
4′-Phosphopantetheine and long acyl chain-dependent interactions are integral to human mitochondrial acyl carrier protein function†
 ‡*a
Renea
Gooch,
d
‡*a
Renea
Gooch,
d
Abstract
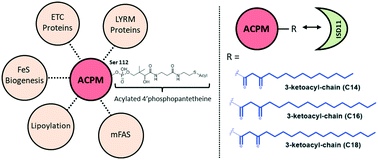
The mitochondrial acyl carrier protein (human ACPM, yeast Acp1) is an essential mitochondrial protein. Through binding of nascent acyl chains on the serine (S112)-bound 4′-phosphopantetheine (4′-PP) cofactor, ACPM is involved in mitochondrial fatty acid synthesis and lipoic acid biogenesis. Recently, yeast Acp1 was found to interact with several mitochondrial complexes, including the iron–sulfur (Fe–S) cluster biosynthesis and respiratory complexes, via the binding to LYRM proteins, a family of proteins involved in assembly/stability of complexes. Importantly, the interaction of LYRM proteins with Acp1 was shown to be essential in maintaining integrity of mitochondrial complexes. In human, recent structures show that ACPM binding to LYRM proteins involves acyl chains attached to the 4′-PP cofactor. Here, we performed an detailed characterization of the mitochondrial interactome of human ACPM by mass spectrometry (MS) and demonstrate the crucial role of the 4′-PP cofactor in most of ACPM interactions. Specifically, we show that ACPM interacts with endogenous Fe–S cluster complex components through binding of the LYRM protein ISD11/LYRM4. Using knockdown experiments, we further determine that ACPM is essential for the stability of mitochondrial respiratory complexes I, II and III, as well as the Fe–S cluster biosynthesis complex. Finally, using native MS and a top-down MS approach, we show that C14, C16 and C18 3-keto-acyl chains on ACPM are implicated in binding to ISD11 through analysis of the recombinant ACPM–ISD11 complex. Taken together, our data provide novel understanding of the role of 4′-PP- and long acyl chains-dependent interactions in human ACPM function.
- This article is part of the themed collection: Molecular Pharmacology
 Please wait while we load your content...
Please wait while we load your content...