
A sample-to-answer, portable platform for rapid detection of pathogens with a smartphone interface†‡
 *acd
*acd
Abstract
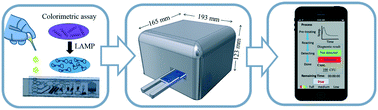
Emerging and re-emerging infectious diseases pose global threats to human health. Although several conventional diagnostic methods have been widely adopted in the clinic, the long turn-around times of “gold standard” culture-based techniques, as well as the limited sensitivity of lateral-flow strip assays, thwart medical progress. In this study, a smartphone-controlled, automated, and portable system was developed for rapid molecular diagnosis of pathogens (including viruses and bacteria) via the use of a colorimetric loop-mediated isothermal amplification (LAMP) approach on a passive, self-driven microfluidic device. The system was capable of 1) purifying viral or bacterial samples with specific affinity reagents that had been pre-conjugated to magnetic beads, 2) lysing pathogens at low temperatures, 3) executing isothermal nucleic acid amplification, and 4) quantifying the results of colorimetric assays for detection of pathogens with an integrated color sensor. The entire, 40 min analytical process was automatically performed with a novel punching-press mechanism that could be controlled and monitored by a smartphone. As a proof of concept, the influenza A (H1N1) virus and methicillin-resistant Staphylococcus aureus bacteria were used to characterize and optimize the device, and the limits of detection were experimentally found to be 3.2 × 10−3 hemagglutinating units (HAU) per reaction and 30 colony-forming units (CFU) per reaction, respectively; both such values represent high enough sensitivity for clinical adoption. Moreover, the colorimetric assay could be both qualitative and quantitative for detection of pathogens. This is the first instance of an easy-to-use, automated, and portable system for accurate and sensitive molecular diagnosis of either viruses or bacteria, and it is envisioned that this smartphone-controlled apparatus may serve as a platform for clinical, point-of-care pathogen detection, particularly in resource-limited settings.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

