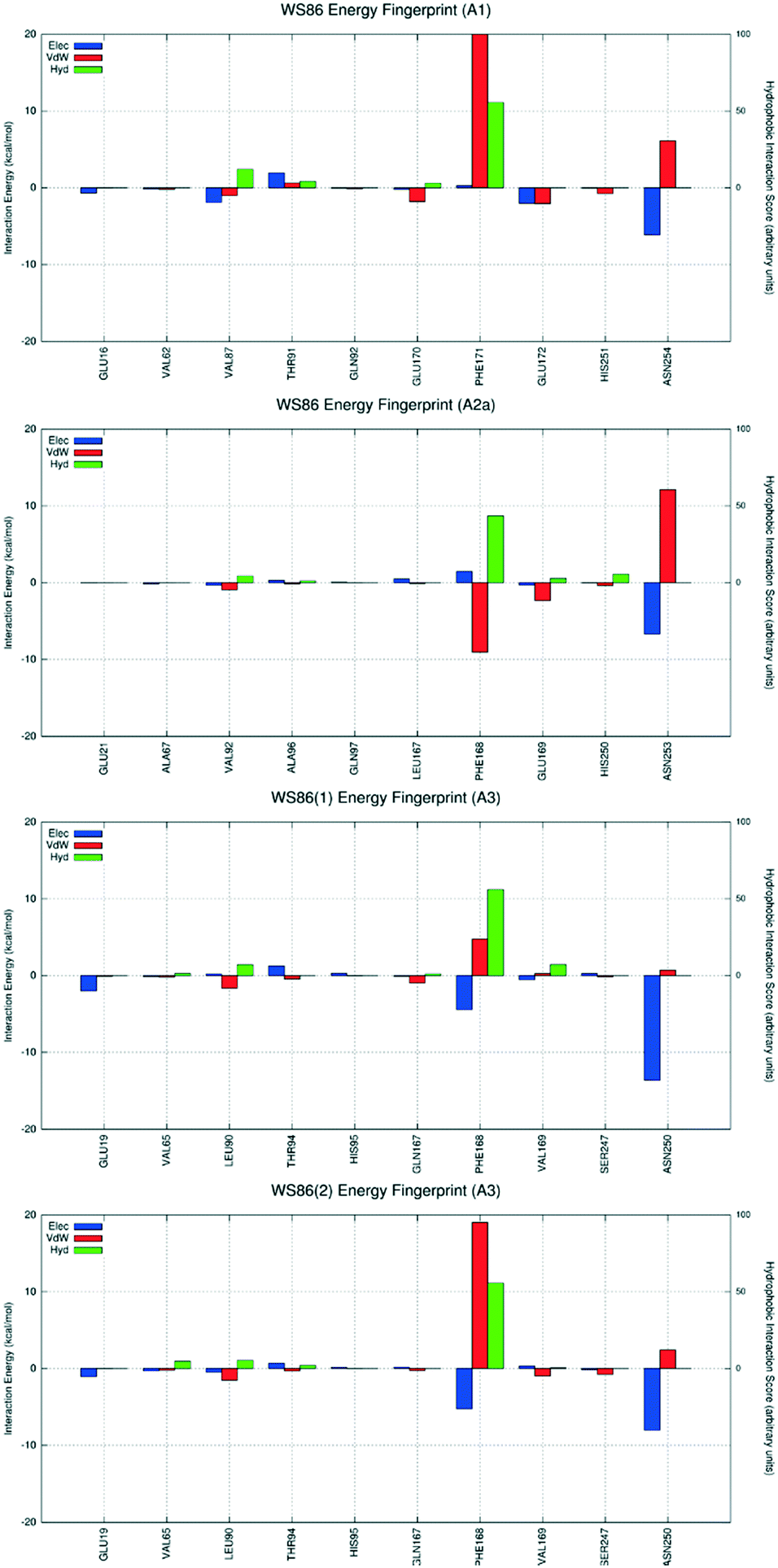 | ||
| Fig. 11 Interaction energy histograms of compound 2 (WS86) for conserved and non-conserved residues of any receptor subtype. BM1down and BM2 are reported for the hA3AR subtype. | ||
Considering the appreciable hA3AR affinity, all SuMD simulations have been performed using this specific receptor subtype. Five independent supervised simulations have been carried out starting from an unbound state in which the ligand has been randomly positioned 40 Å far away from the center of the orthosteric hA3AR binding cavity.
As expected, and already reported in previous studies,26 each SuMD trajectory describes a specific ligand–receptor recognition pathway (see Fig. 12, Panel A) and allows the ligand to sit in different regions of the orthosteric binding site (bound state) and adopt different binding poses (see Fig. 12, Panel B). Even if the different bound states obtained by SuMD simulations are not perfectly superimposable to those suggested by docking experiments, we emphasize that the possibility to have a multiple binding mode for a single ligand is clearly shown in the results of the SuMD trajectory analysis. Particularly interesting is the result extrapolated from Replica 5, in which the observed bound state is close to the one described as BM2, as shown in Video S4.† As previously reported, also for derivative 2, the different SuMD trajectories described the presence of several meta-binding sites (energetically stable binding sites spatially distinct from the orthosteric site) with an energy profile (Fig. 12, Panel A) that smoothly decreases from an average value 〈E1〉 of −30 kcal mol−1 (for distances >10 Å) to an average value 〈E2〉 of −50 kcal mol−1 (〈E2〉 ≈ 2 〈E1〉), in a distance window between 2 and 10 Å. This behavior of the interaction energy profile is typical of a ligand–receptor recognition process characterized by a reasonably high kon value.
 | ||
| Fig. 12 A) Total compound 2 (WS86)-protein interaction energy vs. distance from the binding site, for each SuMD simulation, represented by different colors. Average energies are reported for particular distance ranges. B) Visual indicative inspection of final states as vdW surfaces within the binding site. Peculiar regions of the transmembrane receptor are reported. | ||
As already anticipated, different bound states obtained by SuMD simulations are not perfectly superimposable to the poses suggested by docking experiments, even if in Replica 5 the SuMD bound state is close to the one described as BM2. Starting from the Replica 5 final bound state, a non-biased molecular dynamics simulation has been performed to analyse the time-evolution (stability) of this specific bound state. Surprisingly, after 250 ns of simulation, the ligand flips its original position assuming a new configuration very similar to the BM3 detected in docking simulations, as shown in Fig. 13, and more energetically favored. This evidence indicates that the accommodation of the furyl ring down, compared to the extracellular side, is the most favored one.
 | ||
| Fig. 13 Compound 2 (WS86)-protein interaction energy vs. simulation time of the SuMD complex (binding mode 2). Total energy is decomposed in electrostatic (Elec, blue) and van der Waals (vdW, red) energy. Chemical structures of BM2 and BM3 are also reported. | ||
The analysis of both supervised and non-biased MD trajectories showed that the adaptive positioning of the ligand inside the orthosteric binding site is strictly related to the simultaneous conformational variation of the side chains of the two amino acids Phe168 (EL2) and Trp243 (6.48) or, in other words: the final ligand pose depends on the reshaping of the binding cavity (induce fit) caused by the side chains of the two amino acids Phe168 (EL2) and Trp243 (6.48). Moreover, considering that all SuMD simulations have been carried out with a sodium ion and its first hydration shell in the hA3AR structure, it is interesting to note that both the sodium ion and its coordinated water molecules are also involved in this transition (Video S4†).
In conclusion, the combination of molecular docking and dynamics (supervised and non-biased) simulations can be considered an interesting computational strategy to improve the descriptive capacity of the interaction modality of this new series of AR antagonists. In particular, SuMD can facilitate the interpretation at a molecular level of the possible ligand–receptor recognition pathways bridging the stability of the canonical orthosteric binding (thermodynamic binding propensity) with the ease of accessing the orthostatic site (kinetic binding propensity).
Experimental
Chemistry
Synthesis of compound 26 is reported in the literature.20 Mono-Boc-protected-diamines were synthesized following the procedure reported by Onajole et al.27:methanol 1) to assess the disappearance of the starting material, then the solvent was removed under reduced pressure and the residue was purified by flash chromatography (ethyl acetate 9.5:MeOH 0.5) affording the desired compound.
N′-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)-hydrazine carboxylic acid tert-butyl ester (2). Yield 90%; white solid; mp 160 °C; 1H-NMR (200 MHz, CDCl3) δ: 1.49 (9H, s), 4.13 (3H, s), 6.59 (1H, dd, J = 2, J = 4), 6.81 (1H, bs), 7.21 (1H, d, J = 4), 7.62 (1H, d, J = 2), 7.90 (1H, bs), 8.16 (1H, s). HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C16H18N8O3: 393.1394, found: 393.1398.
[2-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-ethyl]-carbamic acid tert-butyl ester (3). Yield 85%; white solid; mp 217 °C; 1H-NMR (200 MHz, CDCl3) δ: 1.43 (9H, s), 3.50–3.55 (2H, m), 3.76–3 (2H, m), 4.11 (3H, s), 4.96 (1H, bs), 6.58–6.65 (2H, m), 7.20 (1H, d, J = 4), 7.62 (1H, d, J = 2), 8.10 (1H, s). HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C18H22N8O3: 421.1707, found: 421.1709.
[3-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino) propyl]-carbamic acid tert-butyl ester (4). Yield 66%; white solid; mp 217 °C; 1H-NMR (200 MHz, CDCl3) δ: 1.46 (9H, s), 1.93 (2H, quint, J = 6), 3.27 (2H, bs), 3.75 (2H, q, J = 6), 4.11 (3H, s), 5.11 (1H, bs), 6.53–6.59 (2H, m), 7.21 (1H, d, J = 4), 7.62 (1H, d, J = 2), 8.08 (1H, s). 13C NMR (101 MHz, CDCl3) δ 192.40, 156.64, 155.89, 147.23, 145.65, 144.67, 144.48, 143.07, 126.90, 114.68, 112.46, 109.11, 79.71, 77.48, 77.16, 76.84, 40.73, 38.80, 37.84, 29.99, 28.55. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C19H24N8O3: 435.1864, found: 435.1862.
[4-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-butyl]-carbamic acid tert-butyl ester (5). Yield 79%; white solid; mp 167 °C; 1H-NMR (200 MHz, CDCl3) δ: 1.44 (9H, s), 1.60–1.84 (4H, m), 3.14–3.26 (2H, m), 3.48–3.80 (2H, m), 4.10 (3H, s), 4.59 (1H, bs), 6.31 (1H, bs), 6.59 (1H, dd, J = 2, J = 4), 7.21 (1H, d, J = 4), 7.62 (1H, d, J = 2), 8.08 (1H, s). ES-MS (methanol): m/z 449.47 [M + Na]+. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C20H26N8O3: 449.2020, found: 449.2021.
[5-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-pentyl]-carbamic acid tert-butyl ester (6). Yield 82%; white solid; mp 119 °C; 1H-NMR (200 MHz, CDCl3) δ: 1.43–1.82 (15H, m), 3.08–3.18 (2H, m), 3.69 (2H, dd, J = 7, J = 14), 4.11 (3H, s), 4.54 (1H, bs), 6.27 (1H, bs), 6.60 (1H, dd, J = 2, J = 4), 7.22 (1H, d, J = 4), 7.63 (1H, d, J = 2), 8.09 (1H, s). ES-MS (methanol): m/z 463.2 [M + Na]+. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C21H28N8O3: 463.2177, found: 463.2179.
(2-{2-[2-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-ethoxy]-ethoxy}-ethyl)-carbamic acid tert-butyl ester (7). Yield 55%; sticky foam; 1H-NMR (200 MHz, CDCl3) δ: 1.41 (9H, s), 3.36 (2H, bs), 3.56–3.66 (6H, m), 3.80–3.92 (4H, m), 4.11 (3H, s), 5.14 (1H, bs), 6.59 (1H, dd, J = 2, J = 4), 6.62 (1H, bs), 7.23 (1H, d, J = 4), 7.62 (1H, d, J = 2), 8.11 (1H, s). HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C22H30N8O5: 509.2231, found: 509.2233.
[3-(2-{2-[3-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-propoxy]-ethoxy}-ethoxy)-propyl]-carbamic acid tert-butyl ester (8). Yield 67%; pale pink solid; mp 69 °C; 1H-NMR (200 MHz, CDCl3) δ: 1.44 (9H, s), 1.68–1.82 (2H, m), 1.96–2.15 (2H, m), 3.24 (2H, bs), 3.51–3.86 (14H, m), 4.12 (3H, s), 5.00 (1H, bs), 6.60 (1H, dd, J = 2, J = 4), 6.84 (1H, bs), 7.22 (1H, d, J = 4), 7.62 (1H, d, J = 2), 8.08 (1H, s). HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C26H38N8O6: 581.2807, found: 581.2805.
{4-[(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-methyl]-cyclohexylmethyl}-carbamic acid tert-butyl ester (9). Yield 79%; pale yellow solid; mp 65 °C; 1H-NMR (200 MHz, CDCl3) δ: 1.43–1.80 (19H, m), 2.95–3.08 (4H, m), 4.10 (3H, s), 4.56 (1H, bs), 6.35 (1H, bs), 6.59 (1H, dd, J = 2, J = 4), 7.20 (1H, d, J = 4), 7.62 (1H, d, J = 2), 8.07 (1H, s). C24H32N8O3. M.W.: 480.56. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C24H32N8O3: 503.2490, found: 503.2494.
4-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)-piperazine-1-carboxylic acid tert-butyl ester (10). Yield 63%; pale yellow solid; mp 112 °C; 1H-NMR (200 MHz, CDCl3) δ: 1.52 (9H, s), 3.68–3.70 (4H, m), 3.85–3.99 (4H, m), 4.15 (3H, s), 6.59 (1H, dd, J = 2, J = 4), 7.23 (1H, d, J = 4), 7.64 (1H, d, J = 2), 8.19 (1H, s). HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C20H24N8O3: 447.1864, found: 447.1866.
:methanol 1, the desired compound doesn't move). The solvent was then removed under reduced pressure and the desired compounds (11–19) were filtered off.
N′-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)-hydrazinium; chloride (11). Yield 65%; yellow solid; mp 192 °C; 1H-NMR (200 MHz, CDCl3–D2O) δ: 4.13 (3H, s), 4.77–4.83 (2H, bm), 6.59 (1H, dd, J = 2, J = 4), 7.21 (1H, d, J = 4), 7.63 (1H, d, J = 2), 8.13 (1H, s). 13C NMR (101 MHz, DMSO-d6) δ 155.71, 153.57, 149.37, 145.53, 144.97, 143.31, 126.62, 112.81, 112.28, 98.34, 40.22. HRMS (ESI-QTOF): m/z [M + H]+ calcd for C11H10N8O: 271.1050, found: 271.1045.
2-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-ethyl-ammonium; chloride (12). Yield 62%; pale yellow solid; mp 212 °C; 1H-NMR (200 MHz, D2O) δ: 3.31–3.40 (2H, m), 3.79–3.83 (2H, m), 3.92 (3H, s), 6.56 (1H, bs), 6.86 (1H, bs), 7.59 (1H, bs), 7.97 (1H, s). HRMS (ESI-QTOF): m/z [M + H]+ calcd for C13H14N8O: 299.1363, found: 299.1367.
3-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-propyl-ammonium; chloride (13). Yield 75%; pale yellow solid; mp 198 °C; 1H-NMR (200 MHz, D2O) δ: 2.06 (2H, m), 3.12 (2H, t, J = 7), 3.56 (2H, t, J = 7), 3.93 (3H, s), 6.57 (1H, dd, J = 2, J = 4), 6.94 (1H, d, J = 4), 7.59 (1H, d, J = 2), 8.00 (1H, s). HRMS (ESI-QTOF): m/z [M + H]+ calcd for C14H16N8O: 313.1520, found: 313.1521.
4-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-butyl-ammonium; chloride (14). Yield 69%; pale brown solid; mp 224 °C; 1H-NMR (200 MHz, D2O) δ: 1.72–1.88 (4H, m), 3.05 (2H, t, J = 7), 3.43 (2H, t, J = 7), 3.92 (3H, s), 6.59 (1H, dd, J = 2, J = 4), 6.92 (1H, d, J = 4), 7.62 (1H, d, J = 2), 8.01 (1H, s). HRMS (ESI-QTOF): m/z [M + H]+ calcd for C15H18N8O: 327.1676, found: 327.1674.
5-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-pentyl-ammonium; chloride (15). Yield 73%; pale yellow solid; mp 126 °C; 1H-NMR (200 MHz, D2O) δ: 1.41–1.56 (2H, m), 1.70–1.74 (4H, m), 3.03 (2H, t, J = 7), 3.37 (2H, t, J = 7), 3.91 (3H, s), 6.56 (1H, dd, J = 2, J = 4), 6.88 (1H, d, J = 4), 7.58 (1H, d, J = 2), 7.95 (1H, s). HRMS (ESI-QTOF): m/z [M + H]+ calcd for C16H20N8O: 341.1833, found: 341.1833.
2-{2-[2-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-ethoxy]-ethoxy}-ethyl-ammonium; chloride (16). Yield 61%; sticky foam; 1H-NMR (200 MHz, D2O) δ: 3.16 (2H, bs), 3.74–3.87 (10H, m), 3.96 (3H, s), 6.61 (1H, dd, J = 2, J = 4), 6.96 (1H, d, J = 4), 7.64 (1H, d, J = 2), 8.07 (1H, s). HRMS (ESI-QTOF): m/z [M + H]+ calcd for C17H22N8O3: 387.1888, found: 387.1885.
3-(2-{2-[3-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ylamino)-propoxy]-ethoxy}-ethoxy)-propyl-ammonium; chloride (17). Yield 56%; sticky foam; 1H-NMR (200 MHz, D2O) δ: 1.87–1.99 (4H, m), 3.05 (2H, t, J = 7), 3.55–3.75 (14H, m), 3.97 (3H, s), 6.62 (1H, dd, J = 2, J = 4), 6.99 (1H, d, J = 4), 7.65 (1H, d, J = 2), 8.08 (1H, s). HRMS (ESI-QTOF): m/z [M + H]+ calcd for C21H30N8O4: 459.2463, found: 459.2464.
N-((4-(Aminomethyl)cyclohexyl)methyl)-2-(furan-2-yl)-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-ammonium; chloride (18). Yield 66%; sticky foam; 1H-NMR (200 MHz, D2O) δ: 1.78–1.82 (10H, m), 2.84–3.18 (4H, m), 3.78 (3H, s), 6.34 (1H dd, J = 2, J = 4), 6.58 (1H, d, J = 4), 7.36 (1H, d, J = 2), 7.73 (1H, s). HRMS (ESI-QTOF): m/z [M + H]+ calcd for C19H24N8O: 381.2146, found: 381.2148.
4-(2-Furan-2-yl-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)-piperazin-1-ium; chloride (19). Yield 67%; white solid; mp > 300 °C; 1H-NMR (200 MHz, D2O) δ: 3.46–3.54 (4H, m), 3.98–4.14 (7H, m), 6.63 (1H, dd, J = 2, J = 4), 7.06 (1H, d, J = 4), 7.68 (1H, d, J = 2), 8.21 (1H, s). 13C NMR (101 MHz, DMSO-d6) δ 154.35, 154.14, 150.41, 145.74, 145.27, 145.22, 126.02, 112.37, 112.16, 98.86, 44.59, 41.98, 40.09. HRMS (ESI-QTOF): m/z [M + H]+ calcd for C15H16N8O: 325.1520, found: 325.1521.
:1 methanol as an eluent. The solvent was removed under reduced pressure and the residue was purified by preparative TLC in the same solvent dissolving the residue in a little amount of methanol and the final compounds (20–25) were obtained as orange solids.
5-(2-(2-(Furan-2-yl)-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)hydrazinecarbothioamido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid (20). Yield 63%, orange solid, mp 180 °C (methanol); 1H NMR (500 MHz, DMSO-d6) δ: 3.78 (3H, s), 6.44 (4H, bs), 6.57 (1H, s), 6.75 (2H, bs), 6.85 (1H, s), 7.32 (1H, d, J = 7.3), 7.72 (2H, m), 8.05 (2H, m), 8.11 (1H, s), 10.83 (1H, bs), 12.45 (1H, bs). UV: λmax 525. Fluorescence: λem 541. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C32H21N9O6S: 682.1228, found: 682.1224.
5-(3-(3-((2-(Furan-2-yl)-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)amino)propyl)thioureido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid (21). Yield 54%, orange solid, mp > 250 °C (methanol); 1H NMR (500 MHz, DMSO-d6) δ: 1.97 (m, 2H), 3.51–3.70 (4H, m), 3.99 (3H, s), 6.30 (4H, m), 6.68 (2H, d, J = 8.3), 6.71 (1H, s), 7.10 (1H, d, J = 7.6), 7.19 (1H, s), 7.67 (1H, s), 7.92 (1H, s), 8.06 (1H, s), 8.18 (1H, s), 8.45–8.54 (2H, m), 10.17 (1H, bs). UV: λmax 518. Fluorescence: λem 541. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C35H27N9O6S: 724.1697, found: 724.1699.
5-(3-(4-((2-(Furan-2-yl)-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)amino)butyl)thioureido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid (22). Yield 56%, orange solid, mp > 250 °C (methanol); 1H NMR (500 MHz, DMSO-d6) δ: 1.67 (m, 2H), 1.70–1,77 (2H, m), 3.48–3.66 (4H, m), 4.00 (3H, s), 6.53 (2H, d, J = 8.3), 6.57–6.66 (4H, m), 6.72 (1H, s), 7.13 (1H, d, J = 8.3), 7.20 (1H, d, J = 3.2), 7.77 (1H, d, J = 8.2), 7.92 (1H, s), 8.02 (1H, t, J = 5.6), 8.27 (1H, s), 8.55 (1H, s), 8.59 (1H, s), 10.36 (1H, bs). UV: λmax 521. Fluorescence: λem 537. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C36H29N9O6S: 738.1854, found: 738.1854.
5-(3-(5-((2-(Furan-2-yl)-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)amino)pentyl)thioureido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid (23). Yield 49%, orange solid, mp 220 °C (methanol); 1H NMR (500 MHz, DMSO-d6) δ: 1.37–1.47 (2H, m), 1.65–1.76 (2H, m), 3.49 (4H, m), 4.00 (3H, s), 6.37–6.54 (4H, m), 6.64 (2H, d, J = 9), 6.71 (1H, dd, J = 4, J = 2), 7.06 (1H, d, J = 8), 7.19 (1H, d, J = 4), 7.75 (1H, d, J = 8), 7.92 (1H, s), 7.98 (1H, t, J = 7), 8.24 (1H, s), 8.45–8.68 (2H, m), 10.50 (1H, bs). UV: λmax 522. Fluorescence: λem 541. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C37H31N9O6S: 752.2010, found: 752.2011.
5-(3-(2-(2-(2-((2-(Furan-2-yl)-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)amino)ethoxy)ethoxy)ethyl)thioureido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid (24). Yield 43%, orange solid, mp 216 °C (methanol); 1H NMR (500 MHz, DMSO-d6) δ: 3.30 (m, 2H), 3.60 (6H, m), 3.67 (4H, m), 4.00 (3H, s), 6.37–6.54 (4H, m), 6.63 (2H, d, J = 7.7), 6.71 (1H, s), 7.12 (1H, d, J = 8.1), 7.19 (1H, d, J = 2.9), 7.72 (1H, s), 7.83 (1H, s), 7.92 (1H, s), 8.17 (1H, s), 8.25 (1H, s), 8.55 (1H, s), 10.11 (1H, bs). UV: λmax 522. Fluorescence: λem 538. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C38H33N9O8S: 798.2065, found: 798.2068.
5-(3-(3-(2-(2-(3-((2-(Furan-2-yl)-8-methyl-8H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)amino)propoxy)ethoxy)ethoxy)propyl)thioureido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid (25). Yield 53%, orange solid, mp 245 °C (methanol); 1H NMR (500 MHz, DMSO-d6) δ: 1.74 (2H, m), 1.92 (2H, m), 2.89–3.30 (m, 16H), 4.00 (3H, s), 6.27 (4H, m), 6.57–6.88 (3H, m), 7.04 (1H, s), 7.19 (1H, s), 7.68 (1H, s), 7.92 (1H, s), 8.14 (1H, s), 8.50 (1H, s), 10.31 (1H, bs). UV: λmax 512. Fluorescence: λem 540. HRMS (ESI-QTOF): m/z [M + Na]+ calcd for C42H41N9O9S: 870.2640, found: 870.2643.
Pharmacology
All pharmacological methods followed the procedures as described earlier.22,28–32 In particular, two slight different protocols were used for compounds 2–19 (ref. 28–31) and for compounds 20–25.22,32 For radioligand binding at the hA1, hA2A and hA3 ARs, [3H]CCPA or [3H]R-PIA, [3H]NECA or [3H]CGS21680 and [3H]HEMADO or [125I]I-AB-MECA were used, respectively. Radioligand binding at hA2BARs is problematic as no high-affinity radioligand agonist is commercially available for this subtype.33 Therefore, inhibition of NECA-stimulated adenylyl cyclase activity was determined as a measurement of potency of compounds, following a well-known procedure.28Computational studies
The MOE suite34 was exploited in order to perform general molecular modeling operations. All preliminary computations were carried out on a 12 CPU (Intel® Xeon® CPU E5-1650 3.80 GHz) Linux workstation. SuMD and MD simulations were performed exploiting the ACEMD engine35 on NVIDIA drivers: NVIDIA GTX 980Ti and NVIDIA GTX 980. The CHARMM36/CHARMM36 general force field (CGenFF) combination was adopted. The GOLD37 suite and goldscore scoring function were used as the docking program. 3D structures of the ligands were built by the MOE-builder tool. Ionization states were predicted using the MOE-protonate 3D tool. Tautomers and atom hybridization were checked. Structures were minimized by the MMFF94x until the root mean square (RMS) gradient fell below 0.05 kcal mol−1 A−1. For each ligand, 20 docking simulation runs were performed on each receptor subtype, searching on a sphere of 20 Å radius, centered on backbone nitrogen of the conserved Asn (6.55). Ligand and protein partial charges were calculated by means of the MMFF94 and AMBER14:EHT38 force fields, respectively.The hA2A and hA1 AR coordinates were retrieved from the Protein Data Bank39 using the crystal structures coded as 3PWH40 and 5UEN,41 respectively. Given the lack of any crystallographic structures for hA3AR, a homology model was used. The model was built using 5UEN as a template, as reported in our recently submitted work,42 after a docking/structural based model assessment. According to our recently published work,25 all molecular docking studies have been carried out with a sodium ion and its first hydration shell in all adenosine receptor structures.
The Ballesteros–Weinstein43 numbering system is used sometimes to indicate conserved residues. IE electrostatic and van der Waals energy contributions to the binding energy were calculated by MOE, together with per residue electrostatic and hydrophobic interactions. Per-residue information was reported in the so-called “Interaction Energy Fingerprints”: they consist of heat maps reporting the strength of the interaction of each residue (x-axis) and each ligand (y-axis) according to a colorimetric scale going from blue to red for negative to positive values in the case of electrostatic contributions and from white to dark green for low to high values for hydrophobic contributions. Interactions of most relevant residues were also reported by histograms, whose height is proportional to the strength of the interaction. Plots were generated using Gnuplot 4.6.44 2D chemical structures in figures were drawn with Marvin Sketch.45 Molecular graphics were performed with the UCSF Chimera package,46 except Fig. 12B, which was obtained using the VMD program (version 1.9.3).47 The in-house MMsDocking video maker tool was exploited to produce videos showing the docking poses, per residue hydrophobic and electrostatic contributions for selected residues, experimental binding data, and scoring values. Representations of docking poses were produced using the UCSF Chimera package, 2D depictions were constructed by the cheminformatics toolkit RDKit,48 and the heat maps were obtained by Gnuplot 4.6; in the end, videos were mounted using MEncoder.
Ligand 3 force field parameters for MD simulations were initially retrieved from the Paramchem web service and then deeply optimized in concordance with CGenFF, at the MP2/6-31G* level of the theory49 by using Gaussian 09 (ref. 50) and RESP partial charges. Systems were embedded in a 1-palmitoyl-2oleyl-sn-glycerol-3-phospho-choline (POPC) lipid bilayer, according to the pre-orientation provided by the Orientations Proteins in Membrane (OPM) database51 and by using the VMD membrane builder plugin. Lipids within 0.4 Å from the protein were removed and TIP3P52 model water molecules were added to solvate the system by means of Solvate1.0.53 System charge neutrality was reached by adding Na+/Cl− counterions to a final concentration of 0.154 M. Equilibration was performed through a three-step procedure. In the first one, 1500 conjugate-gradient minimization steps were applied in order to reduce the clashes between protein and lipids. Then, a 5 ns long MD simulation was performed in the NPT ensemble, with a positional constraint of 1 kcal mol−1 Å−2 on ligand, protein, and lipid phosphorus atoms. During the second stage, 10 ns MD simulation in the NPT ensemble was performed constraining all the protein and ligand atoms but leaving the POPC residues free to diffuse in the bilayer. In the last equilibration stage, positional constraints were applied only to the ligand and protein backbone alpha carbons for further 5 ns MD simulation. All the MD simulations were performed using: (1) an integration time step of 2 fs; (2) the Berendsen barostat54 in order to maintain the system pressure at 1 atm; (3) the Langevin thermostat55 to maintain the temperature at 310 K with a low dumping of 1 ps−1; (4) the M-SHAKE algorithm56 to constrain the bond lengths involving hydrogen atoms; and (5) a long-range cutoff of 10 Å. According to the SuMD approach,57,58 the timescale needed to reproduce complete intermolecular complex formations is in the range of nanoseconds, instead of hundreds of nanoseconds or microseconds usually necessary with unsupervised MD. Sampling is gained by applying a tabu-like algorithm to monitor the distance between the centers of mass of the ligand and the binding site, during short unbiased MD simulations. SuMD considers the ligand atoms and the atoms of user-defined protein residues to monitor the distance between the centers of mass of the binder and the binding site. A series of 600 ps unbiased MD simulations are performed and after each simulation, the distance points collected at regular time intervals are fitted into a linear function. If the resulting slope is negative the next simulation step starts from the last set of coordinates and velocities produced, otherwise the simulation is restarted by randomly assigning the atomic velocities. Short simulations are perpetuated under the supervision until the distance between the ligand and the binding site goes below 5 Å, then, the supervision is disabled and a classical MD simulation is performed. For the orthosteric center of mass, we considered hA3AR residues N250, F168, H272, and S247.
NAMD energy Plugin 1.4 (ref. 59) was used to calculate ligand–protein interaction energies.
Conclusions
A series of pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidines bearing diaminoalkyl chains at the 5 position (2–19) was developed to target ARs after conjugation with the desired function-bearing moiety. Compounds 2–19 showed different binding profiles at hA1, hA2A and hA3 ARs. In particular, compound 4 and compound 15 are the most potent compound towards hA3 and hA2A ARs, respectively. Docking simulations suggested four different binding modes for synthesised compounds at the considered ARs. The preference for one of these binding modes is regulated by the substitution at the 5 position. SuMD analysis on hA3AR confirmed the existence of multiple binding modes and that sodium and its hydration molecules are involved in the transition from one pose to another. An example of application of these compounds was reported. In particular, conjugation with fluorescein isothiocyanate led to dual, but weak, hA2A and hA3 AR fluorescent ligands. Docking revealed that the insertion of the bulky fluorescent moiety on the PTP scaffold forced the ligand to adopt the BM2, in which the linker oriented the fluorophore to the extracellular side of the receptor, and could be the reason for the weak AR interaction. Notably, combination of molecular docking with SuMD techniques helped to better understand the SAR of these derivatives. This can facilitate the future development of a second generation of more potent and selective conjugable compounds with different linkers, and, in addition, the inclusion of an alternative scaffold to PTP is not excluded.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KAJ thanks the NIDDK Intramural Res. Program (ZIA DK3117) for support. SF, SM and GS thank the Italian Ministry for University and Research for support.Notes and references
- L. A. Stoddart, L. E. Kilpatrick, S. J. Briddon and S. J. Hill, Neuropharmacology, 2015, 98, 48–57 CrossRef CAS PubMed.
- D. E. Sosnovik and R. Weissleder, Curr. Opin. Biotechnol., 2007, 18, 4–10 CrossRef CAS PubMed.
- B. Nabi, S. Rehman, S. Khan, S. Baboota and J. Ali, Brain Res. Bull., 2018, 142, 384–393 CrossRef CAS PubMed.
- M. Srinivasarao and P. S. Low, Chem. Rev., 2017, 117, 12133–12164 CrossRef CAS PubMed.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- R. Morphy and Z. Rankovic, J. Med. Chem., 2005, 48, 6523–6543 CrossRef CAS PubMed.
- D. K. Tosh, L. S. Yoo, M. Chinn, K. Hong, S. M. Kilbey, M. O. Barrett, I. P. Fricks, T. K. Harden, Z. G. Gao and K. A. Jacobson, Bioconjugate Chem., 2010, 21, 372–384 CrossRef CAS PubMed.
- F. Ciruela, V. Fernández-Dueñas and K. A. Jacobson, Neuropharmacology, 2015, 98, 58–67 CrossRef CAS PubMed.
- M. Markovic, S. Ben-Shabat, S. Keinan, A. Aponick, E. M. Zimmermann and A. Dahan, Pharmaceutics, 2018, 10, 1–17 CrossRef PubMed.
- J.-F. Chen, H. K. Eltzschig and B. B. Fredholm, Nat. Rev. Drug Discovery, 2013, 12, 265–286 CrossRef CAS PubMed.
- J. Sawynok, Neuroscience, 2016, 338, 1–18 CrossRef CAS PubMed.
- A. Guerrero, Curr. Med. Chem., 2018, 25, 3597–3612 CrossRef CAS PubMed.
- B. Hocher, Kidney Int., 2010, 78, 438–445 CrossRef CAS PubMed.
- B. Cacciari, G. Spalluto and S. Federico, Mini-Rev. Med. Chem., 2018, 18, 1168–1174 CrossRef CAS PubMed.
- G. Haskó, B. Csóka, Z. H. Németh, E. S. Vizi and P. Pacher, Trends Immunol., 2009, 30, 263–270 CrossRef PubMed.
- S. L. Cheong, S. Federico, G. Venkatesan, A. L. Mandel, Y.-M. Shao, S. Moro, G. Spalluto and G. Pastorin, Med. Res. Rev., 2013, 33, 235–335 CrossRef CAS PubMed.
- S. Redenti, A. Ciancetta, G. Pastorin, B. Cacciari, S. Moro, G. Spalluto and S. Federico, Curr. Top. Med. Chem., 2016, 16, 3224–3257 CrossRef CAS PubMed.
- B. Cacciari, C. Bolcato, G. Spalluto, K. Klotz, M. Bacilieri, F. Deflorian and S. Moro, Purinergic Signalling, 2007, 3, 183–193 CrossRef CAS PubMed.
- S. L. Cheong, A. Dolzhenko, S. Kachler, S. Paoletta, S. Federico, B. Cacciari, A. Dolzhenko, K. Klotz, S. Moro, G. Spalluto and G. Pastorin, J. Med. Chem., 2010, 53, 3361–3375 CrossRef CAS PubMed.
- S. Federico, A. Ciancetta, D. Sabbadin, S. Paoletta, G. Pastorin, B. Cacciari, K. N. Klotz, S. Moro and G. Spalluto, J. Med. Chem., 2012, 55, 9654–9668 CrossRef CAS PubMed.
- S. Federico, S. Redenti, M. Sturlese, A. Ciancetta, S. Kachler, K.-N. Klotz, B. Cacciari, S. Moro and G. Spalluto, PLoS One, 2015, 10, e0143504 CrossRef PubMed.
- E. Kozma, T. S. Kumar, S. Federico, K. Phan, R. Balasubramanian, Z.-G. Gao, S. Paoletta, S. Moro, G. Spalluto and K. A. Jacobson, Biochem. Pharmacol., 2012, 83, 1552–1561 CrossRef CAS PubMed.
- K. A. Jacobson, S. K. Thatikonda, E. E. Kozma, G. Spalluot, S. Moro and S. Federico, US2013/0190335A1, 2013, p. 32.
- E. Kozma, P. S. Jayasekara, L. Squarcialupi, S. Paoletta, S. Moro, S. Federico, G. Spalluto and K. A. Jacobson, Biochem. Pharmacol., 2013, 23, 26–36 CAS.
- E. Margiotta, G. Deganutti and S. Moro, J. Comput.-Aided Mol. Des., 2018, 32, 1337–1346 CrossRef CAS PubMed.
- A. Ciancetta, D. Sabbadin, S. Federico, G. Spalluto and S. Moro, Trends Pharmacol. Sci., 2015, 36, 878–890 CrossRef CAS PubMed.
- O. K. Onajole, P. Govender, T. Govender, G. E. M. Maguire and H. G. Kruger, Struct. Chem., 2009, 20, 1067–1076 CrossRef CAS.
- K.-N. Klotz, J. Hessling, J. Hegler, C. Owman, B. Kull, B. B. Fredholm and M. J. Lohse, Naunyn-Schmiedeberg's Arch. Pharmacol., 1998, 357, 1–9 CrossRef CAS PubMed.
- A. De Lean, A. A. Hancock and R. J. Lefkowitz, Mol. Pharmacol., 1982, 21, 5–16 CAS.
- K. N. Klotz, G. Cristalli and M. Grifantini, J. Biol. Chem., 1985, 260, 14659–14664 CAS.
- S. Federico, S. Redenti, M. Sturlese, A. Ciancetta, S. Kachler, K.-N. Klotz, B. Cacciari, S. Moro and G. Spalluto, PLoS One, 2015, 10, e0143504 CrossRef PubMed.
- Z. G. Gao, J. B. Blaustein, A. S. Gross, N. Melman and K. A. Jacobson, Biochem. Pharmacol., 2003, 65, 1675–1684 CrossRef CAS PubMed.
- S. Hinz, W. M. Alnouri, U. Pleiss and C. E. Müller, Purinergic Signalling, 2018, 14, 223–233 CrossRef CAS PubMed.
- Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, 6Montreal, QC, H3A 2R7 (Canada), 2017 Search PubMed.
- M. J. Harvey, G. Giupponi and G. D. Fabritiis, J. Chem. Theory Comput., 2009, 5, 1632–1639 CrossRef CAS PubMed.
- A. D. MacKerell, N. Banavali and N. Foloppe, Biopolymers, 2000–2001, 56, 257–265 Search PubMed.
- GOLD - The Cambridge Crystallographic Data Centre (CCDC). https ://www.ccdc.cam.ac.uk/solutions/csd-disco very/compo nents/gold/. Accessed 27 Dec 2017 (Genetic Optimization for Ligand Docking, version 5.4.2).
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- A. S. Doré, N. Robertson, J. C. Errey, I. Ng, K. Hollenstein, B. Tehan, E. Hurrell, K. Bennett, M. Congreve, F. Magnani, C. G. Tate, M. Weir and F. H. Marshall, Structure, 2011, 19, 1283–1293 CrossRef PubMed.
- A. Glukhova, D. M. Thal, A. T. Nguyen, E. A. Vecchio, M. Jörg, P. J. Scammells, L. T. May, P. M. Sexton and A. Christopoulos, Cell, 2017, 168, 867–877 CrossRef CAS PubMed.
- E. Margiotta and S. Moro, Appl. Sci., 2019, 9, 821 CrossRef.
- J. A. Ballesteros and H. Weinstein, Methods Neurosci., 1995, 25, 366–428 CAS.
- Gnuplot homepage. http://gnuplot.sourceforge.net/, Accessed 27 Dec 2018.
- MarvinSketch. https://docs.chemaxon.com/displ ay/docs/MarvinSketch+Home. Accessed 27 Dec 2018.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed , 27–28.
- RDKit: Cheminformatics and Machine Learning Software, Available from: http://www.rdkit.org Search PubMed.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503–506 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. A. Lomize, A. L. Lomize, I. D. Pogozheva and H. I. Mosberg, Bioinformatics, 2006, 22, 623–625 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- H. Grubmuller and V. Groll, Available online: http://www.mpibpc.mpg.de/grubmueller/solvate.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- R. J. Loncharich, B. R. Brooks and R. W. Pastor, Biopolymers, 1992, 32, 523–535 CrossRef CAS PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. A. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- D. Sabbadin and S. Moro, J. Chem. Inf. Model., 2014, 54, 372–376 CrossRef CAS PubMed.
- A. Cuzzolin, M. Sturlese, G. Deganutti, V. Salmaso, D. Sabbadin, A. Ciancetta and S. Moro, J. Chem. Inf. Model., 2016, 56, 687–705 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Videos concerning docking poses, electrostatic and hydrophobic contributions of peculiar residues and results of SuMD for synthesised compounds. See DOI: 10.1039/c9md00014c |
| ‡ Current affiliation: Sygnature Discovery, Nottingham, UK. |
| This journal is © The Royal Society of Chemistry 2019 |